

Probing of the pH-Dependent Redox Mechanism of a Biologically Active Compound, 5,8-Dihydroxynaphthalene-1,4-dione
Shamsa Munir A , Afzal Shah A C , Usman Ali Rana B , Imran Shakir B ,A Department of Chemistry, Quaid-i-Azam University, Islamabad 45320, Pakistan.
B Deanship of Scientific Research, College of Engineering, PO Box 800, King Saud University, Riyadh 11421, Saudi Arabia.
C Corresponding author. Email: afzals_qau@yahoo.com
Australian Journal of Chemistry 67(2) 206-212 https://doi.org/10.1071/CH13373
Submitted: 16 July 2013 Accepted: 5 September 2013 Published: 20 September 2013
Abstract
The redox behaviour of a potential anticancer organic compound, 5,8-dihydroxynaphthalene-1,4-dione (DND), was investigated in 1 : 1 buffered aqueous ethanol using cyclic, differential pulse, and square wave voltammetry. The redox processes were found to occur in a pH-dependent diffusion-controlled manner. Presence of an α-hydroxyl group stabilised semiquinone radical of DND, formed by the gain of 1 e– and 1 H+, prevented the second step reduction, which is in contrast to the general mechanism previously reported for quinines in protic and aprotic media. In addition, our results supported an independent oxidation and reduction process. Square wave voltammetry provided evidence about the reversible and quasi-reversible nature of oxidation and reduction peaks. Based on the voltammetric results, the electrode reaction mechanism of DND was proposed. Parameters including pKa, transfer coefficient, diffusion coefficient, and electron transfer rate constant were evaluated. The values of pKa obtained from cyclic voltammetry and ultraviolet-visible spectroscopy not only agreed with each other, but also with reported values of structurally related compounds evaluated by other techniques.
Introduction
Quinones are widespread in nature as they have a vital role in many biological electron transfer processes including respiration and photosynthesis. These compounds are bestowed with fungicidal, antibacterial, and anticancer activities.[1,2] Fungal diseases are classified as superficial and systemic depending upon the affected areas of the body. Most antifungal drugs suppress the growth of fungal reservoirs but in turn the fungal strains develop resistance. Hence, efficacious antimycotic agents are required. Naphthoquinones, being fungicidal and anticancerous, are gaining growing attention in this regard. Quinones, being the defensive chemicals of various plants, are commonly used for the study of oxidative stress. These compounds are important to scientists because many chemotherapeutic agents contain the quinone functionality. The biological importance of quinones is linked with their ability to accept electrons to form radical anions or dianions. The redox properties of these compounds are governed by the electron-donating or electron-withdrawing substituents attached to the basic framework.[3] In biological systems, quinone toxicity is associated with reduction of the quinone moiety to the semiquinone radical, which in turn results in the reduction of oxygen thus converting it to the superoxide anion radical.[4]
Naphthoquinones are used in the preparation of dyes as these pigments typically display variable colours with the change of pH.[5] Naphthoquinone pigments employed in cosmetics are of dual importance because of their stability as a colouring agent and broad range of activities such as antiphlogistic, antibacterial, and antiviral.[6,7] Ultraviolet-visible (UV-Vis) spectroscopy is commonly employed for the study of naphthoquinones as these compounds have intense colours.[8]
Since quinones and naphthoquinones are important in redox systems, study of their redox behaviour could be very helpful in understanding the mechanism of action in compounds such as anti-tumour agents. The voltammetric behaviour of quinones including naphthoquinones has been extensively studied in non-aqueous and unbuffered aqueous media.[9–11] However, the detailed electrochemical fate of these compounds in aqueous media of different pH is yet to be explored. Inspired by the wide applications of this class of compound, the redox mechanism of the biologically important agent naphthoquinone 5,8-dihydroxynaphthalene-1,4-dione (DND), unexplored in protic solvent, was studied using three electrochemical techniques over a wide range of pH. Contrary to previous studies of closely related compounds in aprotic solvents,[12–15] the results of our experiments evidenced the independent nature of oxidation and reduction.
Experimental
Materials and Reagents
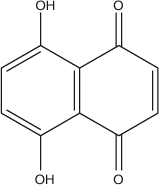
In this work, 5,8-dihydroxynaphthalene-1,4-dione (Scheme 1) from Sigma was used without further purification. A 2.5 mM stock solution was prepared in analytical grade ethanol and stored at 25°C. Working solutions of the compound were prepared in 50 % aqueous ethanol solvent. All supporting electrolytes whose composition is given in Table 1 were prepared using analytical grade reagents and doubly distilled water. Microvolumes were measured using EP-10 and EP-100 Plus Motorized Microlitre Pipettes (Rainin Instrument Co. Inc., Woburn, USA). All experiments were carried out at room temperature (25 ± 1°C).
|
|
Equipment and Measurements
Voltammetric experiments were performed using µAutolab running with GPES 4.9 software (Eco-Chemie, The Netherlands). A glassy carbon electrode (GCE) with an electroactive area of 0.071 cm2 was used as working electrode. A Pt wire and saturated calomel electrode (SCE) were employed as counter and reference electrodes. Before every electrochemical experiment, the surface of the GCE was polished with alumina powder followed by thorough rinsing with distilled water. For reproducible experimental results, the clean GCE was placed in supporting electrolyte solution and cyclic voltammetry (CV) was conducted until a voltammogram with a steady-state baseline was obtained. All voltammetric experiments were conducted under a high purity argon atmosphere. Differential pulse voltammetry (DPV) was carried out at 10 mV s–1 scan rate. For square wave voltammetry (SWV) the experimental conditions were 20 Hz frequency with 5 mV potential increments, corresponding to an effective scan rate of 100 mV s–1. Absorption spectra were recorded on a Shimadzu 1601 spectrophotometer and the pH measurements were carried out with a Crison micro pH 2001 pH-meter with an Ingold combined glass electrode.
Results and Discussion
Cyclic Voltammetry of 5,8-Dihydroxynaphthalene-1,4-dione (DND)
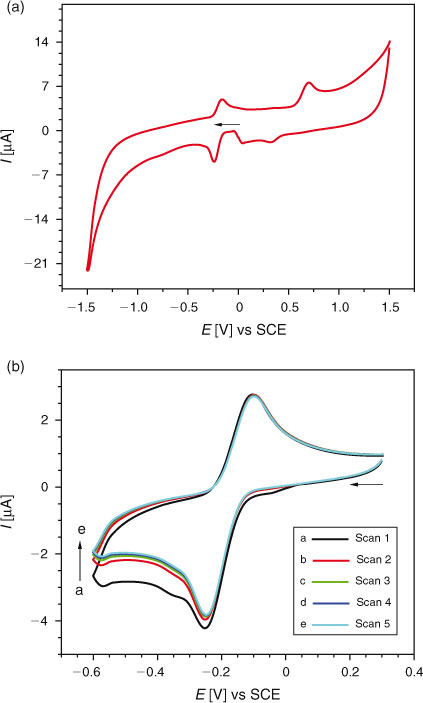
CV of 0.2 mM DND at a GCE was carried out between +1.5 and –1.5 V. As shown in Fig. 1a, a reduction peak at –0.25 V and an oxidation peak at –0.16 V were observed. In the positive scan, an oxidation peak was also observed at +0.69 V. Peak clipping experiments showed oxidation and reduction of DND to occur independently. In peak clipping experiments, the voltammograms are recorded by starting the potential scan from different initial values. When the potential was scanned from 0 to –1.5 V and back to 0 V, a single reduction peak at –0.25 V was observed in the forward scan with corresponding oxidation peak at –0.16 V in the backward scan. The peak at –0.16 V did not appear when the potential was scanned from –1.5 to 0 V. Hence, this peak is due to the oxidation of the reduction product formed at –0.25 V. Similarly the scanning of potential from 0 to +1.5 V and back to 0 V resulted in the appearance of an oxidation peak at +0.69 V in the forward direction and a small reduction peak of the oxidation product at +0.33 V in the backward direction. The peak at +0.33 V was not evidenced when the potential was scanned between +1.5 and 0 V. Hence, this peak is due to the reduction of the oxidation product of DND formed at +0.69 V. These experiments indicate the independent nature of oxidation and reduction of DND due to the presence of oxidisable and reducible electrophores in its structure. Hence, further cyclic electrochemical experiments of DND oxidation and reduction were carried out separately.
|
Five successive potential scans of 0.2 mM solution of DND are shown in Fig. 1b. No decrease in current intensity of the peak at –0.16 V with successive scans inferred the reduction product not to adsorb at the electrode surface.
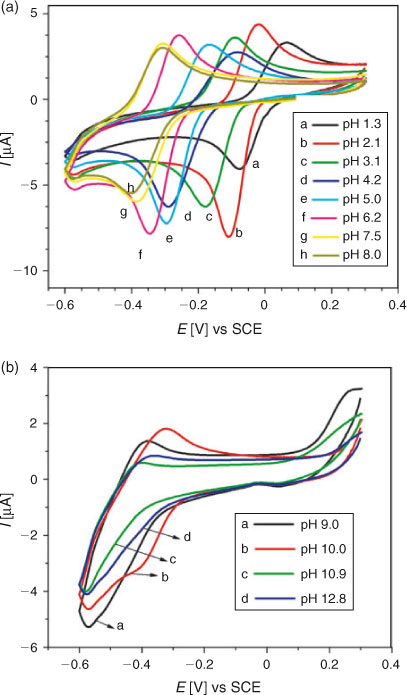
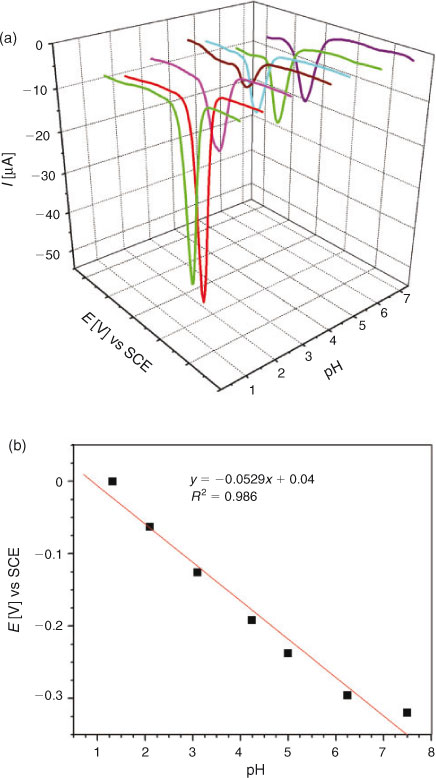
For examining the effect of pH on the reduction mechanism of DND, CVs were recorded at different pH ranging from 1.3 to 12.8 using 0.2 mM solution of the analyte. Before every experiment, the solutions were well saturated with argon to avoid the interference of oxygen reduction. Fig. 2a demonstrates the cathodic shifting of the reduction peak with increasing pH, indicating facile reduction of DND at low pH values. In strongly alkaline conditions cathodic peaks (Fig. 2b) were no more recognisable thus suggesting high pH media to be unfavourable for the reduction process of DND. A plot of peak potential as a function of pH followed a linear trend according to the equation Epc/V = –0.033 – 0.052pH (pH 1.3 to 8.0). The slope of 52 mV pH–1 unit is close to the theoretical value of 59[16] indicating the reduction of DND to occur by the involvement of 1 e and 1 H+.
|
The effect of scan rate on peak current and potential of DND was examined at pH 4.2. With increasing scan rate, the peak potential of DND could no t remain at the same position thus indicating the quasi-reversible nature of the redox process. A plot of peak current vs square root of scan rate gave a straight line with slope equal to 2.08 × 10–5 A/(V s–1)1/2. The linearity of the plot evidenced the reduction process to be limited by diffusion.[16] The diffusion controlled nature of the process was also supported by a linear log Ipc vs log ν plot with a slope of 0.46, which is within experimental error of the theoretical value of 0.5 for a diffusion‐controlled process.[17] A diffusion coefficient of 3.73 × 10–5 cm2 s–1 was evaluated using Epc – Epc/2 = 77 mV, αcn = 0.623 and n = 1 (determined from DPV).
The heterogeneous electron transfer rate constant was determined to obtain insights on the kinetics of the redox process. The Nicholson method was employed to evaluate the value of ksho. Using the transfer parameter, Ψ = 0.048 for 290 mV peak separation, ksho was found to be 7.15 × 10–3 cm s–1. This value is in the range of a quasi-reversible electrode process.[18,19]
For the study of electro-oxidation of DND, its cyclic voltammogram was first recorded in a medium buffered at pH 4.2 between –0.1 to 1.4 V with a starting potential of 0 V. As shown in Fig. 3, a well defined oxidation peak at +0.72 V and a small reduction peak at +0.33 V were observed. Fig. 3 also displays CVs recorded at pH between 1.3 and 12.9. With the rise in pH, peak potential shifted in the negative direction indicating facile electron abstraction in alkaline conditions. As shown in Fig. 3c, a new oxidation peak in the potential range of 0.51 to 0.59 V appeared when the pH was between 9.0 and 12.9. This is attributed to the oxidation of the hydroxy-adduct of DND. The adduct formation of some other hydroxyl quinones is also reported in the literature.[20] It can be seen in Fig. 3a, b that the profiles of DND curve are the same in the pH range of 1.3 to 6.2, indicating that the oxidation of DND and reduction of its oxidation product follow the same reaction mechanism in these pH media. However, with the increase of pH, the oxidation potential (Epa) changed its location towards less positive values. The shift of Epa to less positive values indicates an increase in the nucleophilicity of DND and that its antioxidant activity is favoured at higher pH. Moreover, in solutions of pH ≥ 7.5, the reduction peak of DND was not observed in the reverse scan due to the instability of the DND oxidation product resulting in the appearance of voltammograms showing an irreversible oxidation process.
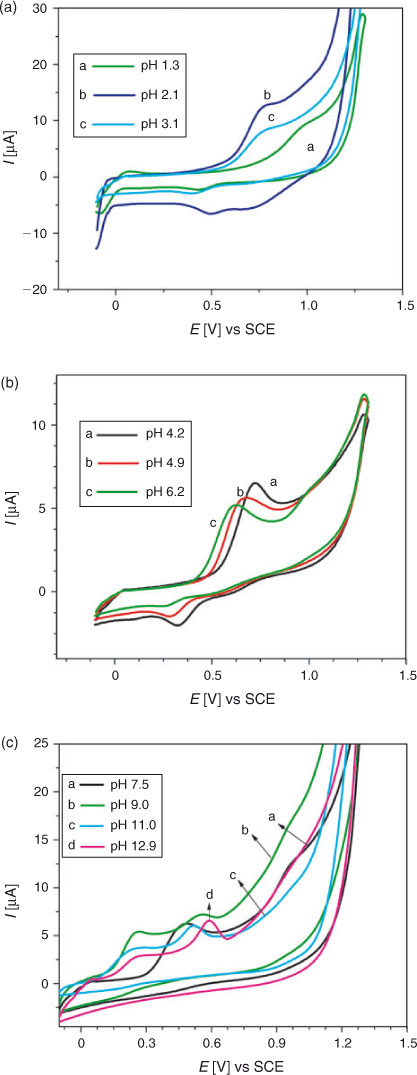
|
The Epa vs pH plot (Fig. 4), with a slope of 59 mV per pH unit, suggested the involvement of the same number of electrons and protons. pKa (the pH above which the compound exhibits only electron transfer reaction) showing chemical protonation-deprotonation was found to be 8.83. The value is well within the range of pKas reported for several other compounds structurally related to DND.[21–23] At pH 9.0–12.9, oxidation of DND occurred by the abstraction of electron only. Hence EC (electron transfer reaction followed by chemical reaction )/CE (chemical reaction followed by electron transfer reaction ) mechanism switched to EE (electron transfer reaction followed by electron transfer reaction) mechanism after pKa.
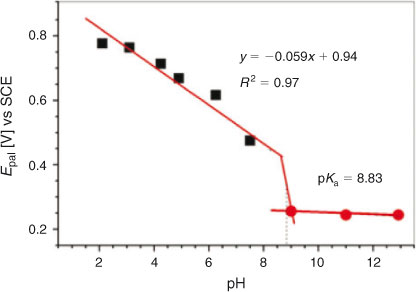
|
Differential Pulse Voltammetry
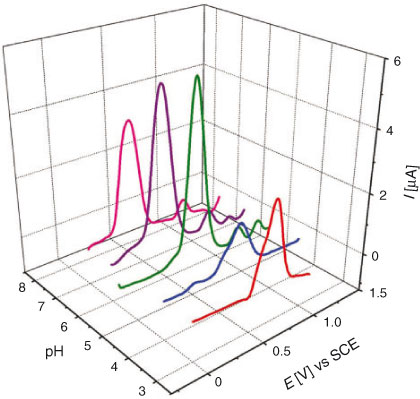
DPV was performed to determine the number of electrons involved in the oxidation and/or reduction process and for correlation of the results obtained from cyclic voltammetry. Similar to CV, DPV was also carried out separately for oxidation and reduction processes. DP voltammograms of 0.2 mM solution of DND recorded in different supporting electrolytes of pH 1.3 to 7.5 can be seen in Fig. 5a. The electro-reduction of DND was found to occur by the gain of a single electron as evidenced by the half peak width (W1/2) value of 98 mV, which is close to the theoretical value of 90 mV.[20] The Epc vs pH plot (Fig. 5b) showed a slope of 53 mV per pH unit, which supported the CV results of DND reduction to occur by the involvement of the same number of electrons and protons. DP voltammograms for the oxidation of DND are displayed in Fig. 6. A broad anodic peak with W1/2 = 191 mV at pH 4.2 suggested the overlapping of peaks corresponding to two one-electron transfer oxidation processes. Similar to CV, other oxidation peaks at high positive potential corresponding to the oxidation of a hydroxy adduct and enolic form of DND also appeared in DPV.
|
|
Square Wave Voltammetry of DND
SWV of a 0.2 mM DND solution was performed separately for oxidation and reduction regions to study the electrochemical reversibility of electrode processes. Similar to CV, SWV results (Fig. 7a) showed a quasi-reversible nature of DND reduction. However, a dramatically different situation from CV was observed when the oxidation of DND was examined. Unlike cyclic voltammetry, DND signalled a reversible anodic peak as demonstrated by the square wave voltammograms shown in Fig. 7b. This behaviour can be explained by the high speed analysis and ability of SWV to record the forward and backward current components in only one scan. The electroactive oxidised product of DND is not fully captured in the time domain of CV due to time lag in the recording of forward and backward scans.
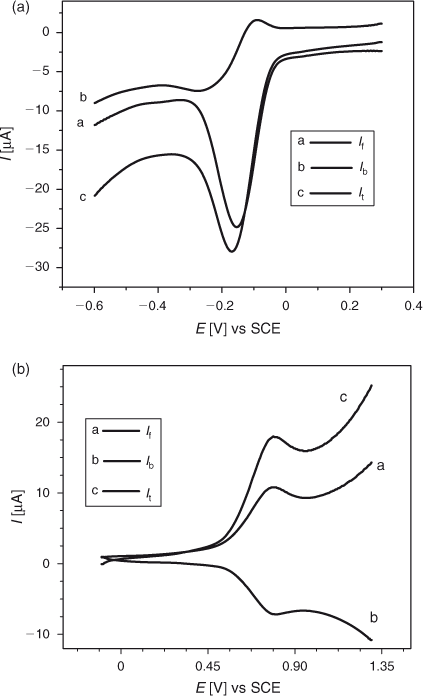
|
Proposed Redox Mechanism
The independent nature of DND oxidation and reduction was evidenced by all three voltammetric techniques employed in this work. On the basis of the results obtained from CV, SWV, and DPV the following electrode reaction mechanism was proposed:
Reduction
The W1/2 value close to 90 mV (from DPV) and slope of 52 mV pH–1 of Epc vs pH plot (CV results) suggested the reduction of DND to occur by the involvement of one electron and one proton. The proposed reduction mechanism in the pH interval 1.3–8.0 is depicted in Scheme 2. Addition of a single electron and a single proton results in the formation of a semiquinone radical stabilised by hydrogen bonding of the hydroxyl group of DND, which makes the second reduction step difficult and hence an additional reduction peak is not observed in the potential window of GCE. Stabilisation of semiquinone has been reported in literature for α-hydroxy naphthoquinones using aprotic media such as DMSO where the second reduction step occurs at potentials more negative than –0.6 V.[24,25]
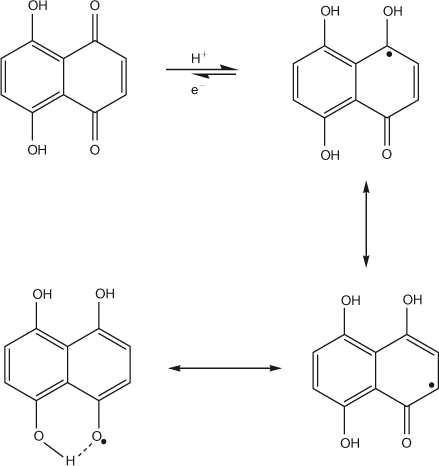
|
Oxidation
The Epa vs pH plot shows oxidation of DND to occur by the transfer of equal numbers of electrons and protons. W1/2 value of 191 mV predicted the overlapping of two 1e– transfer steps suggesting that the energy difference between removal of the first and second electron is not enough to resolve them as separate peaks. Based on these results, the oxidation mechanism of DND presented in Scheme 3 was proposed. The reason why DND oxidation could not proceed via a 2 e–‐2 H+ process in a single step is that the stabilisation of the 1e– oxidation product by six resonance structures causes the abstraction of the second electron to occur at a higher potential, resulting in overlapping of two oxidation peaks rather than a single peak.
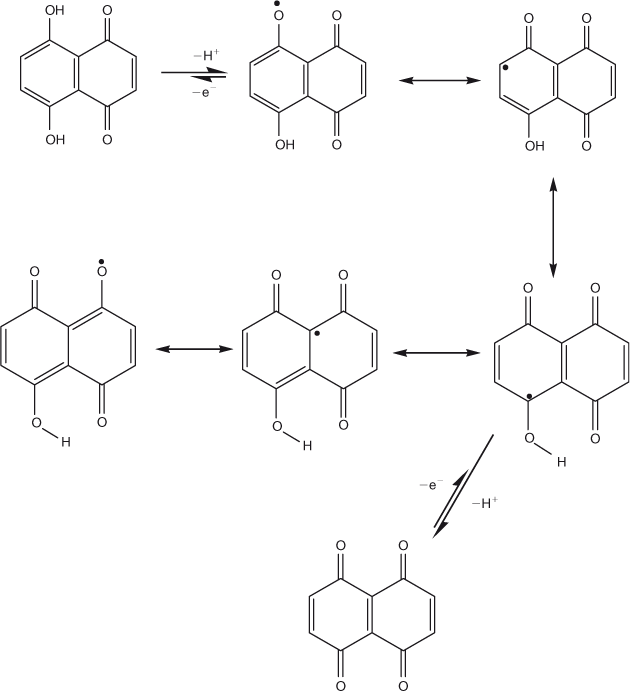
|
Electronic Absorption Spectroscopy
UV-Vis spectroscopy of a 0.125 mM solution of DND was carried out to understand its electronic absorption behaviour in the pH range 1.2–12.9. The spectrum of DND displayed in Fig. 8 showed a broad band of overlapping peaks in the range of 400–560 nm consistent with the reddish pink colour of the solution and a rather sharp band at 270 nm attributed to π→π* transitions of the quinonoid structure. Drastic changes in the spectral characteristics were noticed with increasing pH. All peaks were slightly dislocated bathochromically as the pH changed from 1.2 to 4.2. In the pH regime 7.5–12.9 the solution changed its colour from light violet to royal blue with a correspondingly larger bathochromic shift. The reason for such a spectral response can be attributed to deprotonation of DND thus resulting in a negative charge on the oxygen atom, with correspondingly higher electronic density for the π→π* transition.
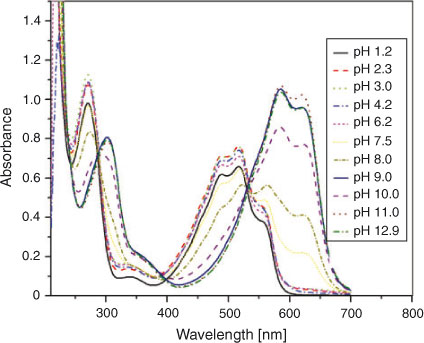
|
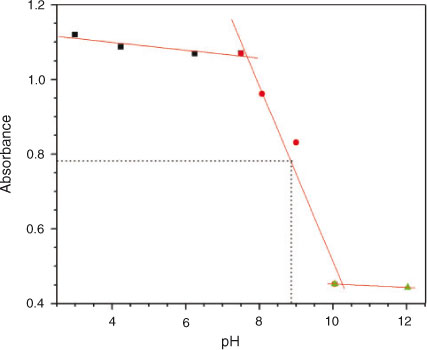
From acidic to basic pH, absorption maxima related to π→π* transition of the sharp band decreased while the broad band absorbance (n→π*) increased. The former is associated with the quinonoid structure including C=O groups while the higher absorbance intensities at 270 nm could be attributed to the fact that doubly bonded oxygen atoms are stabilised at lower pH values. In fact the compound can exist in keto and enol tautomeric forms with the keto form being more stable in acidic media.[22] As the pH of the medium is increased, the percentage of enolic form increases and hence a decrease in the intensity at 270 nm is observed with the concomitant increase in the intensity of the broad band at 400–560 nm. The bathochromic shift at higher pH values can be explained in terms of deprotonation phenomenon. The deprotonation of the enolic form of DND takes place in a stepwise manner giving monoprotonated and dideprotonated forms. The appearance of three isosbestic points in the electronic absorption spectra of the compound obtained in different pH media suggests the presence of neutral, monoprotonated, and dideprotonated forms. The pKa of DND with a value of 8.89 was evaluated from the plot of absorbance vs pH (Fig. 9) at 268 nm owing to the pronounced variation of absorbance at this wavelength. The pKa is in very good agreement with 8.83, obtained from CV. Our results justify the validity of Idriss and Saleh’s research work who determined a pKa value (8.2) of DND by potentiometric titration.[24]
|
Conclusion
The voltammetric behaviour of 5,8-dihydroxynaphthalene-1,4-dione (DND) in buffered aqueous ethanol media was found quite different from usual protic and aprotic solvents. DND was found to oxidise and reduce independently at a glassy carbon electrode in the pH range 1.3–12.9. The results of UV-Vis spectroscopy revealed the occurrence of DND in keto and enol forms. Reduction of DND, involving the gain of electron and proton, resulted in the formation of a semiquinone radical that was stabilised by intramolecular hydrogen bonding as evidenced by the absence of a second reduction peak. A broad oxidation peak in the positive realm helped to conclude that the hydroxyl groups (at positions 5 and 8) of DND to oxidise in two one-electron transfer pH-dependent processes. The detailed electrochemical probing enabled us to propose the redox mechanism of DND and thus predicted the possible pathway of biochemical actions of this class of compounds in biological systems where its properties are completely attributed to redox cycling.
Acknowledgements
The authors gratefully acknowledge the financial support of Quaid-i-Azam University, Higher Education Commission Islamabad, Pakistan, and Deanship of Scientific Research at King Saud University through the research group project no RGP-VPP-312.
References
[1] A. K. Boudalis, X. Policand, A. Sournia-Saquet, B. Donnadieu, J. P. Tuchagues, Inorg. Chim. Acta 2008, 361, 1681.| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXislyqtLw%3D&md5=29e7df3bdd33a4074c428e4b2c689942CAS |
[2] V. K. Tandon, R. B. Chhor, R. V. Singh, S. Raib, D. B. Yadava, Bioorg. Med. Chem. Lett. 2004, 14, 1079.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXhsFemt7s%3D&md5=1c8a0f1830a4af9df4a5c497fcc90163CAS | 14980639PubMed |
[3] P. Zuman, Substituent Effects in Organic Polarography 1967 (Plenum Press: New York, NY).
[4] P. S. Guin, S. Das, P. C. Mandal, Int. J. Electrochem. Sci. 2008, 3, 1016.
| 1:CAS:528:DC%2BD1cXhtVCju7vM&md5=0f8d7eab574379fea8bc6fc209a1d4bbCAS |
[5] Y. N. Hu, Z. H. Jiang, K. S. Y. Leung, Z. Z. Zhao, Anal. Chim. Acta 2006, 577, 26.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XoslGgtLw%3D&md5=79a1dcca2698d5b5b67d48fd3dca3e4cCAS |
[6] H. Zhou, J. Wang, B. Yea, J. Anal. Chem. 2010, 65, 749.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXotlykur0%3D&md5=55a8d3d0b0e78c4b241de7fa4b45285bCAS |
[7] H. Gershon, Can. J. Microbiol. 1975, 21, 1317.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE28Xht1eitA%3D%3D&md5=5ddd67e8eb9987bb522bcd2ae9793ceaCAS | 241479PubMed |
[8] S. V. Rodrigues, L. M. Viana, W. Baumann, Anal. Bioanal. Chem. 2006, 385, 895.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XmslSlsrg%3D&md5=d37be8c8d074ac0aa4db2ada47bfe85cCAS | 16791570PubMed |
[9] R. J. Forster, J. P. O’Kelly, J. Electroanal. Chem. 2001, 498, 127.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXhsVentLY%3D&md5=9cef16028b39416ecc18bf832932bd0dCAS |
[10] Y. Tang, Y. Wu, Z. Wang, J. Electrochem. Soc. 2001, 148, E133.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXivVahurY%3D&md5=2ddc925e44c9ecf090736f50e2fd3f96CAS |
[11] M. Quan, D. Sanchez, M. F. Wasylkiw, D. K. Smith, J. Am. Chem. Soc. 2007, 129, 12847.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhtFShtbnJ&md5=585bfa98a22bf025328bb4a0dd85e86bCAS | 17910453PubMed |
[12] N. Gupta, H. Linschitz, J. Am. Chem. Soc. 1997, 119, 6384.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXktlKjtro%3D&md5=1722067c772f20d318970b8c02ec6b09CAS |
[13] M. D. Stallings, M. M. Morrison, D. T. Sawyer, Inorg. Chem. 1981, 20, 2655.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL3MXks12rtLg%3D&md5=75dd1c5bc63f5827eaaa2d3b9db69d1eCAS |
[14] M. Shamsipur, A. Siroueinejad, B. Hemmateenejad, A. Abbaspour, H. Sharghi, K. Alizadeh, S. Arshadi, J. Electroanal. Chem. 2007, 600, 345.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXpslGntA%3D%3D&md5=71846338efa11c0f17cf244fd94dd8c1CAS |
[15] M. W. Lehmann, D. H. Evans, J. Electroanal. Chem. 2001, 500, 12.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXhslelsr4%3D&md5=c9192cdcc40348d06dbea3d3f98c84e3CAS |
[16] E. Nosheen, A. Shah, A. Badshah, Z. Rehman, H. Hussain, R. Qureshi, S. Ali, M. Siddiq, A. M. Khan, Electrochim. Acta 2012, 80, 108.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xht1GisLfE&md5=454ca1f8eed40c7f9f7586df0183d2eeCAS |
[17] S. S. Kalanur, J. Seetharamappa, U. Katrahalli, P. B. Kandagal, Int. J. Electrochem. Sci. 2008, 3, 711.
| 1:CAS:528:DC%2BD1cXlvFejsrw%3D&md5=2c8a0b83cce6a1fe5a8d1b70d718a9ddCAS |
[18] A. J. Bard, L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, 2nd edn 2001 (John Wiley & Sons, Inc.: New York, NY).
[19] C. M. A. Brett, A. M. O. Brett, Electrochemistry: Principles, Methods and Applications 1993 (Oxford University Press: Oxford).
[20] M. A. B. H. Susan, M. Begum, Y. Takeoka, M. Watanabe, J. Electroanal. Chem. 2000, 481, 192.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXhtV2jt7c%3D&md5=56dd87a4f900ae7bef236b7890f71096CAS |
[21] D. Y. Chung, E. H. Lee, J. Ind. Eng. Chem. 2006, 12, 962.
| 1:CAS:528:DC%2BD28XhtlSjtbrM&md5=f6d4ac16983b7bfc14562ed748cf3bbfCAS |
[22] S. Gallot, O. Thomas, Fresenius’ J. Anal. Chem. 1993, 346, 976.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3sXmsFKitLg%3D&md5=0861b7edb47605d00eeda4dc43da338cCAS |
[23] S. Riahi, S. Eynollahi, M. R. Ganjali, Int. J. Electrochem. Sci. 2009, 4, 1128.
| 1:CAS:528:DC%2BD1MXhtVKltL%2FM&md5=48f039a02d07a788cfeb97d062ddfd45CAS |
[24] K. A. Idriss, M. M. S. Saleh, Monatsh. Chem. 1993, 124, 1089.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2cXhvVWqtr0%3D&md5=e0bcf41fd762528cd9f6857de65b4391CAS |
[25] J. Gendell, W. R. Miller, G. K. Fraenkel, J. Am. Chem. Soc. 1969, 91, 4369.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaF1MXksFGgur4%3D&md5=8bc23c90d03b3bbac9237244db1104b9CAS |