

Perspectives for Photobiology in Molecular Solar Fuels
Kastoori Hingorani A and Warwick Hillier A BA Research School of Biology, Australian National University, Canberra, ACT 0200, Australia.
B Corresponding author. Email: warwick.hillier@anu.edu.au
Australian Journal of Chemistry 65(6) 643-651 https://doi.org/10.1071/CH12096
Submitted: 14 February 2012 Accepted: 11 April 2012 Published: 22 May 2012
Abstract
This paper presents an overview of the prospects for bio-solar energy conversion. The Global Artificial Photosynthesis meeting at Lord Howe Island (14–18 August 2011) underscored the dependence that the world has placed on non-renewable energy supplies, particularly for transport fuels, and highlighted the potential of solar energy. Biology has used solar energy for free energy gain to drive chemical reactions for billions of years. The principal conduits for energy conversion on earth are photosynthetic reaction centres – but can they be harnessed, copied and emulated? In this communication, we initially discuss algal-based biofuels before investigating bio-inspired solar energy conversion in artificial and engineered systems. We show that the basic design and engineering principles for assembling photocatalytic proteins can be used to assemble nanocatalysts for solar fuel production.
Introduction
Civilization on Earth has been gradually advancing, and with this has come not only discovery and enlightenment but also an increasing dependence on energy. The energy consumption rates underpin lifestyles and affluence and afford luxuries in terms of personalised transport, climate control and food that can be sourced out of season and shipped in from afar. In 2010, estimated energy consumption on Earth globally was 16.3 terawatts (TW) of energy.[1] This energy burn rate† is the joule per second power consumption that is sustained continually 24/7 all year. The discovery of the Edison light bulb in 1879 was a practical and commercial success,[2] and perhaps in many ways the onset of significant increases in energy demand. A century after this invention, a standard feature in the industrialised world was the 100-W incandescent bulb. Remarkably today, a line of these 100-W bulbs operating edge-to-edge stretched from the earth to the moon would require 0.57 TW of power. The 2010 global energy burn rate is sufficient to encircle the moon 14 times with light bulbs.
The global energy production is derived primarily from fossil-fuel supplies. These products derive from eons-past photosynthesis and are finite. The world energy consumption contributes to anthropogenically generated carbon emissions, and in 2010, atmospheric CO2 averaged 389 ppm.[1] Predicted increases in global energy from projected increases in both population and gross domestic product (GDP) indicate that global energy demand will be >30 TW by 2050. It is reasoned that most of the alternative energy technologies currently available may not be sufficient to supply such a growing global energy demand.[3,4] Renewable energy sources such as wind are possible, but TW quotients of energy are very significant indeed.
Solar Energy and Photobiology
The potential scope of solar power on Earth is substantial as terawatt power quotients are received hourly. The practical challenge is that the solar energy arrives in relatively low density and therefore requires extended arrays for capture.[5] In this paper, we will discuss two possible photosynthetic solutions (Fig. 1). The first approach for solar energy conversion is the use of natural photosynthetic organisms such as cyanobacteria or algae to function as molecular factories (Fig. 1). These organisms are self-replicating and can also be genetically reprogrammed to produce molecular fuels such as hydrocarbons for biodiesel.[6] In many regards, these organisms may well hold significant projections for future global farming activities in the coming decades. A second approach outlined in this paper is artificial photosynthesis, whereby protein scaffolds localise pigments to capture solar energy in artificial photosynthetic reaction centres. The photochemistry of a reaction centre can generate electricity[7] or preferably a molecular fuel with the incorporation of an enzymatic catalyst (Fig. 1). Artificial photosynthesis systems that are biologically inspired present enzymatic solutions to catalysis with several advantages: use of Earth-abundant metals, and operation at neutral pH, ambient temperature and ambient pressures. There is considerable effort invested in hybridising these catalysts to photocatalysts as the catalytic reactions are also efficient in terms of thermodynamics.[8,9]
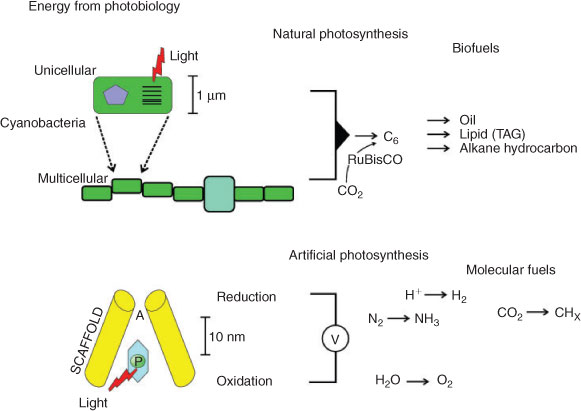
|
The most unique feature of photosynthetic organisms is the reaction centre (RC) that functions as the ‘engine-room’ for driving unfavoured reactions by extracting work from a photon.[10,11] All natural biological reaction centres function essentially the same way, with only minor variations in cofactors. In its simplest form, a reaction centre holds a chromophore pigment (P) such that the excited state (P*) can interact with a nearby acceptor molecule (A) and undergo electron transfer, forming a charge-separated state (P+/A–).[12] The structures of the biological reaction centres are essentially the same: a series of transmembrane proteins that localise the P/A pair across the membrane; a central C2-symmetric core that leads to bifurcated electron transfer pathways in some systems. The development of only a very limited number of reaction centres has had a profound influence on life on Earth, and today they sustain complex life on the planet.[10,13]
Engineered Photosynthesis for Algal Biofuels
Global photosynthesis accounts for ~200 × 1015 g of carbon fixed per annum. At the domestic level, ‘energy farming’ is already being applied to photosynthetic organisms to make biofuels and operates as a multimillion dollar business. Various biofuel products exist but are ultimately underpinned by plant productivity and efficiency. The biomass accumulation in crops (offset against food production[14]) is limited by net photosynthetic efficiencies of 1–2 %.[15,16] The so-called third-generation biofuels derived from photosynthetic microbes probably raise the photosynthetic efficiency to ~5 %, and although this intrinsic efficiency of photosynthesis is not higher than terrestrial plants, yield increases can be offset by increased culture density and lighting in photobioreactors.[17,18]
The microbial world has utilised solar energy conversion for some 3 billion years. The first organisms to reap the free energy of the photon were photosynthetic bacteria, but some 2.5 billion years ago, oxygenic photoautotrophs began to oxidize water as the source of electrons and molecular oxygen accumulated as a by-product. These organisms because of their unlimited supply of water went on to colonise the planet, terraforming Earth with an aerobic and oxidative atmosphere. The first oxygenic photoautotrophs to do this were cyanobacteria, and over a billion years later, eukaryotic algae appeared with larger cells and more complex cellular organisation. The biochemical revolution that was photosynthesis enabled reductive reactions to fix CO2 (carbon dioxide) into sugars, carbohydrates and storage molecules such as neutral lipids, triacylglycerol (TAG) and diacylglycerol (DAG). For most of these organisms, the storage of these carbon sources is sparse as accumulation is compensated by cell replication and division. However, some organism provide natural factories for biofuels and biodiesel.[19] Under conditions of nutrient stresses (low N, P or Si) or temperature extremes, natural cells go into overdrive and produce many hundred-fold increases in storage molecules. The highlights are microalgae stains such as Botryococcus braunii that secrete long-chain hydrocarbons >C30–36 outside the cell under biochemical stress.[20] The green algae Chlamydomonas reinhardtii will accumulate up to 70 % dry weight lipid,[21] and Chlorella sp. can also accumulate significant lipid under mixotrophic conditions.[22] Saline and hypersaline strains such as Dunaliella salina will accumulate significant lipid content and also a variety of diatoms.[23,24] The cyanobacteria typically don’t accumulate significant quantities of storage products but their natural diversity can be readily re-engineered via genetic modification (GM). There are several examples of engineering cyanobacteria to make ethanol,[25] sucrose[26] and the volatile C4 isobutyraldehyde[27] and C5 isoprene.[28] Various strains have also been engineered to produce hydrogen.[29]
Algal fuels are often considered as providing a ‘drop-in fuel’. This is in reference to the biochemical reactions that generate long-chain hydrocarbons > C8, and by inference require minimal downstream refining before use. Ethanol derived from fermentation can be a biofuel (typically a fuel additive), whereas the longer-chain lipids and alkanes from cyanobacteria and algae are better suited for biodiesel and aviation fuels. The calculated per annum biofuel yields of algae are of the order of 3000 gallons per acre, or ~3.5 L m–2. Genetic engineering of these organisms might yield something of the order of 15–20 L m–2 on a per annum basis. At present, there is tremendous work with biotechnology companies trying to leverage algal biofuels as drop-in fuels with a CO2-neutral footprint. Commercial strategies are closely guarded but intellectual property claims for using halophilic or thermophilic cultivars containing increased carbon transporters, tailored metabolite profiles and excretion pathways for lipids or alkanes are being promoted. In the coming years, there will be a new era of energy biotechnology and this will revolutionise photobiology, as these systems are the conduits for converting solar energy into chemical fuel.
Engineering Proteins for Artificial Photosynthesis
Interestingly, the concept of engineering photosynthesis on the nano scale is also being pursued. Molecular engineering of specific photosynthetic reactions offers catalytic simplification and also potential improvement in overall solar energy conversion efficiency. A practical limitation with the natural in vivo system is the intrinsic overall 1–2 % efficiency of plant photosynthetic solar conversion.[15,16] There are various issues relating to this; foremost, the photosynthetic organism has evolved to reproduce itself, not to store and secrete reduced carbon compounds. The photochemical reaction centres also physically turn over in a matter of hours, so need continual protein resynthesis and repair cycles, and CO2 fixation by the RuBisCO enzyme has a competing oxygenation reaction. Both these and additional biochemical constraints of physiology limit the overall photochemical efficiency.
The alternative is to use biotechnology for protein engineering to emulate the key reactions of photosynthesis. Protein engineering has been widely recognised in industrial catalysis, medicine and pharmaceuticals, and agriculture, and only more recently has been explored in the topic of developing novel artificial photosynthetic systems.[30–32] Protein engineering has also played a decisive role in unravelling natural processes by making molecular perturbations of reactions or enzymatic catalytic sites. Extensive progress in recombinant biotechnology and computational biology presents this as the mainstream approach today for exploiting the extraordinary properties of proteins.
When it comes to applying protein engineering, there are essentially two main approaches: (1) directed evolution of existing functional proteins, and (2) rational design of natural and novel proteins. Directed evolution, also referred to as ‘in vitro evolution’, involves generating desirable properties in proteins not found in nature. The process of directed evolution conventionally entails generating molecular diversity through random mutagenesis and in vitro recombination followed by high-throughput selection.[33,34] The need for effective selection markers for specific photocatalytic properties makes it challenging to apply this approach to engineering proteins for artificial photosynthesis.[35]
Rational design, however, involves an engineering approach to protein design, often in combination with molecular modelling of the protein structure. The approach leads itself to redesigning existing proteins for a change (or improvement) of function[30,36] or to approach this with a fresh palette and to create de novo proteins with novel protein sequences.[37–40] The present authors’ interest lies in engineering proteins through rational design to create nature-inspired photocatalytic proteins for artificial photosynthesis. The advantage of this over a purely de novo approach is that highly evolved proteins with intrinsic structural robustness can be finessed via molecular engineering to take on new chemistries. There are two features specifically required to assemble a photocatalytic protein: (1) selection and incorporation of pigments/chromophores for light capture and to initiate photochemistry, and (2) the choice and placement of redox-active intermediates for electron transfer to convert primary charge separation into redox activity.
Light Harvesting
The concept of light capture is important for chromophores as there is no perfect single molecule that absorbs all visible light. To do so, it would need to be black. Instead, natural systems utilise different pigments with different site energies and absorption, and in effect create arrays with multiple interacting chromophores that function as excitonically coupled micrometre structures. These antennae deliver photons into reaction centres where photochemical work is ultimately extracted. The natural antenna solutions are diverse, with differing chromophores (chlorophylls a–f; bacteriochlorophylls a–g, carotenoids and xanthophyll) and differing protein scaffolds.[41,42]
For natural photosynthesis, the antennae arrays function as dynamically changing structures that adjust to light intensity and quality, and function specifically to dramatically increase the absorption cross-section of the individual reaction centre. In the mainstream oxygenic photohetertrophs (cyanobacteria, algae and plants), chlorophyll a (Chl) is the principal pigment for light capture and charge separation. However, from a protein engineering perspective, Chl is difficult to utilise as it is hydrophobic, forms aggregated clusters in solution, suffers from instability outside a protein environment and has a tendency to be readily oxidised. Prospects of engineering of alternative chromophores do exist and have been explored as photo-active pigments (Fig. 2).

|
Ruthenium (Ru) pyridines have sufficient potential to oxidise water and have long-lived excited states.[43] The practical application, however, with utilising ruthenium complexes as photosensitisers for artificial photosynthesis is that these compounds are highly valuable in test systems, but the scarcity and high cost of the Ru element make such examples unlikely to be suitable for large-scale implementation.[44] Flavins have also been identified as potential chromophores for light-induced electron transfer in engineered proteins (Fig. 2). These generate a highly oxidising triplet state on irradiation with blue light.[45] Metal-substituted porphyrins can also be used for this purpose. Porphyrins are more stable than chlorophylls and cheaper than ruthenium. Zinc-substituted porphyrins that have one amino acid axial ligand and exhibit pentacoordinate geometry also prove suitable to assemble artificial photosynthetic reaction centres[46,47] (Fig. 2).
Principles of Electron Transfer
Within photosynthetic proteins, the photochemical event leads to the generation of an excited state. The excited state is either transferred via long-range electron transfer (ET) to an acceptor molecule, or is lost as a wasted recombination reaction. In proteins, productive electron transfer is a tunnelling reaction between a reactant and product state. These reactant and product states represent the electron donors and electron acceptor cofactors before and after electron tunnelling. When it comes to designing and emulating electron-transfer pathways in proteins, the rationale is one of distance between and choice of cofactor. Electron-transfer rates were outlined initially by Marcus in the 1950s and 60s.[48] The classical Marcus electron-transfer theory describes the dependence of electron-transfer rates on an electronic coupling term, reorganisation energy (λ) and driving force (ΔG). However, from the Marcus framework, a simplified model for long-range electron transfer in proteins has been proposed by Dutton and Moser.[49–51] The model is based on a rationalisation of a close correlation of electron-transfer rate versus distance. The rate of pure electron-transfer tunnelling reactions in their analysis can be approximated by the following equation
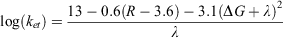
where ket (s–1) is the electron transfer rate, R (Å) is the edge-to-edge distance between redox cofactors (the 3.6 represents van der Waals contacts), and ΔG and λ (eV) are the free energy and reorganisation energy terms respectively. The three parameters, distance (R), driving force (ΔG) and the reorganisation energy (λ), modulate rates of electron transfer ( ). From a review of numerous biological proteins, it was found that almost all productive single-electron transfer reactions lie between R ~ 4 and 14 Å,[40,49] although there appear to be some unique examples of rapid electron transfer over long distances (>30 Å) in proteins.[52]
). From a review of numerous biological proteins, it was found that almost all productive single-electron transfer reactions lie between R ~ 4 and 14 Å,[40,49] although there appear to be some unique examples of rapid electron transfer over long distances (>30 Å) in proteins.[52]
A paradigm in biological systems is that long-range electron transfer is mediated via redox chains[51,53]; i.e. several redox cofactors are needed to propagate electron transfer from the photocatalyst to the ultimate terminal oxidant and reductant. Practical steps in building an electron-transfer pathway are design selection of cofactors based on redox potential to define the overall ΔG. Modulating the redox potentials, and thereby the ΔG, can be achieved by varying the protein surrounds of cofactors by mutagenesis.[54,55] When designing proteins, it is optimal to bury both electron donors and acceptors within the artificial system. Electron acceptors in solution have large reorganisation energies associated with electron transfer and thus may have reduced rates of electron transfer. Burying these cofactors at optimal distances within a protein matrix is ideal for effective electron-transfer rates.
Electron-Transfer Donor and Acceptors
For an artificial photosynthesis reaction, an abundant substrate electron donor needs to be identified. It is possible to use an anode (electrode) if electrical potentials alone are to be harnessed. In the biological photosynthetic reaction centres, two examples exist for electron-transfer donors. One is a mobile cytochrome electron-carrier involved in mediating electrons in a cyclic reaction. The second example is in Photosystem II (PSII), which is uniquely capitalised on the catalytic oxidation of water. The oxidation of water was a significant solution as it is abundant, and the chemistry in PSII is quite unique, involving a cluster of four manganese ions and one calcium ion structurally arranged as an Mn4CaO5 cluster.[56] The Mn ions undergo redox cycling and catalyse the concerted four-electron oxidation of two water molecules. A nearby tyrosine D1-Y161 (YZ) functions as the redox-active electron intermediate between the Mn4CaO5 cluster and the light-activated pigment P680+. The whole reaction is optimised for ~103 turnovers per second, with low driving force, and such efficiency has not yet been achieved with synthetic catalysts.[57,58] Similarly, in other oxidoreductases, electron donors involve metal clusters, redox-active amino acids and haem-cytochromes. Yet for many chemists, in finding an abundant renewable terminal oxidant, it is hard to overlook water.
The prospects for electron acceptors in proteins are similarly numerous. Quinones are a common intermediate in natural oxidoreductase systems and feature prominently in the Type II photosynthetic reaction centres that include PSII. Yet engineering a hydrophobic quinone binding pocket in proteins is challenging as the binding domain has to stabilise a hydrophobic molecule with non-covalent interactions. An alternative approach relies on covalent attachment of quinones on protein scaffolds and de novo proteins via linkage to a free sulfhydro group (–SH) through a sulfur addition reaction.[59] Cysteine residues can be utilised for this purpose. Other variants of this theme use thiol addition reactions and addition/elimination reactions between the cysteine sulfur and thioether quinones.[60] Additional electron acceptors commonly utilised for such studies to bind to synthetic photosensitisers are methyl viologen and cobalt(iii) pentamine.[61]
Artificial Photosynthesis – Modification of Natural Proteins
The biochemical reactions of life were established ~3.5 billion years ago and from this ancient linage, abundant protein structures exist today (structures can be found in the protein data bank at www.rcsb.org). Through the course of evolution, most enzymes have been optimised for improved kinetic or thermodynamic performance. Using such a library of natural proteins, many protein scaffolds are available to explore, and those that can be (1) expressed in bacteria, (2) don’t require post-translational modification, or (3) don’t require chaperone-assisted folding and assembly can be considered as viable targets for protein re-engineering.
Modification of amino acids (removal, replacement or deletion) is achieved with mutagenic primers using the polymerase chain reaction (PCR). Systems are constantly improving, but it is possible to modify them by mutagenesis (up to three amino acids in the same reaction). Increasingly, it is also possible to purchase modified DNA sequences for a whole gene and therefore make substantial changes to the protein amino acid sequence.
Re-engineered natural proteins for photoactivity include using apo-myoglobin to study the in vitro binding of the protopheophorbide a derivatives of Chl[62] and binding of Chl (and derivatives) to the water-soluble Chl protein (WSCP).[63] Ruthenium complexes have also been widely linked to protein scaffolds to initiate photochemical electron-transfer reactions mimicking a photosynthetic reaction centre.[64–66] There are also examples of attached Ru complexes involving iron–sulfur proteins and myoglobin.[67,68] Photoactive porphyrins have been studied in b-type cytochromes when haem is first removed[69] and then replaced with zinc and iron porphyrins.[70,71] In apo-cytochrome b562, binding of zinc-chlorin e6 (ZnCe6) has been studied as ZnCe6 has a long-lived excited state.[72] Flavin derivatives have also been covalently bound to surface cysteine residues of cytochrome c.[73]
Modifications to existing photosynthetic proteins have also been investigated to examine the ‘evolutionary argument’. The purple bacterial reaction centre (BRC) from Rhodobacter sphaeroides is an ancestor of the plant PSII. The reaction centres P870 and P680 operate with different pigments and potential, yet Kalman and coworkers[74,75] re-engineered the BRCs to increase the midpoint potential and, after the addition of tyrosine amino acids, photooxidise them.[76,77] The group also further paralleled PSII evolution by introducing carboxylate acid residues at mirror positions in the BRC and binding of mononuclear Mn to function as an electron donor.[78] This work with the BRC has gone the furthest in reprogramming photosynthetic function on the donor side. On the acceptor side, the first successful quinone binding to a modified non-photosynthetic protein scaffold was shown by Hay et al. in 2004.[46] This was done by introducing a cysteine residue within the hydrophobic core of cyt b562 and covalently linking various quinone variants through a sulfur addition reaction to the quinone ring. The modified cyt b562 was probably the first example of an introduced donor–acceptor pair in a natural protein.
Modified Bacterioferritin as a Photoactive Reaction Centre
An approach initiated by T. Wydrzynski was to explore a non-photoactive protein scaffold as a framework to introduce the redox-active cofactors similar to the PSII reaction centre in a simple in vitro model system. The protein used for this purpose is a haem-containing ferritin found in Escherichia coli called bacterioferritin (BFR).[79] First identified from its optical spectrum as cytochrome b1 in 1934 by Keilin,[80] BFR is a soluble non-toxic iron storage and detoxification protein.[81,82] The expressed protein assembles into a hollow, almost spherical shell-like nanostructure (~8 nm internal diameter) of 24 subunits configured as a dodecameric structure (Fig. 3a). Each subunit in its oligomeric form is a four-helix bundle with a molecular mass of ~18.5 kDa.[83,84] The four helices align antiparallel to each other with a small fifth helix at the C-terminus that aligns almost perpendicularly to the rest of the bundle (Fig. 3b).[85–87] Each of the four-helix-bundle subunits of BFR contains a dinuclear metal binding site. The site binds two FeII atoms with histidine (H54 and H130) and glutamate (E51, E18, E127 and E94) residues as capping ligands. The two atoms are connected by the two bridging carboxylate groups (E51 and E127) (Fig. 4a). Bacterioferritins are uniquely different to other ferritins in possessing an intrinsic haem group that is non-covalently bound at the interface between each of the two four-helix bundle subunits. This symmetrical inter-subunit haem-binding site is highly hydrophobic, and a single methionine residue from each monomer provides a pair of axial ligands to the metal centre of iron-protoporphyrin IX in the assembled BFR dimer (Fig. 4b).[86]
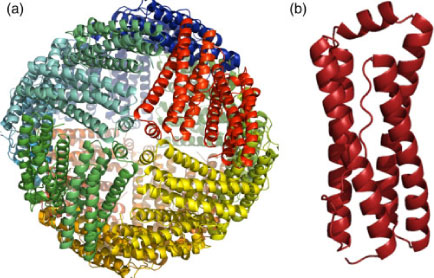
|
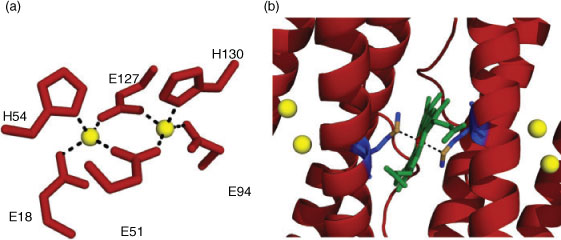
|
These inherent properties of E. coli BFR present it as a good scaffold for reverse-engineering PSII RC reactions. It is also a novel way to explore the complex pathways of light-driven electron transfer in an artificial PSII analogue system. The design features of BFR that make it a suitable starting point for multistep protein engineering work are (i) the dinuclear site provides binding ligands for other class II metals such as Mn; (ii) the native haem in the hydrophobic pocket of the protein can be removed[69] and replaced with a photosensitive porphyrin, in this case a chlorin (Ce6); and (iii) the presence of tyrosine residues proximal to the bound cofactors, which can be utilised as efficient electron-tunnelling intermediates. We have demonstrated through spectroscopy and calorimetry that apo-BFR (haem and Fe removed) can bind Mn in place of Fe and zinc-chlorin e6 (ZnCe6) in place of haem. We have also shown that in the presence of weakly coupled bound MnII,II at the metal site, light-activation of the ZnCe6 induces an electron-transfer reaction oxidising the inherent tyrosine residues.[79,88] There are seven tyrosine residues per BFR monomer and therefore fourteen possible tyrosine residues per homodimer that might be redox-active. Our current focus is on understanding the electron transfer/tunnelling pathways through the redox active tyrosine residue(s) in this artificial system.[88]
Artificial Photosynthesis – ‘De Novo’ Design Approach
The design and synthesis of de novo proteins involve construction of a novel polypeptide sequence. Some groups refer to them as ‘maquettes’, likening them to architectural models or sculptures. Usually, there is no comparison with existing proteins in nature, but from this scaffold, function is added.[37,89] In general, research in de novo protein design and synthesis is directed by the following basic principles[37,90]:
-
Simple design – making for a small, versatile and robust polypeptide sequence that is generated through either chemical synthesis or by recombinant expression;
-
Stable protein folding – the peptide design must contain elements in the heptad repeat (for α-helices) to stabilise a desired fold thermodynamically by incorporating amino acids that favour specific chemical interactions energetically;
-
Cofactor binding sites – inserting amino acids to bind protein cofactors such as metals and porphyrin groups. These may correspond to catalytic sites for enzyme design or may facilitate folding of polypeptides to oligomeric structures;
-
Structural information – it is important to resolve the structure of designed novel proteins as minor changes can alter the original design;
-
Functional characterisation – test of the folded polypeptide for its function. This is often associated with redesigning (adding or deleting amino acids) of the de novo sequence to improve functional efficiency.
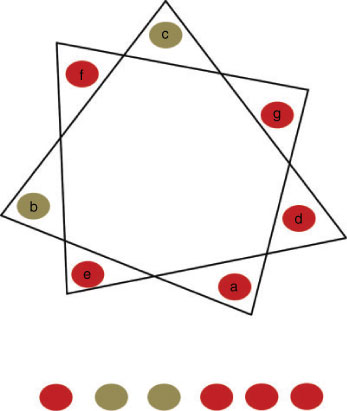
Perhaps the only real limitation with de novo maquettes is that their size limits the available dimensions of the protein scaffold. The initial de novo protein design success was developed by DeGrado and coworkers in the 1980s. The first product was a peptide synthesised as a self-folding amphiphilic four-helix bundle.[91] The methodology used for assembling α-helical bundles is now termed as ‘binary patterning’ and the principle is based on the folding pattern of secondary structures in native four-helix bundles. The α-helix has 3.6 residues per turn, resulting in a two-turn heptad (almost seven residues) repeat (Fig. 5). Alternating peptide sequences of hydrophilic and hydrophobic residues can be designed to form a tetrameric bundle with a hydrophobic core driven by hydrophobic sequestration. In the past two decades, there have been remarkable advances in de novo peptide design to generate correctly folded proteins with increased functional complexity.[33,92,93] These maquettes are usually small, with a size of ~16 kDa.[94] There are several examples of using de novo α-helical bundles as scaffolds for haem binding via bis-histidine ligation[95,96] and the development of multi-haem polypeptides to perform long-range ET between cofactors.[94] A recent study demonstrated binding of a water-soluble bacteriochlorophyll derivative to a de novo designed haem-binding four-helix bundle.[97] There are also other examples of Chl binding to synthetic peptides,[98–100] photo-active pigments such as Ru-pyridines, flavins and light-sensitive porphyrins[47,101–103] (Fig. 2).
|
Metal binding sites engineered in de novo proteins in early designs incorporated zinc and mercury.[104,105] Subsequently, the ‘Duo Ferri’ (DF) series of maquettes was developed to mimic di-iron proteins. The DF maquettes bind two iron atoms and will also bind other metals ions (Zn, Co and Mn) with the stoichiometry of two ions per protein.[106,107] A four-helix bundle was also designed to incorporate an iron–sulfur cluster (Fe4S4) in its hydrophobic core in a recent de novo protein engineering effort.[108] In an alternative approach, βαβ structures computationally designed were shown to bind mononuclear zinc ions.[109] Quinone binding to these novel proteins was shown in one example, where a three-helix bundle was designed with a cysteine to link 2,6-dimethylbenzoquinone (DMBQ).[110] These examples provide hope for incorporating metal clusters and various electron acceptors such as quinones in photocatalytic electron transfer in proteins through de novo engineering approach.
It could potentially be argued that structural and functional aspects of natural proteins are representative of ‘evolutionary baggage’. Enzymes such as RuBisCO or the photosynthetic and respiratory electron-transfer proteins are not perfect, yet are essentially conserved with inbuilt limitations. Thus, revisiting enzymatic function using de novo design is a way of potentially optimising chemical and redox reactions. Using this approach, only the amino acids that are essential for the protein’s function are incorporated and the role of each amino acid is defined by the engineer. It also provides the opportunity for exploring protein folding and function in ways that are not present in nature.[93,111] In the alternative approach of modifying natural proteins, the advantages lie in the remarkable inherent features of selected proteins that may facilitate redesigning with minimal modification.[36] Another highly valuable attribute of manipulating naturally occurring proteins is the ease of incorporating the modified variants into biological systems through recombinant technology, and therefore the possibility of immediate in vivo characterisation.
Looking to the immediate future, the prospect of engineering proteins from natural sources or de novo design to provide artificial photosynthesis is probably not a commercially reality. Small amounts of material are currently made in vitro. However, it may be expected that in vivo production will become achievable in the coming decade owing to developments in the field of synthetic biology. This approach will enable expression of protein-based catalysts, and then the full in vivo assembly of cofactors for solar-driven catalysis. The prospects for assembling chromophores and cofactors into the scaffold could even use non-standard amino acids to expand the ligation – imagine a protein zipper with novel amino acids!
Concluding Remarks
Global Artificial Photosynthesis (GAP) targets to resolve a future global ‘energy problem’ by creating novel technologies inspired from the extremely efficient photosynthetic reactions. These systems would likely be based on whole organisms, either algal-based or bacterial, and use sunlight to generate storable, transportable chemical fuels. The capabilities of GAP would be underpinned by molecular engineering and provide for solar farming (Table 1). It is interesting to return to Thomas Edison who once said, ‘I would put my money on the sun and solar energy. What a source of power! I hope we don’t have to wait until oil and coal run out until we tackle that’.[112] He also stated, ‘Anything that won’t sell, I don’t want to invent. Its sale is proof of utility, and utility is success.’ This puts another emphasis on the GAP product: what is the utility of these technologies to the end consumer? Oil extraction rates alone of liquid fuels are 150000 L s–1 globally and this translates to enormous financial revenue from the origin of extraction through the refining and distribution networks. Can GAP compete and be implemented in the developing world where the uptake of energy is at its greatest? Not now certainly, but in the future, the bio-based solar fuels can be produced by farming techniques. The inroads to this are carbon tax credits, emission regulations and discovery of new science. There is willingness for a ‘green’ planet, but at the end of the day, financially unburdening energy usage for the end user will be essential.

|
Acknowledgements
The authors are grateful to Tom Wydrzynski for much of the pioneering work and to Brendon Conlan for stimulating discussions. Support for this work was provided by the Australian Research Council FT0990972.
References
[1] International Energy Agency, World Energy Outlook Report 2010, http://www.iea.org (accessed February 2012).[2] T. A. Edison (US Patent Office) 1880 (USA).
[3] T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets, D. G. Nocera, Chem. Rev. 2010, 110, 6474.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtl2lsb3I&md5=438de29bdee38432d249bcc4efea7a26CAS |
[4] D. J. MacKay, Sustainable Energy – Without the Hot Air 2009 (UIT: Cambridge, UK).
[5] R. E. Blankenship, D. M. Tiede, J. Barber, G. W. Brudvig, G. Fleming, M. Ghirardi, M. R. Gunner, W. Junge, D. M. Kramer, A. Melis, T. A. Moore, C. C. Moser, D. G. Nocera, A. J. Nozik, D. R. Ort, W. W. Parson, R. C. Prince, R. T. Sayre, Science 2011, 332, 805.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXlslylsLk%3D&md5=7fef5a2be0a1e90cdea051d4e8001fa9CAS |
[6] C. Yusuf, Biotechnol. Adv. 2007, 25, 294.
| Crossref | GoogleScholarGoogle Scholar |
[7] R. Das, P. J. Kiley, M. Segal, J. Norville, A. A. Yu, L. Y. Wang, S. A. Trammell, L. E. Reddick, R. Kumar, F. Stellacci, N. Lebedev, J. Schnur, B. D. Bruce, S. G. Zhang, M. Baldo, Nano Lett. 2004, 4, 1079.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXktValsLc%3D&md5=09459ff10b3f1300af8d5009a52b1941CAS |
[8] C. E. Lubner, R. Grimme, D. A. Bryant, J. H. Golbeck, Biochemistry 2010, 49, 404.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhs1Wqsr3O&md5=ff61e1a2f34a06ee7dc5351979a08871CAS |
[9] H. Krassen, A. Schwarze, ACS Nano 2009, 3, 4055.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsV2htbfM&md5=a09dc8b6d631c227217de0728117194dCAS |
[10] W. Hillier, G. T. Babcock, Plant Physiol. 2001, 125, 33.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXjslymt7o%3D&md5=cec68f5d73586f0b67386f674ebeaf1bCAS |
[11] M. F. Hohmann-Marriott, R. E. Blankenship, Annu. Rev. Plant Biol. 2011, 62, 515.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXnslansLo%3D&md5=44ce45cfdca08957d9a4b41a509faef5CAS |
[12] D. Noy, C. C. Moser, P. L. Dutton, BBA-Bioenergetics 2006, 1757, 90.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XisF2nsb8%3D&md5=bcb25eb8cc687aa145f71b9aa7c45760CAS |
[13] R. E. Blankenship, H. Hartman, Trends Biochem. Sci. 1998, 23, 94.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXisVOms74%3D&md5=fe6fc94aefa1b453e1d38d12b3767ee4CAS |
[14] D. Graham-Rowe, Nature 2011, 474, S6.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXnvVektL0%3D&md5=d5433258406de64a983c284ab698520aCAS |
[15] D. A. Walker, J. Appl. Phycol. 2009, 21, 509.
| Crossref | GoogleScholarGoogle Scholar |
[16] X. G. Zhu, S. P. Long, D. R. Ort, Annu. Rev. Plant Biol. 2010, 61, 235.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXnslSjsL8%3D&md5=b576d4c7e235db3bf0343c697b5a1ddbCAS |
[17] C. U. Ugwu, H. Aoyagi, H. Uchiyama, Bioresour. Technol. 2008, 99, 4021.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXjtl2guro%3D&md5=3a49591e68e8bf56001046212304a267CAS |
[18] M. Janssen, J. Tramper, L. R. Mur, R. H. Wijffels, Biotechnol. Bioeng. 2003, 81, 193.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXht1Wktw%3D%3D&md5=842441869807c19a858b2ff73e842f29CAS |
[19] J. Sheehan, T. Dunahay, J. Benemann, P. Roessler, A Look Back at the U.S. Department of Energy’s Aquatic Species Program — Biodiesel from Algae 1998, p. 296 (National Renewable Energy Laboratory: Golden, CO).
[20] E. Casadevall, D. Dif, C. Largeau, C. Gudin, D. Chaumont, O. Desanti, Biotechnol. Bioeng. 1985, 27, 286.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2MXhsVOms7w%3D&md5=e1602a22e22d2fbafaef3d364b2a1ffeCAS |
[21] G. O. James, C. H. Hocart, W. Hillier, H. Chen, F. Kordbacheh, D. Price, M. A. Djordjevic, Bioresour. Technol. 2011, 102, 3343.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXks1Cktw%3D%3D&md5=0f1d238aa4be1819c87cd1b647ef89cbCAS |
[22] T. Heredia-Arroyo, W. Wei, B. Hu, Appl. Biochem. Biotechnol. 2010, 162, 1978.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXht12qtr3J&md5=191ae0f3cdb13fec26c6dfae812e6acdCAS |
[23] T. G. Tornabene, G. Holzer, S. L. Peterson, Biochem. Biophys. Res. Commun. 1980, 96, 1349.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL3cXmtVWjtrY%3D&md5=794034040457ae119df6e15bbeaf0e67CAS |
[24] T. C. Peeler, M. B. Stephenson, K. J. Einspahr, G. A. Thompson, Plant Physiol. 1989, 89, 970.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL1MXhs12qtLw%3D&md5=97f6934eed0dc4477a5036df7c21abe4CAS |
[25] M. D. Deng, J. R. Coleman, Appl. Environ. Microbiol. 1999, 65, 523.
| 1:STN:280:DC%2BD2critVKgsA%3D%3D&md5=386753f339aaf1f61381d6ac898037ceCAS |
[26] D. R. Nobles, R. M. Brown, Cellulose 2008, 15, 691.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtVartL3L&md5=8d335331289a551fd208c019500cfcfbCAS |
[27] S. Atsumi, W. Higashide, J. C. Liao, Nat. Biotechnol. 2009, 27, 1177.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVWlsbrF&md5=beb46bcc4206a7319c2589bbcc8a0cc7CAS |
[28] P. Lindberg, S. Park, A. Melis, Metab. Eng. 2010, 12, 70.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVajtrnN&md5=bd0ace65ae3d85b82261bfb6741ddf18CAS |
[29] M. L. Ghirardi, M. C. Posewitz, P. C. Maness, A. Dubini, J. P. Yu, M. Seibert, Annu. Rev. Plant Biol. 2007, 58, 71.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXnsVahs78%3D&md5=7e4d2cb22adf5575c5fbdd42066b75d9CAS |
[30] M. Leisola, O. Turunen, Appl. Microbiol. Biotechnol. 2007, 75, 1225.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXmvVKnt7Y%3D&md5=2a28d6925b30e7ca42e534242e19aa3aCAS |
[31] R. L. Koder, P. L. Dutton, Dalton Trans. 2006, 3045.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XlvVOkt74%3D&md5=a3a6b55e4fdc1242387e29ebac218b6aCAS |
[32] J. Barber, Chem. Soc. Rev. 2009, 38, 185.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhsFWjtL3F&md5=8520e2df19ce1a690b6b38d14084c912CAS |
[33] G. Saab-Rincon, B. Valderrama, Antioxid. Redox Signal. 2009, 11, 167.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhsFWqu7bF&md5=aa680d41e9495cba64ab8b74e07f279eCAS |
[34] A. V. Shivange, J. Marienhagen, H. Mundhada, A. Schenk, U. Schwaneberg, Curr. Opin. Chem. Biol. 2009, 13, 19.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXlsVGjs7o%3D&md5=45ee61f55b6256f53ff4b72c279824ecCAS |
[35] S. Lutz, Curr. Opin. Biotechnol. 2010, 21, 734.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsVals7%2FI&md5=9a97fefc488b2e3a6de7b854922bfb21CAS |
[36] T. Wydrzynski, W. Hillier, B. Conlan, Photosynth. Res. 2007, 94, 225.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhtl2ks7fP&md5=d684f6bc946ce782b376efd2052c78ebCAS |
[37] W. F. DeGrado, C. M. Summa, V. Pavone, F. Nastri, A. Lombardi, Annu. Rev. Biochem. 1999, 68, 779.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXlvFajt7o%3D&md5=1337d5bd74867af5df3f0aff7c590d6bCAS |
[38] J. Kaplan, W. F. DeGrado, Proc. Natl. Acad. Sci. USA 2004, 101, 11566.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXntVektb8%3D&md5=fb3477a93480baba74e73e5fb751d941CAS |
[39] B. R. Gibney, Y. Isogai, F. Rabanal, K. S. Reddy, A. M. Grosset, C. C. Moser, P. L. Dutton, Biochemistry 2000, 39, 11041.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXlslSksbY%3D&md5=bc3dc8641722facb635d27e680c54012CAS |
[40] C. C. Page, C. C. Moser, X. X. Chen, P. L. Dutton, Nature 1999, 402, 47.
| 1:CAS:528:DyaK1MXntlChurw%3D&md5=7627b0012fb0836203e4a283639ac0c9CAS |
[41] A. M. Collins, J. Wen, R. E. Blankenship, in Molecular Solar Fuels 2012, pp. 85–106 (Eds T. Wydrzynski, W. Hillier) (The Royal Society of Chemistry: Cambridge, UK).
[42] J. Kargul, J. Barber, in Molecular Solar Fuels 2012, pp. 107–42 (Eds T. Wydrzynski, W. Hillier) (The Royal Society of Chemistry: Cambridge, UK).
[43] M. Borgstrom, N. Shaikh, O. Johansson, M. F. Anderlund, S. Styring, B. Akermark, A. Magnuson, L. Hammarström, J. Am. Chem. Soc. 2005, 127, 17504.
| Crossref | GoogleScholarGoogle Scholar |
[44] J. Murray, in Molecular Solar Fuels 2012, pp. 408–425 (Eds T. Wydrzynski, W. Hillier) (The Royal Society of Chemistry: Cambridge, UK).
[45] A. Losi, W. Gartner, Photochem. Photobiol. 2011, 87, 491.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXmvFersLg%3D&md5=64c32fa0c0c7ef3e0ebad3e5880afd10CAS |
[46] S. Hay, B. B. Wallace, T. A. Smith, K. P. Ghiggino, T. Wydrzynski, Proc. Natl. Acad. Sci. USA 2004, 101, 17675.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXjtVShtA%3D%3D&md5=52ef3a5e6f6a3fb8b8248474b3dc670dCAS |
[47] A. R. Razeghifard, T. Wydrzynski, Biochemistry 2003, 42, 1024.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXhs1ansA%3D%3D&md5=e8c88bc3b1b5e440277264e8062b4cc8CAS |
[48] R. A. Marcus, N. Sutin, BBA-Bioenergetics 1985, 811, 265.
| 1:CAS:528:DyaL2MXltFygs78%3D&md5=f61543f08eb2de7738179f6072924728CAS |
[49] C. C. Moser, J. M. Keske, K. Warncke, R. S. Farid, P. L. Dutton, Nature 1992, 355, 796.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK38XhsVKhu78%3D&md5=a3439db0169b941752f2819bc690724aCAS |
[50] M. M. Sheehan, L. A. Solomon, J. L. R. Anderson, P. L. Dutton, C. C. Moser, BBA-Bioenergetics 1797, 2010, 62.
[51] C. C. Page, C. C. Moser, P. L. Dutton, Curr. Opin. Chem. Biol. 2003, 7, 551.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXot12hs7w%3D&md5=0a548fa2f128c99ecdbec01ad3c39ef5CAS |
[52] H. B. Gray, J. R. Winkler, Proc. Natl. Acad. Sci. USA 2005, 102, 3534.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXisVOgsrg%3D&md5=eabeb3384d942c4595eb44c1a47a3667CAS |
[53] C. C. Moser, J. L. R. Anderson, P. L. Dutton, BBA-Bioenergetics 2010, 1797, 1573.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXptVWhtr8%3D&md5=b77b9f77a4ca37b3fad49cb6d4a0b1d8CAS |
[54] A. L. M. Haffa, S. Lin, E. Katilius, J. C. Williams, A. K. W. Taguchi, J. P. Allen, N. W. Woodbury, J. Phys. Chem. B 2002, 106, 7376.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XkvVGms70%3D&md5=657e616010b2385d785803b9b1faa108CAS |
[55] J. C. Williams, R. G. Alden, H. A. Murchison, J. M. Peloquin, N. W. Woodbury, J. P. Allen, Biochemistry 1992, 31, 11029.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK38XmsVyksLY%3D&md5=646bfba595281946be88e9c4dcf87226CAS |
[56] K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber, S. Iwata, Science 2004, 303, 1831.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXitFehtbs%3D&md5=031fd950c39696f610bd2aa132128886CAS |
[57] W. Hillier, J. Messinger, in Photosystem II: The Light-Driven Water: Plastoquinone Oxidoreductase 2005, pp. 567–608 (Eds T. Wydrzynski, K. Satoh) (Springer: Dordrecht, The Netherlands).
[58] W. Lubitz, E. J. Reijerse, J. Messinger, Energ. Environ. Sci. 2008, 1, 15.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtV2htrvE&md5=b3cb1a4cb93656028c8b994f205f19a8CAS |
[59] E. Redfearn, Biochemistry of Quinones 1965, pp. 149–81 (Academic Press: New York, NY).
[60] W. W. Li, J. Heinze, W. Haehnel, J. Am. Chem. Soc. 2005, 127, 6140.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXjtFSjtb4%3D&md5=944caa1d2fccea334100248b53e1b909CAS |
[61] M. Sykora, K. A. Maxwell, J. M. DeSimone, T. J. Meyer, Proc. Natl. Acad. Sci. USA 2000, 97, 7687.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXkvFCntrs%3D&md5=885ec84db562af3918feac229ff0e402CAS |
[62] S. Proll, B. Wilhelm, B. Robert, H. Scheer, BBA-Bioenergetics 2006, 1757, 750.
| Crossref | GoogleScholarGoogle Scholar |
[63] K. Schmidt, C. Fufezan, A. Krieger-Liszkay, H. Satoh, H. Paulsen, Biochemistry 2003, 42, 7427.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXjvFOit7Y%3D&md5=b384171cade84dcf9d84a3a89b6796c9CAS |
[64] D. G. Nocera, J. R. Winkler, K. M. Yocom, E. Bordignon, H. B. Gray, J. Am. Chem. Soc. 1984, 106, 5145.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2cXltVGqt7w%3D&md5=e9e291fd486a6abe7a8011367baeb9e1CAS |
[65] J. R. Winkler, D. G. Nocera, K. M. Yocom, E. Bordignon, H. B. Gray, J. Am. Chem. Soc. 1982, 104, 5798.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL38XlsFygsb0%3D&md5=c9b222416d58f987c6760954a40f87feCAS |
[66] K. M. Yocom, J. B. Shelton, J. R. Shelton, W. A. Schroeder, G. Worosila, S. S. Isied, E. Bordignon, H. B. Gray, Proc. Natl. Acad. Sci. USA 1982, 79, 7052.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL3sXhtlaisg%3D%3D&md5=3cb4caa1fdf1a32a1970ccd935c3a4f5CAS |
[67] E. Babini, I. Bertini, M. Borsari, F. Capozzi, C. Luchinat, X. Y. Zhang, G. L. C. Moura, I. V. Kurnikov, D. N. Beratan, A. Ponce, A. J. Di Bilio, J. R. Winkler, H. B. Gray, J. Am. Chem. Soc. 2000, 122, 4532.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXis1ahur0%3D&md5=698f734f0955aa3c7282618cd88d24b8CAS |
[68] M. J. Bjerrum, D. R. Casimiro, I. J. Chang, A. J. Di Bilio, H. B. Gray, M. G. Hill, R. Langen, G. A. Mines, L. K. Skov, J. R. Winkler, D. S. Wuttke, J. Bioenerg. Biomembr. 1995, 27, 295.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXosVGns7k%3D&md5=83a4db578a27fdd62b94ce0e92e1bb2eCAS |
[69] F. W. J. Teale, Biochim. Biophys. Acta 1959, 35, 543.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaF3cXhslChsw%3D%3D&md5=b2bc14eeff25d94e7d2b1e02f8a253c4CAS |
[70] J. L. McGourty, S. E. Petersonkennedy, W. Y. Ruo, B. M. Hoffman, Biochemistry 1987, 26, 8302.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL1cXhsl2lsA%3D%3D&md5=50997de2e63b869dce7163b1b19da6abCAS |
[71] K. P. Simolo, G. L. McLendon, M. R. Mauk, A. G. Mauk, J. Am. Chem. Soc. 1984, 106, 5012.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2cXkvVGrsrs%3D&md5=0b9d6d1fda236c9a0c0018579965a60fCAS |
[72] S. Hay, T. Wydrzynski, Biochemistry 2005, 44, 431.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXhtVejsbfK&md5=557a9904a7a54dfc92075abc70dc696fCAS |
[73] M. B. Twitchett, J. C. Ferrer, P. Siddarth, A. G. Mauk, J. Am. Chem. Soc. 1997, 119, 435.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXksVCjsQ%3D%3D&md5=c1c2630d0787c7413b19562c1dab7d9fCAS |
[74] L. Kalman, R. LoBrutto, J. P. Allen, J. C. Williams, Biochemistry 2003, 42, 11016.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXmslCnurc%3D&md5=695a3b2b3b2d5dc95cc3aaa2ff2eb7deCAS |
[75] L. Kalman, J. C. Williams, J. P. Allen, Photosynth. Res. 2008, 98, 643.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhsVOgt7fL&md5=e1502cae2956b69ba8048cf55ade62f9CAS |
[76] L. Kalman, J. Williams, J. Allen, in Photosystem II: The Light-Driven Water: Plastoquinone Oxidoreductase 2005, pp. 715–727 (Eds T. Wydrzynski, K. Satoh) (Springer: Dordrecht, The Netherlands).
[77] L. Kalman, R. LoBrutto, J. P. Allen, J. C. Williams, Nature 1999, 402, 696.
| Crossref | GoogleScholarGoogle Scholar |
[78] M. Thielges, G. Uyeda, A. Camara-Artigas, L. Kalman, J. C. Williams, J. P. Allen, Biochemistry 2005, 44, 7389.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXjsFCmtb4%3D&md5=3aacf38a7cd3c203b235aadf52727769CAS |
[79] B. Conlan, N. Cox, J.-H. Su, W. Hillier, J. Messinger, W. Lubitz, P. L. Dutton, T. Wydrzynski, BBA-Bioenergetics 2009, 1787, 1112.
| 1:CAS:528:DC%2BD1MXnsFOktb8%3D&md5=d9ee746d3256963389f7879e3c579d0dCAS |
[80] D. Keilin, Nature 1934, 133, 290.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaA2cXisF2htA%3D%3D&md5=aea46498c0d2ba50ffaa30a875134adcCAS |
[81] J. Yariv, A. J. Kalb, R. Sperling, E. R. Bauminger, S. G. Cohen, S. Ofer, Biochem. J. 1981, 197, 171.
| 1:CAS:528:DyaL3MXls1CisL8%3D&md5=2b878a16fd4bf1550c7b0a0215997240CAS |
[82] J. M. A. Smith, G. C. Ford, P. M. Harrison, J. Yariv, A. J. Kalb, J. Mol. Biol. 1989, 205, 465.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL1MXhtlSkurs%3D&md5=27f517f3158bd69b10408be65b83d5edCAS |
[83] S. C. Andrews, J. M. A. Smith, J. R. Guest, P. M. Harrison, Biochem. Biophys. Res. Commun. 1989, 158, 489.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL1MXlt12iu7Y%3D&md5=7af54b12a18c0ad03240adfe4333aac6CAS |
[84] G. C. Ford, P. M. Harrison, D. W. Rice, J. M. A. Smith, A. Treffry, J. L. White, J. Yariv, Philos. T. Roy. Soc. B 1984, 304, 551.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2cXhslSjsL4%3D&md5=14343874c4afa0c7abaa067d50b38f14CAS |
[85] R. R. Crichton, J. P. Declercq, BBA-Gen. Subjects 2010, 1800, 706.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXnvVyhtLc%3D&md5=2caf24f6871f269397b93d984bb50264CAS |
[86] F. Frolow, A. J. Kalb, J. Yariv, Nat. Struct. Biol. 1994, 1, 453.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXhvF2gs7c%3D&md5=61ede2df3f0883cbfd6d85f719cc175aCAS |
[87] A. Dautant, J. B. Meyer, J. Yariv, G. Precigoux, R. M. Sweet, A. J. Kalb, F. Frolow, Acta Crystallogr. D 1998, 54, 16.
| Crossref | GoogleScholarGoogle Scholar | 1:STN:280:DyaK1M%2FkvFeitg%3D%3D&md5=0ce7110a7a1a4c6c911697a9c0000507CAS |
[88] K. Hingorani, B. Conlan, W. Hillier, T. Wydrzynski, Aust. J. Chem. 2009, 62, 1351.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXht1Kis7%2FI&md5=8c44ce4950da979b0f7c6e2d84744158CAS |
[89] Y. Lu, N. Yeung, N. Sieracki, N. M. Marshall, Nature 2009, 460, 855.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXpvFCkt7g%3D&md5=e26858012ac5926b22e9436f17a9f724CAS |
[90] R. L. Koder, J. L. R. Anderson, L. A. Solomon, K. S. Reddy, C. C. Moser, P. L. Dutton, Nature 2009, 458, 305.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXjtlynsbw%3D&md5=1b273fabe580e9879e00da4c3d86fa4eCAS |
[91] S. P. Ho, W. F. Degrado, J. Am. Chem. Soc. 1987, 109, 6751.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2sXlvFyqs7g%3D&md5=6421128a9930ca083aaa51d67dbee713CAS |
[92] B. R. Gibney, P. L. Dutton, Adv. Inorg. Chem. Rad. 2000, 51, 409.
| Crossref | GoogleScholarGoogle Scholar |
[93] B. A. Smith, M. H. Hecht, Curr. Opin. Chem. Biol. 2011, 15, 421.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXntV2lu7w%3D&md5=617d82eb04b0d49e9eef2556a547123cCAS |
[94] D. E. Robertson, R. S. Farid, C. C. Moser, J. L. Urbauer, S. E. Mulholland, R. Pidikiti, J. D. Lear, A. J. Wand, W. F. DeGrado, P. L. Dutton, Nature 1994, 368, 425.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXmt1Sh&md5=c85c878f56bf5d5ef299c319931b2b10CAS |
[95] C. T. Choma, J. D. Lear, M. J. Nelson, P. L. Dutton, D. E. Robertson, W. F. Degrado, J. Am. Chem. Soc. 1994, 116, 856.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2cXisVGktb8%3D&md5=40457674d66974593fc2b60618cbeb5cCAS |
[96] C. J. Reedy, B. R. Gibney, Chem. Rev. 2004, 104, 617.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXmtFalsg%3D%3D&md5=7a51c2cfd9b900424e647f36ea8a774fCAS |
[97] I. Cohen-Ofri, M. van Gastel, J. Grzyb, A. Brandis, I. Pinkas, W. Lubitz, D. Noy, J. Am. Chem. Soc. 2011, 133, 9526.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXmslGmsLc%3D&md5=157bfe9a52b2aba442f1367ff6beab8fCAS |
[98] D. Noy, P. L. Dutton, Biochemistry 2006, 45, 2103.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XmvVWkuw%3D%3D&md5=071777533fe37335ee290da956cced7eCAS |
[99] H. K. Rau, H. Snigula, A. Struck, B. Robert, H. Scheer, W. Haehnel, Eur. J. Biochem. 2001, 268, 3284.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXktlSgsbk%3D&md5=8f7509ebbf303a9452e5cf18d75b3310CAS |
[100] L. L. Eggink, J. K. Hoober, J. Biol. Chem. 2000, 275, 9087.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXitlCrsbo%3D&md5=9d45782205ca9ae9a29399e66095e976CAS |
[101] N. De Jonge, H. K. Rau, W. Haehnel, Z. Phys. Chem. 1999, 213, 175.
| 1:CAS:528:DC%2BD3cXhvFehtrY%3D&md5=fc1bc8e12ab8e64fd1e88779566bf330CAS |
[102] R. E. Sharp, C. C. Moser, F. Rabanal, P. L. Dutton, Proc. Natl. Acad. Sci. USA 1998, 95, 10465.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXlvFWgtrk%3D&md5=be00818e0d8de804431a04b916080842CAS |
[103] A. Mennenga, W. Gartner, W. Lubitz, H. Gorner, Phys. Chem. Chem. Phys. 2006, 8, 5444.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28Xht1antrfM&md5=df8cd00390bcef46faf2da4eb382f7fbCAS |
[104] L. Regan, N. D. Clarke, Biochemistry 1990, 29, 10878.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3cXmt1Klt7s%3D&md5=e2e9db6afa2ba8c9da9df9a75978ba6cCAS |
[105] G. R. Dieckmann, D. K. McRorie, D. L. Tierney, L. M. Utschig, C. P. Singer, T. V. Ohalloran, J. E. Penner-Hahn, W. F. DeGrado, V. L. Pecoraro, J. Am. Chem. Soc. 1997, 119, 6195.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXktlels70%3D&md5=fea9d38b63ea842f3352ac17c79be049CAS |
[106] R. Torres Martin de Rosales, M. Faiella, E. Farquhar, L. Que, C. Andreozzi, V. Pavone, O. Maglio, F. Nastri, A. Lombardi, J. Biol. Inorg. Chem. 2010, 15, 717.
| Crossref | GoogleScholarGoogle Scholar |
[107] A. Lombardi, C. M. Summa, S. Geremia, L. Randaccio, V. Pavone, W. F. DeGrado, Proc. Natl. Acad. Sci. USA 2000, 97, 6298.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXktFajsr8%3D&md5=8e140382f02d5f573ebdfaf175ce9339CAS |
[108] J. Grzyb, F. Xu, L. Weiner, E. J. Reijerse, W. Lubitz, V. Nanda, D. Noy, BBA-Bioenergetics 2010, 1797, 406.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtlegt7c%3D&md5=c38109d069e9fd2dc0d3a1370ce5c841CAS |
[109] C. Zhu, C. Zhang, H. Liang, L. Lai, Protein & Cell. 2011, 2, 1006.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XmsVSrsg%3D%3D&md5=c484d0224bdd9dd9eb99bd8b851fd9b9CAS |
[110] S. Hay, K. Westerlund, C. Tommos, J. Phys. Chem. B 2007, 111, 3488.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXis1ansr0%3D&md5=d0ec93c61fd522cbb3e3851e6648d80cCAS |
[111] P. L. Dutton, C. C. Moser, Faraday Discuss. 2011, 148, 443.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsFKju7rF&md5=efe94f4afdf90126838bd8a1853c402cCAS |
[112] J. Newton, Uncommon Friends: Life with Thomas Edison, Henry Ford, Harvey Firestone, Alexis Carrel and Charles Lindbergh 2001 (Mariner Books: San Diego, CA)
[113] C. Branden, J. Tooze, Introduction to Protein Structure 1998 (Garland Publishing, Inc.: New York).
† TW is tera (1012) watt (from Joule per second) and in 2010, the global consumption of 16.36 TW of energy was equivalent to 516 EJ or 1.3×104 Mtoe (megaton of oil equivalent).