

Engineering Enzymes for Energy Production
David L. Ollis A B , Jian-Wei Liu A and Bradley J. Stevenson AA Research School of Chemistry, Australian National University, Canberra, ACT 0200, Australia.
B Corresponding author. Email: ollis@rsc.anu.edu.au
Australian Journal of Chemistry 65(6) 652-655 https://doi.org/10.1071/CH11452
Submitted: 30 November 2011 Accepted: 15 February 2012 Published: 24 April 2012
Abstract
Harvesting the energy of sunlight can be achieved with a variety of processes and as one becomes obsolete, others will need to be developed to replace it. The direct conversion of sunlight into electrical energy could be used to provide power. Energy could also be obtained by combusting hydrogen produced by splitting of water with sunlight. None of these direct approaches will entirely satisfy the entire energy needs of a modern economy and the conversion of biological materials into liquid fuels for transport and other applications may prove to be important for tomorrow’s energy needs. In fact, biofuels such as bioethanol and biodiesel are already used in many countries. However, the long-term viability of these fuels depends on the efficiency of the processes used to produce them. We outline here a method by which ethanol can be produced using enzymes that can be optimized for this purpose.
The depletion of fossil fuels and the growing concern with climate change have stimulated the search for new ways of harvesting the energy of sunlight. There are several options available that have recently received a great deal of attention; the direct conversion of sunlight into electrical energy is one, and a process that uses sunlight to split water into hydrogen and oxygen is another. However, these endeavours suffer from several practical problems – some being efficiency, cost and energy storage. Photosynthetic organisms have managed to overcome many of the difficulties that chemists and engineers currently face and they did so by processes that utilize enzymes that were produced by evolution. This short manuscript explores the idea that enzymes can be evolved to solve current energy dilemmas – not all, but perhaps some.
In the short term, we cannot hope to evolve all the apparatus necessary for photosynthesis. In any case, photosynthesis is a modular process, so that we can focus attention on different parts of this complex machinery. The light-harvesting and the processes responsible for water splitting are separated from the proteins that are responsible for the catalytic fixation of CO2. The latter process is extremely complex and results in the production of compounds that enable energy to be conveniently stored so that it can be retrieved at a later time. These processes are only possible through the use of efficient and highly regulated enzymes. One way of gaining access to the energy reserves of nature is to modify enzymes so that compounds useful to man can be generated from biological materials.
There are two principal biomaterials that can be used to produce fuels.[1] Fats and oils are one type that are used by organisms to store energy for relatively long periods of time. They are triacyl-esters of glycerol that can be converted to biodiesel by transesterification processes catalyzed by enzymes or chemical means. Biodiesel production can use oils that are produced specifically for this purpose or from food industry waste. The other principal type of biomaterial is carbohydrate and makes up most of the chemical energy produced by photosynthesis. A long-standing example of converting carbohydrate into biofuel is the fermentation of sucrose from sugarcane into ethanol for use as transport fuel in Brazil.[2] However, attempts to emulate this ‘first-generation’ biofuel production with corn starch have put biofuels in direct competition with global food supply.[3]
In order to replace fossil fuels with biofuels sustainably, the biomaterial feedstock needs to be supplied in parallel with food. Ideally, structural carbohydrates present in agricultural residues or domestic waste would be converted to biofuel. Although this offers a plentiful renewable feedstock, it consists of recalcitrant polymers such as cellulose, hemicellulose and lignin. Converting lignocelluloses to useful chemical energy requires combinations of physical, chemical or biological treatments. The challenge for contemporary biotechnology is to find feedstocks and treatments that are commercially viable.
Metabolic engineering aims to employ the catalytic efficiency of enzymes together with the economy of microbes to produce biofuel sustainably. This aspect of biotechnology redirects the biochemical pathways of an organism by removing or adding enzymes by disabling or inserting corresponding genes. For example, the model bacterium, Escherichia coli, has been engineered to secrete cellulases to hydrolyze cellulose and hemicellulose for use in biofuel production.[4] The research of Bokinsky et al. tested three sets of additional enzymes for biofuel synthesis: one for butanol as a petrol substitute, another set for biodiesel, and a third for pinene as jet fuel substitute. Biotechnologists have engineered microbes for other potential biofuels including: hydrogen,[5] branched alcohols,[6] biodiesel[7] and terpene-based chemicals.[8,9] In each case, the rate of biofuel production is far from ideal, typically measured in milligrams per litre of culture. In short, although it is possible to produce biofuels from a variety of material, the efficiency of these processes is in general not high. Small increments in efficiency translate into reductions in cost. Costs determine the viability of a process and whether or not it will be adopted on a large scale. So technologies that can be used to increase the efficiency of a process are clearly of considerable importance to those interested in the production of biofuels.
Genetically modified organisms use enzymes to greatly accelerate chemical reactions to produce biofuels. Many aspects of these metabolically engineered microbes can be optimized in order to reach viable biofuel production. One key aspect is optimizing the enzymes for high catalytic activity and stability in the reaction environment – either extracellular or in a foreign organism. Although it is most efficient to carry out biofuel production using living organisms, the process of testing enzymes for optimal activity is best carried out in a test tube.
But, you might ask, why do we need to improve enzymes? After all, enzymes have evolved over millions of years – they should be perfect. In fact, in many ways they are perfect – they are perfectly suited to the needs of the host organism. Enzymes evolve in organisms to suit the needs of the organism – and no more. If the organism only lives for a short time then the enzymes need not be particularly stable. If a metabolic intermediate is present in small quantities, then the enzymes involved in its production and utilization need only be present at low levels – they do not have to be particularly soluble nor do they have to be easily expressed. Perhaps more importantly, enzymes may be highly regulated so that their activities can be switched on and off to satisfy the needs of the cell. All of these properties present problems for the practical utilization of enzymes. These difficulties can be overcome by rational methods that rely on a detailed knowledge of structure and an understanding of how proteins function. Alternatively, methods akin to natural evolution can be used to improve enzyme properties. These processes have been referred to as ‘directed evolution’ or ‘directed molecular evolution’ and can be divided into several stages – as can Darwinian evolution. Genetic diversity is generated in the first stage and favourable characteristics are selected in the second. Variants with desirable attributes serve as the parents for subsequent generations. The change in any new generation may be small, but significant improvements can be observed over several generations. The difference between directed molecular evolution and Darwinian evolution is really one of focus. In directed molecular evolution, attention is focussed on a single gene whereas in natural evolution, it is the whole organism that is of interest. It should be noted that bacterial geneticists have been generating mutants for many years. These experiments were used to gain a better understanding of the metabolic processes in living cells and were not aimed at particular genes. It was not till the advent of polymerase chain reaction (PCR) that it became reasonable to generate large libraries of randomly mutated copies of a single gene. The process is generally referred to as error-prone PCR (epPCR). A specified error rate per gene can be obtained by adjusting experimental conditions of the epPCR experiment. Also, the epPCR parameters can be adjusted to give ‘DNA shuffling’, a process that mimics recombination used in nature to generate genetic diversity. The mechanics of library generation are more involved than the description presented here and are elaborated on elsewhere.[10–12] The more difficult aspect of directed evolution is not generating diversity, but rather, it is the selection of favourable traits.
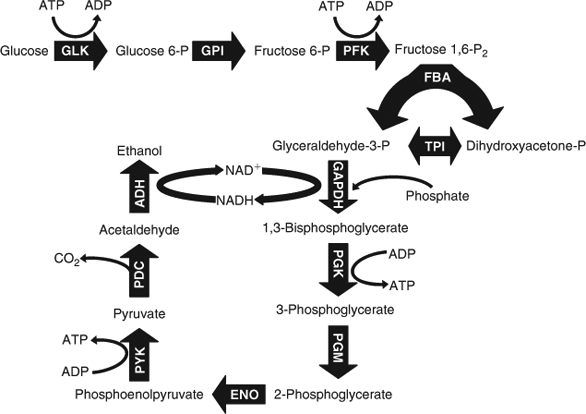
In nature, the collection of enzymes used to convert simple sugars like glucose to pyruvate are referred to as the glycolytic enzymes. Pyruvate is usually converted to CO2 in the citric acid cycle, but under oxygen-limiting conditions, it can be converted to other compounds – lactate in the case of oxygen-starved muscle tissues or ethanol in the case of yeast deprived of oxygen. Glycolysis is a central metabolic pathway in most organisms and the production of many biofuels depends on access to glycolytic intermediates. Like many enzymatic pathways, glycolysis is highly regulated and suited to the needs of the organism. If the organism has an adequate supply of energy, then it will downregulate glycolysis – not really a desirable attribute for a biotechnologist interested in optimizing the production of a product that depends on an efficient glycolytic pathway. Our work with glycolysis should be seen as an example of how a pathway can be modified to increase production and not just as a way of producing ethanol.
An inspection of the glycolytic pathway shown in Fig. 1 reveals that it generates ATP and requires that the NAD+ co-factor be recycled. The relevant enzymes were obtained by simply isolating their genes from E. coli and using bacterial expression systems to produce the proteins with a polyhistidine tag to simplify their purification. With one exception, sufficient activity for each of the enzymes necessary to convert glucose to ethanol was obtained.[13] The exception was pyruvate decarboxylase (PDC) – the enzyme that converts pyruvate to acetaldehyde so that it can be subsequently converted to ethanol. This enzyme is not encoded in the E. coli genome and an alternative source of the enzyme had to be identified. Eventually, the gene for yeast PDC was obtained and expressed in E. coli.
|
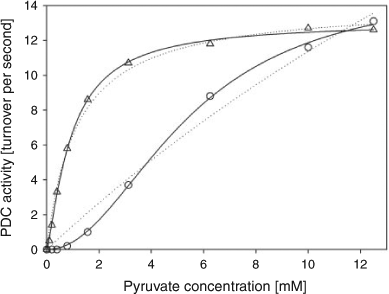
Although yeast PDC could be isolated in large quantities, its use proved to be problematic. All the glycolytic enzymes could be combined with glucose and co-factors in vitro, but the production of ethanol was not observed. The problem appeared to be in the latter half of the glycolytic pathway. The NAD+ co-factor was converted to NADH, but its subsequent oxidation was not observed. A consideration of the enzyme properties suggested that the most likely cause of the problem was PDC. Yeast only produces alcohol in the absence of oxygen and only when its supply of ATP is low and there is a build-up of pyruvate. In other words, yeast only produces alcohol when it is required and it minimizes production at other times. To achieve this end, PDC is tightly regulated – it is a good example of a cooperative enzyme. Its activity does not follow simple Michaelis–Menten kinetics, with the curve of initial velocity versus substrate concentration taking a hyperbolic form. Rather, the dependence of initial velocity on substrate concentration is a sigmoidal curve. At low substrate concentrations, little substrate is converted to product. The properties of yeast PDC are ideally suited to its use in yeast, but they are not suited for the rapid production of ethanol in practical applications. Can PDC be evolved so that it is more active at low substrate concentrations? This is typical of the type of problem encountered in developing enzymes for practical applications.
PDC activity was enhanced with five cycles of directed evolution.[14] Library generation involved alternate rounds of epPCR and DNA shuffling. The error rate varied during the course of the work but was usually held to an average of about five base changes per gene. Library sizes were typically of the order of 10000 and were screened in two stages. A high-throughput screen using a plate scanner was used to identify mutants with enhanced activities – typically ~200 mutants were selected. A secondary screen was used to confirm the activity of the mutants and to provide data to select mutants to serve as parents for the next generation. Typically, the genes of 10 mutants were isolated and used to produce the next generation. The activity of the mutant PDC enzymes was not measured directly – their activity was coupled to that of alcohol dehydrogenase that could be monitored using changes in the spectral properties of its cofactor NADH. The genes of mutant enzymes were sequenced to determine the sequence changes that were responsible for increased activity. Mutations were observed throughout the gene and were of two types – silent and expressed. The silent mutations changed a base codon but did not change the amino acid type. These mutations occurred at random throughout the gene as would be expected. These changes do not result in change in activity – they give an indication that the gene is being randomly mutated. The expressed mutations resulted in changes in amino acid type and were concentrated in two of the three domains of the protein that formed the active site of the enzyme. Many mutations were observed through the course of the experiments, but very few survived to the final rounds of evolution – consistent with the idea that only favourable mutations would be retained. As the experiments progressed, specific mutations were found to occur in most of the genes – the evolutionary process converged. It should be noted that the end point of an evolutionary process depends on the screening conditions.
Mutant libraries were screened at low substrate concentration. The resulting mutant proteins were expressed, purified and kinetically characterized. It was noted that these new enzymes exhibited reduced levels of cooperativity, and in some cases, the proteins behaved like Michaelis–Menten enzymes, as can be seen in Fig. 2. Although the activities of the mutant enzymes were increased at low substrate concentrations, there was little if any enhancement at saturating levels of substrate. The increase in activity was in part due to the way the enzyme bound its substrate, but it was also due to increased stability of the enzyme. Why did the stability of PDC increase?
|
Our experiments with PDC involved the use of E. coli – an enteric bacterium that grows optimally at 37°C. As a consequence, genetic manipulations were done at a temperature that was optimal for E. coli. Yeast, however, evolved in an environment with a much lower temperature than E. coli and as a consequence, its proteins were not optimized for activity at 37°C. By evolving yeast PDC in E. coli, mutants with improved stability were obtained. Improved stability gave rise to increased activity.
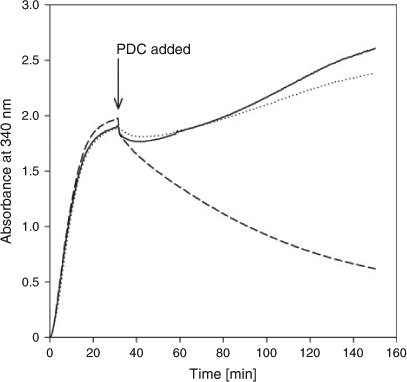
So, did the mutant proteins actually enhance glycolysis in our in vitro system? We added 11 glycolytic enzymes, the necessary substrates and cofactors into a cuvette and measured the absorption of 340-nm light (A340). Once the reaction was started by adding glucose, everything was present to produce ethanol except PDC. A340 increases rapidly owing to the reduction of NAD+ to NADH, but NADH can only be oxidized back to NAD+ by the reduction of acetaldehyde (the product of PDC) to ethanol. Fig. 3 shows that the addition of a mutant (4S25)[14] PDC to the in vitro glycolysis system resulted in a decrease in A340. This evidence of ethanol production was not seen when adding native PDC. In this case, the use of an evolved enzyme produced a dramatic increase in ethanol production. The percentage increase due to the use of a modified enzyme is difficult to determine because the production of ethanol with the native PDC was so low that it was difficult to measure.
|
Most biotechnology problems can be solved in a variety of ways and the example presented here is no exception. A change in the assay conditions could have resulted in ethanol production with the native enzyme. Alternatively, ethanol could have been produced without directed evolution by selecting a PDC from another organism.[15] However, the work presented here does show that the properties of an enzyme can be tailored to fit the needs of a particular process. Clearly, more than one enzyme would need to be modified if glycolysis were to be optimized for industrial applications, so the work presented here provides a start to a much longer process.
References
[1] S. N. Naik, V. V. Goud, P. Rout, A. K. Dalai, Renew. Sustain. Energy Rev. 2010, 14, 578.| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsF2isbvF&md5=2be3aa51dbb97c958fc713d475d6aa34CAS |
[2] J. Goldemberg, Biotechnol. Biofuels 2008, 1, 6.
[3] A. Banerjee, Dev. Change 2011, 42, 529.
| Crossref | GoogleScholarGoogle Scholar |
[4] G. Bokinsky, P. P. Peralta-Yahya, A. George, B. M. Holmes, E. J. Steen, J. Dietrich, T. Soon Lee, D. Tullman-Ercek, C. A. Voigt, B. A. Simmons, J. D. Keasling, Proc. Natl Acad. Sci. USA 2011, 108, 19949.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhs12gtbjK&md5=008101a7ceba0314aa021f4a0ec809faCAS |
[5] T. Maeda, V. Sanchez-Torres, T. K. Wood, Appl. Microbiol. Biotechnol. 2007, 77, 879.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhtlaqtbzF&md5=aecf6922e9d693f6c1c40e668f36dc39CAS |
[6] S. Atsumi, T. Hanai, J. C. Liao, Nature 2008, 451, 86.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtlSltQ%3D%3D&md5=fea562f8730dcab077ebabe71dd9c005CAS |
[7] E. J. Steen, Y. Kang, G. Bokinsky, Z. Hu, A. Schirmer, A. McClure, S. B. Del Cardayre, J. D. Keasling, Nature 2010, 463, 559.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXht1Slu70%3D&md5=19c35a19e87f8ca491a616c32d55fddcCAS |
[8] C. Wang, S. H. Yoon, A. A. Shah, Y. R. Chung, J. Y. Kim, E. S. Choi, J. D. Keasling, S. W. Kim, Biotechnol. Bioeng. 2010, 107, 421.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtFSisr7I&md5=37c20a65ad5b4099453a86bf16f73346CAS |
[9] P. P. Peralta-Yahya, M. Ouellet, R. Chan, A. Mukhopadhyay, J. D. Keasling, T. S. Lee, Nat. Commun. 2011, 2, 483.
| Crossref | GoogleScholarGoogle Scholar |
[10] F. H. Arnold, A. A. Volkov, Curr. Opin. Chem. Biol. 1999, 3, 54.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXht1ektLc%3D&md5=3314941ebc71832c0a1226879b6c4a04CAS |
[11] N. J. Turner, Nat. Chem. Biol. 2009, 5, 567.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXoslentrk%3D&md5=c24bbe9b0a46793cb3eea9ad8d545348CAS |
[12] C. J. Jackson, E. M. J. Gillam, D. L. Ollis, in Comprehensive Natural Products Chemistry II: Modern Methods in Natural Product Chemistry (Eds L. N. Mander, H.-W. Liu) 2010, Vol. 9, Ch. 20, pp. 723–749. (Elsevier: Oxford, UK).
[13] B. J. Stevenson, J.-W. Liu, P. W. Kuchel, D. L. Ollis, J. Biotechnol. 2012, 157, 113.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhvVCqtg%3D%3D&md5=963f3b2b248a5c7a01165c509cf93072CAS |
[14] B. J. Stevenson, J.-W. Liu, D. L. Ollis, Biochemistry 2008, 47, 3013.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXht1Omtro%3D&md5=a72000e50994b69c8469444e6092d0feCAS |
[15] L. O. Ingram, T. Conway, D. P. Clark, G. W. Sewell, J. F. Preston, Appl. Environ. Microbiol. 1987, 53, 2420.
| 1:CAS:528:DyaL2sXmtFaqsLc%3D&md5=26dc7b7fd834056e6348ae8325986e76CAS |