

The Chemical Problem of Energy Change: Multi-Electron Processes
Joseph L. Hughes A B and Elmars Krausz AA Research School of Chemistry, Australian National University, Canberra, ACT 0200, Australia.
B Corresponding author. Email: hughes@rsc.anu.edu.au

Joseph Hughes completed his PhD in the group of Professor Elmars Krausz at the Research School of Chemistry, Australian National University, in 2006. He has held postdoctoral positions in France at CEA Saclay and École Polytechnique. His recent research interests lie in light-induced processes and mechanisms of solar energy conversion. |

Elmars Krausz, a graduate of the University of Sydney, is currently Group Leader at the Research School of Chemistry Australian National University. He has worked in key areas of chemical spectroscopy and photophysics in the condensed phase, most recently concentrating on fundamental processes in natural and artificial photosynthesis. |
Australian Journal of Chemistry 65(6) 591-596 https://doi.org/10.1071/CH12105
Submitted: 17 February 2012 Accepted: 5 March 2012 Published: 10 May 2012
Abstract
This special issue is focussed on arguably the most important fundamental question in contemporary chemical research: how to efficiently and economically convert abundant and thermodynamically stable molecules, such as H2O, CO2, and N2 into useable fuel and food sources. The 3 billion year evolutionary experiment of nature has provided a blueprint for the answer: multi-electron catalysis. However, unlike one-electron transfer, we have no refined theories for multi-electron processes. This is despite its centrality to much of chemistry, particularly in catalysis and biology. In this article we highlight recent research developments relevant to this theme with emphasis on the key physical concepts and premises: (i) multi-electron processes as stepwise single-electron transfer events; (ii) proton-coupled electron transfer; (iii) stimulated, concerted, and co-operative phenomena; (iv) feedback mechanisms that may enhance electron transfer rates by minimizing activation barriers; and (v) non-linearity and far-from-equilibrium considerations. The aim of our discussion is to provide inspiration for new directions in chemical research, in the context of an urgent contemporary issue.
Urgency: The Chemical Problem of Energy Change
The urgent development and deployment of sustainable, whole-of-life-cycle carbon-neutral energy sources are now well recognized. A rigorous discussion of this subject is complex, and ongoing, attested by this special Aust. J. Chem. issue, but arguably the most significant point is that we require cheap sustainable fuel sources derivable from abundant materials. World energy usage currently divides ~80 : 20, fuel/electricity,[1] and critically we must acknowledge that most electricity is generated by burning fuel. Whilst cheap electricity may correspondingly allow low-cost fuel production, the economic and socio-political reality for the developing world, and those yet to enter it, dictates that inexpensive small-scale portable solar-to-fuel energy conversion devices are a necessity. Nocera has expanded upon this theme in some detail.[2,3] Effective multi-electron processes are well recognized as crucial to achieving this goal.[2,4–6] In this highlight, we argue that the convergence of these urgent contemporary issues should provide a profound inspiration to move beyond traditional thinking in chemistry and promote fertile new directions in chemical research. As this report is not intended as an exhaustive review, our discussion and examples are naturally limited and biased toward ideas we feel are particularly noteworthy for any dialogue they may inspire in generating new research directions.
We Understand Single-Electron Transfer Processes
Fundamentally, our understanding of single electron transfer processes arises from an empirical concept that links the dependency of rate constants, with thermodynamic and molecular dynamic parameters, i.e. Gibbs (ΔG°) and reorganizational (λ) energies. Marcus Theory was initially formulated to deal with weak donor–acceptor interactions and quasi-equilibrium situations (outer-sphere electron transfer) and has successfully been applied to many electron transfer problems in chemistry and biology. There continues to be sophisticated extensions and generalizations of the theory to deal with stronger resonance interactions including strong-overlap (inner-sphere) electron transfer,[7–9] and vibronic coupling[9,10] where electron and nuclear motions cannot be separated; recently this theme has incorporated hydrogen atom, proton, and hydride transfer.[11]
However, even with its great success, some electron transfer problems have struggled to reach a consensus through advancements of the Marcus Theory framework. Our favourite example is the primary photo-energetics of charge separation Photosystem II where difficulty arises in physically explaining the ultrafast kinetics and energy-trapping events,[12–14] not to mention the catalytic four-electron water oxidation chemistry.[15,16] This example also highlights our theme, the chemical problem of energy change.
We Don’t Understand Multiple-Electron Transfer Processes
Here our use of the term ‘multiple-electron transfer’ refers to a situation where discrete, stepwise single electron transfer events may not provide the most suitable and/or useful description of the chemistry. This might be due, for example, to simultaneous transfer of two (or more) electrons, or due to the strong coupling of sequential electron transfer events. Two-electron mixed valency complexes[4,17,18] may be an example of the former. Physically, strong coupling refers to situations where particle motions are not separable, such as vibronic coupling,[9,10] where electronic motion cannot be thought of as independent from nuclear motion. This highlight serves to briefly expand upon this theme in the context of multiple electron transfer for the purpose of inspiring further discussion and initiating new research directions.
There are no refined theories for multiple electron transfer processes.[4,6,19] Instead, adaptations of Marcus Theory and its extensions are often applied.[19] However, in all cases each single electron transfer step can be inverted (reversible) and the role of the medium is typically passive (e.g. non-interacting harmonic oscillators), acting as a thermal bath providing a Boltzmann distribution of fluctuations necessary to prepare the donor–acceptor states for isoenergetic electron transfer. This allows the separation of donor–acceptor and medium variables, resulting in a set of linear equations describing the system.
This also raises the question of the applicability of thermodynamics and equilibrium statistics when electron transfer can proceed as fast as 20 fs.[20] Furthermore, strong and specific molecular interactions occur during multiple electron transfer events relevant to catalysis, most obviously to substrate binding and bond breaking events. These situations are at odds with the original Marcus Theory premises of near-equilibrium statistics and weak donor–acceptor interaction. These issues are appreciated by treatments that deal with stronger resonance interaction, such as H+/H/H– transfer mentioned above,[11] and for example non-statistical dynamical effects incorporating quantum mechanical tunnelling and non-equilibrium solvation.[21,22]
Within ‘classical’ linear electron transfer theories, the efficient net transfer of multiple electrons can only proceed via discrete stepwise mechanisms. The implications are that for catalysis close to the overall thermodynamic potential of the reaction, classical (single-) electron transfer theories require comparable or more favourable conditions for every intermediate species, including compatible reorganization energies (Scheme 1). Tributsch raised and expanded upon these points 14 years ago.[23]
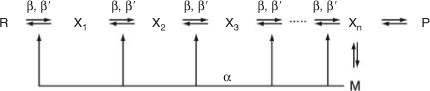
|
Today, multiple electron transfer events typically continue to be examined solely within such linear, stepwise frameworks, even those suggested to be ‘simultaneous’.[19] This mindset has great consequence for the rational design of multi-electron catalysts and photo-catalysts, where success, gauged by high efficiency and turnover number, is often plagued by recombination reactions and undesired side-reactions due to reactive intermediates. These events are inescapable within such model frameworks; the result is a necessary delicate balance of kinetics and thermodynamic molecular architecture, a criterion not desirable for a robust, high turnover number catalyst.
What Might be Important for Multiple-Electron Transfer Processes?
What phenomena beyond such ‘traditional’ thinking might be important for new approaches to multi-electron processes? As an example, we note that Tributsch has developed a phenomenological model[23–28] of multiple electron transfer that has several key physical distinctions to Marcus Theory and its extensions. We highlight some features of this model here.
(i) Feedback: Feedback mechanisms originating from autocatalytic events near the end of an electron transfer chain can improve the rate of electron transfer nearer to the beginning (Scheme 2). In systems with kinetic irreversibility (forward rate larger than back reaction), as the feedback parameter is increased, the rate of electron transfer increases exponentially.[24,27] Feedback results in the system being driven far from equilibrium.
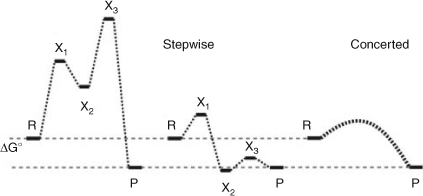
|
(ii) Non-linearity: Positive and negative feedback loops mathematically lead to coupled equations, which could lead to either linear or non-linear solutions. For cooperative phenomena (e.g. synchronous electron transfer), autocatalytic feedback must be initiated via a kinetically non-linear step.[24,25,27] This is in contrast to a kinetically linear feedback relation (Scheme 2), which can only result in accelerated individual electron transfer rates, i.e. stimulated electron transfer.[24,25,27]
(iii) Cooperativity: Electrons are not transferred independently, but rather in a synchronized manner. Such a mechanism can be realized by applying the slaving principle,[29] where at a bifurcation point the change of sign of one eigenvalue of the kinetic problem leads to a reduction from all the degrees of freedom to one degree of freedom (the reaction coordinate). This slowest mode then slaves all the other modes, leading to a non-linear system where all the kinetic events occur in synchrony (simultaneously). There are no formally discrete steps, but rather one overall non-linear kinetic equation.
(iv) No formal intermediates: A result of cooperative electron transfer is a breakdown of the concept of activation energies.[26,27] Intermediates cannot be assigned specific individual activation energies; instead they are part of one cooperatively acting complex. Multi-electron transfer near the overall thermodynamic potential for the reaction becomes possible via non-linear autocatalytic feedback.
(v) Export of entropy: Reduction in the degrees of freedom for a catalytic event equate to the export of entropy from the active site, at the expense of net overall entropy increase. Chemically, this means the creation of a highly ordered transition complex, which is not expected to occur easily close to equilibrium, but is possible far from equilibrium within the scope of irreversible thermodynamics.
The obvious question to ask is whether there exist clear examples where such phenomena are necessarily invoked to model multiple-electron transfer events; they are not required for long-range (outer sphere) single electron transfer where Marcus Theory has generally proven to be a sufficient description. So as to argue in the affirmative, we first introduce a brief but pertinent discussion of proton-coupled electron transfer.
Proton-Coupled Electron Transfer
With a history over a century old, there have been substantial advancements in the field of proton-coupled electron transfer (PCET) in very recent years.[30] Theories are emerging[11] where many of the phenomena discussed above are recognized as essential in describing the relevant molecular mechanisms. We briefly discuss the correspondence between some concepts discussed above for concerted multiple electron transfer and our formative molecular understanding of PCET. Indeed, it is now apparent that many multi-electron redox transformations are dependent on PCET, particularly in biology.[30]
PCET can be treated as subsequent, discrete electron and proton transfer events, or as a genuine concerted process where both the electron and proton are transferred simultaneously. Concerted PCET has now been described (or suggested) in many systems[30] including, among others, photosynthetic reaction centres,[31] intervalence charge transfer complexes,[32] electrochemical systems[33] and via photo-induced PCET in Ru-polypyridyl–Tyrosine(Tryptophan) complexes.[34]
Mayer[35] has outlined the energetic advantage of a concerted PCET event over stepwise electron transfer and proton transfer, and stressed the relative importance of various thermochemical parameters for understanding such reactions. Chemically, this becomes critical when large changes in redox potential occur upon protonation/deprotonation and large changes in pKa occur upon oxidation/reduction. In the terminology used here, this process would be considered a feedback mechanism. Furthermore, we would suggest that if electron and proton motions occur simultaneously, then these motions must be coupled.
A defining feature of concerted PCET mechanisms is where no stable intermediate arises from either a single electron or single proton transfer event.[11] This is similar to a consequence of cooperativity resulting from a feedback kinetic non-linearity that has driven the system far from equilibrium; i.e. where intermediates cannot be ascribed specific activation energies.[23,24,26,27]
In the context of our current highlight, we note that PCET is still commonly (only?) treated via extensions of Marcus theory,[11] i.e within a linear quasi-equilibrium framework, which cannot fundamentally allow a genuine multi-electron process. The coupling of proton and electron motions allows the concerted transfer of both; to our knowledge formalisms based on Marcus Theory do not (yet) allow for the coupled motion of two electrons.
How Should We Experimentally Verify New Phenomena in Multi-Electron Transfer?
Is it necessary to move beyond discrete, uncoupled, stepwise electron transfer events in describing and predicting the behaviour of systems that undergo multiple changes in redox state? In other words, are there examples of the phenomena we have discussed where ‘classical’ linear electron transfer theories built on quasi-equilibrium statistics might not be a sufficient description? To stimulate discussion of this question, we mention here some examples, old and new, of the phenomena outlined above that may be pertinent to mutli-electron transfer processes.
Cooperativity
Cooperative systems in chemistry are known, where the textbook examples are the Belousov–Zhabotinsky chemical reactions that oscillate due to autocatalytic feedback and remain far from equilibrium for significant time periods. Oscillatory behaviour in electrochemistry associated with corrosion and passivation is also well known.[36,37] Far more extensively, however, cooperativity is well recognized in the energetics and mechanisms of enzyme function,[38] where microscopic descriptions of the roles that such phenomena play are far from complete.[39]
For a more recent example from a very different perspective, we note that collective effects in interfacial charge transfer have recently been observed in a hybrid organic–inorganic system by measuring fluctuations of transistor conductivity.[40] Operation at temperatures in the vicinity of a structural order–disorder transition of molecular bridges was suggested to explain electron transfer cooperativity; the initial electron transfer event triggers a structural change that accelerates the rate of further electron transfer events. Significantly, a non-Gaussian distribution of current fluctuations was observed at 300 K, while at 80 K there was a Gaussian distribution.
Order Increase (Entropy Export)
A striking feature of the phenomenological model for multi-electron transfer is the creation of a highly ordered activation complex and the export of entropy during the stimulated or cooperative catalytic event far from equilibrium. In other words, energy is drawn from the medium for the creation of ‘electronic-vibrational order’ at the active site, where improved electron transfer rates result from a better management of entropy, i.e. via the slaving principle, all the degrees of freedom are reduced to one degree of freedom. This is a substantial contrast to Transition State Theory, for example, where the degrees of freedom for transition state are reduced by one compared with the reactants/product. Clearly, experimental approaches to measure local, transient entropy changes would provide insight on the applicability of such concepts.
Calorimetric techniques have been used to measure the enthalpy changes during the photochemical cycle of bacteriorhodopsin, where a significant transient entropy decrease of at least 125 cal mol–1 K–1 was found[41] and suggested to indicate a major increase in molecular order. This has been discussed[42] in terms of self-organization, autocatalytic processes, and entropy export relevant to the themes of cooperative multi-electron transfer presented in our highlight.
Time-resolved photoacoustic measurements can provide information on molecular volume changes including conformational change or electrostriction, enthalpy changes and entropy changes in photochemical reactions.[43–45] Similar data can be obtained via the method of photothermal beam deflection.[46] Enthalpy changes of Photosystem II water oxidation reactions[44,46] and charge separation in synthetic carotene–porphyrin–acceptor triads[45] have been directly measured by such techniques, where significant entropic contributions are suggested to contribute to the reactions. As noted by Hou et al.[44] the Gibbs free energy term in applications of Marcus Theory typically do not account for such entropic components.
Precise structural arrangement of precursor complexes is a feature of concerted PCET processes, compared with outer-sphere electron transfer of structure-less spherical reactants. For example, this theme was recently suggested by Savéant and coworkers in a comparative investigation[33] of electrochemical v. homogeneous kinetics for phenol oxidation with water and hydrogen phosphate. Structural pre-arrangement is also a central tenet of enzyme mechanisms[39] where active sites increase the probability of orientation-specific molecular collisions required for substrate reaction; that is, they increase electronic-vibrational order.
Feedback Non-Linearity
Feedback-associated kinetic non-linearity of the electron transfer reaction is another clear outcome of co-operative multi-electron transfer. However, there are relatively few well-defined electron transfer reactions with higher order kinetics. Fukuzumi and coworkers[47,48] provided some of the first observations of ‘self-promoted’ electron transfer from metal complexes to organic substrates that displays second and third order kinetic behaviour. Such reactions have subsequently been termed metal-ion-coupled (or -ion-mediated) electron transfer.[49,50] The electron transfer rates from the donor are accelerated due to complexation of a redox-inactive metal with the radical anion of the substrate, significantly reducing the redox potential of the latter.
This latter phenomenon has also recently been shown[51] for the metal-ion-coupled reduction of high-valent metal-oxo species, where the electron transfer rate increased by a factor of 108 in the presence of Sc3+. Such metal-ion-coupled electron transfer reactions have very recently been studied[52] in the context of stepwise v. concerted mechanisms, analogous to PCET.
While these examples are only one-electron transfer reactions, we note that several key features of the phenomenological models for stimulated[25,26] and co-operative[24,26,27] electron transfer are observed. We add that to our knowledge, such ion-mediated reactions have not yet been studied for multi-electron transfer systems, and the significance of such new research for the themes in this highlight present a possible new avenue for chemical research. In this context, and as highlighted by Fukuzumi and coworkers,[51] we note that although Ca2+ is known[53,54] to be crucial for catalytic function of the water oxidation complex (Mn4Ca) in Photosystem II, the precise functional role of Ca2+ is not yet clarified.[15,16]
A Two-Electron Photo-Catalyst: Work in Progress
The catalytic mechanism of the two-electron Jacobsen–Katsuki epoxidation catalyst (Mn–Salen)[55] is not fully understood, although commonly assumed to involve a MnV–oxo species. Clear identification of any catalytically relevant MnV–oxo species has proved elusive, however (see Herrero et al.[56] and references therein). Herrero et al.[56] reported a molecular photosensitizer–catalyst construct (Scheme 3), where a RuII–polypyridyl photosensitizer was covalently attached to a MnIII–Salen moiety. An ultimate aim of this research is to employ laser pulses to achieve quantitative temporal control of stepwise catalyst activation, allowing access to catalytically relevant intermediates. Okamoto et al.[57] have also identified such an approach for the study of multi-electron catalysis, and Sjodin et al.[34] have employed a similar molecular architecture using a RuII–polypyridyl photosensitizer for the study of photo-initiated PCET.
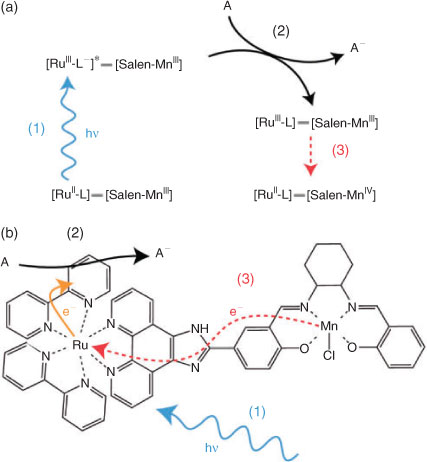
|
With this photosensitizer–catalyst we have demonstrated light-induced activation of the MnIII–Salen catalytic moiety via intramolecular electron transfer from MnIII to RuIII.[56] The RuIII is generated following photo-excitation of the [RuII–polypyridyl] MLCT transition(s), and subsequent oxidative quenching, via sacrificial electron acceptor, of the excited state [RuIII–polypyridyl–]* complex.
New data is presented in Fig. 1 that shows spectra and room temperature kinetics obtained using a custom-built CCD-based spectrograph[58] of the formation and decay of an absorption feature with a peak at ~680 nm that is assigned[56] to the photo-generated MnIV. Kinetic traces (d), (e), and (f) were obtained with continuous illumination corresponding to ~0.3, 0.03, and 0.003 absorbed photons per centre per second (see caption for details).
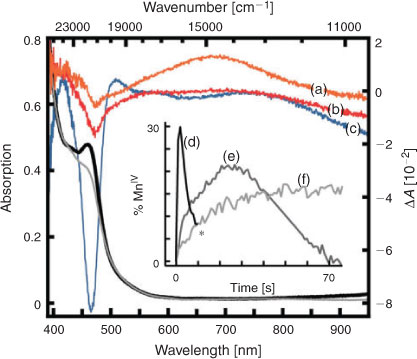
|
While this research is only in its formative stages, the point we would like to make here is that we have found no physically meaningful linear kinetic scheme that is able to globally fit this data. This photosensitizer–catalyst system presents an ideal case for investigating some of the ideas we have highlighted in this article.
Outlook
We have placed our commentary in the context of an urgent contemporary issue that necessitates advances in chemical research, yet has far wider implications: energy change. Several phenomena have been highlighted that suggest the hypothesis of cooperative, concerted or stimulated multi-electron transfer warrants rigorous investigation to stimulate new avenues for chemistry research. As a summarizing remark on this theme, we take lead from the thermodynamic advantage of concerted PCET[35] and generalize that if the energy barrier to producing stepwise single electron transfer intermediate states is sufficiently greater than another reaction pathway that involves the simultaneous or cooperative transfer of two electrons, then the latter will be the preferred pathway. This is essentially the point made by Tributsch,[23,26,27] from which we have taken much inspiration for our commentary.
Theories of multi-electron transfer need to be developed that are beyond a simple phenomenological description. They need to extend to the microscopic level as theories of PCET have, and continue to be developed. Additionally, only dedicated and targeted experimentation will be able to verify whether such theories are indeed necessary in the description of chemical phenomena. However, we note that new insights are rapidly emerging and suggest the possibility that concerted processes may be a rather important general phenomenon in chemistry. For example, a recent report has indicated that electron and proton transfer are both concerted with the cleavage of bonds between heavy atoms.[59]
Acknowledgements
This work was supported by an Australian Research Council Discovery Grant, DP 110104565.
References
[1] World Key Energy Statistics 2011, International Energy Agency, http://www.iea.org.[2] N. S. Lewis, D. G. Nocera, Proc. Natl. Acad. Sci. USA 2006, 103, 15729.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XhtFymtbrJ&md5=e4252961598780ced30bcd2df19c9437CAS |
[3] D. G. Nocera, ChemSusChem 2009, 2, 387.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXmtlCju78%3D&md5=6da08e70820241e9c47a378939b84c66CAS |
[4] J. L. Dempsey, A. J. Esswein, D. R. Manke, J. Rosenthal, J. D. Soper, D. G. Nocera, Inorg. Chem. 2005, 44, 6879.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXhtVGis77P&md5=1000e4bc38384a8f0586954fd90cefa9CAS |
[5] H. Tributsch, Electrochim. Acta 2007, 52, 2302.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXktlOruw%3D%3D&md5=5462e11d66fd9f6993888ee9de15016eCAS |
[6] D. G. Nocera, Inorg. Chem. 2009, 48, 10001.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtFKktLrP&md5=dab2a773bcbbb1bf5e6f63e44277b4eaCAS |
[7] S. V. Rosokha, J. K. Kochi, Acc. Chem. Res. 2008, 41, 641.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXktFSgsro%3D&md5=7c9d1207cb4cc4267a3059dbf367cba8CAS |
[8] S. M. Hubig, R. Rathore, J. K. Kochi, J. Am. Chem. Soc. 1999, 121, 617.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXis1Gquw%3D%3D&md5=c4aa68cefcae1d4d8e9531acfcaaf111CAS |
[9] K. Y. Wong, P. N. Schatz, S. B. Piepho, J. Am. Chem. Soc. 1979, 101, 2793.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE1MXktlyktrc%3D&md5=5f2f073dc9c03d21b1e378f7b198859cCAS |
[10] S. B. Piepho, P. N. Schatz, Group Theory in Spectroscopy with Applications to Magnetic Circular Dichroism, 1983 (Wiley-Interscience: New York).
[11] S. Hammes-Schiffer, A. A. Stuchebrukhov, Chem. Rev. 2010, 110, 6939.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtlyhtbzN&md5=bb18f93f356bb51078c4b5de56a18a6dCAS |
[12] E. Krausz, J. L. Hughes, P. Smith, R. Pace, S. Peterson Årsköld, Photochem. Photobiol. Sci. 2005, 4, 744.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXpt1aku7Y%3D&md5=2d917b5359c96e9dd6f7c8fafe44f95eCAS |
[13] T. Renger, E. Schlodder, ChemPhysChem 2010, 11, 1141.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXlt1agsbo%3D&md5=ec75475b0bc08c586a2aef4ade8c8a8eCAS |
[14] T. Renger, E. Schlodder, J. Photochem. Photobiol. B 2011, 104, 126.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXntFWjs7w%3D&md5=cd5e82111469d217ba9ad02d1ab18c4bCAS |
[15] Y. Umena, K. Kawakami, J. R. Shen, N. Kamiya, Nature 2011, 473, 55.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXkslCmtLg%3D&md5=f7b48594fae4a870a2b3d1c7ed10df43CAS |
[16] Y. L. Pushkar, J. Yano, K. Sauer, A. Boussac, V. K. Yachandra, Proc. Natl. Acad. Sci. USA 2008, 105, 1879.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXitl2ntL8%3D&md5=7989d9d4ad23464d20e8e92637293cefCAS |
[17] T. G. Gray, D. G. Nocera, Chem. Commun. 2005, 1540.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXjsVaru70%3D&md5=09d54fc19ba63f27fcdcc10349a1aa5fCAS |
[18] T. S. Teets, T. R. Cook, B. D. McCarthy, D. G. Nocera, Inorg. Chem. 2011, 50, 5223.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXlvFWjt70%3D&md5=c9766a5888885bdac195cedb26c15c3eCAS |
[19] E. Gileadi, J. Electroanal. Chem. 2002, 532, 181.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XntFyhtbs%3D&md5=cb529b24edecb5a0194d7527535957d6CAS |
[20] T. Hannappel, B. Burfeindt, W. Storck, F. Willig, J. Phys. Chem. B 1997, 101, 6799.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXltFyntbg%3D&md5=762dd7cdb69d3d69ad90207da7852a4cCAS |
[21] J. Z. Pu, J. L. Gao, D. G. Truhlar, Chem. Rev. 2006, 106, 3140.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28Xntlagt7w%3D&md5=9421fcdbb2d1033d0fa05e00d26d4978CAS |
[22] G. K. Schenter, B. C. Garrett, D. G. Truhlar, J. Phys. Chem. B 2001, 105, 9672.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXms1Ohu7o%3D&md5=6403402b33e811376cc483e1d749963aCAS |
[23] H. Tributsch, L. Pohlmann, Science 1998, 279, 1891.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXhvFKns7g%3D&md5=2a8146ac843f7e16463ab6efc4a8c892CAS |
[24] L. Pohlmann, H. Tributsch, J. Theor. Biol. 1992, 156, 63.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK38XkvVGgtLg%3D&md5=16cd23236ad575f5e1ab7abdff7735a5CAS |
[25] L. Pohlmann, H. Tributsch, J. Theor. Biol. 1992, 155, 443.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3sXlsVChtQ%3D%3D&md5=2c90f210a4bb8662f7b14ae6d60be75aCAS |
[26] H. Tributsch, J. Electroanal. Chem. 1992, 331, 783.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK38Xltl2lsr0%3D&md5=5d1a48e1f53de9db9da26ec9cf722305CAS |
[27] H. Tributsch, L. Pohlmann, Chem. Phys. Lett. 1992, 188, 338.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK38XhsF2rt74%3D&md5=09669d42a9a9086299e7b14c55f4847aCAS |
[28] H. Tributsch, L. Pohlmann, J. Electroanal. Chem. 1995, 396, 53.
| Crossref | GoogleScholarGoogle Scholar |
[29] H. Haken, Synergetics, An Introduction, 1983 (Springer: Berlin).
[30] Chem. Rev. 2010, 110, special issue on proton-coupled electron transfer.
[31] P. Faller, C. Goussias, A. W. Rutherford, S. Un, Proc. Natl. Acad. Sci. USA 2003, 100, 8732.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXlvVyitr0%3D&md5=d877767b267d9ae7c6c181f216ffa3ceCAS |
[32] R. Balasubramanian, G. Blondin, J. C. Canales, C. Costentin, J.-M. Latour, M. Robert, J.-M. Savéant, J. Am. Chem. Soc. 2012, 134, 1906.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XpsVOlsg%3D%3D&md5=9ea193756105cb0574951e1d577cd1ddCAS |
[33] C. Costentin, V. Hajj, C. Louault, M. Robert, J. M. Saveant, J. Am. Chem. Soc. 2011, 133, 19160.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsVCgt7vK&md5=d415255715b4434c328057a1fbee27c5CAS |
[34] M. Sjodin, S. Styring, H. Wolpher, Y. H. Xu, L. C. Sun, L. Hammarstrom, J. Am. Chem. Soc. 2005, 127, 3855.
| Crossref | GoogleScholarGoogle Scholar |
[35] J. J. Warren, T. A. Tronic, J. M. Mayer, Chem. Rev. 2010, 110, 6961.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXht1GqsbvN&md5=d40acf19cdfaffc1c7dd444bc053e19eCAS |
[36] S. Cattarin, S. Flechter, C. Pettenkofer, H. Tributsch, J. Electrochem. Soc. 1990, 137, 3484.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3MXhtlyluw%3D%3D&md5=3096b88d04da37d899c3613b1d188a24CAS |
[37] J. Wojtowicz, in Modern Aspects of Electrochemistry, 1972 (Eds J. O. M. Bockris, B. E. Conway) (Plenum Press: New York).
[38] D. A. Kraut, K. S. Carroll, D. Herschlag, Annu. Rev. Biochem. 2003, 72, 517.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXntFSgtbw%3D&md5=91e9b9910a6ed652122e7425dca793ecCAS |
[39] G. G. Hammes, S. J. Benkovic, S. Hammes-Schiffer, Biochemistry 2011, 50, 10422.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtlOgu7rL&md5=4c4fa709c3abc177c34da60bfc238cf5CAS |
[40] Y. Paltiel, G. Jung, T. Aqua, D. Mocatta, U. Banin, R. Naaman, Phys. Rev. Lett. 2010, 104, 016804.
| 1:STN:280:DC%2BC3c3jslOgtQ%3D%3D&md5=2bda5654616c58ac993d85bf62a60edaCAS |
[41] D. R. Ort, W. W. Parson, Biophys. J. 1979, 25, 355.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE1MXhtFKksb4%3D&md5=7cc614d8309be6adcff43f90f5ec749cCAS |
[42] H. Tributsch, L. Pohlmann, J. Theor. Biol. 1996, 178, 17.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK28XhslynsLc%3D&md5=7fd0958ec9798c5aedb946348b101b5eCAS |
[43] S. E. Braslavsky, G. E. Heibel, Chem. Rev. 1992, 92, 1381.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK38Xls1Smsbc%3D&md5=f2795d15efd605ed23ddf9b19a0ef1f5CAS |
[44] H. J. M. Hou, D. Mauzerall, J. Photochem. Photobiol. B 2011, 104, 357.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXntFWjsbY%3D&md5=8c9b4e23e3d49777e792b98509a09764CAS |
[45] A. C. Rizzi, M. van Gastel, P. A. Liddell, R. E. Palacios, G. F. Moore, G. Kodis, A. L. Moore, T. A. Moore, D. Gust, S. E. Braslavsky, J. Phys. Chem. A 2008, 112, 4215.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXksVOksbY%3D&md5=3190ae79c74ce0ec711240d541fd685aCAS |
[46] R. Krivanek, H. Dau, M. Haulmann, Biophys. J. 2008, 94, 1890.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXit1yqu74%3D&md5=521b9ceb8dc9aa4e33de33102cacc334CAS |
[47] K. Okamoto, S. Fukuzumi, J. Am. Chem. Soc. 2003, 125, 12416.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXnsVehsbw%3D&md5=e03c3576bd55f6ce7c789a8ee151b74cCAS |
[48] J. Yuasa, T. Suenobu, S. Fukuzumi, J. Am. Chem. Soc. 2003, 125, 12090.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXnt1OmsLY%3D&md5=b5addb9a70af60c0654a1c143f3a9f96CAS |
[49] J. S. Park, E. Karnas, K. Ohkubo, P. Chen, K. M. Kadish, S. Fukuzumi, C. W. Bielawski, T. W. Hudnall, V. M. Lynch, J. L. Sessler, Science 2010, 329, 1324.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtFajs7rO&md5=70fc9767e48fef005a7abc2a5696ff71CAS |
[50] S. Fukuzumi, K. Ohkubo, Coord. Chem. Rev. 2010, 254, 372.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsFyjsbrP&md5=868b9738180d008d80a7ee51d571e7a9CAS |
[51] Y. Morimoto, H. Kotani, J. Park, Y. M. Lee, W. Nam, S. Fukuzumri, J. Am. Chem. Soc. 2011, 133, 403.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsFKjtbfN&md5=fa11e39ddb9544b100875f78b0cb977fCAS |
[52] T. Kawashima, K. Ohkubo, S. Fukuzumi, Phys. Chem. Chem. Phys. 2011, 13, 3344.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsFymsLc%3D&md5=fc403c0febc84614323be48e01b1778dCAS |
[53] J. S. Vrettos, D. A. Stone, G. W. Brudvig, Biochemistry 2001, 40, 7937.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXktFekuro%3D&md5=993632a29787098e74038ee46c487bfaCAS |
[54] G. Hendry, T. Wydrzynski, Biochemistry 2003, 42, 6209.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXjtlehtb0%3D&md5=f753abe7f72b559f1464408dc21971d4CAS |
[55] T. Katsuki, Coord. Chem. Rev. 1995, 140, 189.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXks1Kgsrw%3D&md5=9311283a0074782a0143f4ed2223ef79CAS |
[56] C. Herrero, J. L. Hughes, A. Quaranta, N. Cox, A. W. Rutherford, W. Leibl, A. Aukauloo, Chem. Commun. 2010, 46, 7605.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXht1ClsLzI&md5=3773ebd17ad98d250a60c14a79100510CAS |
[57] A. Okamoto, R. Nakamura, H. Osawa, K. Hashimoto, J. Phys. Chem. C 2008, 112, 19777.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhsVShsLrM&md5=03f4962ef7401d4cae017aee3543c27bCAS |
[58] R. Steffen, K. Jackman, E. Krausz, Meas. Sci. Technol. 2008, 19, 075601.
[59] C. Costentin, V. Hajj, M. Robert, J. M. Saveant, C. Tard, Proc. Natl. Acad. Sci. USA 2011, 108, 8559.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXntVGqtL4%3D&md5=bb49085a03b1be046a3bf8a58c80377aCAS |