

Syntheses, Crystal Structures, and Properties of Six Coordination Complexes Based on a Newly Designed Mercapto-thiadiazole Ligand
Lu-Lu Wei A , Lin-Ke Li A B , Li-Yan Fan A , Chang-Hong Wang A and Hong-Wei Hou A BA The College of Chemistry and Molecular Engineering, Zhengzhou University, Zhengzhou 450001, China.
B Corresponding authors. Email: lilinke@zzu.edu.cn; houhongw@zzu.edu.cn
Australian Journal of Chemistry 67(2) 241-249 https://doi.org/10.1071/CH13417
Submitted: 14 August 2013 Accepted: 16 September 2013 Published: 17 October 2013
Abstract
Six coordination complexes were solvothermally synthesised: a 3D framework [Cd(tmtt)2]n (1), 2D architectures [Zn(tmtt)2]n (2) and [Pb(tmtt)2]n (3), 1D chain structures [Ni(tmtt)2·(H2O)2]n (4) and [Co(tmtt)2·(H2O)2]n (5), and a mononuclear structure [Hg(tmtt)2] (6). The complexes, based on self-assembly of different metal ions with a newly designed mercapto-thiadiazole ligand tmttH (tmttH = 5-[(1H-1,2,4-triazol-1-yl)methyl]-1,3,4-thiadiazole-2(3H)-thione), were characterised by single crystal X-ray diffraction analyses. Crystal structure analyses reveal that complex 1 exhibits a four-fold interpenetrating 3D framework with {64.82} topology based on two kinds of right-handed single-helical chains, 2 displays a bilayer structure, 3 presents a crown-shaped network, 4 and 5 show 1D double–helical chain structures, and 6 is a mononuclear structure. Moreover, the thermal stabilities of crystalline samples 1–6 have been investigated, and the luminescent properties of complexes 1, 2, 3, 6, and the free ligand have been studied. The results of photoluminescent measurements illustrate that 2 and 3 may serve as excellent candidates for potential photoactive materials.
Introduction
The construction of metal–organic frameworks (MOFs) with metal ions and multifunctional organic ligands is currently a hot research topic that covers diverse areas of chemical science, with a particular emphasis on crystal engineering amongst other areas. The intense interest in building MOFs is driven by their intrinsic architectural beauty and aesthetically pleasing structures, as well as potential applications such as catalysis, molecular magnets, photoluminescence, ion exchange, adsorption, and phase separation.[1] In general, the self-assembly of MOFs are frequently influenced by many factors,[2,3] and greatly depends on the selection of organic ligands with appropriate functional groups which can extend the metal ions in a predetermined fashion, to produce the target network-based materials with specific structures and properties.[4] Minor structural changes of a ligand such as the angle, distance, and relative orientation of the donor or functional groups can also result in variation of the coordination framework.[5] Therefore, it is still a synthetic challenge to predict and control the synthesis of final products at this stage.
Recently, the selection of thiol and nitrogen-containing ligands (e.g. sulfhydryl imidazole, sulfhydryl triazole, and sulfhydryl pyridine ligand) have aroused increasing interest in the self-assembly of MOFs.[6–8] As a five-membered nitrogen containing heterocycle, neutral triazole or its deprotonated anion contains three potential metal binding sites, which can form a MOF through multiple bonding interactions, and can exhibit diverse coordination modes under different synthetic conditions.[9] Thus, triazole and its derivatives have been broadly used for the construction of frameworks and their properties have been examined.[10–13]
Our recent findings show that ligands based on mercapto-thiadiazole are promising for constructing MOFs.[14] As an extension of this work and in order to elucidate details of the assembly process of this kind of ligand with different metal ions, we herein disclose a flexible, functionalised ligand containing both triazole and mercapto-thiadiazole groups, namely, 5-[(1H-1,2,4-triazol-1-yl)methyl]-1,3,4-thiadiazole-2(3H)-thione (tmttH, Scheme 1), which is expected to be a versatile bridge in forming complexes. In this paper, a series of new complexes with a variety of zero- to three-dimensional frameworks were synthesised successfully under solvothermal conditions, namely, a four-fold interpenetrating 3D framework [Cd(tmtt)2]n (1), a 2D bilayer [Zn(tmtt)2]n (2), a 2D crown-shaped network [Pb(tmtt)2]n (3), 1D double–helical chain structures [Ni(tmtt)2·(H2O)2]n (4) and [Co(tmtt)2·(H2O)2]n (5), and a mononuclear structure [Hg(tmtt)2] (6). Single-crystal X-ray diffraction data reveal that the ligand tmttH has strong coordinative abilities. The structural analyses of these complexes are discussed in detail. In addition, the thermal stabilities of 1–6 and solid-state fluorescence properties of 1, 2, 3, 6, and the ligand have been investigated in detail.
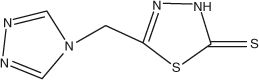
|
Results and Discussion
Structure Descriptions
[Cd(tmtt)2]n 1
Single-crystal X-ray structural analysis revealed that complex 1 crystallised in the asymmetrical trigonal space group P3(1)21, and exhibits a four-fold interpenetrating 3D framework based on two distinct types of right-handed single-helical chains with {64.82} topology.
As shown in Fig. 1a, the CdII centre is four-coordinated by two N atoms from triazole rings and two S atoms from sulfhydryl groups of four different ligands to form a distorted tetrahedral coordination geometry. The Cd–N bond lengths are 2.290(3) Å, the Cd–S bond lengths are 2.5160(3) Å, and the angles of S2B-Cd1-N1A and N1A-Cd1-N1 are 106.54° and 86.71°, respectively, which are comparable to previously reported values.[15]

|
In the asymmetric unit, the ligands exhibit a monodentate coordination mode. The combination of the thiadiazole and triazole rings results in twisting across the C–N single bond, giving rise to (Cd–tmtt)n helical chains. Along the a direction, neighbouring metal centres are bridged together by completely deprotonated ligands, affording two kinds of extraordinary right-handed single-helical chains with Cd⋯Cd distances ranging from 11.2921 to 26.4803 Å. The screw axes of these helices are all parallel to the a axis. As shown in Fig. 1b, these chains display a ‘∞’ shape and the pitches are all 15.2884 Å. These different types of neighbouring helix chains are arranged along the a axis by using CdII cations as hinges to give rise to a 2D network. Therefore, these layers are further connected by ligands to form a 3D framework with two kinds of open channels. The large vacancies can facilitate high-fold interpenetration.[16] Thus, the network incorporates three frameworks, directly resulting in a four-fold interpenetrating 3D architecture (Fig. 1c). It is noted that the nonchiral ligand and metal ion cannot produce a helical structure that is exclusively right- or left-handed, resulting in a racemic mixture of right- and left-handed helical products.[17] Therefore, a 3D framework with the same helicity is quite rare and the unusual complex 1 may have autoresolved itself into separate right-handed crystals from the solution. To fully understand the network structure of 1, a topological approach was applied to simplify the 3D coordination framework. The coordinated CdII can be reduced to four-connected nodes, and the ligand can be simplified as one linker, respectively. Therefore, the structure of 1 becomes a four-connected net with the symbol of {64.82} (Fig. 1d).
[Zn(tmtt)2]n 2
Single-crystal X-ray diffraction analysis indicates that complex 2 is a 2D bilayer motif.
As shown in Fig. 2a, the coordination environment of 2 is a distorted tetrahedron with two nitrogen atoms (N1 and N6) from triazole rings, and one nitrogen atom (N5A) and one sulfur atom (S4A) from mercapto-thiadiazole groups of four different ligands. The Zn–N bond lengths vary from 2.014(2) to 2.024(3) Å, and the Zn–S bond length is 2.3082(12) Å, which are within normal values or close to those observed for N-heterocyclic-based ZnII complexes.[18] In the asymmetric unit, the tmttH ligands have two different distortions: in one the dihedral angle between the triazole and thiadiazole rings from one ligand, which the S atom acts as coordination donor, is 52.8°, while in the other it is 58.7°.

|
In complex 2, all ligands adopt the same bidentate coordination mode and each ligand acts as a linker using its nitrogen atom from the triazole ring and sulfur atom from the thiadiazole group to coordinate with two ZnII ions in the b direction. In this way, the ZnII cations are connected by tmtt, affording infinite parallel 1D channels. Interestingly, each channel is enclosed by two intertwined helices with the same handedness (Fig. 2b). The separation of the Zn⋯Zn ions bridged by the ligand are 9.5262 and 10.9699 Å. In addition, the neighbouring metal centres are bridged by two nitrogen atoms from the triazole ring and the thiadiazole group of one ligand along the a direction. Therefore, a 2D bilayer motif (as shown in Fig. 2c) is built up from the same right-handed double-helical chains with a pitch of 15.5202 Å. Even though many complexes containing the 2D meso-helical sheet consisting of alternating left- and right-handed helical chains have been reported,[19–22] a 2D helical sheet with only one type of helical chain is quite rare.[23] Interestingly, the right-handed double-helical chains, from the two nets, revolve around the same axis. Further topological analyses performed by the OLEX program indicate that the framework of 2 could also be classified as a four-connected topology with symbol of 6.6.6(2).6.6.8(2)-dia, if the four-coordinated ZnII can be reduced to four-connected nodes, and the ligand can be simplified as one linker (Fig. 2c).
[Pb(tmtt)2]n 3
Complex 3 crystallises in a monoclinic system with space group C2/c and exhibits an infinite crown-shaped 2D network. As shown in Fig. 3a, the coordination environment around the PbII ion can be described as a distorted octahedral coordination geometry. Each PbII ion is six-coordinated with two Ntri (tri = triazole) atoms, two Ntdz (tdz = thiadiazole) atoms, and two sulfur donors from four ligands. The Ntri and S donors from mercapto-thiadiazole ligands exhibit bidentate chelation, while the other Ntri is monodentate. The triazole and thiadiazole rings in the same ligand are distorted with dihedral angles of 91.2° and 88.8°. The lead(ii) ion has a lone pair of electrons which can be stereochemically active and can have important effects on the structure of its complexes. Although not obvious, the presence of the active lone pair could explain the highly asymmetric arrangement of the N-donors; Pb–N bond lengths range from 2.738(10) to 2.812(13) Å, which are reasonable compared with the values in reported octahedral PbII complexes.[24,25] The Pb–S bond lengths are 2.991(4) Å, which are slightly shorter than those incomplexes [Pb(L6)2]·0.5H2O·0.3MeOH (HL6 = 4-ethyl-2,5-dihydro-3-methyl-5-oxo-1H-pyrazole-1-carbothiamide) and [Pb(L7)2]·0.5H2O·0.3MeOH (HL7 = N-ethyl-2,5-dihydro-3-methyl-5-oxo-1H-pyrazole-1-carbothiamide).[26,27]
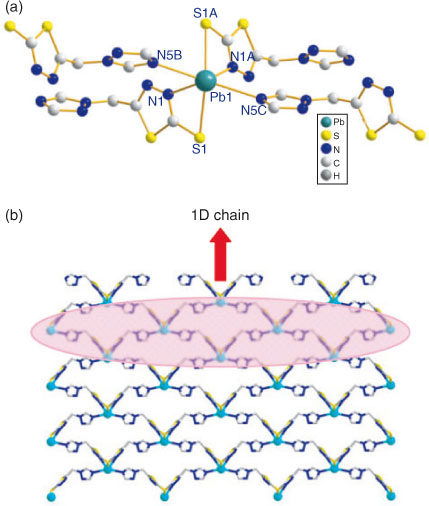
|
The PbII cations are bridged by ligands to yield one-dimensional zigzag chains along the a direction. Pb⋯Pb⋯Pb angles are 127.7° and the Pb⋯Pb distances are 11.172(2) Å, respectively. The connection of two contiguous PbII ions gives rise to an extended crown-shaped 2D layer (Fig. 3b) along the ab plane. All the PbII ions in the layer are strictly coplanar. The grid motif (34-membered metallocyclic rings) has dimensions of 20.055(4) × 9.8530(2) Å (diagonal distances).
[Ni(tmtt)2·(H2O)2]n 4 and [Co(tmtt)2·(H2O)2]n 5
Single-crystal X-ray diffraction analysis reveals that complexes 4 and 5 are isomorphic, containing divalent metal ions with the same stereo-chemical preferences, crystallising in the triclinic system with space group P-1. In these two complexes, the coordination environments of the central metal ions are almost consistent with each other. Complex 4 is representative for further discussion.
Complex 4 exhibits a charming one dimensional double–helical chain structure. As shown in Fig. 4a, each NiII atom is six-coordinated by four nitrogen atoms (N1, N5A, N10A, and N6) from four different ligands and two oxygen atoms (O1 and O2) from two coordinated water molecules, leading to a distorted octahedral geometry. Atoms N6, N5A, N1, and O1 form the equatorial plane (the mean deviation from the plane is 0.0017 Å) while atoms N10A and O2 occupy the axial positions. It is interesting to find that all the N-Ni-N, N-Ni-O, and O-Ni-O bond angles are close to 90° or 180°. The Ni–O and Ni–N distances are in the range 2.113(2)–2.130(2) Å and 2.064(2)–2.137(2) Å (Table 1), which are close to those observed previously in related NiII complexes.[28,29] In 5 (Fig. 5), the Co–O and Co–N bonds span the range of 2.113(3)–2.165(3) and 2.100(3)–2.186(3) Å; and N-Co-N, N-Co-O, and O-Co-O bond angles are close to 90° or 180° as well, which are in good agreement with the bond lengths and bond angles observed in other CoII complexes.[30]

|

|
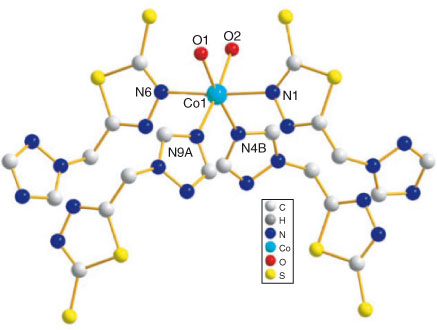
|
In 4, all ligands adopt the same μ2-bridging coordination mode to bridge two NiII ions, with metal separations of 9.1095 and 9.2434 Å. (For 5, the separation of Co⋯Co are 9.3719 and 9.2054 Å). The units are linked to each other through the triazole and thiadiazole nitrogen atoms of the ligands to form a 1D double-ribbon chain comprising two intertwined infinite helical chains with different handedness and alternating 16-membered metallocycles (Fig. 4b). The 16-membered ring involving {Ni2N8C6} have the approximate ring sizes of 5.4484 × 5.5146 Å2. (For 5, {Co2N8C6} have approximate ring sizes of 6.8493 × 5.4524 Å2). Along the bc plane, adjacent chains are connected by intermolecular C–H⋯O (C⋯O = 3.484 Å) hydrogen bonds (Fig. S2), and C–H⋯S (C⋯S = 3.670 Å) hydrogen bonds along the a direction (Fig. S3). Thus, adjacent parallel double–helical chains are extended by these interactions to generate a 2D network. The 3D framework of 4 is illustrated in Fig. S4. It should be pointed out that the hydrogen bonds could have a positive effect on the final structure of the complexes 4 and 5.
[Hg(tmtt)2] 6
X-ray crystallographic analysis shows that 6 crystallises in the monoclinic space group P2(1)/c, and the mononuclear two-coordinated environment around the HgII is shown in Fig. 6. The HgII ion, lying at the crystallographic centre of symmetry, is strongly coordinated by two sulfur atoms from two sulfhydryl groups, forming a linear HgS2 coordination geometry. In the structure, S-Hg-S lies in a straight line and the Hg–S distance of 2.362(2) Å is shorter than that found in [HgI2(imtH2)] (imtH2 = 1,3-imidazole-2-thione) which is 2.460(2) Å.[31] The thiadiazole (the mean deviation from the plane is 0.003 Å) and triazole (the mean deviation from the plane is 0.0034 Å) rings in the same ligand are distorted, with a dihedral angle of 60.9°. As illustrated in Fig. S5, [Hg(tmtt)2] units are connected by Hg⋯N (2.858 Å) interactions, which are less than the sum of the van der Waals radii (3.1 Å)[32] and can be considered as relatively strong interactions, forming a 1D chain structure. Adjacent chains are arranged in a parallel fashion by intermolecular weak interactions (Hg⋯Hg = 4.421 Å; Hg⋯S = 3.344 Å) to generate a two-dimensional network extended along the bc plane, further resulting in a 3D supramolecular architecture (Fig. S6).
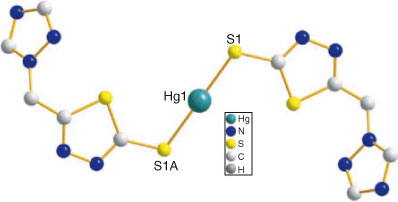
|
Thermogravimetric Analyses
To characterise the polymers more fully in terms of thermal stability, thermogravimetric analyses of the complexes 1–6 were investigated by thermal gravimetric (TG) and differential scanning calorimetry (DSC). The decomposition steps of the complexes with the corresponding weight losses are illustrated in Figs S7–S13.
TG data (Fig. S8) shows that 1 is stable in the solid state in air up to ~304°C, above which its decomposition is detected. There is a two-step weight loss in the range of 304–720°C, corresponding to the decomposition of ligands. Finally, a plateau region is observed after 720°C. A white powdered residue of CdO (observed 25.78 %; calculated 25.26 %) remains. Correspondingly, the DSC curve shows clearly two strong exothermic peaks at 325°C and 680°C, and a weak endothermic peak at 300°C, respectively.
TG data shows that polymer 2 is stable without weight loss until 300°C (see Fig. S9). The DSC curve shows two very strong exothermic peaks at 360°C and 840°C, and two weak endothermic peaks at 350°C and 850°C, respectively. The TG data shows that there is a two-step weight loss in the range of 300 to 820°C, which is in agreement with the decomposition of ligands. A white powdered residue of ZnO (observed 17.91 %; calculated 17.57 %) remains approaching 820°C.
For polymer 3 (Fig. S10), the first and second weight loss steps are from 280 to 350°C and from 350 to 575°C, respectively. The total weight loss of both steps is 57.45 %, which is attributed to the loss of ligands. Finally, a plateau region is observed from 575 to 750°C. The remaining weight of 37.35 % corresponds to the percentage (calculated 37.00 %) of the Pb and O components, indicating that the final product is PbO. Correspondingly, the DSC curve shows clearly one strong exothermic peak at 575°C and a weak exothermic peak at 315°C, and one quite weak endothermic peak at 295°C, respectively.
TG data of polymer 4 (Fig. S11) shows that there is a three-step weight loss in the range of 150–540°C. The first weight loss is from 150 to 200°C, corresponding to the loss of coordinated water molecules. The second and third weight loss steps are from 200 to 535°C, which correspond to the loss of ligands. A green powdered residue of NiO (observed 15.61 %; calculated 15.22 %) remains approaching 700°C. The DSC curve shows two very weak exothermic peaks at 240°C and 350°C, one strong exothermic peak at 515°C, and one weak endothermic peak at 200°C.
As for polymer 5 (Fig. S12), the thermal stability is similar to that of 4. The first weight loss is from 115 to 175°C, corresponding to the loss of coordinated water molecules. The second and third weight loss steps are from 175 to 700°C, corresponding to the loss of the ligands. The remaining weight of 15.82 % corresponds to the percentage (calculated 15.26 %) of Co and O components, indicating that the final product is CoO. The DSC curve also shows two very weak exothermic peaks at 310°C and 600°C, one strong exothermic peak at 580°C, and one weak endothermic peak at 185°C.
In the TG curve of complex 6 (Fig. S13), there is no weight loss until 145°C. Then there is a three-step weight loss in the range of 145–725°C, corresponding to the decomposition of ligands. Finally, a plateau region is observed from 725 to 800°C. In contrast to complexes 1–5, the remaining weight of 0 % corresponds to the volatilisation of Hg vapour. Apparently, we can find that there is one weak endothermic peak at 745°C and one strong endothermic peak at 265°C, and four weak exothermic peaks at 265, 305, 450, and 720°C, respectively.
Photoluminescent Properties
It is well known that coordination polymers constructed by d10 and s2 metal centres and conjugated organic linkers are promising candidates for photoactive materials, with potential applications such as chemical sensors and in photochemistry.[33] These crystalline solids usually display controllable photoluminescence properties.[34,35] Therefore, the solid-state luminescent properties of complexes 1–3 and 6 were investigated at room temperature together with the free tmttH ligand for comparison, as shown in Fig. 7. On complexation of the ligand with different metal ions, different luminescences were observed in the solid state. The free tmttH ligand exhibits a broad weak luminescent emission band with two emission peaks at 358 and 410 nm (λex = 290 nm), respectively, which can be ascribed to the π–π* and/or n–π* transitions as previously reported.[36] Very weak emission bands at 475 and 600 nm (λex = 290 nm) for complex 1, and intense bands at 630 nm (λex = 250 nm) for complex 2 and 465 and 585 nm (λex = 310 nm) for complex 3 were observed, respectively. Since the ZnII and CdII ions are difficult to be oxidised or reduced, the emission bands of 1 and 2 may be attributed mainly to ligand-to-ligand charge transfer (LLCT), which is mixed with metal-to-ligand charge transfer (MLCT) and ligand-to-metal charge transfer (LMCT).[37] As shown in Fig. 7, the luminescent intensity of complex 2 is almost sixty-fold larger than that of complex 1. A possible reason is that nitrogen atoms from the thiadiazole rings coordinate to the metal ions in 2, which can increase the overall conjugation of the entire system and reduces the loss of energy via radiationless decay intraligand emission from the excited state.[38] For 3, the luminescent emissions can be attributed to a metal-centred transition involving the s and p metal orbitals, which has been commonly observed in other s2-metal complexes.[39,40] Compared with 2, the central metal ions of 3 may be joined together by nitrogen and terminal sulfur atoms of the ligands, which cannot produce a large conjugated system, resulting in the reduction of the luminescence intensity. Complex 6 displays very weak luminescence with almost no emission, which is attributed to the heavy-atom effect (the graph of 6 is omitted).[41]

|
Conclusions
Six new coordination complexes based on the newly designed mercapto-thiadiazole ligand tmttH have been successfully synthesised under solvothermal conditions. Their molecular structures have been characterised by single-crystal X-ray diffraction, elemental analysis, thermal analysis, and IR spectroscopy. The strong coordination ability and different coordination modes of the ligand tmttH was confirmed from both theoretical and experimental aspects. It was found that the structural types of complexes 1–6 are strongly influenced by the reaction factors including ligands and metal ions amongst other things. In addition, the luminescence properties of complexes are not only influenced by the ligand, but depend on the structures of complexes and their central metal ion. These observations indicate that 2 and 3 may serve as excellent candidates for potential photoactive materials.
Experimental
Materials and General Methods
The ligand tmttH was prepared according to a literature procedure.[42] Other chemicals were commercially available and used without further purification.
Elemental analyses (C, H, and N) were carried out on a Carlo-Erba1106 elemental analyser. IR data were recorded on a BRUKER TENSOR 27 spectrophotometer as KBr pellets in the 400–4000 cm–1 region. TG-DSC measurements were performed by heating the crystalline samples from 20 to 800°C at a rate of 10°C min–1 in air on a Netzsch STA 409PC differential thermal analyser. Fluorescence spectra were obtained at room temperature by an F-4500 fluorescence spectrophotometer.
Preparation of [Cd(tmtt)2]n 1
tmttH (0.0079 g, 0.04 mmol) in DMF (1 mL) was added to a solution of CdCl2·2H2O (0.0045 g, 0.02 mmol) in water (4 mL). The mixture was then placed in a Teflon-lined stainless steel vessel (20 mL) and heated at 110°C for 36 h, followed by slowly cooling to room temperature at a rate of 5°C h–1. Yellow transparent block crystals of complex 1 (56 % yield) suitable for X-ray diffraction were separated. (Found: C 23.43, H 1.64, N 27.82. C10H8 CdN10S4 requires C 23.60, H 1.57, N 27.54 %). νmax (KBr)/cm–1 3432 (s), 1636 (w), 1516 (m), 1480 (w), 1368 (w), 1350 (s), 1287 (m), 1216 (m), 1160 (w), 1128 (s), 1062 (s), 1041 (s), 987 (m), 942 (w), 784 (w), 664 (m), 476 (w).
Preparation of [Zn(tmtt)2]n 2
An ethanolic solution (1 mL) of tmttH (0.0079 g, 0.04 mmol) was added dropwise to a solution of Zn(NO3)2·6H2O (0.0059 g, 0.02 mmol) in H2O (4 mL), then placed in a Teflon-lined stainless steel vessel (20 mL). The mixture was heated at 110°C for 3 days, then cooled to room temperature at a rate of 5°C h–1. Yellow, transparent block-shaped crystals of complex 2 (65 % yield) were obtained. (Found: C 26.17, H 1.60, N 30.22. C10H8N10S4Zn requires C 26.03, H 1.74, N 30.37 %). νmax (KBr)/cm–1 3447 (s), 1634 (w), 1524 (m), 1420 (w), 1357 (m), 1280 (s), 1214 (m), 1164 (w), 1126 (s), 1103 (s), 993 (s), 909 (w), 883 (w), 790 (w), 660 (m), 484 (w).
Preparation of [Pb(tmtt)2]n 3
A mixture of Pb(OAc)2 (0.0075 g, 0.02 mmol), tmttH (0.0079 g, 0.04 mmol), ethanol (3 mL), and H2O (2 mL) were added to a Teflon-lined stainless steel vessel, and the resulting mixture was heated at 100°C for 36 h, before cooling to room temperature at a rate of 5°C h–1. Yellow block crystals of complex 3 (55 % yield) were isolated. (Found: C 20.06, H 1.48, N 23.93. C10H8N10PbS4 requires C 19.90, H 1.33, N 23.22 %). νmax (KBr)/cm–1 3444 (s), 1635 (w), 1503 (s), 1466 (w), 1426 (w), 1319 (s), 1273 (m), 1225 (w), 1128 (s), 1085 (s), 1046.18(s), 1008 (m), 967 (m), 673 (m), 614 (w), 553 (w).
Preparation of [Ni(tmtt)2·(H2O)2]n 4
A methanolic solution (3 mL) of tmttH (0.0079 g, 0.04 mmol) was added dropwise to a solution of Ni(NO3)2·6H2O (0.0058 g, 0.02 mmol) in H2O (2 mL), then placed in a Teflon-lined stainless steel vessel (20 mL). The resulting mixture was heated at 105°C for 36 h, followed by slowly cooling to room temperature at a rate of 5°C h–1. Pure green crystals of complex 4 (60 % yield) were collected. (Found: C 24.58, H 2.31, N 28.69. C10H12N10NiO2S4 requires C 24.45, H 2.44, N 28.54 %). νmax (KBr)/cm–1 3125 (s), 3103 (s), 1770 (s), 1679 (m), 1573 (w), 1472 (w), 1528 (m), 1312 (s), 1298 (s), 1207 (m), 1127 (s), 1094 (s), 1012 (m), 994 (m), 887 (w), 673 (s), 494 (w).
Preparation of [Co(tmtt)2·(H2O)2]n 5
Complex 5 was prepared following the same synthetic procedure as that of 4 using Co(NO3)2·6H2O as metal salt. The resulting product were pure red block crystals (62.5 % yield). (Found: C 24.59, H 2.28, N 28.67. C10H12CoN10O2S4 requires C 24.45, H 2.44, N 28.54 %). νmax (KBr)/cm–1 3396 (s), 1774 (w), 1625 (w), 1526 (m), 1431 (w), 1351 (w), 1310 (s), 1297 (s), 1206 (m), 1128 (s), 1091 (s), 1011 (m), 993 (m), 939 (w), 673 (s), 473 (w).
Preparation of [Hg(tmtt)2] 6
In a Teflon-lined steel vessel, a solution containing a methanol (3 mL) solution of tmttH (0.0079 g, 0.04 mmol) was mixed with a solution of Hg(OAc)2 (0.0063 g, 0.01 mmol) in H2O (2 mL). The resulting mixture was heated at 105°C for 36 h, then cooled to room temperature at a rate of 5°C h–1, from which yellow massive crystals of complex 6 were produced (65 % yield). (Found: C 21.39, H 1.31, N 25.05. C20H16Hg2N20S4 requires C 21.58, H 1.44, N 25.18 %). νmax (KBr)/cm–1 2973 (s), 1504 (s), 1430 (m), 1354 (s), 1278 (m), 1210 (m), 1156 (w), 1127 (s), 1337 (s), 1210 (m), 1156 (w), 1127 (s), 790 (w), 766 (m), 659 (s), 627 (w), 535 (w).
X-ray Crystallographic Study
Crystal data collection and refinement parameters for complexes 1–6 are given in Table 1. Single-crystal X-ray data of 1, 2, 3, 5, and 6 were collected on a Rigaku Saturn 724 CCD diffractometer with graphite monochromatic Mo-Kα radiation (λ = 0.71073 Å); for 4, single-crystal X-ray data were collected on a Rigaku D/max-3B diffractometer with graphite monochromatic Cu-Kα radiation (λ = 1.5406 Å). Single crystals were selected and mounted on a glass fibre. The data were collected at a temperature of 291(3) K and corrected for Lorentz-polarisation effects. A correction for secondary extinction was applied. The structures were solved by direct methods and expanded using Fourier techniques. All of the non-hydrogen atoms were refined anisotropically. The hydrogen atoms were included but not refined. The final cycle of full-matrix least-squares refinement was based on observed reflections and variable parameters. All calculations were performed using the SHELXL-97 crystallographic software package.[43] Selected bond lengths and bond angles are tabulated in Table S1 (Supplementary Material). CCDC numbers 945292–945297 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Supplementary Material
The optimised geometries and non-bonding orbital charge distributions of the free ligand and the TG and DSC signal curves for complexes 1–6 are available on the Journal’s website.
Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (Nos. 20801048, 20971110, and J1210060), Program for New Century Excellent Talents of Ministry of Education of China (NCET-07–0765), and the Outstanding Talented Persons Foundation of Henan Province.
References
[1] (a) P. Dechambenoit, J. R. Long, Chem. Soc. Rev. 2011, 40, 3249.| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXmsVWkurY%3D&md5=c82b44ef6f6af9c37fb4211b73290e98CAS | 21298169PubMed |
(b) H.-L. Jiang, Q. Xu, Chem. Commun. 2011, 47, 3351.
| Crossref | GoogleScholarGoogle Scholar |
(c) Z.-J. Lin, T.-F. Liu, X.-L. Zhao, R. Cao, Cryst. Growth Des. 2011, 11, 4284.
| Crossref | GoogleScholarGoogle Scholar |
(d) L. Hou, W.-J. Shi, Y.-Y. Wang, Y. Guo, C. Jin, Q.-Z. Shi, Chem. Commun. 2011, 47, 5464.
| Crossref | GoogleScholarGoogle Scholar |
(e) Z.-Z. Lu, R. Zhang, Y.-Z. Li, Z.-J. Guo, H.-G. Zheng, J. Am. Chem. Soc. 2011, 133, 4172.
| Crossref | GoogleScholarGoogle Scholar |
(f) I. Eryazici, O. K. Farha, O. C. Compton, C. Stern, J. T. Hupp, S. T. Nguyen, Dalton Trans. 2011, 40, 9189.
| Crossref | GoogleScholarGoogle Scholar |
(g) M. V. Lucky, S. Sivakumar, M. L. P. Reddy, A. K. Paul, S. Natarajan, Cryst. Growth Des. 2011, 11, 857.
| Crossref | GoogleScholarGoogle Scholar |
(h) F. Nouar, J. Eckert, J. F. Eubank, P. Forster, M. Eddaoudi, J. Am. Chem. Soc. 2009, 131, 2864.
| Crossref | GoogleScholarGoogle Scholar |
(i) H.-X. Deng, S. Grunder, K. E. Cordova, C. Valente, H. Furukawa, M. Hmadeh, F. Gándara, A. C. Whalley, Z. Liu, S. Asahina, H. Kazumori, M. O’Keeffe, O. Terasaki, J. F. Stoddart, O. M. Yaghi, Science 2012, 336, 1018.
| Crossref | GoogleScholarGoogle Scholar |
[2] L.-L. Song, Q.-H. Jin, L.-N. Cui, C.-L. Zhang, Inorg. Chim. Acta 2010, 363, 2425.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXpt1amtb8%3D&md5=a3214d48ea70d09617ecf2e0150e84feCAS |
[3] I. T. Ahmed, J. Chem. Eng. Data 2003, 48, 272.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXivFajuw%3D%3D&md5=8d1003a7e944e8017b27fedf85c2b235CAS |
[4] M. Du, Q. Wang, C.-P. Li, X.-J. Zhao, J. Ribas, Cryst. Growth Des. 2010, 10, 3285.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXms1Shsrg%3D&md5=7225f37c176995ac4cc1f08fd7d292ebCAS |
[5] (a) P.-P. Liu, A.-L. Cheng, Q. Yue, N. Liu, W.-W. Sun, E.-Q. Gao, Cryst. Growth Des. 2008, 8, 1668.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXkvVKgt7c%3D&md5=a056f796ce8a177d2d82cc792bcc72e5CAS |
(b) T.-L. Hu, R.-Q. Zou, J.-R. Li, X.-H. Bu, Dalton Trans. 2008, 1302.
| Crossref | GoogleScholarGoogle Scholar |
[6] (a) J.-S. Hu, L.-F. Huang, X.-Q. Yao, L. Qin, Y.-Z. Li, Z.-J. Guo, H.-G. Zheng, Z.-L. Xue, Inorg. Chem. 2011, 50, 2404.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhs1ahsbY%3D&md5=0c6cd6df4ffbb139c28e9d8769c5f6cdCAS |
(b) D. P. Halbach, C. G. Hamaker, J. Organomet. Chem. 2006, 691, 3349.
| Crossref | GoogleScholarGoogle Scholar |
[7] C.-L. Ma, J.-H. Zhang, R.-F. Zhang, Polyhedron 2004, 23, 1981.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXltlyrtLw%3D&md5=5d4bb9f155126794e130af616cffde5fCAS |
[8] M. Ghassemzadeh, S. Bahemmat, M. Mahmoodabadi, B. Rezaii-Rad, H. H. Monfared, E. Mottefakeri, B. Neumuller, Polyhedron 2010, 29, 3036.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXht12ns7nE&md5=3f26e65af818d2615a00c448eafee7ccCAS |
[9] Q. Q. Liang, Z. Y. Liu, E. C. Yang, X. J. Zhao, Z. Anorg. Allg. Chem. 2009, 635, 2653.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhs1WnsrfN&md5=5be52ef8920b20619ef94376b9dc127cCAS |
[10] J.-P. Zhang, Y.-Y. Lin, X.-C. Huang, X.-M. Chen, J. Am. Chem. Soc. 2005, 127, 5495.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXis1ahsL8%3D&md5=908971e8177ad138325689c2494dd16eCAS | 15826187PubMed |
[11] W. Ouellette, M.-H. Yu, C. J. O’Connor, D. Hagrman, J. Zubieta, Angew. Chem. 2006, 45, 3497.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XlsVKjsbk%3D&md5=5aea6d9308d22a85a7c22b594de98287CAS |
[12] W. Ouellette, A. V. Prosvirin, V. Chieffo, K. R. Dunbar, B. Hudson, J. Zubieta, Inorg. Chem. 2006, 45, 9346.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XhtVyntb%2FO&md5=3be4701879ddd1442a8bdfc69723cff1CAS | 17083234PubMed |
[13] Q.-G. Zhai, X.-Y. Wu, S.-M. Chen, C.-Z. Lu, W.-B. Yang, Cryst. Growth Des. 2006, 6, 2126.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28Xot1Chu7s%3D&md5=c3810fb520a4a921ed82309573f35446CAS |
[14] J.-J. Zhao, L.-K. Li, C.-H. Wang, W.-J. Li, R.-N. Wang, X.-F. Zheng, H.-W. Hou, Inorg. Chem. Commun. 2012, 20, 205.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XnslGquro%3D&md5=1282292c889632759cc8d1bc4431621eCAS |
[15] K.-B. Chew, M. T. H. Tarafder, K. A. Crousea, A. M. Ali, B. M. Yamin, H.-K. Fun, Polyhedron 2004, 23, 1385.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXjs1ersr8%3D&md5=61bb98711f89f7fc82bfb251d83ee501CAS |
[16] (a) J. Yang, J.-F. Ma, Y.-Y. Liu, S. R. Batten, CrystEngComm 2009, 11, 151.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVCqsw%3D%3D&md5=0f307ebbaaf655a94ad1f9adca97fa02CAS |
(b) Y. Ma, A.-L. Cheng, J.-Y. Zhang, Q. Yue, E.-Q. Gao, Cryst. Growth Des. 2009, 9, 867.
| Crossref | GoogleScholarGoogle Scholar |
(c) Y. Qi, Y.-X. Che, J.-M. Zheng, Cryst. Growth Des. 2008, 8, 3602.
| Crossref | GoogleScholarGoogle Scholar |
(d) Y.-Q. Wei, Y.-F. Yu, K.-C. Wu, Cryst. Growth Des. 2007, 7, 2262.
| Crossref | GoogleScholarGoogle Scholar |
[17] L. Yi, X. Yang, T.-B. Lu, P. Cheng, Cryst. Growth Des. 2005, 5, 1215.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXht1aqtLw%3D&md5=28b86c6f1366e39fbc9c303ed33e5896CAS |
[18] G. A. Farnum, J. S. Lucas, C.-Y. Wang, R. L. LaDuca, Inorg. Chim. Acta 2011, 368, 84.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXisVyrt7k%3D&md5=6db96326e95214b29ff4c018e1f50da3CAS |
[19] L.-F. Ma, L.-Y. Wang, Y.-Y. Wang, CrystEngComm 2009, 11, 109.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVCrtw%3D%3D&md5=f72d7a83ab8907aeea3674dc90922c2dCAS |
[20] L.-F. Ma, L.-Y. Wang, Y.-Y. Wang, Inorg. Chem. 2009, 48, 915.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXmsVeq&md5=9303877402e44a18d0a19195676bd295CAS | 19117421PubMed |
[21] G.-L. Wen, Y.-Y. Wang, H. Wang, J. Mol. Struct. 2009, 928, 125.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXms1els7o%3D&md5=990dc8b2150b717c8b2c271e9fb4123eCAS |
[22] Z. Dong, W.-H. Zhang, Y.-Y. Wang, Chin. Sci. Bull. 2009, 54, 4285.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXltlansw%3D%3D&md5=687ef0332f0781c12cfdb9aa48314495CAS |
[23] (a) J.-Q. Liu, Y.-Y. Wang, L.-F. Ma, CrystEngComm 2008, 10, 1123.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtV2rtbrF&md5=7241f92f3d7e2daf371ace2bbe03f601CAS |
(b) L. Yi, X. Yang, T.-B. Lu, P. Cheng, Cryst. Growth Des. 2005, 5, 1215.
| Crossref | GoogleScholarGoogle Scholar |
[24] S. Hino, M. Olmstead, A. D. Phillips, R. J. Wright, P. P. Power, Inorg. Chem. 2004, 43, 7346.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXotlaqsrs%3D&md5=498f059ee4397957fde650e1e3038587CAS | 15530084PubMed |
[25] Y.-Z. Yuan, J. Zhou, X. Liu, L.-H. Liu, K.-B. Yu, Inorg. Chem. Commun. 2007, 10, 475.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXitl2gu7g%3D&md5=4ec5a8f6fb85846bd87eb22c7002ed2aCAS |
[26] J. S. Casas, E. E. Castellano, J. Ellena, Polyhedron 2007, 26, 4228.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXpvVCjsbw%3D&md5=6acca73b2b6ef06d486f43aefee60dfbCAS |
[27] A. R. Mahjoub, A. Morsali, Polyhedron 2002, 21, 1223.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XksFKrtLg%3D&md5=558d1b1e2940fb044804943a1c8ea24bCAS |
[28] L. Qin, J.-S. Hu, L.-F. Huang, Y.-Z. Li, Z.-J. Guo, H.-G. Zheng, Cryst. Growth Des. 2010, 10, 4176.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXptFyjt74%3D&md5=b397a06ed43780c6f996b9f835269894CAS |
[29] D. Maity, S. Chattopadhyay, A. Ghosh, Inorg. Chim. Acta 2011, 365, 25.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhs1Wjsb3P&md5=3be0b4a8d5c7476cdef7e5ebdc3531f1CAS |
[30] L.-F. Song, C.-H. Jiang, C.-L. Jiao, Cryst. Growth Des. 2010, 10, 5020.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtleitbvP&md5=2815c09da70e5fcc6e7b00cee45224a7CAS |
[31] Z. Popović, D. Matković-Čalogović, Z. Soldin, G. Pavlović, N. Davidović, D. Vikić-Topić, Inorg. Chim. Acta 1999, 294, 35.
| Crossref | GoogleScholarGoogle Scholar |
[32] A. J. Canty, G. B. Deacon, Inorg. Chim. Acta 1980, 45, L225.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2cXhvFyjurc%3D&md5=7170c42883c447bc72a009c5fd2086a7CAS |
[33] (a) J. E. McGarrah, Y. J. Kim, M. Hissler, R. Eisenberg, Inorg. Chem. 2001, 40, 4510.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXlsVSrsLw%3D&md5=2ae8ce5ec07bd0be44a20b8ce341a97fCAS | 11511190PubMed |
(b) G. De Santis, L. Fabbrizzi, M. Licchelli, A. Poggi, A. Taglietti, Angew. Chem. 1996, 35, 202.
| Crossref | GoogleScholarGoogle Scholar |
[34] (a) S.-L. Zheng, X. M. Chen, Aust. J. Chem. 2004, 57, 703.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXmsFSksr8%3D&md5=2b33f536ee6ba29a7158f5dd32a89ddaCAS |
(b) S.-L. Zheng, J.-H. Yang, X.-L. Yu, X.-M. Chen, W.-T. Wong, Inorg. Chem. 2004, 43, 830.
| Crossref | GoogleScholarGoogle Scholar |
[35] M. D. Allendorf, C. A. Bauer, R. K. Bhakta, R. J. T. Houk, Chem. Soc. Rev. 2009, 38, 1330.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXkvVamurg%3D&md5=b9af394ddf4e2d27aa4e63c2518eef04CAS | 19384441PubMed |
[36] (a) W.-Q. Kan, Y.-Y. Liu, J. Yang, Y.-Y. Liu, J.-F. Ma, CrystEngComm 2011, 13, 4256.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXnsFKqt7s%3D&md5=78c783631a0adc4409cc91f15a5dbe70CAS |
(b) Y.-Y. Liu, C. Chen, J.-F. Ma, J. Yang, CrystEngComm 2012, 14, 6201.
| Crossref | GoogleScholarGoogle Scholar |
[37] (a) L.-F. Ma, L.-Y. Wang, J.-L. Hu, Y.-Y. Wang, G.-P. Yang, Cryst. Growth Des. 2009, 9, 5334.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtlKms7zF&md5=c641f2d962c83dfcbdb63a60e80be54bCAS |
(b) Y. Gong, J. Li, J. Qin, T. Wu, R. Cao, J. Li, Cryst. Growth Des. 2011, 11, 1662.
| Crossref | GoogleScholarGoogle Scholar |
[38] C.-Z. Mei, W.-W. Shan, B.-T. Liu, Spectrochim. Acta [A] 2011, 81, 764.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtF2rtr%2FO&md5=6e22f0817e3c7e1f7742e6942ba9fa61CAS |
[39] G.-P. Yang, L. Hou, Y.-Y. Wang, Y.-N. Zhang, Q.-Z. Shi, S. R. Batten, Cryst. Growth Des. 2011, 11, 936.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjtVKhsLc%3D&md5=f9bddaaba402226fe16a8e462639d626CAS |
[40] Y.-Y. Zhang, S.-X. Liu, K.-K. Du, M.-X. Xue, Inorg. Chem. Commun. 2010, 13, 641.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXlsVGktbs%3D&md5=bd6564d5358e025f075ae49f7b20db67CAS |
[41] (a) C. Seward, J. Chan, D. Song, S.-N. Wang, Inorg. Chem. 2003, 42, 1112.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXlsFKgtA%3D%3D&md5=77a03d6242560e291761b89169bed75bCAS | 12588146PubMed |
(b) R. S. Drago, Physical Methods in Chemistry 1977, Ch 5 (Saunders: Philadelphia).
(c) T. C. Werner, W. Hawkins, J. Facci, R. Torrisi, T. Trembath, J. Phys. Chem. 1978, 82, 298.
| Crossref | GoogleScholarGoogle Scholar |
(d) M. C. Aragoni, M. Arca, F. Demartin, F. A. Devillanova, F. Isaia, A. Garau, V. Lippolis, F. Jalali, U. Papke, M. Shamsipur, L. Tei, A. Yari, G. Verani, Inorg. Chem. 2002, 41, 6623.
| Crossref | GoogleScholarGoogle Scholar |
(e) Y.-Y. Zhang, S.-X. Liu, K.-K. Du, M.-X. Xue, Inorg. Chem. Commun. 2010, 13, 64.
[42] Z.-Q. Hu, Y.-X. Yang, Y.-Q. Shang, K. Zhou, L.-Z. Xu, Acta Crystallogr. 2006, E62, o3457.
[43] G. M. Sheldrick, SHELX-97, Program for the Solution and Refinement of Crystal Structures, 1997 (University of Göttingen, Germany).