

A Snapshot of Ionic-Liquid-Tagged Proline-Based Organocatalysts
Hannah L. Brozinski A , Joshua P. Delaney A and Luke C. Henderson A B CA Strategic Research Center for Chemistry and Biotechnology, Deakin University, Pigdons Road, Geelong, Vic. 3216, Australia.
B Institute for Frontier Materials, Deakin University, Pigdons Road, Geelong, Vic. 3216, Australia.
C Corresponding author. Email: luke.henderson@deakin.edu.au
Australian Journal of Chemistry 66(8) 844-847 https://doi.org/10.1071/CH13218
Submitted: 29 April 2013 Accepted: 27 June 2013 Published: 31 July 2013
Abstract
This highlight focuses on the developments in ionic-liquid (IL)-tagged proline-based organocatalysts. An overview of catalyst structure and application to asymmetric transformations is provided, and a representative synthesis of an IL-tagged organocatalyst is also discussed.
Introduction
Organocatalysis has become recognised over the past decade as a legitimate means of installing chirality into complex organic systems. The exponential growth within this field, first developed by List and Barbas et al. in 2000, and popularised by MacMillan and Jørgenson, has led to organocatalysis being considered the ‘third pillar’ of asymmetric synthesis.[1–3] Most organocatalysts which operate by an enamine intermediate are based around a proline motif. A range of structural elaborations of the l-proline unit have been undertaken to decrease catalyst loadings and improve stereoinduction. These aspects of organocatalysis have been extensively reviewed elsewhere.[4–9] Concurrent with the increasing structural complexity of such catalysts is the growing need to recycle and reuse them in a cost efficient manner. To this end there have been reports of organocatalysts supported on polymers,[10] mesoporous silica,[11] and gold nanoparticles.[12]
Discussion
Conducting organocatalysed reactions within IL reaction media has seen some success for the reuse of catalysts, although reports of catalyst leaching (loss of the catalyst during product extraction or IL purification) has hampered progress.[13,14] Throughout the past five years several groups have gone to further lengths to facilitate efficient reuse of their catalysts by incorporating an IL-tag(s) directly onto organocatalyst scaffolds. A question to be asked is: ‘what advantages do ionic liquid organocatalysts possess over other, more established, organocatalysts?’ One answer lies in that this approach offers a robust alternative to traditional solid-support strategies. This is due to the pendant IL groups offering an opportunity to customise properties such as solubility, melting point, and catalytic activity, while still providing the mechanistic and kinetic advantages of a homogenous catalytic system. The ability to tailor the aqueous versus organic solubility (or even selective solubility in organic solvents) of IL-tagged organocatalysts makes them highly suitable for recovery and reuse. From a mechanistic standpoint, Zlotin has stated that ‘they [IL-tagged organocatalysts] create the hydrophobic environment of the enamine-type transition state, which resembles a hydrophobic pocket of aldolases, which is essential for efficient reaction stereocontrol’.[15]
The first report of an IL-tagged organocatalyst based on the proline scaffold was by Miao and Chan in May 2006,[16] although there was a near concurrent report of a pyrrolidine-based organocatalyst reported by Luo et al.[17] in April of the same year. The catalyst design adopted by Chan (1, Fig. 1) incorporated an imidazolium group (a typical IL moiety) at the 4 position on trans-4-hydroxy-l-proline, leaving the α-amino acid portion of the scaffold intact. The approach of using the 4-hydroxy moiety as a handle to install the IL-tag has become a mainstay within this field, with most compounds conforming to the general structure 2 (Fig. 1).
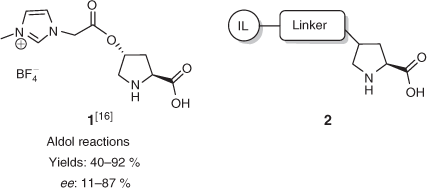
|
Since that initial work, there have been several reports of this class of catalyst, although major contributions to this field have emerged primarily from the groups of Zlotin,[15,18–22] Lombardo,[23,24] and Liebscher.[14,25,26] Organocatalyst 3 (Fig. 2), synthesised by Lombardo et al.[23] was used in an array of asymmetric aldol reactions. In each case a small amount of water (~1.2 equivalents with respect to the aldehyde) was required to achieve high catalyst turnover numbers (TON, e.g. 930) and products in very high enantiopurity (>97 % for 20 examples). This high TON was attributed to a ‘cis-effect’ where the imidazolium cation and Tf2N– anion are positioned on the same face of the enamine as the incoming electrophile. This conformation encourages electrostatic stabilisation of the transition state, providing a rate enhancement. A variation on 3, whereby the IL unit is directly bound to the 4-position of 4-hydroxyproline, was similarly employed by Zhu et al.[27] in the α-amination of aldehydes.

|
Organocatalysts 4a–c (Fig. 2), based on the α,α-bis-aryl scaffold pioneered by Jørgenson et al.[28] were developed and used to catalyse the Michael addition of nitromethane to a range of substituted cinnamaldehydes[18] in aqueous methanol (4 % H2O v/v). These studies echoed the findings by Lombardo et al. with water being required for high conversions and enantioselectivities. Under these conditions 4b performed the best (>99 % conversion and 94 % enantiomeric excess, ee) of the three catalysts, activities that are consistent with the non-IL-tagged analogues reported by Jørgenson et al.[28] Interestingly, 4a also performed well under these conditions (>99 % conversion and 70 % ee) which is in contrast to most reports of this catalyst when used with aldehydes, as it is well known to form a catalytically inactive hemi-aminal species.[28] This discrepancy was not commented on by the authors but is an aspect that deserves further investigation.
Catalysts 5a and 5b (Fig. 2), synthesised by Zlotin and co-workers,[22] were employed in an aqueous aldol reaction in high catalyst loading (30 mol-%). It was found that 5a (incorporating the BF4– anion) was water soluble and unable to facilitate the reaction, while 5b formed aqueous emulsions and furnished the desired aldol products in very high yield and optical purities, demonstrating the dramatic implications carried by the chosen anionic species. This study also demonstrated the reuse of the organocatalyst system by selective solubilisation of the products and reactants in diethyl ether,[22] leaving the catalyst and the aqueous phase which could be reused by the addition of more starting materials. Employing this recycling protocol, 5b was reused five times without loss of yield or stereoselectivity.
The 1,2,3-triazole-derived IL-tagged organocatalysts 6 and 7 (Fig. 2) were developed by Liebscher et al.[25] taking advantage of well established copper-catalysed azide–alkyne cycloaddition (CuAAC) protocols.[20] Of the two organocatalysts, 6 performed better than 7 in the aldol reaction, typically giving yields, diastereomeric excess (de), and ee values over 90 % for several cyclohexanone/benzaldehyde combinations. Unfortunately, application of 7 to the Michael addition of ketones to nitrostyrenes afforded products in low enantiopurity (typically <30 % ee). Attempts to reuse 7, by selective solubilisation, resulted in substantial losses in ee with each successive reuse.
Catalyst 8 (Fig. 2) is the most structurally complex IL-tagged catalyst to date[21] and draws on recent catalyst design strategies to develop scaffolds which bear more than one ‘catalytic unit’.[29–33] In that study, a 10 mol-% catalyst loading in the presence of water (100 equivalents with respect to the aldehyde) furnished aldol products in excellent enantioselectivity and very high yield (96 % ee, 95 : 5 anti : syn, 99 % yield).[21] The proposed role of water in the emulsion was to reinforce important hydrophobic effects which enhance stereoselectivity within the transition state, although removal of water from the reaction mixture had a minor influence on the diastereomeric ratio (dr) (89 : 11 anti : syn) only. Despite this inconsistency, under the optimal conditions 8 was a suitable catalyst for cyclic ketones paired with activated aldehydes although it did not perform well when employing electron rich aldehydes as the electrophile, or when any aldehyde was employed as the nucleophile.[12] Catalyst 8 was recycled 15 times using a cyclohexanone/4-nitrobenzaldehyde aldol reaction, with a negligible change observed for the dr, ee, and yield after the 15th reuse.
As with most molecules a plethora of potential syntheses are possible, several of which have been successfully employed to install IL moieties onto the proline scaffold, and so an exhaustive analysis of these syntheses cannot be provided here. Through examination of several published synthetic strategies, several common features become apparent and as such will be discussed in the context of the scheme provided (Scheme 1). The synthesis illustrated in Scheme 1 shows a method employed by Lombardo et al.[23] to access imidazolium-tagged organocatalyst 3. A common starting point for the installation of IL tags is the esterification of the 4-hydroxy group with a halogenated carboxylic acid. A Mitsonobu reaction is then employed to install the desired cis-stereochemistry in order to exploit the previously discussed ‘cis-effect’. This same transformation can also be done using common coupling agents (N,N′-dicyclohexylcarbodiimide (DCC), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI), 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU), etc.) to retain the preinstalled trans-stereochemistry.
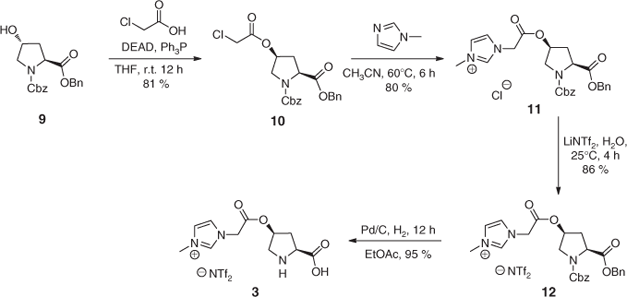
|
The installation of the pendant imidazolium group is undertaken by reaction of 10 (bearing the primary alkyl halide) with an N-alkylimidazole reagent. The alkylation depicted in Scheme 1 employs N-methyl imidazole, but variations of this moiety are often used to tailor the solubility and activity of the final catalyst. An example of this is provided by Zlotin and co-workers whereby N-dodecyl imidazole was introduced in order to increase the lipophilicity of the corresponding organocatalyst.[22]
Anion metathesis is most commonly carried out before the nucleophilic nitrogen is deprotected, affording 12 in high yield and purity. This process is typically performed by dissolving the imidazolium salt (in this case 11) in water followed by the addition of an excess of an organic soluble salt such as KPF6 or NaBF4, followed by extraction into an organic solvent. A common means of ensuring complete halogen anion removal is by the addition of silver salts such as AgBF4 which cause the precipitation of AgX salts.
The nucleophilic nitrogen is, to the best of our knowledge, always revealed by catalytic hydrogenolysis of either a benzyl or carboxybenzyl (Cbz) protecting group. The universal reliance on these two similar protecting groups presumably arises due to the mild conditions and the heterogeneous catalyst used in the deprotection procedure. In the case of tert-butoxycarbamates, the standard removal conditions (20 % trifluoroacetic acid (TFA) in CH2Cl2) would invariably result in anion scrambling to some extent.
The representative synthesis described in Scheme 1 concerns IL-tagged organocatalysts based on the l-proline structure that forms the basis of this highlight. Proline-derived organocatalysts are just one class of the many organocatalytic scaffolds that have been successfully elaborated with IL moieties, examples of other IL-tagged organocatalysts are highlighted in Fig. 3.

|
Conclusion
The evolution of IL-tagged organocatalysts has already begun with ionic functionalities becoming an increasingly prevalent feature of organocatalyst design. So far, IL-tagged organocatalysts have been applied only to routine chiral transformations (e.g. aldol, Mannich, Michael reactions, etc.), although they show great promise as a reusable means to install asymmetry within organic molecules. Future directions for this field would be the application of IL-tagged catalysts to cascade and domino processes with the intent of accessing increasingly sophisticated chiral scaffolds. Current preliminary results from this field so far are furnishing catalysts which possess ever greater recyclability and the potential to catalyse a growing pool of asymmetric transformations.
Acknowledgements
The authors thank Deakin University, the SRC for Chemistry and Biotechnology for their continued financial support. they also thank the Australian Government for APA scholarships to both HB and JD.
References
[1] B. List, R. A. Lerner, C. F. Barbas, J. Am. Chem. Soc. 2000, 122, 2395.| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXhtlGqurk%3D&md5=79c01b9061838d376912945d02063796CAS |
[2] K. L. Jensen, G. Dickmiess, H. Jiuang, Ł. Albrecht, K. A. Jørgensen, Acc. Chem. Res. 2012, 45, 248.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtVemu7zO&md5=9e109cda77fde8bd5f29287eb7d6f06fCAS | 21848275PubMed |
[3] K. A. Ahrendt, C. J. Borths, D. W. C. MacMillan, J. Am. Chem. Soc. 2000, 122, 4243.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXisVGjurw%3D&md5=207e90af843218e8f49b7858feceae6bCAS |
[4] J. Mlynarski, B. Gut, Chem. Soc. Rev. 2012, 41, 587.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhvFGqtg%3D%3D&md5=b2c286c6512e84b3758118e0bed19115CAS | 21796310PubMed |
[5] B. List, Chem. Rev. 2007, 107, 5413.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhsVSqsL3I&md5=73bc90d788d020084092d2bbb84ee877CAS |
[6] Z. Shao, H. Zhang, Chem. Soc. Rev. 2009, 38, 2745.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtVSlsLfJ&md5=c4d48e4680e383601c79637f92de17beCAS | 19690751PubMed |
[7] G. Guillena, C. Nájera, D. J. Ramón, Tetrahedron Asymmetr. 2007, 18, 2249.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXht1Smt7zO&md5=68f13af560b65f4d54ef8b324205f464CAS |
[8] J. Mlynarski, J. Paradowska, Chem. Soc. Rev. 2008, 37, 1502.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXoslOgtLk%3D&md5=4a700d5a5cb309e0d4fe3e58cbf9b247CAS | 18648676PubMed |
[9] S. Mukherjee, J. W. Yang, S. Hoffmann, B. List, Chem. Rev. 2007, 107, 5471.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhsVSqsLrP&md5=de6f212aaf83049fbc4137ea5c52ce86CAS | 18072803PubMed |
[10] T. E. Kristensen, T. Hansen, Eur. J. Org. Chem. 2010, 3179.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXmvFejtLs%3D&md5=c2622164623c2b63218156ec53cf4ea4CAS |
[11] J. Yan, L. Wang, Chirality 2009, 21, 413.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXltFaqsL0%3D&md5=90dabbfd6275565ca10e5ca5c106e29eCAS | 18570310PubMed |
[12] A. V. Malkov, M. Figlus, G. Cooke, S. T. Caldwell, G. Rabani, M. R. Prestly, P. Kocovský, Org. Biomol. Chem. 2009, 7, 1878.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXksV2itr0%3D&md5=205183cccd4fd0e8c2f042245a6f3e45CAS | 19590783PubMed |
[13] R. Sebesta, I. Kmentov, S. Toma, Green Chem. 2008, 10, 484.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXltFyjsrg%3D&md5=d7640a004b45b49a2c7eea48933d955cCAS |
[14] S. S. Khan, J. Shah, J. Liebscher, Tetrahedron 2011, 67, 1812.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhvFChu7w%3D&md5=d440e35a08782397f6853600cc859f97CAS |
[15] D. E. Siyutkin, A. S. Kucherenko, S. G. Zlotin, Tetrahedron 2010, 66, 513.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsFGntb3J&md5=247c72299ec44aa4be518367db92ee63CAS |
[16] W. Miao, T. H. Chan, Adv. Synth. Catal. 2006, 348, 1711.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XovFyms74%3D&md5=984390a4a6bf3dad5c8098d305785255CAS |
[17] X. Ding, W. Tang, C. Zhu, Y. Cheng, Adv. Synth. Catal. 2010, 352, 108.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtFanu7s%3D&md5=d3cce4824b0b14cc7840ab1f88ef2e3aCAS |
[18] O. V. Maltsev, A. S. Kucherenko, I. P. Beletskaya, V. A. Tartakovsky, S. G. Zlotin, Eur. J. Org. Chem. 2010, 2927.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXmt1eju7c%3D&md5=cd72b529fe53d353266e598779a059bbCAS |
[19] O. V. Maltsev, A. S. Kucherenko, S. G. Zlotin, Eur. J. Org. Chem. 2009, 5134.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXht1KjurjL&md5=bcfc028f1f6541be2360d21cdb9c69deCAS |
[20] O. V. Maltsev, A. S. Kucherenko, A. L. Chimishkyan, S. G. Zlotin, Tetrahedron Asymmetr. 2010, 21, 2659.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsFehurzK&md5=cdfb434fe8806d17a84ab16e0b6702edCAS |
[21] S. V. Kochetkov, A. S. Kucherenko, G. A. Kryshtal, G. M. Zhdankina, S. G. Zlotin, Eur. J. Org. Chem. 2012, 7129.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xhs1agsbzP&md5=c2ab396ea66d4a23d7b9089c7a17fcebCAS |
[22] D. E. Siyutkin, A. S. Kucherenko, M. Struchkova, S. G. Zlotin, Tetrahedron Lett. 2008, 49, 1212.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXosFansA%3D%3D&md5=7fe415da9fafef93c16053f9d3530e02CAS |
[23] M. Lombardo, S. Easwar, F. Pasi, C. Trombini, Adv. Synth. Catal. 2009, 351, 276.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhvVWktL0%3D&md5=974211a7a9f1dc90d24b9fb178e05bccCAS |
[24] M. Lombardo, S. Easwar, F. Pasi, C. Trombini, Adv. Synth. Catal. 2007, 349, 2061.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhtVels7bP&md5=009c03be6b48208c93db1b91e082e1efCAS |
[25] J. Shah, S. S. Khan, H. Blumenthal, J. Liebsher, Synthesis 2009, 23, 3975.
[26] S. S. Khan, J. Shah, J. Liebsher, Tetrahedron 2010, 66, 5082.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXntVyqurk%3D&md5=bc854b28dfa1b9fbdbb799fd06287a47CAS |
[27] X. Ding, H.-L. Jiang, C.-J. Zhu, Y.-X. Cheng, Tetrahedron Lett. 2010, 51, 6105.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtlentrvJ&md5=a3c391024babf5d308062a8ea914f94dCAS |
[28] J. Franzen, M. Marigo, D. Fielenbach, T. C. Wabnitz, A. Kjærsgaard, K. A. Jørgensen, J. Am. Chem. Soc. 2005, 127, 18296.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXht1KktrjF&md5=09a0d6133e55e14c98944f796d33b1ecCAS | 16366584PubMed |
[29] J. P. Delaney, L. C. Henderson, Adv. Synth. Catal. 2012, 354, 197.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xmt1Krsw%3D%3D&md5=2410fe4a000c0a9ec3b99caa26adba9aCAS |
[30] J. P. Delaney, L. C. Henderson, Int. J. Mol. Sci. 2011, 12, 9083.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XptF2juw%3D%3D&md5=2dfb5f52ed27bd7d8617e58215039e7fCAS | 22272120PubMed |
[31] J. P. Delaney, H. L. Brozinski, L. C. Henderson, Org. Biomol. Chem. 2013, 11, 2951.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXmtVWksL4%3D&md5=e791a0b5d807ad496f9b2707a8ed13b3CAS | 23455677PubMed |
[32] S. Samanta, J. Liu, R. Dodda, C.-G. Zhao, Org. Lett. 2005, 7, 5321.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXhtVKmtb%2FP&md5=5d4921f1a6f49088d49f593fa96f88ceCAS | 16268568PubMed |
[33] G. Guillena, M. C. Hita, C. Najera, Tetrahedron Asymmetr. 2006, 17, 1493.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28Xms1aitL0%3D&md5=01bd354d92ad58d9f5efb53530df9cb7CAS |
[34] A. S. Kucherenko, D. E. Siyutkin, A. G. Nigmatov, A. O. Chizhov, S. G. Zlotin, Adv. Synth. Catal. 2012, 354, 3078.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xhs1ags7vO&md5=31a814eca2bbad1a86aabafb18be38d9CAS |
[35] D. E. Siyutkin, A. S. Kucherenko, S. G. Zlotin, Tetrahedron 2009, 65, 1366.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXptl2gtA%3D%3D&md5=6af2da3081f1bbcc13ff49b155424975CAS |
[36] Z.-L. Shen, K. K. K. Goh, C. H. A. Wong, W.-Y. Loo, Y.-S. Yang, J. Lu, T.-P. Loh, Chem. Commun. 2012, 48, 5856.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xnt1ert7k%3D&md5=9d1317f4df547484d60cb0463c66ba7aCAS |