

Structure, Dynamics, and Function in the Major Light-Harvesting Complex of Photosystem II
Gabriela S. Schlau-Cohen A B C and Graham R. Fleming A B DA Department of Chemistry, University of California, Berkeley, CA 94720, USA.
B Physical Biosciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA.
C Current address: Department of Chemistry, Stanford University, Stanford, CA 94305, USA.
D Corresponding author. Email: grfleming@lbl.gov
Australian Journal of Chemistry 65(6) 583-590 https://doi.org/10.1071/CH12022
Submitted: 18 January 2012 Accepted: 7 March 2012 Published: 3 May 2012
Abstract
In natural light-harvesting systems, pigment-protein complexes (PPC) convert sunlight to chemical energy with near unity quantum efficiency. PPCs exhibit emergent properties that cannot be simply extrapolated from knowledge of their component parts. In this Perspective, we examine the design principles of PPCs, focussing on the major light-harvesting complex of Photosystem II (LHCII), the most abundant PPC in green plants. Studies using two-dimensional electronic spectroscopy (2DES) provide an incisive tool to probe the electronic, energetic, and spatial landscapes that enable the efficiency observed in photosynthetic light-harvesting. Using the information about energy transfer pathways, quantum effects, and excited state geometry contained within 2D spectra, the excited state properties can be linked back to the molecular structure. This understanding of the structure-function relationships of natural systems constitutes a step towards a blueprint for the construction of artificial light-harvesting devices that can reproduce the efficacy of natural systems.
Introduction
Sunlight is a ubiquitous source of renewable energy, powering the growth of natural systems. Biomimetic devices offer one approach to harvest, store, and use solar power for the world’s energy needs.[1] The first step in the construction of such devices is extracting a blueprint from natural systems through an understanding of photosynthetic function. In the primary steps of photosynthetic light-harvesting, photoenergy is converted to chemical energy with near unity quantum efficiency.[2] This process occurs by spatial transport of photoenergy through networks of pigment-protein complexes (PPCs) to a central location, known as the reaction centre, where the excitation drives a chain of electron transfer events. The structure that gives rise to this photoenergy to photochemical energy conversion in green plants, the Photosystem II supercomplex, is shown in Fig. 1a, with the primary antenna PPC, the major light-harvesting complex of Photosystem II (LHCII), shown in Fig. 1b.[3–5] Within the photosynthetic apparatus, the PPCs are found in a specific arrangement within a lipid bilayer membrane. Within a PPC, pigments are found within a surrounding protein matrix in fixed positions and relative orientations, which dictate the electrodynamic interactions, or Coulombic couplings, between pigments and with the surrounding protein matrix.[6] These couplings, in turn, govern the resultant efficient, unidirectional, and controllable energy flow.

|
There are two driving forces behind the design of the photosynthetic apparatus: 1) the sun is a dilute energy source (with maximum intensity ~1 kW m–2); and 2) on a nanosecond timescale, photoenergy can be lost as fluorescence or through triplet formation, which in turn can give rise to deleterious photoproducts, such as singlet oxygen. As will be discussed in this manuscript, these two driving forces both require a high density of pigments. Thus, they lead to dense packing of PPCs within the membrane (80 % of the area of the thylakoid membrane is comprised of PPCs)[7] and dense packing of pigments within PPCs (pigments are held by the protein matrix in densities of up to 1 M within PPCs).[8] It is notable that at concentrations of chlorophyll in solution much lower than that of photosynthetic complexes the fluorescence yield begins to decrease due to quenching effects.[9] Here, the relative orientations and spacings of the pigments are designed to prevent quenching effects. There are functional benefits from the dense packing. First, as the combined excited state transition dipole moments of some PPCs, such as LHCII, are close to isotropic,[2,10,11] the density of pigments allows for maximal absorption of all polarizations of incident sunlight. The directions of the excited state transition dipole moments are known within the chlorophyll molecule, which in turn are known from the structural model from X-ray crystallography. Second, the high density has the effect of giving rise to strong coupling between pigments, which leads to ultrafast energy transfer between them, allowing the excitation to reach the reaction centre before other photochemical processes can occur.
The most abundant PPC in green plants, LHCII, binds >50 % of the world's chlorophyll, and is the antenna system where most initial absorption events occur. As shown in Fig. 1b, LHCII has a trimeric structure, with 14 chlorophyll per monomer, 8 chlorophyll-a (Chl-a) and 6 chlorophyll-b (Chl-b).[10] These chlorophylls are held in a closely-packed arrangement within the protein matrix, with few-angstrom distances between pigments and between pigments and the surrounding protein. These small distances give rise to strong pigment–pigment and pigment–protein couplings that are similar in magnitude to each other, and to kBT at room temperature (200 cm–1).[6] The pigment–pigment and pigment–protein couplings modify the electronic and dynamical functions of the individual molecules, and in this way produce emergent properties different from those of the individual pigments. Because the combination of multiple couplings determines the observed behaviours, the contribution of each molecule is often obscured. An understanding of the function of LHCII as the site of initial absorption, and how its architecture is optimized for this function, requires characterizing the electronic structure, how the individual molecules produce this electronic structure, and determining the pathways of energy flow through time and space.[6,13–17]
Two-dimensional electronic spectroscopy (2DES) has provided a valuable tool to study structure-function relationships within condensed phase molecular systems.[16,18–20] 2DES correlates excitation and emission energies as a function of a delay time between excitation and emission events. In these spectra, the excited state couplings, energies, geometries, and dynamics can be observed with femtosecond temporal resolution. In this work, we enumerate a series of characteristics of photosynthetic systems seen using 2DES on LHCII, and speculate on their utility. Building on extensive previous theoretical and experimental work on LHCII,[13,21–26] the excited state energies,[27] short-time dynamics,[12] and angles between individual excited states[11] were observed. This information, in combination with theoretical work, informs on how the molecular structure produces the electronic structure. Observing these structure-function relationships provides a further step, adding to the substantial body of knowledge from previous work, towards understanding the design principles behind photosynthetic PPCs. Thus, this information can be used as a guide towards a blueprint for artificial photosynthetic systems.[28,29]
Electronic and Molecular Structure
Excited State Geometry
The spectroscopically observable excited states are the energy eigenstates, or excitons, of the systems.[6] These are delocalized states consisting of linear combinations of the excited states of the uncoupled chlorophyll, where the delocalization is driven by the couplings between the chlorophylls. Thus, the excitons have energies, positions, and orientations of their transition dipole moments that differ from those of the individual chlorophyll. The excited state transition dipole moments of the individual chlorophyll are the Qy, or S0 → S1, transitions.[2,30,31] This is illustrated for three coupled chlorophylls within the structural model of monomeric LHCII in Fig. 2a and b. The excited state transition dipole moments are shown for the individual chlorophyll in the absence of coupling and for the delocalized excited states that result from the presence of coupling. These are calculated using the Hamiltonian as described elsewhere.[11,12] As evidenced from the differences in directions and lengths of the transition dipole moments between the two panels, the excited states do not have a clear correspondence with the molecular structure. Determining the spatial position of the delocalized states within the molecular structure, or the contributions of individual chlorophylls, requires knowledge of the chlorophyll–chlorophyll coupling and of the transition energies of the uncoupled chlorophyll, known as the site energies. The chlorophyll–chlorophyll couplings can be calculated from the distances and relative orientations of the chlorophylls, which can be extracted from the structural model from X-ray crystallography. The site energies, however, are difficult to determine. They depend on various types of interaction with the surrounding protein matrix, such as electrodynamic interactions with the backbone of the α-helix and with side chains, differences in Mg2+ ligation, conformational changes of the porphyrin ring.[32–34] The site energies are difficult to calculate theoretically because many of these interactions have similar strengths; they are also difficult to determine experimentally because the energy of each eigenstate depends on multiple uncoupled chlorophyll, each with its own coefficient to describe its contribution. The precision of the experiment makes this inversion prohibitively difficult. In order to characterize the excited state manifold, the angle between excited state transition dipole moments is a much more sensitive probe than the energies of the excited states, because the orientation scales non-linearly with the properties (energy and couplings) of the individual chlorophyll, whereas the energy scales roughly linearly with the energy of the coupled chlorophyll.[11] For example, energetic shifts within the inhomogeneous linewidth appear as large rotations of the direction of the transition dipole moments.
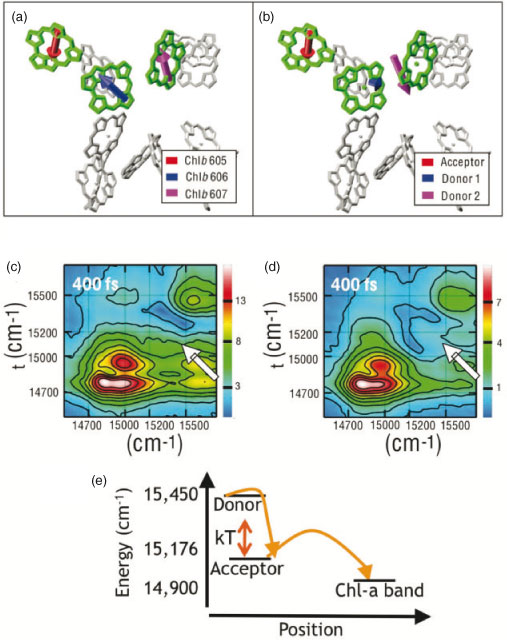
|
2D spectroscopy can be used to determine the angles between excited state transition dipole moments.[35] 2D spectra are plotted as a function of both excitation and emission for a given waiting time. Along the diagonal, when excitation and emission are equal, features mostly correspond to linear absorption and reveal the energies of the excited states. Off the diagonal, when excitation and emission are not equal, peaks indicate coupling at zero delay and show energy transfer through the appearance and evolution with delay time of cross-peaks. 2D spectra are generated through interactions with four electric fields. The variety of apparatuses used for 2D spectroscopy, and the apparatus used for this work,[36] have been well described elsewhere.[12,20,37–42] In the experiments described here, there are four laser pulses incident on the sample, where the first three pulses generate a non-linear (3rd order) response, which is heterodyne detected in the frequency domain through interferometry with the fourth pulse, the local oscillator.[37,43] The delay between the first and second pulses, the two responsible for excitation of the system, is known as the coherence time, τ, and the delay between the third pulse and signal emission is known as the rephasing time, t. The 2D spectra are recorded for a series of delay times, known as the waiting time, T, between the second and third pulses, or between excitation and emission events. Two characteristics of 2DES enable access to the angle between pairs of excited state transition dipole moments: 1) isolated cross-peaks corresponding to a single energy transfer step, due to resolution along both excitation and emission energy; and 2) independent control over the polarization of the four incident laser pulses. Each peak within a 2D spectrum is scaled based on orientational effects, meaning the scaling factor depends on the angles between the polarizations of the incident laser pulses in the laboratory frame and the angles between the excited state transition dipole moments in the molecular frame.[16,44,45] By varying the angle between pulse polarizations in the experiment and monitoring the effect on amplitude of a given cross-peak, the angle between excited state transition dipole moments of the donor and acceptor can be determined.
This technique was applied to LHCII in order to determine the angle between two of the excited states shown in Fig. 2a.[11] Two absolute value, non-rephasing (where non-rephasing refers to the ordering of pulses 1 and 2[35]) spectra of the Qy region (S0 to S1 transition) of LHCII are shown in Fig. 2c and d, under the all-parallel, (0, 0, 0, 0), and cross-peak-specific, (π/3, –π/3, 0, 0), polarizations respectively. While both spectra have diagonal peaks in the Chl-a (14750 and 14900 cm–1) and Chl-b (15450 cm–1) bands, and show many similar cross-peaks, one cross-peak shows much larger relative amplitude under the cross-peak-specific polarization. The cross-peak showing energy transfer from the Chl-b band to the region between the Chl-a and Chl-b bands, highlighted with an arrow in the spectra, increases in relative amplitude in the polarized spectrum. Because the orientational prefactor under the cross-peak-specific polarization is at its maximum when the transition dipole moments are perpendicular, the enhancement indicates that the two states involved in the molecular process which gives rise to this cross-peak, the donor and acceptor states, must have an angle between their transition dipole moments close to 90°.
One notable trait of LHCII that emerges upon examination of the direction of the excited state transition dipole moments is that they have been optimized towards more isotropic absorption within spectral regions. The position of the chlorophylls is close to isotropic, thus optimizing the system to absorb incoherent sunlight.[10] Even more notable is the isotropic absorption within all spectral regions. This suggests that the couplings and the site energies, discussed in the subsequent section, are such that, according to the working model, for the distribution of strength of the transition dipole moments between the two halves of the Chl-a band, the average standard deviation of the transition dipole moment strength of all three directions is only 0.33.[11]
Transition Energies Tuned by Protein Environment
The addition of a new observable, the angle between excited state transition dipole moments, provides a new constraint to the information already provided by resolving across both excitation and emission energies: this allows determination of the site energies, and thus accesses the final variable in the description of how the molecular structure produces the electronic structure. For the energy transfer step shown by the cross-peak highlighted in Fig. 2c and d, the chlorophylls that contribute to the donor and acceptor states can be determined based on a working model of the site-basis contributions to the excited states.[12,15] By constraining the two donor Chl-bs, Chl-b 606 and Chl-b 607 (where the numbering is taken from the X-ray crystallography structure), to the range of energies observed spectroscopically, and then adding the new criterion that one excited state must have an angle close to 90° with the acceptor state, the site energies of Chl-b 606 and Chl-b 607 can be bounded. They were found to be 15630–15710 cm–1 for Chl-b 606 and 15680–15760 cm–1 for Chl-b 607.
As can be understood by the differences in these values, the protein environment tunes the transition energies of the chlorophylls. Introducing this variation serves to allow chlorophyll to absorb over a broader range of the solar spectrum and to introduce energy gradients between the chlorophyll, which is partially responsible in driving energy flow through the PPCs. In this way, nature is able to be parsimonious in its production of types of pigments, while still having the advantages of multiple different transition energies.
Directional Energy Flow
The excited state manifold produces a series of energy transfer steps that funnel the excitation to an exit site for transfer towards the reaction centre. These relaxation steps are facilitated by the spatial overlap of the excited states, which is determined by the Chl–Chl coupling, and by the Chl–protein coupling.[46,47] The ability of 2DES to simultaneously resolve excitation energy, emission energy, and delay time between excitation and emission events provides a tool to survey the excitation energy transfer pathways that gives a more detailed picture of the dynamics of LHCII than was previously available.
Real, 2D relaxation (the combined rephasing and nonrephasing contributions at T > 0) and non-rephasing spectra of the Qy region of LHCII are shown in Fig. 3 with all-parallel pulse polarization, (0, 0, 0, 0). Energy transfer processes are shown by the appearance and evolution of cross-peaks, which are indicated by arrows in Fig. 3, right. The series of spectra illustrates three distinct timescales of energy transfer processes.[12] As observed in Fig. 3a and b, energy transfers within all spectral regions within the first 100 fs. As shown in the spectra for these two waiting times, multiple energy transfer processes occur on slightly different timescales within 100 fs. Second, as shown in Fig. 3c and d, energy transfers on several hundred femtosecond timescales, and finally, as shown in Fig. 3e, on a picosecond timescale.
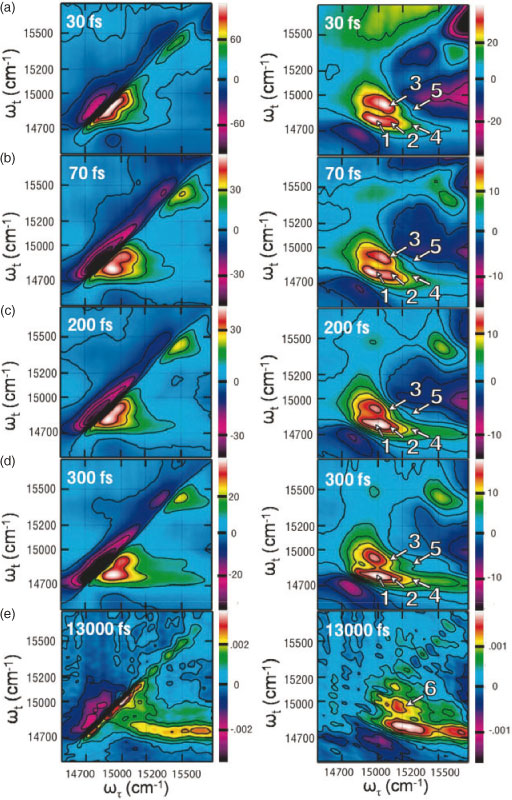
|
In combination with modelling of the electronic couplings based on the structural model from X-ray crystallography, these energy transfer pathways can be mapped back onto the molecular structure, giving a picture of energy flow through LHCII, which can then be studied to understand the functionality behind the dynamics.[12,24] The results from 2DES[12] and previous experimental and theoretical efforts[15,24] are summarized in Fig. 4. There are two important attributes of these relaxation pathways, both of which have implications for the efficacy of the energy transfer processes. First, energy transfers on a variety of timescales, from sub-100 fs to picoseconds. This combination of timescales may aid unidirectional energy transfer. This is illustrated for one particular relaxation pathway in Fig. 2e, where the dynamics show the energy transfer steps studied to determine the site energies described in the previous section. Here, the first step is driven by strong pigment-pigment coupling, and thus the excitation energy relaxes across an energy gap larger than kBT at room temperature. The large energy gap helps prevent thermal backflow of excitation energy. This step is followed by energy transfer to a weakly coupled state close in energy. The weak coupling precludes a long-lived superposition, and thus prevents coherent backflow within a PPC. Therefore, the range of pigment-pigment couplings, as seen in the presence of a range of energy transfer timescales, prevents multiple types of backtransfer and so aids in driving unidirectional excitation energy transfer within a PPC. This discussion highlights the mechanisms by which excitations preferentially populate the low energy states of LHCII without getting trapped in local minima. Overall, the energy flow occurs within the context of a steep funnel in individual, monomeric LHCII complexes, where excited state energies spans >800 cm–1, or more than four times kBT at room temperature. However, for monomer–monomer energy transfer within LHCII, or PPC–PPC energy transfer within the PSII supercomplex the energy landscape is much flatter, and reversible energy transfer is indeed most likely important for these steps. Within LHCII, reversible energy transfer among the low energy Chl-a states most probably facilitates equilibration within the monomers of trimeric LHCII[48] and positions the excitation to transfer to neighbouring complexes.

|
The second feature of the energy transfer pathways shown in Fig. 4 is independent of where the initial absorption occurs, energy relaxes through the excited state manifold to preferentially populate two low energy Chl-a states. According to the model, both of these states are localized on the external portion of the complex, thus positioning the excitation to transfer to neighbouring complexes in order to move towards the reaction centre. Because there are two states that are close in energy in this trimer of chlorophyll, the excitation can transfer easily between them. Therefore, there are two potential donors for transfer to neighbouring PPCs. Each donor is optimized to transfer energy in certain directions, that is to acceptors at optimal orientations with respect to the donor, depending on the value of its transition dipole moment.[12] In combination, these states can transfer energy nearly isotropically within the membrane plane. Thus, energy can transfer from LHCII to nearby complexes no matter what the relative positions of the two PPCs. This functionality is especially important for LHCII, as it is situated within an array of antenna PPCs in the membrane.[3] Additionally, this flexibility makes the system more robust to variations in the relative positions of the PPCs.
Quantum Coherence
The many ultrafast energy transfer steps within LHCII, described in the previous section, arise from the strong pigment–pigment coupling between these chlorophyll. This strong coupling also means that a classical picture of an excitation hopping from excited state to excited state breaks down. Instead, recent work has observed quantum coherence, showing that the coupling is strong enough that the excitation oscillates between excited states.[27,49–52] Essentially, the coupling between chromophores, which drives electronic coherence, is stronger than the coupling to the surrounding protein environment. Thus, the system maintains a superposition state, meaning that there is phase coherence between two states, for longer than the timescale of many of the energy transfer steps.[53,54] Because the strong coupling is a by-product of the dense packing required for optimal absorption of solar energy, the quantum coherence is also a result of this density.
While previous experiments had suggested the presence of quantum coherence in photosynthetic systems, 2DES is newly able to directly access these signals. Because 2DES is a phase-sensitive measurement, in all-parallel spectra quantum coherences appear as intensity oscillations with frequencies corresponding to the energy gap between the two states in the superposition. Alternately, a coherence-specific-polarization sequence, (π/4, –π/4, π/2, 0), isolates the signal corresponding to quantum coherence and only contains features that arise from quantum coherence.[55] Thus, the evolution with waiting time of peaks in spectra taken under this polarization sequence indicate the evolution of quantum coherences. Integrating spectra taken under this polarization sequence shows the evolution of the total coherence with waiting time. The decay of quantum coherence in the system produces the decay of intensity. Fitting this decay can quantify the timescales of quantum coherence. In LHCII, there is a fast decay component (~50 fs), corresponding to the short-time effects from ultrafast experiments, and a longer time component (~700 fs), corresponding to the quantum coherences that may play a role in energy transfer.[56] The observation of this second, long-lived timescale of quantum coherence means that quantum coherence lasts longer than many energy transfer steps. The pigments must be strongly coupled, which in turn means they must be closely spaced, to ensure this long-lived quantum coherence.
The observation of quantum coherence within photosynthetic PPCs[27,49–52] has led to speculation that the excitation may maintain phase coherence as it travels through the molecular structure,[53,57–60] and that this behaviour may contribute to the efficiency of photosynthetic light-harvesting. This speculation has led to a significant theoretical effort around quantifying potential contributions of quantum coherence to the efficiency of photosynthetic light-harvesting. Oscillations of an excitation between excited states may have functional benefits, such as avoiding trap states. The ability to oscillate between two states could prevent the excitation from getting stuck in local minima, making light harvesting robust with respect to spatial and energetic disorder resulting from different protein conformations, pigment positions and temperature.
Conclusions and Outlook
Using 2DES to study LHCII, the most abundant antenna complex in natural photosynthesis, several characteristics of photosynthetic PPCs have been observed: 1) as a result of the dense packing of pigments, there is strong electronic coupling, which can be detected through ultrafast energy transfer and quantum coherence; 2) a range of coupling strengths to ensure unidirectional energy transfer without trapping in local minima; 3) optimizing input/output states by position and level of delocalization. In combination with theoretical work, these characteristics can be identified, and their molecular origin can be assigned. Essentially, these characteristics arise from the choice of pigment density, which provides an important tool in overcoming both the paucity of photons and giving rise to energy transfer steps from couplings that produce ultrafast, directional energy transfer towards the reaction centre. The observed functional benefits for natural light-harvesting systems means these characteristics should be included in any blueprint for a light-harvesting device.[28]
Many different approaches have already been applied to replicating photosynthetic light-harvesting, including genetic manipulation of natural systems,[61,62] incorporating biological components into artificial devices,[63,64] synthetic biomimetic devices,[65–67] and artificial mixed biological/inorganic devices.[68] In developing an understanding of the structure-function relationships that underlie photosynthetic light-harvesting, more and more of the principles of natural systems can be incorporated into artificial devices in order to optimize their function to meet growing energy needs.[69]
Acknowledgements
This work was supported by the Director, Office of Science, Office of Basic Energy Sciences, of the USA Department of Energy under Contract DE-AC02–05CH11231 and by the Chemical Sciences, Geosciences and Biosciences Division, Office of Basic Energy Sciences, USA Department of Energy under contract DEAC03–76SF000098. G.S.S.-C. is grateful for an American Fellowship from the American Association of University Women.
References
[1] R. E. Blankenship, D. M. Tiede, J. Barber, G. W. Brudvig, G. Fleming, M. Ghirardi, M. R. Gunner, W. Junge, D. M. Kramer, A. Melis, T. A. Moore, C. C. Moser, D. G. Nocera, A. J. Nozik, D. R. Ort, W. W. Parson, R. C. Prince, R. T. Sayre, Science 2011, 332, 805.| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXlslylsLk%3D&md5=7fef5a2be0a1e90cdea051d4e8001fa9CAS |
[2] R. E. Blankenship, Molecular Mechanisms of Photosynthesis, 2002 (Blackwell Publishing: Oxford).
[3] J. P. Dekker, E. J. Boekema, BBA-Bioenergetics 2005, 1706, 12.
| 1:CAS:528:DC%2BD2cXhtFGht77J&md5=0faa83b1aa067447c8a89177543fbd2dCAS |
[4] J. Nield, J. Barber, BBA-Bioenergetics 2006, 1757, 353.
| 1:CAS:528:DC%2BD28Xmsl2jsb4%3D&md5=6cc5d788a3ca042933b20220944cbf5aCAS |
[5] S. Caffarri, R. Kouil, S. Kereche, E. Boekema, R. Croce, EMBO J. 2009, 28, 3052.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXpvFCrtLo%3D&md5=9d91d4294c37f43e9ff2631ed8b280c6CAS |
[6] H. van Amerongen, L. Valkunas, R. van Grondelle, Photosynthetic Excitons, 2000 (World Scientific: Singapore).
[7] H. Kirchhoff, Trends Plant Sci. 2008, 13, 201.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXlvVWgtL0%3D&md5=20c6887bb41247a1d8885243e372ee17CAS |
[8] P. Jordan, P. Fromme, H. Witt, O. Klukas, W. Saenger, N. Krauß, Nature 2001, 411, 909.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXkvVWhs78%3D&md5=1506f78aea17e4f9f2bc4dbc635defaeCAS |
[9] G. Beddard, G. Porter, Nature 1976, 260, 366.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE28XktlSktbs%3D&md5=10593416b1de513a6da875665c825b7cCAS |
[10] Z. F. Liu, H. C. Yan, K. B. Wang, T. Y. Kuang, J. P. Zhang, L. L. Gui, X. M. An, W. R. Chang, Nature 2004, 428, 287.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXitFCntbc%3D&md5=dc220abbcad5e1363d76885afecfc505CAS |
[11] G. S. Schlau-Cohen, T. R. Calhoun, N. S. Ginsberg, M. Ballottari, R. Bassi, G. R. Fleming, Proc. Natl. Acad. Sci. USA 2010, 107, 13276.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXht1GrurvE&md5=425a055a1ef0d03d60afb713fe08ccfeCAS |
[12] G. S. Schlau-Cohen, T. R. Calhoun, N. S. Ginsberg, E. L. Read, M. Ballottari, R. Bassi, R. van Grondelle, G. R. Fleming, J. Phys. Chem. B 2009, 113, 15352.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtlWrurfK&md5=e7c5a27324b5e135ce24101adade6b28CAS |
[13] R. Remelli, C. Varotto, D. Sandonà, R. Croce, R. Bassi, J. Biol. Chem. 1999, 274, 33510.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXns1ejs7w%3D&md5=7340034df047330d3cda3abedde50683CAS |
[14] H. van Amerongen, R. van Grondelle, J. Phys. Chem. B 2001, 105, 604.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXptVWhsrs%3D&md5=b0a12d7ff5998673f6821995d5421be3CAS |
[15] R. van Grondelle, V. I. Novoderezhkin, Phys. Chem. Chem. Phys. 2006, 8, 793.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XhtFymur0%3D&md5=841a1eb3329aedb9e680ca4333542678CAS |
[16] G. Schlau-Cohen, A. Ishizaki, G. Fleming, Chem. Phys. 2011, 386, 1.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXpvVyltbk%3D&md5=896824ad95e894b3da0ca65501fe879bCAS |
[17] V. Novoderezhkin, R. van Grondelle, Phys. Chem. Chem. Phys. 2010, 12, 7352.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXotlWms7o%3D&md5=b9b34b9eea60a927acd3ff289c63b877CAS |
[18] D. M. Jonas, Annu. Rev. Phys. Chem. 2003, 54, 425.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXntFSgsLs%3D&md5=16ebab243298c129f89af85a262cb592CAS |
[19] M. Cho, Two-Dimensional Optical Spectroscopy, 2009 (CRC Press: Boca Raton, FL).
[20] G. S. Schlau-Cohen, J. M. Dawlaty, G. R. Fleming, IEEE J. Sel. Top. Quant. Elect. 2012, 18, 283.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XjtFaitr4%3D&md5=2d3537fab5de0f22fb1f433111a3ebeaCAS |
[21] R. Agarwal, B. P. Krueger, G. D. Scholes, M. Yang, J. Yom, L. Mets, G. R. Fleming, J. Phys. Chem. B 2000, 104, 2908.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXhs1Wkurs%3D&md5=75b4a6f3950a93572cdde0f9ca6abd05CAS |
[22] J. M. Salverda, M. Vengris, B. P. Krueger, G. D. Scholes, A. R. Czamoleski, V. Novoderezhkin, H. van Amerongen, R. van Grondelle, Biophys. J. 2003, 84, 450.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXisVSmsA%3D%3D&md5=b7f773188f33affc3172e3503a47a71eCAS |
[23] V. Novoderezhkin, M. Palacios, H. van Amerongen, R. van Grondelle, J. Phys. Chem. B 2004, 108, 10363.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXks1agtbo%3D&md5=c4904ca5bb05aa2fec06c1c01a07568cCAS |
[24] V. I. Novoderezhkin, M. A. Palacios, H. van Amerongen, R. van Grondelle, J. Phys. Chem. B 2005, 109, 10493.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXjs1SjtLk%3D&md5=8eed60638c1fa43daf0af37f5d9f1e4fCAS |
[25] S. Georgakopoulou, G. van der Zwan, R. Bassi, R. Van Grondelle, H. van Amerongen, R. Croce, Biochemistry 2007, 46, 4745.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXjslWgs7c%3D&md5=85dedfeff154a41c296e53ebfc78ae18CAS |
[26] K. Gibasiewicz, M. Rutkowski, R. Van Grondelle, Photosynthetica 2009, 47, 232.
| Crossref | GoogleScholarGoogle Scholar |
[27] T. R. Calhoun, N. S. Ginsberg, G. S. Schlau-Cohen, Y.-C. Cheng, M. Ballottari, R. Bassi, G. R. Fleming, J. Phys. Chem. B 2009, 113, 16291.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXht1yntLnP&md5=dbb253e65482873ef5b736ae041a757aCAS |
[28] G. Scholes, G. Fleming, A. Olaya-Castro, R. Van Grondelle, Nat. Chem. 2011, 3, 763.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXht1ajurzF&md5=421bf4c2a62e8c4d9d6f91dfe1147472CAS |
[29] G. Fleming, G. Schlau-Cohen, K. Amarnath, J. Zaks, Faraday Discuss. 2012, 155, 27.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xhs1eltLw%3D&md5=1f428125857a6dd733d59b1d71ec55f7CAS |
[30] M. Gouterman, The Porphyrins, 1978, Volume III (Academic Press: New York, NY).
[31] M. Linke, A. Lauer, T. von Haimberger, A. Zacarias, K. Heyne, J. Am. Chem. Soc. 2008, 130, 14904.
| Crossref | GoogleScholarGoogle Scholar |
[32] J. Adolphs, T. Renger, Biophys. J. 2006, 91, 2778.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XhtVGls7vO&md5=8094ac5d2677a62105e43bf75a170f38CAS |
[33] F. Müh, M. Madjet, J. Adolphs, A. Abdurahman, B. Rabenstein, H. Ishikita, E. Knapp, T. Renger, Proc. Natl. Acad. Sci. USA 2007, 104, 16862.
| Crossref | GoogleScholarGoogle Scholar |
[34] F. Muh, M. Madjet, T. Renger, J. Phys. Chem. B 2010, 114, 13517.
| Crossref | GoogleScholarGoogle Scholar |
[35] E. L. Read, G. S. Schlau-Cohen, G. S. Engel, J. Wen, R. E. Blankenship, G. R. Fleming, Biophys. J. 2008, 95, 847.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXotlKhsLY%3D&md5=b62a507d2b839fbe967455d10830e018CAS |
[36] T. Brixner, T. Mancˇal, I. V. Stiopkin, G. R. Fleming, J. Chem. Phys. 2004, 121, 4221.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXmvVCnurs%3D&md5=c42c291dbd0c39c6ab263557d3448e9fCAS |
[37] A. W. Albrecht, J. D. Hybl, S. M. Gallagher Faeder, D. M. Jonas, J. Chem. Phys. 1999, 111, 10934.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXnvV2gs7c%3D&md5=4e66c5ac3b67c8df2ba9791bd3887b81CAS |
[38] M. L. Cowan, J. P. Ogilvie, R. J. D. Miller, Chem. Phys. Lett. 2004, 386, 184.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXhsVKis7c%3D&md5=6780898ce6c69c1deccc1fc0a85098fdCAS |
[39] U. Selig, F. Langhojer, F. Dimler, T. Lohrig, C. Schwarz, B. Gieseking, T. Brixner, Opt. Lett. 2008, 33, 2851.
| Crossref | GoogleScholarGoogle Scholar |
[40] S. H. Shim, M. T. Zanni, Phys. Chem. Chem. Phys. 2009, 11, 748.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXnsVWlsg%3D%3D&md5=5bcb93e0d1f0f10e3a21e807a801ee17CAS |
[41] A. D. Bristow, D. Karaiskaj, X. C. Dai, S. T. Cundiff, Opt. Express 2008, 16, 18017.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhsVOhsb3P&md5=2b8abe19634a682d1ace0f6c4b16eb6eCAS |
[42] A. Nemeth, J. Sperling, J. Hauer, H. F. Kauffmann, F. Milota, Opt. Lett. 2009, 34, 3301.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsF2jtbbM&md5=4941b48cb5c99568fc9079718318e885CAS |
[43] S. Mukamel, Principles of Nonlinear Optical Spectroscopy, 1995 (Oxford University Press: New York, NY).
[44] R. M. Hochstrasser, Chem. Phys. 2001, 266, 273.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXjtFCqtLs%3D&md5=ef6d21340ba269a65743886055697679CAS |
[45] J. Dreyer, A. M. Moran, S. Mukamel, Bull. Korean Chem. Soc. 2003, 24, 1091.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXotFKqt7w%3D&md5=4c2015ec2438b821fee674a11483b647CAS |
[46] A. Ishizaki, G. R. Fleming, J. Chem. Phys. 2009, 130, 234111.
| Crossref | GoogleScholarGoogle Scholar |
[47] A. Ishizaki, T. R. Calhoun, G. S. Schlau-Cohen, G. R. Fleming, Phys. Chem. Chem. Phys. 2010, 12, 7319.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXotlWms7c%3D&md5=d0cf2c30fe9f8c61164dea8810973befCAS |
[48] V. Novoderezhkin, A. Marin, R. van Grondelle, Phys. Chem. Chem. Phys. 2011, 13, 17093.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXht1Slt7nI&md5=2db9c223ca4b4ce35a418e306e2ddc80CAS |
[49] G. S. Engel, T. R. Calhoun, E. L. Read, T.-K. Ahn, T. Mančal, Y.-C. Cheng, R. E. Blankenship, G. R. Fleming, Nature 2007, 446, 782.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXktVeqt78%3D&md5=1c3f3f1e836ddcf37f7dbad06c584e49CAS |
[50] H. Lee, Y.-C. Cheng, G. R. Fleming, Science 2007, 316, 1462.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXmtFSjsbs%3D&md5=71ef643de0b34a5537fb13244988783dCAS |
[51] E. Collini, C. Y. Wong, K. E. Wilk, P. M. G. Curmi, P. Brumer, G. D. Scholes, Nature 2010, 463, 644.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsVygu7o%3D&md5=7bb2bb8d5d43cb7b6b580c410c8877b1CAS |
[52] G. Panitchayangkoon, D. Hayes, K. A. Fransted, J. R. Caram, E. Harel, J. Wen, R. E. Blankenship, G. S. Engel, Proc. Natl. Acad. Sci. USA 2010, 107, 12766.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXpsVagt7g%3D&md5=b335ff4ceb5e8325ae1bf66fd65eff1cCAS |
[53] A. Ishizaki, G. R. Fleming, Proc. Natl. Acad. Sci. USA 2009, 106, 17255.
| Crossref | GoogleScholarGoogle Scholar |
[54] A. Ishizaki, G. R. Fleming, New J. Phys. 2010, 12, 055004.
| Crossref | GoogleScholarGoogle Scholar |
[55] M. T. Zanni, N.-H. Ge, Y. S. Kim, R. M. Hochstrasser, Proc. Natl. Acad. Sci. USA 2001, 98, 11265.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXnt1yqt7Y%3D&md5=c3690d85deab74fa93ddbc632e82ade1CAS |
[56] G. S. Schlau-Cohen, A. Ishizaki, T. R. Calhoun, N. S. Ginsberg, M. Ballottari, R. Bassi, G. R. Fleming, Nat. Chem. 2012, 4, 389.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XksVehtLs%3D&md5=0553986f7675459467e729df4a911a0dCAS |
[57] M. Mohseni, P. Rebentrost, S. Lloyd, A. Aspuru-Guzik, J. Chem. Phys. 2008, 129, 174106.
| Crossref | GoogleScholarGoogle Scholar |
[58] P. Rebentrost, M. Mohseni, A. Aspuru-Guzik, J. Phys. Chem. B 2009, 113, 9942.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXns1Sitrw%3D&md5=99e45e84a17962f819bd6518b93949c1CAS |
[59] M. B. Plenio, S. F. Huelga, New J. Phys. 2008, 10, 113019.
| Crossref | GoogleScholarGoogle Scholar |
[60] A. Olaya-Castro, C. F. Lee, F. F. Olsen, N. F. Johnson, Phys. Rev. B 2008, 78, 085115.
| Crossref | GoogleScholarGoogle Scholar |
[61] A. Melis, Plant Sci. 2009, 177, 272.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXpt1KgsrY%3D&md5=1e6610bab2a1457a8cfafa0d36e4ef8dCAS |
[62] D. Noy, C. Moser, P. Dutton, BBA-Bioenergetics 2006, 1757, 90.
| 1:CAS:528:DC%2BD28XisF2nsb8%3D&md5=bcb25eb8cc687aa145f71b9aa7c45760CAS |
[63] G. Steinberg-Yfrach, P. Liddell, S. Hung, A. Moore, D. Gust, T. Moore, Nature 1997, 385, 239.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXlslWktA%3D%3D&md5=d1f135f7cee09a105f4abdaa4eff0b15CAS |
[64] G. Steinberg-Yfrach, J. Rigaud, E. Durantini, A. Moore, D. Gust, T. Moore, Nature 1998, 392, 479.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXisFent7c%3D&md5=21ce04e9b9eac6e72028e47a4a4d1d61CAS |
[65] Y. Terazono, G. Kodis, P. Liddell, V. Garg, T. Moore, A. Moore, D. Gust, J. Phys. Chem. B 2009, 113, 7147.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXltVCgtr0%3D&md5=eb8f31d6f993af04f99f249feae66030CAS |
[66] M. Kanan, D. Nocera, Science 2008, 321, 1072.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtVSitrbP&md5=4d554995a8442318e37fd3fd4f64344fCAS |
[67] B. Hardin, E. Hoke, P. Armstrong, J. Yum, P. Comte, T. Torres, J. Fréchet, M. Nazeeruddin, M. Grätzel, M. McGehee, Nat. Photonics 2009, 3, 406.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXnvVagt78%3D&md5=5936bb2edca034f6dfb8560826cd7a40CAS |
[68] C. Faulkner, S. Lees, P. Ciesielski, D. Cliffel, G. Jennings, Langmuir 2008, 24, 8409.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXos1ahtr8%3D&md5=9bae57e65982a4c42782deb7d6de35cbCAS |
[69] N. Lewis, D. Nocera, Proc. Natl. Acad. Sci. USA 2006, 103, 15729.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XhtFymtbrJ&md5=e4252961598780ced30bcd2df19c9437CAS |