

Structural Diversity and Properties of Five 3D Metal–Organic Frameworks Based on a Pyridine-substituted Triazolyl Benzoate Ligand
Fen-Jun Jiang A , Ming Zhang B , Xiao-Hua Wei B , Lin-Yan Yang B , Sheng-Yun Liao B , Pei-Yao Du B , Jin-Lei Tian B , Wen Gu B and Xin Liu B CA School of Medicine, Nankai University, Tianjin, China 300071.
B Department of Chemistry, Nankai University, Tianjin, China 300071.
C Corresponding author. Email: liuxin64@nankai.edu.cn
Australian Journal of Chemistry 67(2) 302-310 https://doi.org/10.1071/CH13458
Submitted: 31 August 2013 Accepted: 16 October 2013 Published: 28 November 2013
Abstract
Three new 3D metal–organic frameworks, namely {[Zn(L)2]}n (2), {[Cu(L)3H2O]3H2O}n (4), and {[Ag(L)]3H2O}n (5) where HL = 4-[3-methyl-5-(pyridin-4-yl)-1,2,4-triazol-4-yl]benzoate, have been synthesized by reaction of the HL ligand and ZnII, CuII, and AgI salts under similar experimental conditions. By introducing the secondary ligand terephthalic acid (H2bdc), another two new compounds {[Zn1.5L2(bdc)0.5]3H2O}n (1) and {[Cu(L)(bdc)0.5H2O]}n (3) with different 3D structures were obtained. Compound 1 possesses a three-fold interpenetrating framework, with {32.42.54.62}2{32.42.56.65} topology. To the best of our knowledge, 2 is an unusual example of four-fold framework guest-free metal organic framework material. Compound 3 reveals a seven-connected ose topology; magnetic susceptibility measurements indicate that it has dominating antiferromagnetic couplings between metal centres. Photoluminescence measurements of 1, 2, and 5 in the solid state at room temperature show that all coordination networks exhibit a red shift in the emission spectra, which can be assigned to an intraligand π-π* transition.
Introduction
Microporous materials, such as coordination polymers, so-called metal organic frameworks (MOFs), and microporous organic polymers, have gained increasing interest in the last decade.[1,2] Because of their distinct properties as porous materials, this novel class of hybrid materials not only exhibits a high potential for such applications as gas separation and storage,[3] luminescence,[4] and heterogeneous catalysis,[5] but also for their aesthetic topologies.[6] However, for most MOFs, it is much easier to collapse into non-porous frameworks after guest removal.[7] Therefore, how to retain the pores may be a very challenging task. Although interpenetration in porous MOFs may greatly reduce the volume of pores, it is a very effective chemical approach for retaining pores.[8] Furthermore, excellent gas adsorption capacity has been proved possible for interpenetrating coordination networks. The interpenetration may be able to sustain a framework showing dynamic porosity. For example, the cavity inside an interpenetrating coordination polymer can change by sliding between the individual networks.
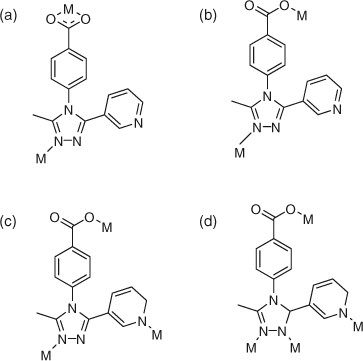
Carboxylate ligands have been extensively studied as polyfunctional linkers for their abundant coordination modes to metal ions[9] allowing various structural topologies. Apart from this, MOFs containing linkers with a combination of both neutral donor groups such as pyridines or 1,2,4-triazoles and charged functional groups such as carboxylates are of interest. Conventionally, an effective strategy for the assembly of variable topological networks and interpenetrating frameworks is to use metal centres with a suitable geometry and connectivity as the nodes along with the rational design of organic spacers.[10] Thus, organic building blocks with a specific geometry, such as linear,[11] tripodal,[12] T-shaped,[13] and rectangular ligands[14], can serve as excellent candidates to construct such frameworks. It is very rare to obtain interpenetrating coordination polymer by using these ligands.[15] Hence, in order to synthesize new coordination polymers, we presented 4-[3-methyl-5-(pyridin-4-yl)-1,2,4-triazol-4-yl] benzoate as ligand with multiple binding sites (Chart 1). On the basis of the ligand 4-[3-methyl-5-(pyridin-4-yl)-1,2,4-triazol-4-yl] benzoate, herein we present five new coordination polymers prepared under similar experimental conditions, namely {[Zn1.5(L)2(bdc)0.5]3H2O}n (1), {[Zn(L)2]}n (2), {[Cu(L)(bdc)0.5H2O]}n (3), {[Cu(L)]3H2O}n (4), and {[Ag(L)]3H2O}n (5) where HL = 4-[3-methyl-5-(pyridin-4-yl)-1,2,4-triazol-4-yl] benzoate and H2bdc = terephthalic acid. Compounds 2, 4, and 5 are linked by L anions; 1 and 5 were obtained through the introduction of the terephthalic acid ligand into the 2 and 4 frameworks. Compound 1–5 have totally different three-dimensional (3D) architectures. Compound 1 possesses a three-fold interpenetrating framework, with {32.42.54.62}2{32.42.56.65} topology. Compound 2 is an unusual stable example of a four-fold framework guest-free MOF material. Compound 3 reveals a seven-connected ose topology. Their crystal structures, topological analyses, magnetic properties, luminescence measurements, and thermal properties were studied in detail.
|
Results and Discussion
Structure of {[Zn1.5(L)2(bdc)0.5]3H2O}n (1)
The structure of 1 is a 3D interpenetrating framework. X-ray analysis reveals that 1 crystallizes in the monoclinic space group C2/c. The asymmetric unit of 1 contains 1.5 independent ZnII cations, two L ligands, 0.5 bdc ligand, and three lattice water molecules. As shown in Fig. 1a, the Zn1II centre is five-coordinated by three O atoms from one L ligand (O5) and one bdc ligand (O3, O4) (Zn1–O = 1.947(4)–2.092(4) Å), and two N atoms (N2, N4D) from another two L ligands (Zn1–N = 2.075(4)–2.082(4) Å) in a distorted trigonal bipyramid coordination geometry. The Zn2II cation is six-coordinated by four carboxylate oxygen atoms from two L anions (Zn2–O = 2.139(5)–2.250(6) Å) and two nitrogen atoms from two L ligands (Zn2–N5 = 2.078(5) Å). The L anion shows μ2-η1 : η1 : η1 :η0 : η0 (Chart 1a) and μ2-η0 : η1 : η1 : η0 : η0 (Chart 1b) coordination modes. The L anions bridge the ZnII cations to form a 2D double chain (Fig. 1b). These chains are further connected by bdc ligands and L ligands to give rise to a pillar–layered 3D framework (Fig. 1c).

|
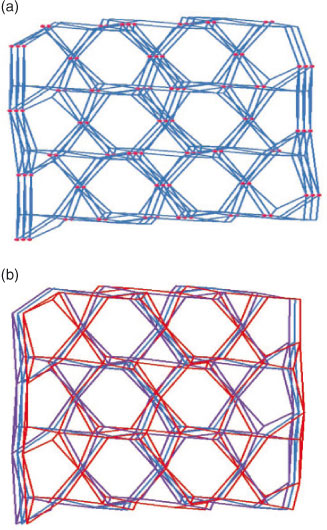
Topologically, the Zn1 cations can be defined as five-connected nodes, the Zn2 cation can be reduced to six-connected nodes, and the L anions and bdc ligands can be considered as linkers. On the basis of this simplification, the structure of 1 can be described as an unusual binodal (5,6)-connected topology; it is a new topology with a point symbol of {32.42.54.62}2{32.42.56.65} (Fig. 2a).
|
Moreover, in 1, there are solvent molecules occupying part of the void space, and another part of the potential voids are filled via mutual interpenetration of three independent equivalent frameworks, that is, the final structure of 1 is a three-fold interpenetrated net (Fig. 2b).
In 1, we used the bdc ligand as a linear ligand to coordinate with ZnII ions, providing a 3D pillar–layer framework. However, without the bdc ligand, what kind of structure would result?
Structure of {[Zn(L)2]}n (2)
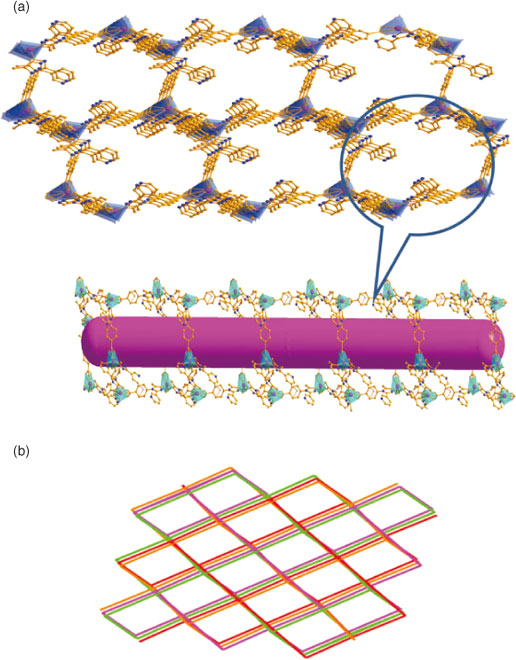
Fortunately, when we only used L as the tripodal ligand to assemble with the Zn ions, 2 was obtained in good yield. As shown in Fig. 3a, the structure of 2 contains one crystallographically unique ZnII cation and two L anions. Each ZnII cation is six-coordinated by four carboxylate oxygen atoms (Zn–O = 2.020(7)–2.283(7) Å) from two L anions and two nitrogen atoms (Zn–N = 2.071(7)–2.074(6) Å) from another two L ligands, forming a distorted ZnN2O4 octahedron (Fig. 3a). Two carboxylic groups and one nitrogen atom link neighbouring ZnII ions to form a 2D layer (Fig. 3b). The L anion bridges the 2D layers in a μ3 : η1 : η1 : η1 : η0 : η0 coordination mode to give rise to a complicated 3D framework (Fig. 4a). It is well known that when coordination polymeric compounds have large holes, the structures usually interpenetrate in order to satisfy close-packing; after carefully inspecting the structure of 2, in the hexatomic ring formed by six Zn ions or six L ligands, the long spacer (the longest Zn–Zn distance is 24.8 Å) in this compound and the void in the lattice is large enough to accommodate another equivalent network, allowing mutual interpenetration of equivalent frameworks. We found that there are four individual 3D networks interpenetrating due to the large void in the single net (Fig. 4b). It is noteworthy that there are still voids although four-fold interpenetration has taken place; the framework of 2 remains 1D open channels, with the dimensions of the window being 24.8 × 22.4 Å. Furthermore, no solvent resides in the void, which means 2 is stable after guest removal.

|
|
For compounds 1 and 2, the same metal ions but different ligand combinations were used. From the structural descriptions above, it can be seen that the HL ligand is a critical factor in the construction of various structures. In this work, two kinds of rigid ligands were used, namely H2bdc and HL, to observe their effect on the assembly of the coordination compounds. Compared with H2bdc, which is a linear ligand, HL is a tridentate triazolyl-type ligand. Ligand H2bdc acting as linker in 1 leads to a 3D three-fold pillar–layered framework; in contrast, 2 features a 3D four-fold framework. H2bdc, with two carboxylic groups extending in opposite directions, is shorter than HL, so the HL serves to increase the length of the links between nodes, which lead to larger free voids. The analyses above strongly demonstrate that the ligand HL is a promising building block for interpenetrating networks.
Structure of{[Cu(L)(bdc)0.5H2O]}n (3)
Single-crystal X-ray analysis of 3 indicates that 3 crystallizes in the P21/C space group. The asymmetric unit of 3 contains one CuII ion, one crystallographically independent L ligand, 0.5 bdc ligand, and one coordination water molecule. CuII is five-coordinated with a slightly distorted trigonal bipyramid environment formed by three carboxylic O atoms from one L ligand (O1), one bdc ligand (O2), and one terminal water molecule (O3), and two N atoms (N1 and N2A) from two different L ligands (Fig. 5a).

|
Interestingly, the CuII ions are bridged by three Ls to adjacent CuII ions and each L ligand links with three CuII ions to afford a 2D reticular structure (Fig. 5b). The 2D network is further connected by bdc ligands into a 3D supramolecular architecture (Fig. 5c). To understand the complicated crystal structure, an analysis of network topology is used. In 3, the CuII can be viewed as seven-connected nodes. Therefore, this structure can be simplified as a seven-connected topological network with the Schläfli symbol of {3^6.4^8.5^7}(Fig. 5d).
Structure of {[Cu(L)]3H2O}n (4) and {[Ag(L)]3H2O}n (5)
Single-crystal X-ray analysis of 4 and 5 indicates that they are isomorphous and crystallize in space group C2/C. Accordingly, the structure of 4 is described representatively here in detail. Single-crystal X-ray analysis shows that compound 4 consists of one CuI ion, one L ligand, and three lattice water molecules. The CuI cation is coordinated by one carboxylate oxygen atom from one L anion, and three nitrogen atoms from three different L ligands in a slightly distorted tetrahedral geometry (Fig. 6a). In addition, the isolated water molecules reside in the interspaces of the neutral channels. The bond lengths are Cu–N(1) = 1.987(3)Å, Cu–N2A = 1.988(3)Å, and Cu–N2B = 1.997(3) Å. Two nitrogen atoms and one oxygen atom of each L ligand link neighbouring CuI ions to form a 1D chain along the b axis (Fig. 6b). These dinuclear compounds are further connected into a (4,4) grid layer by L ions (Fig. 6c). The L ion acts as a pillar connecting these grid layers into a 3D framework with three different kinds of channels along [1 0 0], [0 –1 1], and [0 1 1] (Fig. 6d–f). It appears that, in contrast with other small multitopic ligands such as cyanide and azide, the L anion exhibits a large variety of bridging modes. It should be pointed out that although in compound 4, CuCO3·Cu(OH)2 is used as the starting reactant, the oxidation state of the copper atoms is +1 in the final products. Considering the fact that CuII compounds are usually a green or blue colour, whereas CuI compounds often exhibits a red or yellow colour, it is easily understandable that the HL molecule plays a key role in the formation of CuI compound crystals, in which HL is used as a reductant. Some similar reduction processes have been proposed to occur in the pyridine derivatives under solvothermal conditions.[16]

|
Ligand-directed Assembly for Diverse Structures
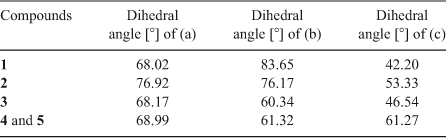
Ligand HL can be regarded as a potential tridentate ligand and reduced to a two-connected linker. As shown in Table 1, the dihedral angles between the pyridyl and phenyl rings vary from 68.02 to 76.92°. HL also adopts different and large dihedral angles between the triazolyl and the phenyl rings, and the pyridyl and the triazolyl rings in compounds 1–5. The results demonstrate that free rotation via the C–N and C–C bonds between aromatic rings allows the rigid ligand to take on appropriate ‘flexibility’.
|
In addition, in the five compounds, the ligand HL exhibits different coordination modes and this leads to the formation of various 3D architectures. Therefore, the rigid ligand can also be employed as a building block to achieve variable topological networks and interpenetrating frameworks.
X-Ray Powder Diffraction Results and Thermal Stabilities
To confirm whether the crystal structures are truly representative of the bulk materials, X-ray diffraction (XRD) experiments were carried out for 1–5. The XRD experimental and computer-simulated patterns of the compounds are shown in the Supporting Information (Fig. S2). The diffraction peaks of the as-synthesized samples are in good agreement with the simulated data, demonstrating the high phase purity of the compounds.
Compounds 1–5 were subjected to thermogravimetric analysis (TGA) to ascertain the stability of their respective supramolecular architectures (Fig. S1, Supporting Information). The TGA curve of 1 shows a weight loss in the temperature range from 26 to 85°C, corresponding to the loss of coordination water molecules, and the anhydrous compound begins to decompose at 243°C, leading to the formation of Zn3O4 as the residue (observed 33.13 %, calculated 33.46 %).The TGA curve of 2 shows that the compound does not lose any weight until 420°C, and then it decomposes at high temperature, suggesting no guest molecules in the lattice, as confirmed by the single-crystal X-ray diffraction analysis. For 3, first, a weight loss of 6.1 % is observed between 40 and 271°C, which can be assigned to the release of the coordinated water molecules (calculated 5.5 %). On further increasing the temperature, the ligand molecules start decomposing, forming an unidentified product. In the CuI and AgI compounds 4 and 5, the guest molecules hosted inside the framework voids could escape on heating, showing similar weight loss for both compounds in the temperature range 69–137°C. The decomposition of the organic components occurs at 224°C.
Luminescent Properties
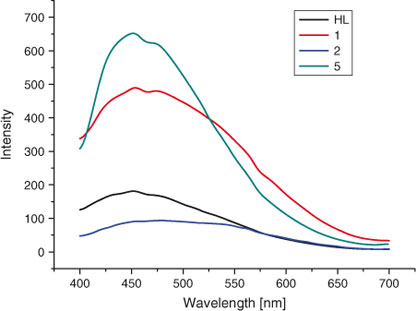
Previous studies have shown that coordination compounds containing ZnII and AgI cations exhibit photoluminescence. Therefore, the photoluminescent properties of compounds 1, 2, 5, and the free ligands were measured in the solid state, as shown in Fig. 7.
|
The emission peak of the free ligand is observed at ~450 nm (λex = 340 nm), resulting from π-π* transitions. For compounds 1, 2, and 5, these peaks are shifted to 458 (λex = 340 nm), 458 (λex = 350 nm), and 459 (λex = 344 nm) respectively. As the Zn2+ and Ag+ ions are difficult to oxidize or reduce due to their d10 configuration, the emissions of all compounds are neither metal-to ligand charge transfer (MLCT) nor ligand-to-metal charge transfer (LMCT) in nature. The red shift of the emission spectra observed in compounds 1, 2, and 5 may be attributed to the deprotonation of the ligands in the compounds. The difference of the intensity may result from different metal centres and conformations of the ligands as well as weak interactions in the interpenetrating crystalline lattice, which may affect the rigidity of the whole network and thus the energy transfer involved in the luminescence.
Adsorption Properties
To further explore the potential properties of the complexes with respect to gas separation, the adsorption isotherms of CO2 for 2 were measured. The adsorption isotherms of CO2 for 2 were measured up to 105 Pa (Fig. S3, Supporting Information). The CO2 uptake value for 2 was 5.89 cm3 g–1 at 273 K. It is worth noting that this storage capacity is far from saturated, which means that uptake can be further maximized at a higher pressure range (restricted by the maximum pressure of the instrument, the CO2 sorption isotherm was recorded only from 0 to 105 Pa in this work).
Magnetic Properties
Variable-temperature magnetic susceptibility measurements were performed on crystalline samples of 3 in the temperature range of 2–300 K with a magnetic field of 1000 Oe. The magnetic susceptibility (χMT) at 300 K is 0.44 electromagnetic units (EMU) K mol–1 for 3, which is higher than the expected value of 0.375 EMU K mol–1 for one CuII ion. With decreasing temperature, the χMT values decrease slowly. Below 100 K, the χMT product decreases more rapidly, reaching 0.12 cm3 K mol–1 Cu at 2 K. The sharp decrease of the χMT value at a very low temperature may be attributed to either a zero-field splitting factor or interdimer antiferromagnetic interactions. The magnetic data in the range of 2–300 K followed the Curie–Weiss law, with a Curie constant of C = 0.44 cm3 K mol–1 and a Weiss constant of θ = –18.2 K (Fig. S4 in the Supporting Information). The decrease of the χMT value and the negative θ value can be induced by antiferromagnetic coupling interactions within and between dimeric units as well as strong spin orbit coupling.
Experimental
Materials and General Methods
All reagents and solvents for synthesis and analysis were commercially available and used as received. Elemental analyses for C, H, and N were carried out on a Model 2400 II Perkin–Elmer elemental analyser. The XRD intensities were measured on a Rigaku D/max-IIIA diffractometer (Cu Kα, λ = 1.54056 Å). The crystalline powder samples were prepared by crushing the crystals and scanned from 3° to 60° with a step of 0.1° s–1. TGA experiments were performed in flowing air on a Netzsch TG 209 instrument with a heating rate of 10°C min1. Near-infrared spectra were recorded on an Edinburgh FLS-920P spectrophotometer. All chemicals were commercially available and were used as received without further purification. The ligands H(Me-3py-pba) and H(Me-4py-pba) were prepared according to Sharga et al.[17]
Magnetic Study
The magnetic susceptibility measurements of the polycrystalline samples were measured over the temperature range of 2–300 K with a Quantum Design MPMS-XL 7 Squid magnetometer using an applied magnetic field of 1000 Oe. Diamagnetic corrections to the observed susceptibilities were made with Pascal’s constants.
X-Ray Structure Determination
Diffraction data for compounds 1–5 were collected with a Bruker Smart Apex CCD instrument with graphite monochromated Mo Kα radiation (λ = 0.71073 Å). The data were collected at 293(2) K. Absorption corrections were made by multiscan methods. The structure was solved by charge flipping methods with the program Olex2 (http://www.olex2.org/content/olex2, accessed 31 October 2013) and refined by full-matrix least-squares methods on all F2 data with Olex2. Non-hydrogen atoms were refined anisotropically. Hydrogen atoms of water molecules were located in a difference Fourier map and refined isotropically in the final refinement cycles. Other hydrogen atoms were placed in calculated positions and refined by using a riding model. The final cycle of full-matrix least-squares refinement was based on observed reflections and variable parameters.
Synthesis of Single Crystals {[Zn1.5(L)2(bdc)0.5]3H2O}n (1)
A steel autoclave with a Teflon vessel was loaded with 42.0 mg (0.15 mmol) HL, 16.6 mg (0.1 mmol) H2bdc, 19.8 mg (0.1 mmol) ZnO·ZnCO3·4H2O (Sigma–Aldrich, American Chemical Society (ACS) 98 %), and 15.0 mL H2O/CH3CH2OH (2 : 1 v/v), sealed and the reaction mixture was heated within 1 h up to 140°C. The temperature was kept constant for 48 h, then the autoclave was cooled slowly to room temperature during a period of 72 h. Compound 1 was obtained as colourless block crystals. νmax (KBr)/cm–1 3424m, 3060w, 1598s, 1551s, 1520s, 1411s, 1310s, 1222m, 1069w, 1024m, 851w, 814m, 790m, 724s. Anal. Calc. for C34 H28 N8O8Zn1.50: C 55.36, H 4.20, N 15.51. Found: C 55.34, H 4.18, N 15.53 %.
Synthesis of Single Crystals of 2, 4, and 5
The compounds 2, 4, and 5 were synthesized following a procedure similar to that exemplified for 2 hereafter. A steel autoclave with a Teflon vessel was loaded with 42.0 mg (0.15 mmol) HL, 19.8 mg (0.1 mmol) ZnO·ZnO3·4H2O (Sigma–Aldrich, ACS +98 %), and 15.0 mL H2O/CH3CH2OH (2 : 1 v/v), sealed and the reaction mixture was heated within 1 h up to 140°C. The temperature was kept constant for 48 h, and then the autoclave was cooled slowly to room temperature during a period of 72 h. The preparation of 4 and 5 was similar to that of 2 except that CuCO3·Cu(OH)2·H2O (0.1 mmol) or AgNO3 was used instead of ZnO·ZnO3·4H2O.
2: νmax (KBr)/cm–1 3645m, 3422s, 1611s, 1478s, 1411s, 851m, 792w, 762m, 587m, 549m. Anal. Calc. for C30H24 N8 O4 Zn: C 50.31, H 4.03, N 11.56. Found: C 50.34, H 4.08, N 11.53 %.
4: νmax (KBr)/cm–1 3645m, 3423s, 1608s, 1385s, 1199m, 1126m, 991m, 874w, 795m, 757m, 704m. Anal. Calc. for CuC15H13N4O5: C 48.59, H 3.53, N 15.27. Found: C 48.53, H 3.65, N 15.23 %.
5: νmax (KBr)/cm–1 3750m, 3420s, 1604s, 1583s, 1384m, 1289m, 1014w, 885w, 797m, 707s, 632m. Anal. Calc. for AgC15H13N4O5: C 42.54, H 3.03, N 13.27. Found: C 42.55, H 3.07, N 13.23 %.
Synthesis of Single Crystals {[Cu(L)(bdc)0.5H2O]}n (3)
The preparation of 3 was similar to that of 1 except that CuCO3·Cu (OH)2·H2O (0.1 mmol) was used instead of ZnO·ZnO3·4H2O. Blue crystals were obtained. νmax (KBr)/cm–1 3799m, 3447s, 1621s, 1458m, 1385s, 1124m, 991w, 875w, 757m, 703m. Anal. Calc. for C19 H17 Cu N4O6: C 49.86, H 3.90, N 16.89. Found: C 49.84, H 3.93, N 16.92 %.
Conclusions
The crystal structures of the coordination complexes of {[Zn1.5L2(bdc)0.5]3H2O}n (1), {[Zn(L)2]}n (2), {[Cu(L)(bdc)0.5H2O]}n (3), {[Cu(L)3H2O]3H2O}n (4), and {[Ag(L)]3H2O}n (5) were determined. Compounds 1–5 show intriguing 3D structures. The structural diversity of the complexes not only demonstrates that HL is a good candidate for the construction of coordination compounds with aesthetic structures but also indicates that the ligand and the central metals play a great role in the formation of the complexes. For 2, TGA measurements show high thermal stability up to 420°C. Therefore, it can be concluded that 2 of high porosity stabilize themselves by multiple interpenetrations. Additionally, their luminescent properties indicate that compounds 1, 2, and 5 may be good candidates for optical materials.
Supplementary Material
XRPD patterns, additional figures and tables, and TGA and X-ray crystallographic files (in CIF format of 1–9) are available on the Journal’s website. Crystallographic data have been deposited with the Cambridge Crystallographic Data Centre, CCDC deposition numbers: 1, 832511; 2, 926769; 3, 926772; 4, 926771 and 5, 948402. Copies of the data can be obtained free of charge from http://www.ccdc.cam.ac.uk/conts/retrieving.html or from the Cambridge Crystallographic Data Centre (12 Union Road, Cambridge, CB2 1EZ, UK; Fax: +44 1223 336033; Email: deposit@ccdc.cam.ac.uk).
Acknowledgement
This work was supported by the National Natural Science Foundation of China (Grant nos 21071083 and 20771062) and the Tianjin Natural Science Foundation (Project no. 08JCZDJC21100).
References
[1] (a) For recent reviews, see: S. Kitagawa, R. Kitaura, S. Noro, Angew. Chem. Int. Ed. 2004, 43, 2334.| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXktFShtLk%3D&md5=501b3993531b6c708f47075e80f1bc45CAS |
(b) G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, Acc. Chem. Res. 2005, 38, 217.
| Crossref | GoogleScholarGoogle Scholar |
(c) D. Bradshaw, J. B. Claridge, E. J. Cussen, T. J. Prior, M. J. Rosseinsky, Acc. Chem. Res. 2005, 38, 273.
| Crossref | GoogleScholarGoogle Scholar |
(d) L. J. Murray, M. Dinca, J. R. Long, Chem. Soc. Rev. 2009, 38, 1294.
| Crossref | GoogleScholarGoogle Scholar |
(e) A. Phan, C. J. Doonan, F. J. Uribe-Romo, C. B. Knobler, M. O. O’Keeffe, M. Yaghi, Acc. Chem. Res. 2010, 43, 58.
| Crossref | GoogleScholarGoogle Scholar |
[2] (a) B. Moulton, J. J. Lu, R. Hajndl, S. Hariharan, M. J. Zaworotko, Angew. Chem. Int. Ed. 2002, 41, 2821.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38Xmt1emsrg%3D&md5=2909b8c1c8a595dcee6e8b5a9a86bfccCAS |
(b) L. Q. Ma, W. B. Lin, Angew. Chem. Int. Ed. 2009, 48, 3637.
| Crossref | GoogleScholarGoogle Scholar |
(c) Y. Liu, J. Eubank, A. Cairns, J. Eckert, V. Ch. Kravtsov, R. Luebke, M. Eddaoudi, Angew. Chem. Int. Ed. 2007, 46, 3278.
| Crossref | GoogleScholarGoogle Scholar |
(d) Q. Fang, G. Zhu, M. Xue, J. Sun, Y. Wei, S. Qiu, R. Xu, Angew. Chem. Int. Ed. 2005, 44, 3845.
| Crossref | GoogleScholarGoogle Scholar |
(e) C. M. Jin, Z. Zhu, Z. F. Chen, Y. J. Hu, X. G. Meng, Cryst. Growth Des. 2010, 10, 2054.
| Crossref | GoogleScholarGoogle Scholar |
(f) M. H. Zeng, Q. X. Wang, Y. X. Tan, S. Hu, H. X. Zhao, L. S. Long, M. J. Kurmoo, J. Am. Chem. Soc. 2010, 132, 2561.
| Crossref | GoogleScholarGoogle Scholar |
[3] (a) M. P. Suh, H. J. Park, T. K. Prasad, D. W. Lim, Chem. Rev. 2012, 112, 782.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhs1GlsbjM&md5=37b56dc1e2f07e8d6e3f8468bd533f14CAS | 22191516PubMed |
(b) J. Liu, P. K. Thallapally, B. P. McGrail, D. R. Brown, Chem. Soc. Rev. 2012, 41, 2308.
| Crossref | GoogleScholarGoogle Scholar |
(c) H. L. Jiang, D. W. Feng, T. F. Liu, J. R. Li, H. C. J. Zhou, J. Am. Chem. Soc. 2012, 134, 14690.
| Crossref | GoogleScholarGoogle Scholar |
(d) Z. J. Zhang, W. Y. Gao, L. Wojtas, S. Q. Ma, M. Eddaoudi, M. J. Zaworotko, Angew. Chem. Int. Ed. 2012, 124, 9464.
| Crossref | GoogleScholarGoogle Scholar |
(e) Y. X. Tan, Y. P. He, J. Zhang, Chem. Commun. 2011, 10647.
| Crossref | GoogleScholarGoogle Scholar |
[4] (a) J. Rocha, L. D. Carlos, F. A. A. Paz, D. Ananias, Chem. Soc. Rev. 2011, 40, 926.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXptFSktA%3D%3D&md5=853a4233cfd90f1de5cd972c3c14ee04CAS | 21180775PubMed |
(b) S. Liu, Z. Xiang, Z. Hu, X. Zheng, D. J. Cao, Mater. Chem. 2011, 21, 6649.
| Crossref | GoogleScholarGoogle Scholar |
(c) B. Chen, L. Wang, Y. Xiao, F. R. Fronczek, M. Xue, Y. Cui, G. Qian, Angew. Chem. Int. Ed. 2009, 48, 500.
| Crossref | GoogleScholarGoogle Scholar |
[5] (a) M. Yoon, R. Srirambalaji, K. Kim, Chem. Rev. 2012, 112, 1196.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsVOlt7rF&md5=576654e72d281875b36f57646a5d5792CAS | 22084838PubMed |
(b) L. Q. Ma, C. Abney, W. B. Lin, Chem. Soc. Rev. 2009, 38, 1248.
| Crossref | GoogleScholarGoogle Scholar |
(c) Y. Liu, W. Xuan, Y. Cui, Adv. Mater. 2010, 22, 4112.
| Crossref | GoogleScholarGoogle Scholar |
(d) P. Y. Wu, C. He, J. Wang, X. J. Peng, X. Z. Li, Y. L. An, C. Y. J. Duan, J. Am. Chem. Soc. 2012, 134, 14991.
| Crossref | GoogleScholarGoogle Scholar |
[6] (a) L.-F. Ma, C.-P. Li, L.-Y. Wang, M. Du, Cryst. Growth Des. 2011, 11, 3309.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXptVKqu78%3D&md5=421c38e5df3bfdd0f7fd136b8e765198CAS |
(b) C. Qin, X.-L. Wang, E.-B. Wang, Z.-M. Su, Inorg. Chem. 2008, 47, 5555.
| Crossref | GoogleScholarGoogle Scholar |
(c) J. H. Cui, Y. Z. Li, Z. J. Guo, H. G. Zheng, Cryst. Growth Des. 2012, 12, 3610.
| Crossref | GoogleScholarGoogle Scholar |
(d) Q.-Y. Yang, K. Li, J. Luo, M. Pana, C.-Y. Su, Chem. Commun. 2011, 4234.
| Crossref | GoogleScholarGoogle Scholar |
(e) C. J. Hansen, S. R. White, N. R. Sottos, J. A. Lewis, Adv. Funct. Mater. 2011, 21, 4320.
| Crossref | GoogleScholarGoogle Scholar |
(f) K. M. Blake, J. S. Lucas, R. L. LaDuca, Cryst. Growth Des. 2011, 11, 1287.
| Crossref | GoogleScholarGoogle Scholar |
(g) K. M. Blake, J. S. Lucas, R. L. LaDuca, Cryst. Growth Des. 2011, 11, 1287.
| Crossref | GoogleScholarGoogle Scholar |
(h) X.-D. Chen, X.-H. Zhao, M. Chen, M. Du, Chem. Eur. J. 2009, 15, 12974.
| Crossref | GoogleScholarGoogle Scholar |
[7] (a) S. Kitagawa, K. Uemura, Chem. Soc. Rev. 2005, 34, 109.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXmvFCrsA%3D%3D&md5=e5b6e7c3d763e0f323d852af291b3c6eCAS | 15672175PubMed |
(b) Z. Guo, H. Wu, S. Gadipelli, T. Liao, Y. Zhou, S. Xiang, Z. Chen, Y. Yang, W. Zhou, M. O’Keeffe, B. Chen, Angew. Chem. Int. Ed. 2011, 50, 3178.
| Crossref | GoogleScholarGoogle Scholar |
(c) B. Chen, S. Xiang, G. Qian, Acc. Chem. Res. 2010, 43, 1115.
| Crossref | GoogleScholarGoogle Scholar |
[8] (a) J. L. C. Rowsell, O. M. J. Yaghi, J. Am. Chem. Soc. 2006, 128, 1304.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XitV2msg%3D%3D&md5=68569d5bab0e4f3c6ae474e2d2607932CAS |
(b) D. Sun, S. Ma, Y. Ke, T. M. Petersen, H.-C. Zhou, Chem. Commun. 2005, 2663.
| Crossref | GoogleScholarGoogle Scholar |
(c) S. Ma, H.-C. J. Zhou, J. Am. Chem. Soc. 2006, 128, 11734.
| Crossref | GoogleScholarGoogle Scholar |
(d) S. Ma, X. Wang, D. Yuan, H.-C. Zhou, Angew. Chem. Int. Ed. 2008, 47, 4130.
| Crossref | GoogleScholarGoogle Scholar |
[9] (a) H. Li, M. Eddaoudi, M. O’Keeffe, O. M. Yaghi, Nature 1999, 402, 276.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXnvFSiuro%3D&md5=96dcac380a1db29dda69820358193182CAS |
(b) G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé, I. Margiolaki, Science 2005, 309, 2040.
| Crossref | GoogleScholarGoogle Scholar |
(c) X.-H. Chang, J.-H. Qin, L.-F. Ma, J.-G. Wang, L.-Y. Wang, Cryst. Growth Des. 2012, 12, 4649.
| Crossref | GoogleScholarGoogle Scholar |
(d) M. Latroche, S. Surblé, C. Serre, C. Mellot-Draznieks, P. L. Llewellyn, J.-H. Lee, J.-S. Chang, S. H. Jhung, G. Férey, Angew. Chem. 2006, 118, 8407.
| Crossref | GoogleScholarGoogle Scholar |
[10] (a) Y. Q. Lan, X. L. Wang, S. L. Li, Z. M. Su, K. Z. Shao, E. B. Wang, Chem. Commun. 2007, 4863.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhsVOgtLzI&md5=d0c20df4e5255abaf2648e44dc7bfc88CAS |
(b) P. K. Chen, S. R. Batten, Y. Qi, J. M. Zheng, Cryst. Growth Des. 2009, 9, 2756.
| Crossref | GoogleScholarGoogle Scholar |
(c) Y. Q. Wang, J. Y. Zhang, Q. X. Jia, E. Q. Gao, C. M. Liu, Inorg. Chem. 2009, 48, 789.
| Crossref | GoogleScholarGoogle Scholar |
(d) L. F. Ma, L. Y. Wang, Y. Y. Wang, S. R. Batten, J. G. Wang, Inorg. Chem. 2009, 48, 915.
| Crossref | GoogleScholarGoogle Scholar |
(e) S. R. Zheng, Q. Y. Yang, R. Yang, M. Pan, R. Cao, C. Y. Su, Cryst. Growth Des. 2009, 9, 2341.
(f) D. R. Xiao, E. B. Wang, H. Y. An, Y. G. Li, Z. M. Su, C. Y. Sun, Chem. Eur. J. 2006, 12, 6528.
| Crossref | GoogleScholarGoogle Scholar |
[11] Z.-X. Li, T.-L. Hu, H. Ma, Y.-F. Zeng, C.-J. Li, M.-L. Tong, X.-H. Bu, Cryst. Growth Des. 2010, 10, 1138.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtlKktLo%3D&md5=3ab5f6e9af5bc3f02a81c4798005b80dCAS |
[12] (a) M. Kondo, T. Yoshitomi, K. Seki, H. Matsuzaka, S. Kitagawa, Angew. Chem. Int. Ed. Engl. 1997, 36, 1725.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXmtVSjtrw%3D&md5=7026310772508aca6dddcc35f3342a81CAS |
(b) K. N. Power, T. L. Hennigar, M. J. Zaworotko, New J. Chem. 1998, 22, 177.
| Crossref | GoogleScholarGoogle Scholar |
(c) C. J. Kepert, M. J. Rosseinsky, Chem. Commun. 1999, 375.
| Crossref | GoogleScholarGoogle Scholar |
(d) Z. Y. Fu, X. T. Wu, J. C. Dai, S. M. Hu, W. X. Du, New J. Chem. 2002, 26, 978.
| Crossref | GoogleScholarGoogle Scholar |
[13] (a) S. R. Zheng, Y. Yang, Y. R. Liu, J. Y. Zhang, Y. X. Tong, C. Y. Zhao, C. Y. Su, Chem. Commun. 2008, 356.
| Crossref | GoogleScholarGoogle Scholar |
(b) X. Y. Cao, J. Zhang, J. K. Cheng, Y. Kang, Y. G. Yao, CrystEngComm 2004, 6, 315.
| Crossref | GoogleScholarGoogle Scholar |
(c) R. Sun, S. N. Wang, H. Xing, J. F. Bai, Y. Z. Li, Y. Pan, X. Z. You, Inorg. Chem. 2007, 46, 8451.
| Crossref | GoogleScholarGoogle Scholar |
[14] Z. Y. Fu, X. T. Wu, J. C. Dai, L. M. Wu, C. P. Cui, S. M. Hu, Chem. Commun. 2001, 1856.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXmvFCitrg%3D&md5=d895e09a7db9a784d2734e7674017b57CAS |
[15] Y.-P. Wu, D.-S. Li, F. Fu, W.-W. Dong, J. Zhao, K. Zou, Y.-Y. Wang, Cryst. Growth Des. 2011, 11, 3850.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXpvFCiurw%3D&md5=331c12fa31c1ff789d4386d358679756CAS |
[16] L. F. Ma, Q. L. Meng, C. P. Li, B. Li, L. Y. Wang, M. Du, F. P. Liang, Cryst. Growth Des. 2010, 10, 3036.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXnsFGit78%3D&md5=cf78b7264e0d41b7722c432b7c843931CAS |
[17] O. V. Sharga, A. B. Lysenko, H. Krautscheid, K. V. Domasevitch, Acta Crystallogr. 2010, C66, m269.