

Towards Hydrogen Energy: Progress on Catalysts for Water Splitting
Gerhard F. Swiegers A D , Douglas R. MacFarlane B D , David L. Officer A D , Amy Ballantyne A , Danijel Boskovic A , Jun Chen A , G. Charles Dismukes C , Graeme P. Gardner C , Rosalie K. Hocking B , Paul F. Smith C , Leone Spiccia B , Pawel Wagner A , Gordon G. Wallace A , Bjorn Winther-Jensen B and Orawan Winther-Jensen BA Intelligent Polymer Research Institute and ARC Centre of Excellence for Electromaterials Science, University of Wollongong, Wollongong, NSW 2522, Australia.
B ARC Centre of Excellence for Electromaterials Science, Monash University, Clayton, Vic. 3800, Australia.
C Department of Chemistry and Chemical Biology, Rutgers, The State University of New Jersey, Piscataway, NJ 08854, USA.
D Corresponding authors. Email: Swiegers@uow.edu.au; Douglas.MacFarlane@monash.edu; DavidO@uow.edu.au

Gerry Swiegers is a Professor in the Intelligent Polymer Research Institute at the University of Wollongong. He is also an adjunct fellow of the ARC-funded Australian Centre for Electromaterials Science (ACES). His interests span bioinspired catalysis. He has been responsible for illuminating important fundamental aspects of chemical and biological catalysis. |

Douglas MacFarlane is an ARC Federation Fellow at Monash University. He is also the program leader of the Energy Program in the ARC-funded Australian Centre for Electromaterials Science (ACES). His research interests include the development of ionic liquids for use in catalysis and energy storage. |

David Officer is Professor of Organic Chemistry in the Intelligent Polymer Research Institute and the ARC Centre of Excellence in Electromaterials Science at the University of Wollongong, Wollongong, Australia (ACES). His research interests are in the areas of porphyrin and conducting polymer chemistry, nanomaterials, solar cells, and artificial photosynthesis. |
Australian Journal of Chemistry 65(6) 577-582 https://doi.org/10.1071/CH12048
Submitted: 30 January 2012 Accepted: 12 March 2012 Published: 27 April 2012
Abstract
This article reviews some of the recent work by fellows and associates of the Australian Research Council Centre of Excellence for Electromaterials Science (ACES) at Monash University and the University of Wollongong, as well as their collaborators, in the field of water oxidation and reduction catalysts. This work is focussed on the production of hydrogen for a hydrogen-based energy technology. Topics include: (1) the role and apparent relevance of the cubane-like structure of the Photosystem II Water Oxidation Complex (PSII-WOC) in non-biological homogeneous and heterogeneous water oxidation catalysts, (2) light-activated conducting polymer catalysts for both water oxidation and reduction, and (3) porphyrin-based light harvesters and catalysts.
Introduction
The splitting of water into hydrogen and oxygen is a process that offers the prospect of a genuinely sustainable energy technology if the source of the energy is derived from sustainable solar, wind, tide, and hydro. The key to an energy efficient water splitting process is catalysts that can carry out the water oxidation and reduction reactions with minimal energy losses. Nature provides important insights in this respect in terms of structures and mechanisms that have evolved to carry out related processes in plants. A particularly key enzyme in this respect is the Photosystem II Water Oxidising Complex (PSII-WOC) of photosynthesis, which facilitates the light-driven oxidation of water (H2O) into dioxygen (O2).[1,2] The PSII-WOC is, at the present time, the only known species capable of catalysing water oxidation in a sustained, efficient and light-driven form. Because of the remarkable versatility and efficiency of enzymes like the PSII-WOC, a key task for chemists is to understand and apply Nature’s catalytic principles in artificial, non-biological systems. Breslow termed this quest ‘biomimetic chemistry’ and defined it as[3]: ‘Imitating the style of enzyme catalysed processes in an effort to achieve some of the advantages, which Nature has realised by the use of enzymes.’ Its application to the specific case of the PSII-WOC is widely spoken of as ‘Artificial Photosynthesis’.
Taking a broader view of the mechanisms utilised by relevant enzymes informs the design of novel inorganic, polymer, and small molecule organic materials that are light harvesting, or catalytic in either or both of water oxidation and reduction. In the various forms that water splitting technology will ultimately take, all of these families of materials have a role to play and all are under active development at Monash and Wollongong Universities (ACES) and their collaborators. This article provides an overview of some of the recent developments in that research.
The Role of the Cubical Architecture of the Photosystem II Water Oxidising Complex in Water Oxidation Catalysis
Fig. 1a depicts a model of the PSII-WOC based on recent X-ray crystal structure data.[4–6] As can be seen, the CaMn3O4 core of the active site comprises a so-called cubane structure in which the Ca and three of the Mn ions are positioned, with the four O atoms, in a cube-like arrangement. A fourth Mn ion lies on the outside of this cube. This structure is conserved in all known photosynthetic organisms, raising questions as to its role and importance. Recent work has shown that several, recently-discovered, active Mn or Co molecular or inorganic water oxidation catalysts employ a cubane core that is structurally virtually identical to the catalytic core present in the PSII-WOC.[7] Not only is the structural motif of the catalytic core of these species a cubane, but the actual physical dimensions of the cubane core also closely match those of the Mn3O4 unit which is capped by the Ca ion in the active site of the PSII-WOC. Examples in this respect include: (1) Nocera’s Co-pi catalyst,[8] (2) Frei and earlier workers’ Co3O4 spinel catalysts (B-site),[9] (3) Spiccia and Hocking’s MnO2 birnessite,[10] (4) Hill’s recently discovered Co-polyoxotungstate catalyst,[11]† (5) Dismuke’s λ-MnO2 spinel (B-site),[12] as well as (6) molecular Mn4O4 and Co4O4 cubanes.[1,2,13]
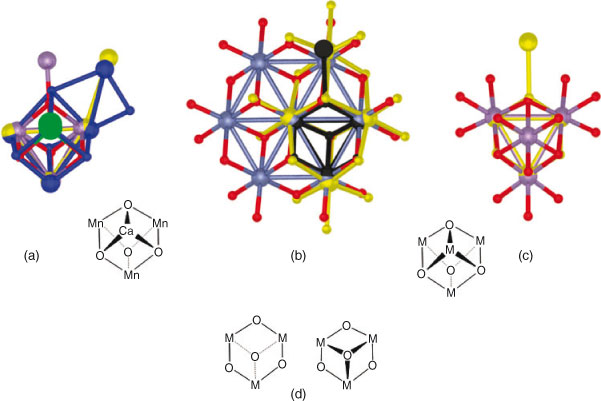
|
Fig. 1 illustrates these similarities. In Fig. 1a, the CaMn4O4 core of the PSII-WOC from the three most detailed single-crystal X-ray crystal structures of this enzyme, are superimposed upon each other. These are: (i) the so-called London structure by Barber, Iwata, and colleagues, which was resolved to 3.5 Å,[4] (ii) the so-called Berlin structure by Loll, Kern, and co-workers, which was resolved to 2.9 Å,[5] and (iii) the so-called Osaka structure by Umena, Kawakami, Shen, and Kamiya, that was resolved to 1.9 Å.[6] The Osaka structure provides the most detailed picture of the PSII-WOC core yet available. As can be seen in Fig. 1a, in all of these X-ray structures the CaMn3O4 core is essentially identical. The only significant difference is in the location of the fourth, outlying, ‘dangler’ Mn ion. While there may have been radiation-induced variations in other parts of the (protein) structure, the CaMn3O4 core is invariant. This is so despite the differences in the technique used and, indeed, in spite of the diversity of the photosynthetic organisms involved.
This lack of evolutionary and structural diversity in the PSII-WOC core cubane arrangement implies that combinatorial biosynthesis in Nature has yielded only this one catalytic structure capable of facilitating sustained water oxidation catalysis. The question arises: why is that the case?
Fig. 1b superimposes the PSII-WOC core as revealed in the London structure[4] upon two recently discovered man-made homogeneous catalysts of water oxidation: Nocera’s Co-pi catalyst[8] and the core of Hill’s Co-polyoxotungstate catalyst.[11] As can be seen, there is a close structural match. In fact, the core M3O4 (M = Co, Mn) structures are essentially identical. Fig. 1c superimposes the PSII-WOC core as seen in the London structure,[4] upon a recently-discovered man-made heterogeneous catalyst of water oxidation: the B-site of Dismukes’ λ-Mn2O4 spinel catalyst.[12] Once again, there is a remarkably close structural match.
In fact, a Root Mean Square (RMS) ‘Goodness-of-Fit’ comparison indicates that there is a greater variation between the London, Berlin, and Osaka structures of the PSII-WOC than there is between the Osaka structure and the man-made catalysts mentioned above.[7] The active site structures of all of the above catalysts very closely match the Mn3O4 core in the PSII-WOC.
Man-made efforts to develop new water oxidation catalysts therefore appear to have inadvertently also converged on a cubane structure that is, in large measure, not only qualitatively but also quantitatively identical to the cubane active site of the PSII-WOC.[7] These apparent commonalities in an otherwise disparate and unconnected range of homogeneous, heterogeneous, and enzymatic catalysts are extraordinary. Human studies appear to be confirming the findings of combinatorial biosynthesis regarding the utility of the cubane structure in water oxidation catalysis.
The commonality may also extend to the actions of these catalysts. For example, several of these catalysts appear to undergo spontaneous dis-assembly and re-assembly of the cubane at an open face (or surface) during catalysis. Fig. 1d depicts the open-face structures that must exist in the Co3O4 and λ-Mn2O4 spinel catalysts.[7] Similar open faces are present in the Co-pi, MnO2 birnessite, and molecular cubane (immediately before O2 formation, according to theoretical studies).[7] Given that Hill’s Co-polyoxotungstate also self-assembles, a library of associated, partially-assembled species must exist in solution; this would undoubtedly include an open-faced arrangement. We should observe also that Hill’s catalyst achieves the highest recorded turnover frequency of any abiological catalyst for catalytic water oxidation: >5 s–1 at pH 8.[11] While short of the turnover frequencies achieved by the PSII-WOC, this nevertheless falls within the range of turnover frequencies typically achieved by enzymes: 1–10000 s–1.
The only reasonable explanation for the fact that all of these heterogeneous, homogeneous, and enzymatic catalysts employ so similar a structure, is that the cubane arrangement is needed to guide the reactant (corner) O atoms along a single, optimum approach trajectory during the formation of the O-O bond of O2. That is, the key commonality in all of these species likely involves a single approach trajectory and this manifests itself physically as a common catalytic structure in all catalytic classes.[14] If this is so, then it implies that the cubane structure must also allow vigorous, albeit constrained motion by the reactant O atoms at the corners of the cube (or open-faced half-cube). In the case of molecular species like the Mn4O4 or Co4O4 molecular cubanes, this would likely involve regular, repeated, conformational motion. However, in heterogeneous species like Co3O4 and the like, such motion would likely involve regular, repeated, oscillatory thermal motion at the surface of the catalyst.
This proposal is considerably strengthened by the finding from Rutgers University that the crystalline spinel LiMn2O4 becomes active in water oxidation only when the Li+ ions are removed, forming the isostructural spinel λ-MnO2.[12] Delithiation presumably imparts freedom of motion to the bridging O atoms essential for catalysis. As can be seen in Fig. 1c, the cubical Mn4O4 units in λ-MnO2 are topologically identical to the Mn4O4 core found in the molecular ‘cubane’ catalysts (all O are dicoordinate).
Most recently a direct comparison of cubic spinel vs layered polymorphs has provided the strongest evidence yet for the functional importance of the B4O4 cube topology for water oxidation.[15] Lithium cobalt oxides occur in two polymorphs of identical composition, Li2Co2O4: cubic spinel (Fd3m) and layered (P3̅m1) (Fig. 2; top), and are used as battery materials due to the high mobility of lithium.[16,17] The spinel polymorph contains Co4O4 cube subunits and forms above 350°C as the exclusive phase at this temperature. However, at temperatures greater than 500°C and longer calcination times, it transforms to the thermodynamically favoured layered polymorph.[18,19] This dichotomy between the two crystal structures made it possible to test the bioinspired hypothesis.[20] The higher calcination temperature and longer reaction times used to form the layered polymorph gives rise to increasing crystallite size.[16] To reduce this difference in particle sizes, the authors adapted a sol-gel method[21] that forms nano-particles (50–100 nm) and limited particle size differences to only two-fold. Powder X-ray diffraction (PXRD) confirmed the crystal structure transformation between 400 (all cubic), 500, 600, and 700°C (all layered), while the rate of oxygen evolution decreased to zero for this range, even when accounting for the surface area.[22]
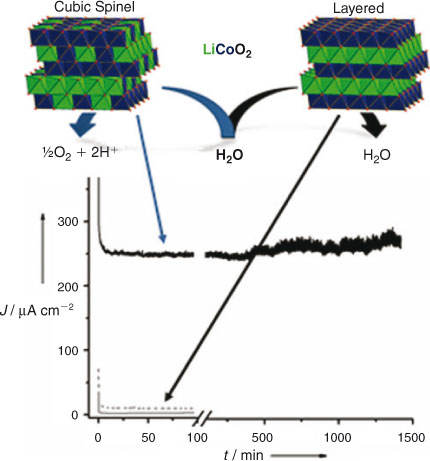
|
The authors showed that this system exhibited the same trend when current density from electrochemical water oxidation was monitored, as follows. Membrane electrode assemblies (MEA’s) were fabricated onto conducting ITO glass by spin coating inks composed of catalyst and proton conduction polymer (neutralised Nafion®).[23] Application of an electrical potential of 1.20 V (vs Ag/AgCl reference) revealed that the membranes containing the cubic, low-temperature (400°C) spinel outperformed the ones containing equal loadings of the layered, high-temperature (700°C) polymorph by greater than 50 fold (>250 μA cm–2 at pH 7), as depicted in Fig. 2. The MEA prepared with the layered material exhibited almost the same current density as the uncatalysed reaction on ITO + Nafion® (control).
Thus, only the cubic, spinel polymorph of the lithium cobalt oxides is able to catalyse water oxidation efficiently, whether assayed in homogeneous solutions using a photochemical assay, or electrolytically in the dark using a composite electrode.
The mechanism was not revealed in these studies; whether the Co4O4 cubes are functioning like the cubical core of photosystem II is still unclear. But the evidence for the intrinsic catalytic activity of the oxo-metallic cubes in water oxidation is now firmly established, both for the solid-state transition metal spinels and the organometallic molecular cubanes. In both cases, the cobalt oxide cubes were considerably more active than the manganese oxide cubes.
These findings have potentially significant implications in biology and biochemistry, as well as in non-biological homogeneous and heterogeneous catalysis. They also buttress the strategy of mimicking enzymes by copying the structures of their active sites whilst ensuring dynamism in catalyst binding and flexing/motion.[14]
Conducting Polymer Electro-Catalysts
Conducting polymers such as poly(ethylenedioxythiophene), PEDOT, and poly(thiophenes) in general, offer an important combination of properties as electro-catalysts. They are moderately electron conductive materials that have well known properties as catalytic electrodes.[24,25] In addition, they can in some cases be intrinsically photo-absorbing and can incorporate other catalysts and sensitisers to further enhance the properties. We have shown,[26] for example, that a composite of PEDOT and polyethylene glycol (PEG) is able to catalyse water reduction to hydrogen at low pH, at rates higher than that of classical electrode materials such as Pt. The role of the PEG component is to ensure facile ion diffusion access to the interior of the polymer material, such that the whole bulk of the electrode layer is active, rather than simply its external surface. The PEG is incorporated during the vapour-phase polymerisation of the conducting polymer and serves to maintain the polymer structure in an open morphology that allows electrolyte access. The conducing polymer material clearly has a role in the mechanism of the reduction reaction beyond simply being a source of electrons. The polymer cycles between redox states, the reduced state being the active site for water or proton reduction. Further work has demonstrated that moving to neutral electrolyte conditions is possible.
Water oxidation has been studied in a similar vein but the family of polymers that are stable at the potentials required for water oxidation is limited. The stability issue relates to irreversible oxidation of the polymer itself; the polythiophenes appear to be sufficiently stable at neutral pH, the polymer oxidation being more or less pH independent while the water oxidation potentials required decrease rapidly with increasing pH.
We have recently shown facile water oxidation on polyterthiophene.[27] Furthermore, we were able to demonstrate enhanced oxidation currents when the electrode was exposed to visible light, indicating that the intrinsic photo-absorption of the polymer enhances activity. The oxidation current-voltage characteristic shifts to lower potentials under exposure to light such that some oxidation current flows below the formal thermodynamic potential for water oxidation at the pH involved. This suggests that the effect of light is to generate a photo-voltage that is additive to the applied external bias.
Taking inspiration from the bulk heterojunction solar cells field, we have developed the concept of a conducting polymer based heterojunction photo-electrocatalyst.[28] This notion involves photo-excitation in one conducting polymer followed by rapid charge separation across the junction such that recombination is suppressed. The effect has been demonstrated in the proton reduction reaction[25] and further work explores the hetero-junction concept for other light enhanced electro-catalysed reactions (B. Kolodziejczyk, D. R. MacFarlane, O. Winther-Jensen, 2012, unpubl. data).
Porphyrins as Light Harvesters and Catalysts
Given the key roles that tetrapyrroles such as chlorophylls play in photosynthetic light harvesting, electron transfer, and mediating water oxidation, it is no surprise that for more than 40 years researchers have been inspired to use synthetic chlorophylls (porphyrins) to develop processes that efficiently harvest light, and produce hydrogen and oxygen from water.[29,30]
Within ACES, we have been at the forefront of developing efficient light harvesting single porphyrins and porphyrin arrays, demonstrating their efficacy in the dye sensitised solar cell (DSSC). In 2007, in collaboration with Professor Michael Grätzel, the inventor of the DSSC, we reported the highest efficiency porphyrin DSSC (7.1 %) using functionalised porphyrins of the type shown in Fig. 3a.[31] Grätzel and his other collaborators have subsequently gone on to show that, under the right set of cell conditions, porphyrins are the most efficient dyes for DSSCs (12.3 % for one sun).[32] We have demonstrated the value of using multiple porphyrins such as that depicted in Fig. 3b for light harvesting, proving that it is possible to harvest light and inject electrons into the device semiconductor from all the porphyrins in a porphyrin array attached to it, emulating the multichlorophyll light harvesting process in photosynthesis.[33]

|
The efficacy of porphyrins as light harvesters, particularly nanostructured porphyrin arrays, has inspired us to investigate their potential to drive water splitting either as the light harvesting component, oxidation or reduction catalyst, or both. Previous work in this area has largely been concerned with using the photoexcited porphyrin to reduce a redox component such as methylviologen, which in turn reduces water (H+) to hydrogen (H2) in the presence of a catalyst like platinum.[34] A sacrificial electron donor or applied potential regenerates the oxidised porphyrin. As in the DSSC, the most commonly used light harvesting porphyrins are zinc porphyrins with a covalent attachment to the electron acceptor in order to ensure good electronic communication and effective charge separation. While some zinc porphyrins have been shown to effectively sensitise H2 evolution from water,[35] water-soluble tin porphyrins (Fig. 3c) have been most studied since, on photoexcitation, they form a relatively stable and long lived radical anion in the presence of an electron donor. The efficacy of such a porphyrin system has been recently demonstrated by Choi and his co-workers who showed that dihydroxytin(iv) tetrapyridylporphyrin in solution with platinum-decorated titanium dioxide nanoparticles and EDTA as a sacrificial electron donor produced H2 across a broad pH range (pH 3–11) under visible light with a turnover number of 410; it was superior to a typical ruthenium complex adsorbed to the titanium dioxide.[36]
While Choi’s work highlights the potential of porphyrins as practical sensitisers for H2 production, it also emphasises the need to use expensive metal catalysts like platinum in any such system and, within ACES, we have been interested to develop catalytic systems that can enhance or even replace platinum. In the former respect, a 7-fold increase in H2 production in 1M acid could be achieved by coating a platinum electrode with polypyrrole containing ferrocene sulfonate as counter-ion.[37] In a more recent study, as described above, it has been shown that in the presence of an ion-coordinating polymer like polyethylene glycol (PEG), the polythiophene conducting polymer, PEDOT, has a comparable performance to platinum for the electrochemical catalytic production of H2 from acidic electrolytes.[26] This presents the opportunity to develop platinum-free photoactive porphyrin-sensitised H2 generation and we are now investigating coupling tin porphyrins to these catalytic systems.
Photosensitisation of water oxidation catalysts has also been achieved albeit with high potential porphyrins.[38] While this is of interest in relation to the photosensitisation of the manganese cubanes that ACES researchers and their collaborators have developed,[20] we have rather focussed our attention on the development of porphyrin-based catalytic systems for water oxidation. In 1994, Naruta et al. reported for the first time oxygen evolution by a four electron oxidation of water using a rigid ortho linked manganese porphyrin dimer.[39] The proximity of the two manganese atoms appeared to promote the formation of a peroxy bridge between the two oxidised manganese species leading to the formation of oxygen. This seemed analogous to the mode of action of the two iron atoms in diferrocene catalysts for hydrogen production that were mirrored by the introduction of ferrocene sulfonate into polypyrrole, as described above.[37] Therefore, in a similar fashion, we electrochemically incorporated monomeric manganese tetrasulfonatophenylporphyrin into polyterthiophene films on indium tin oxide electrodes and demonstrated the production of oxygen from water on illumination of the composite electrode at a significantly reduced overpotential (0.09 V).[40] This overpotential was low enough to allow the production of oxygen from seawater without the formation of chlorine. Given the high density of the porphyrin in the polyterthiophene, oxygen formation appears to arise in a similar fashion to that proposed by Naruta and co-workers[39] for the manganese porphyrin dimer. This result has opened up the possibility of a fully photosensitised porphyrin water oxidation catalyst and we are currently exploring this and other related opportunities.
Conclusion
The focus of the ARC Centre of Excellence for Electromaterials Science research program on a diverse range of electromaterials, from single organic molecules and inorganic complexes through to conducting polymers and nanostructured carbons, has made it a particularly suitable environment within which to develop exciting new materials for water splitting. Thus, the development of metal cubane complexes and subsequently a variety of related metal oxides has led to some very promising water oxidation catalysts that are also helping us to better understand the design of mimics for photosynthetic and other biological catalysts for use in practical devices. As the realisation of practical devices using such catalysts would currently require the use of expensive metals like platinum, we have also made inroads into the development of conducting polymer-based material replacements for platinum. These developments, coupled with our recent successes in the development of light harvesting porphyrins for solar cells and porphyrin catalysts has provided ACES researchers with important new approaches for the attainment of a truly bioinspired artificial photosynthetic water splitting system.
References
[1] W. Ruettinger, C. Campana, G. C. Dismukes, J. Am. Chem. Soc. 1997, 119, 6670.| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXktlKjsbc%3D&md5=128a11932348dada8a9992d4821be67eCAS |
[2] (a) W. Ruettinger, M. Yagi, K. Wolf, S. Bernasek, G. C. Dismukes, J. Am. Chem. Soc. 2000, 122, 10353.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXntVOmurw%3D&md5=2e8eb4f89994b418ec1d9bc5a2c654aaCAS |
(b) M. Yagi, K. V. Wolf, P. J. Baesjou, S. L. Bernasek, G. C. Dismukes, Angew. Chem. Int. Edit. 2001, 40, 2925.
| Crossref | GoogleScholarGoogle Scholar |
[3] (a) R. Breslow, Acc. Chem. Res. 1995, 28, 146.and references therein
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXjvFOmtrc%3D&md5=36e4dcfd884de73966856f77aaabe4c0CAS |
(b) R. Breslow, Chem. Soc. Rev. 1972, 1, 553.
| Crossref | GoogleScholarGoogle Scholar |
[4] K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber, S. Iwata, Science 2004, 303, 1831.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXitFehtbs%3D&md5=031fd950c39696f610bd2aa132128886CAS |
[5] (a) B. Loll, J. Kern, W. Saenger, A. Zouni, J. Biesiadka, Nature 2005, 438, 1040.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXhtlSksrvK&md5=3f186e8cfe16b272f8cbaa8a9d2a7501CAS |
(b) J. Yano, J. Kern, K.-D. Irrgang, M. J. Latimer, U. Bergmann, P. Glatzel, Y. Pushkar, J. Biesiadka, B. Loll, K. Sauer, J. Messinger, A. Zouni, V. K. Yachandra, Proc. Natl. Acad. Sci. USA 2005, 102, 12047.
| Crossref | GoogleScholarGoogle Scholar |
[6] Y. Umena, K. Kawakami, J.-R. Shen, N. Kamiya, Nature 2011, 473, 55.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXkslCmtLg%3D&md5=f7b48594fae4a870a2b3d1c7ed10df43CAS |
[7] G. F. Swiegers, J. K. Clegg, R Stranger, Chem. Sci. 2011, 2, 2254.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXht12isrvL&md5=2bb497c792c79a23dde60032a1b3c096CAS |
[8] M. W. Kanan, D. G. Nocera, Science 2008, 321, 1072.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtVSitrbP&md5=4d554995a8442318e37fd3fd4f64344fCAS |
[9] F. Jiao, H. Frei, Angew. Chem. Int. Edit. 2009, 48, 1841.and references therein
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXjt1Ojurg%3D&md5=b59158f7a83e71611fb56eadfee37230CAS |
[10] R. K. Hocking, R. Brimblecombe, L.-Y. Chang, A. Singh, M. H. Cheah, C. Glover, W. H. Casey, L. Spiccia, Nat. Chem. 2011, 3, 461.
| 1:CAS:528:DC%2BC3MXmtV2htL0%3D&md5=e1a1da8eed8e7449a5936fba7038db72CAS |
[11] Q. Yin, J. M. Tan, C. Besson, Y. V. Geletti, D. Musaev, A. E. Kuznetsov, Z. Luo, K. I. Hardcastle, C. L. Hill, Science 2010, 328, 342.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXks1Sgtbs%3D&md5=31262becc276064b39f67eb7a54d16e6CAS |
[12] D. M. Robinson, Y. B. Go, M. Greenblatt, G. C. Dismukes, J. Am. Chem. Soc. 2010, 132, 11467.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXpsV2ms7o%3D&md5=5bba10d3a2970acbecbbb6d9be882cc3CAS |
[13] (a) N. McCool, D. M. Robinson, J. E. Sheats, G. C. Dismukes, J. Am. Chem. Soc. 2011, 133, 11446.See also;
| 1:CAS:528:DC%2BC3MXovVegsb8%3D&md5=672c446513e213b2f413c709c7a7b3ccCAS |
(b) G. La Ganga, F. Puntoriero, S. Campagna, I. Bazzan, S. Berardi, M. Bonchio, A. Sartorel, M. Natali, F. Scandola, Faraday Discuss. 2012, 155, 177.
| Crossref | GoogleScholarGoogle Scholar |
[14] G. F. Swiegers, Mechanical Catalysis 2008 (John Wiley and Sons: New York, NY).
[15] G. P Gardner, Y. B. Go, D. M. Robinson, P. F. Smith, J. Hadermann, A. Abakumov, M. Greenblatt, G. C. Dismukes, Angew. Chem. Int. Edit. 2012, 51, 1616.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xksl2msQ%3D%3D&md5=0dda1fc0b9376f4ac2506a3b3da998feCAS |
[16] Y. Shao-Horn, S. A. Hackney, C. S. Johnson, A. J. Kahaian, M. M. Thackeray, J. Solid State Chem. 1998, 140, 116.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXmsVyrsb8%3D&md5=e7b29378edc2a3ddf239f3582ec2589eCAS |
[17] S. Choi, A. Manthiram, J. Solid State Chem. 2002, 164, 332.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38Xit1CjsL8%3D&md5=23221049d293bb645fb2daddf3f0e321CAS |
[18] Y. Shao-Horn, S. A. Hackney, A. J. Kahaian, M. M. Thackeray, J. Solid State Chem. 2002, 168, 60.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38Xos1yjs7o%3D&md5=cc719e459bcf3b1204c56156278805abCAS |
[19] H. Gabrisch, R. Yazami, B. Fultz, J. Electrochem. Soc. 2004, 151, A891.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXktVGkt7s%3D&md5=49e15ec7c564f62c11b0a7994d328121CAS |
[20] G. C. Dismukes, R. Brimblecombe, G. A. N. Felton, R. S. Pryadun, J. E. Sheats, L. Spiccia, G. F. Swiegers, Acc. Chem. Res. 2009, 42, 1935.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVSgtb%2FL&md5=7ee1e63bbf20e924e0a4d165bd1f212dCAS |
[21] S. Vivekanandhan, M. Venkateswarlu, N. Satyanarayana, J. Alloy. Comp. 2007, 441, 284.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXmsFCitrs%3D&md5=ec3818e7f94aeaf0cfb8d7af5291adb6CAS |
[22] B. Garcia, P. Barboux, F. Ribot, A. Kahn-Harari, L. Mazerolles, N. Baffier, Solid State Ion. 1995, 80, 111.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXotVegtr4%3D&md5=d5c0c2dd75c31e0f5750fab68433197aCAS |
[23] J. Suntivich, H. A. Gasteiger, N. Yabuuchi, Y. Shao-Horn, J. Electrochem. Soc. 2010, 157, B1263.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXos1GktrY%3D&md5=3b5e0d8ea99e65ad9c0d4c5ced580289CAS |
[24] B. Winther-Jensen, O. Winther-Jensen, M. Forsyth, D. R. MacFarlane, Science 2008, 321, 671.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXptVKnur0%3D&md5=6405cbf71e4202e9333a8633a05a3fd2CAS |
[25] B. Winther-Jensen, D. R. MacFarlane, Energy. Environ. Sci. 2011, 4, 279.
[26] B. Winther-Jensen, K. Fraser, C. Ong, M. Forsyth, D. R. MacFarlane, Adv. Mater. (Deerfield Beach Fla.) 2010, 22, 1727.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXltVyqtL8%3D&md5=fed2082535042c822df0cad820d70908CAS |
[27] O. Winther-Jensen, B. Winther-Jensen, D. R. MacFarlane, Electrochem. Commun. 2011, 13, 307.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjsFentLw%3D&md5=1d5be92d4a66f0ad77ad12c043de1caeCAS |
[28] O. Winther-Jensen, B. Winther-Jensen, D. MacFarlane, Heterojunction Electrocatalyst, PCT patent application 2011.
[29] D. Gust, T. A. Moore, A. L. Moore, Acc. Chem. Res. 2009, 42, 1890.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtlyrtrnN&md5=31f8f2b6799f2b3c146caed304bb4212CAS |
[30] K. Kalyanasundaram, M. Grätzel, Curr. Opin. Biotechnol. 2010, 21, 298.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXmslOht7w%3D&md5=0331f6b70ac7084cd92b8e321d959002CAS |
[31] W. M. Campbell, K. W. Jolley, P. Wagner, K. Wagner, P. J. Walsh, K. C. Gordon, L. Schmidt-Mende, M. K. Nazeeruddin, Q. Wang, M. Grätzel, D. L. Officer, J. Phys. Chem. C 2007, 111, 11760.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXnvVShtbw%3D&md5=3f4f2ef87d01218b1160c84d155ff6dcCAS |
[32] A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin, M. Grätzel, Science 2011, 334, 629.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtlyqu7nI&md5=e133a1a57aae1b4de8c382af9246514dCAS |
[33] A. J. Mozer, M. J. Griffith, G. Tsekouras, P. Wagner, G. G. Wallace, S. Mori, K. Sunahara, M. Miyashita, J. C. Earles, K. C. Gordon, L. Du, R. Katoh, A. Furube, D. L. Officer, J. Am. Chem. Soc. 2009, 131, 15621.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXht1Gju7%2FL&md5=78166dcdbad26fa0a5541a06e880914cCAS |
[34] W. Szulbinski, J. W. Strojek, Inorg. Chim. Acta 1986, 118, 91.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2sXktFeltg%3D%3D&md5=496410b0808644cf357c39aab5a8aa54CAS |
[35] J. Davila, A. Harriman, M. C. Richoux, L. R. Milgrom, J. Chem. Soc. Chem. Commun. 1987, 525.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2sXitFKqs7s%3D&md5=1a6934fbd94d6745089ea03cb91138d4CAS |
[36] W. Kim, T. Tachikawa, T. Majima, C. Li, H.-J. Kim, W. Choi, Energy. Environ. Sci. 2010, 3, 1789.
| 1:CAS:528:DC%2BC3MXivF2ns7o%3D&md5=a8957b277286ecdedf9393f3a6054a08CAS |
[37] J. Chen, J. Huang, G. F. Swiegers, C. O. Too, G. G. Wallace, Chem. Commun. 2004, 308.
[38] G. F. Moore, J. D. Blakemore, R. L. Milot, J. F. Hull, H.-E. Song, L. Cai, C. A. Schmuttenmaer, R. H. Crabtree, G. W. Brudvig, Energy. Environ. Sci. 2011, 4, 2389.
| 1:CAS:528:DC%2BC3MXhtFWrtr3E&md5=07a0b8e1f2374f3a090f0d3b17e64c71CAS |
[39] Y. Naruta, M.-A. Sasayama, T. Sasaki, Angew. Chem. Int. Edit. 1994, 33, 1839.
| Crossref | GoogleScholarGoogle Scholar |
[40] J. Chen, P. Wagner, L. Tong, G. G. Wallace, D. L. Officer, G. F. Swiegers, Angew. Chem. Int. Edit. 2012, 51, 1907.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhtFaisrs%3D&md5=f236c2c4e2527e75d0a88c92b5b60de0CAS |
† Note added in proof: A higher turnover frequency has now been reported in L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet, L. Sun, Nature Chem. 2012, advance article. https://doi.org/ 10.1038/NCHEM.1301