

Adrien Albert Award: How to Mine Chemistry Space for New Drugs and Biomedical Therapies
David WinklerCell Biology Group, Biomedical Materials Program, CSIRO Manufacturing Flagship, Bag 10, Clayton South MDC, Vic. 3169; Monash Institute of Pharmaceutical Sciences, Parkville, Vic. 3052; and Latrobe Institute for Molecular Science, Bundoora, Vic. 3108, Australia. Email: dave.winkler@csiro.au
Australian Journal of Chemistry 68(8) 1174-1182 https://doi.org/10.1071/CH15172
Submitted: 10 April 2015 Accepted: 21 April 2015 Published: 14 May 2015
Abstract
It is clear that the sizes of chemical, ‘drug-like’, and materials spaces are enormous. If scientists working in established therapeutic, and newly established regenerative medicine fields are to discover better molecules or materials, they must find better ways of probing these enormous spaces. There are essentially five ways that this can be achieved: combinatorial and high throughput synthesis and screening approaches; fragment-based methods; de novo molecular design, design of experiments, diversity libraries; supramolecular approaches; evolutionary approaches. These methods either synthesise materials and screen them more quickly, or constrain chemical spaces using biology or other types of ‘fitness functions’. High throughput experimental approaches cannot explore more than a minute part of chemical space. We are nevertheless entering into an era that is data dominated. High throughput experiments, robotics, automated crystallographic beam lines, combinatorial and flow synthesis, high content screening, and the ‘omics’ technologies are providing a flood of data, and efficient methods for extracting meaning from it are essential. This paper describes how new developments in mathematics have provided excellent, robust computational modelling tools for exploring large chemical spaces, for extracting meaning from large datasets, for designing new bioactive agents and materials, and for making truly predictive, quantitative models of the properties of molecules and materials for use in therapeutic and regenerative medicine. We describe these broadly applicable modelling tools and provide examples of their application to serum free stem cell culture, pathogen resistant polymers for implantable devices, new markers and biological mechanisms derived from mathematical analyses of gene array data, and pharmacokinetically important physicochemical properties of small molecules. We also discuss biologically conserved peptide motifs as a design framework for small molecule drugs and give examples of the application of this concept to drug design.
Introduction
This paper recognises the great legacy of Prof. Adrien Albert,† an internationally renowned Australian medicinal chemist[1–4] in whose honour this award is named.
Medicinal chemistry has changed unrecognisably since Albert’s day. The amount of knowledge on the molecular basis for drug action has grown tremendously, and new robotic technology now allows us to synthesise and test millions of potential drugs in a similar time to that required to create a handful of compounds in Albert’s day. In spite of this enormous advantage, many diseases are still lacking drug therapies that are effective, and some diseases have no drug treatments at all. There is simultaneously pressure on drug pipelines, as the cost of inventing new drugs is increasing rapidly ($2.3 billion at last estimate), patent life is limited, and regulatory barriers are constantly increasing. New drugs must be demonstrably better than existing drugs if they are to receive patent protection. Balancing this, the new research fields of nanotechnology, materials science, and regenerative medicine are creating opportunities for medicinal chemists to leverage their skills in novel ways to develop radically different treatments.
Computational modelling and simulation methods are key elements in the effective use of this reservoir of molecular knowledge and dramatic increase in data brought about by automation and new information technology. Detailed understanding of how drugs interact with proteins, cells, organs, and organisms is greatly facilitated by structural biology and molecular modelling methods. These modelling techniques allow researchers to visualise interactions between molecular species and infer and predict their effects on the organism. Likewise, statistical and machine learning methods probe relationships between molecular structure and biological activity for large collections of molecules and can deal with the flood of data generated by modern experimental techniques. These methods provide quantitative predictions of biological activity or other properties for molecules yet to be synthesised or tested. These techniques promise acceleration of the discovery and development of new drugs and medical therapies.
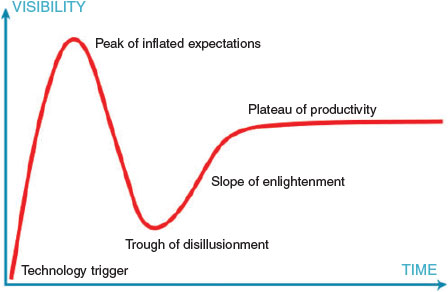
Like most new technologies, computational modelling passed through the ‘hype cycle’ (Fig. 1) where its initially modest capabilities were oversold and some degree of disillusionment resulted.
|
In the past thirty years computational techniques have become much more accurate, robust, and productive through an increased maturity in understanding of how they are best applied, and by the dramatic increases in computational power described by Moore’s Law over the past several decades. The rate of change in this facilitating technology has been staggering – the fastest and most expensive supercomputer in 1990 had the same capabilities as some inexpensive tablet computers in 2015. It has been estimated that by 2020 we will be able to compute 100 million times faster than in 1980.
This paper illustrates some successful applications of computational methods that will be increasingly needed to meet the challenges of financial pressures, an ageing population, increasing lifestyle disease burden, effects of climate change, and the challenges of emerging and important disease threats such as HIV, tuberculosis, and Ebola.
The Threat and Promise of the Vastness of Chemical Space
The automation of medicinal chemistry that occurred in the 1980–1990s, now being leveraged into materials research,[5] has generated wider recognition in the research community of the vastness of chemical space. It is difficult to calculate exactly how many molecules could be synthesised that are stable and obey the rules of chemical reaction and valency, but 1060 for drug-like space and 1080 for chemical space is often nominated.[6] Clearly, no matter how fast and efficient we make automated synthesis and testing we cannot hope to explore spaces this large. Techniques like experimental design, and diversity library methods will allow large spaces to be explored in a very sparse way. Virtual screening and computational design methods that constrain the chemical space by use of protein structural information likewise can identify islands of value in the universe of chemical space possibilities. However, computational modelling and simulation methods provide the only means for exploring significant parts of unconstrained drug-like space to find new therapies.
Although these regions of chemical space that contain valuable new chemotypes are difficult to find, there may be many of them, providing an almost infinite scope‡ for the discovery of new or more effective drug therapies.
Our team has developed effective computational design and modelling techniques over the past 20 years,[7–15] and we have employed them to generate substantial scientific and commercial impact, culminating in more than 20 patents. These methods have been used to design and optimise green pesticides[16–20] in collaboration with Du Pont and Schering Plough, have discovered new peptides and small molecules to control stem cells and cancers,[21–27] are accelerating the development of biomaterials for implantation and stem cell culture,[28–33] have provided new scientific insight into the potential adverse properties of nanomaterials,[34–38] have yielded clinical candidates for Australian SMEs and international companies, and were instrumental in the discovery of antibiotics[39,40] with a novel mode of action for the biotechnology spin off company, Betabiotics. Some of this research is discussed in more detail in the following sections.
Quantitative Structure–Activity Modelling
Increasingly, automated methods are generating large volumes of data on biological or other properties of chemical or materials libraries. These datasets contain very useful information on how the molecular properties modulate the observed, macroscopic properties. Efficient and robust methods for deciphering these relationships are essential if maximum value is to be obtained from such high throughput experiments. One of the most useful ways to achieve this involves statistical and machine-learning methods called quantitative structure–activity relationships modelling (QSAR). This computational tool was developed by Hansch and Fujita[41,42] in the early 1960s to model physicochemical and biological properties of drugs, and has been a mainstay of pharmaceutical discovery since.[43]
In essence the method is deceptively simple. It is a supervised modelling method (i.e. context dependent) that describes the complex relationships between the molecular (microscopic) and physicochemical properties of molecules and their biological (macroscopic) effects:
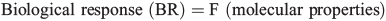
The method involves finding relevant mathematical forms for the microscopic properties of compounds (descriptors) and the optimum (usually nonlinear) complex function:

This function summarises the effects of a range of biological processes occurring between the administration of the drug and its ultimate interaction with the molecular target and the downstream signalling process that this event triggers. QSAR is essentially a kind of complex pattern recognition process. It can accommodate complex ‘models within models’, which is very useful when modelling in vivo properties such as toxicity, carcinogenicity, and mutagenicity. We have developed a suite of robust and predictive QSAR modelling methods over the past 20 years that have been used on a wide range of research problems.
Sparse Learning Methods
It is known that models that are optimally sparse have the greatest ability to predict the properties of new data. The corollary is that models that have too many fitted variables relative to the number of data points (e.g. molecules) will overfit the data and such models will have very little predictive value. We optimised model performance by employing a special type of regularization, a family of methods that control the complexity of models. This utilises Bayesian statistics to generate models that are close to optimal sparsity, and that predict new data as accurately as the errors in the training data allow. We have applied Bayesian regularization to artificial neural networks, learning algorithms that are ‘universal’ approximators capable of fitting any linear or nonlinear function given sufficient training data. Bayesian regularization overcomes the known shortcomings of traditional neural networks and allows them to generate very good predictive models of biological and physical properties, even when the underlying relationships are complex, multidimensional, and nonlinear. The mathematics of Bayesian regularization is challenging and is not repeated here as it is described in numerous publications.[7–9,11–15,44–47]
Bayesian Regularized Neural Networks
We applied these regularization techniques to neural networks in two ways, differing only in the type of Bayesian prior (a probability distribution expressing the uncertainty in a property before evidence is taken into account) that is employed. When a Gaussian prior, 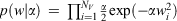 , is used the neural network automatically prunes the weights (the fitted parameters) in the network to an optimum number. Employing a sparsity-inducing Laplacian prior,
, is used the neural network automatically prunes the weights (the fitted parameters) in the network to an optimum number. Employing a sparsity-inducing Laplacian prior, 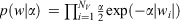 , allows less relevant weights and molecular descriptors to be removed from the model, further improving its predictive power and making interpretation of the relationships between molecular and biological properties simpler.[5,11,14]
, allows less relevant weights and molecular descriptors to be removed from the model, further improving its predictive power and making interpretation of the relationships between molecular and biological properties simpler.[5,11,14]
Feature Selection Using Expectation Maximisation
Combining sparse priors and an expectation maximisation algorithm – an interactive method for finding maximum likelihood estimates of parameters in models – allows the imposition of sparsity control in models by the removal of less relevant attributes of molecules.[15] This can result in extreme sparsity relative to the initial dimensionality of the problem. For example, it is possible to numerically generate thousands of mathematical descriptions of molecular properties. These can often be reduced by 90–99 % using these sparse feature selection methods, initially described by Figeuiredo (Fig. 2).[45] Examples of the ability of these techniques to model extremely diverse chemical datasets, and to discover unexpected mechanisms from large gene expression microarray datasets are summarised below.
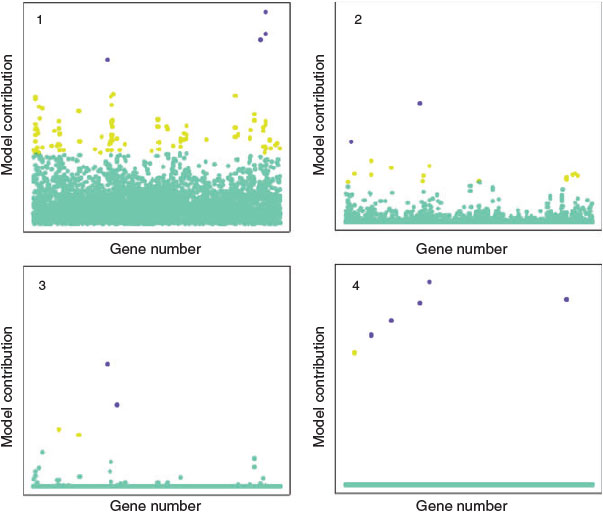
|
Global Prediction of Drug Properties
Sparse modelling methods have successfully generated robust models for predicting physical and biological properties of drugs and other materials that have very large domains of applicability. The domain of applicability is the region of chemical and property space for which the model predictions are valid. For example, farnesyl transferase inhibition and selectivity against geranyl-geranyl transferase of ~2000 molecules were modelled using these sparse, nonlinear methods.[49] The methods were subsequently applied to the modelling and prediction of the ability of stem cell bioreactors to generate expanded populations of cells using data collated from 300 experiments from multiple research groups. These models identified the most important parameters for driving cell expansion down particular lineage pathways.[25] More recently, the Bayesian regularized neural networks and sparse feature selection methods generated global models of aqueous solubility of small organic molecules, based on a very chemically diverse training set of ~5000 drugs displaying over 13 orders of magnitude differences in solubility. This property is one of the most important for predicting the likely pharmacokinetic performance of candidate drugs. The model predicted aqueous solubility within a factor of 3 (logS ± 0.6).[50] Flash point is another very important physical property for materials used in engineering applications. Bayesian regularized neural networks were used to successfully generate a model for this property using a training set of ~10000 molecules spanning 1000 K in flash point and a large range of chemical diversity.[51] The model could predict the flash point of compounds in a test set, data not used to construct the model, within a standard error of ±17 K.
Discovery of Novel Biomarkers and Mechanisms of Action by Sparse Feature Selection
The very sparse feature selection methods described above have also been applied to gene expression microarray data. Two long-standing research problems were resolved: an explanation for the mechanism by which strontium ion induces mesenchymal stem cells (MSCs) to differentiate towards bone; and the discovery of biomarkers that identify symmetric versus asymmetric division of stem cells.
Strontium ranelate (Protelos™) 1 (Chart 1) is a drug approved in the EU for the treatment and prevention of osteoporosis – strontium ion is the active component. It reduces risk of vertebral and non-vertebral fractures in post-menopausal women and can be useful in replacing bone lost due to drug treatment for cancer. Strontium ion’s mechanism of action is not fully understood, but it is thought to upregulate differentiation of osteoprogenitors or to directly stimulate bone formation.[52,53] Autefage et al. designed experiments that exposed MSCs to various levels of strontium ion and measured changes in gene expression over time. We analysed the microarray data using the sparse Bayesian feature selection methods to identify genes of highest relevance to the strontium-induced bone formation. The genes identified by feature selection unexpectedly identified a previous unrecognised fatty acid and steroid pathway, subsequently validated by real-time polymerase chain reaction and other biochemical experiments.[33]
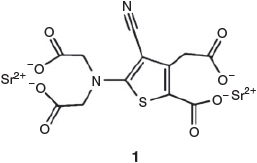
|
There is a long-standing unmet clinical need for biomarkers that can identify whether distributed stem cells (DSCs) in tissues divide symmetrically or asymmetrically. DSCs are essential for tissue maintenance and repair but they are difficult to identify and count. We combined a sparse feature selection method with combinatorial molecular expression data to identify such DSC biomarkers. Our analysis identified reduced expression of the histone H2A variant H2A.Z as a biomarker of DSC asymmetric self-renewal (Fig. 3).[54] The biomarkers arising from this research provided the intellectual property for the establishment of the company, Asymmetrex.

|
Self-Assembling Amphiphilic Drug Delivery Systems
Amphiphilic lyotropic liquid crystalline materials spontaneously self-assemble into a variety of nano-containers with important applications in drug delivery and diagnostic imaging. There is a paucity of knowledge on the effect of the incorporated drug on the resulting nanostructures and predicting these effects is widely considered intractable. We used sparse modelling methods to predict the effects of concentration and molecular properties of 10 common drugs, and temperature on the resulting nanostructures using data obtained from high throughput data synthesis and small-angle X-ray scattering (SAXS) characterisation experiments.[56] The models predicted the type and coexistence of multiple nanophases (Fig. 4), important because only some of these are suitable for drug delivery. The model further predicted the nanophases resulting from incorporation of eleven new drugs in a blind test of the prediction ability of the model. Subsequent synchrotron experiments showed that the nanophases resulting from incorporation of these drugs were predicted with accuracies of 85–91 %, depending on the amphiphile used to encapsulate them.[30]

|
Subsequent work extended these nanophase prediction models to include the effects of time, and the presence of components of crystallisation screens used to stabilise membrane-bound proteins for X-ray structural biology studies. We predicted the existence of individual nanophases with accuracies of 98–99 % and the complex coexistence of multiple phases to a similar accuracy using nonlinear models.[57] From the models we could identify which crystallisation screen components were most relevant to the temporal evolution of individual mesophases. We also studied the effect of amphiphile structure on the formation of mesophase formation. Again using high throughput synthesis and assessment data we could make quantitative predictions of the effects of amphiphile structure and crystallographic screen components on the formation of mesophases.[58]
Cellular Attachment to Polymer Library Surfaces
Bacterial adhesion and growth on biomaterial surfaces such as joint prostheses, heart valves, shunts, vascular and urinary catheters, intraocular lenses, and similar implants is a serious problem in health care. The discovery of polymers that resist the attachment of the most important pathogens would greatly ameliorate these problems. Hook et al. developed methods for synthesising large polymer libraries on slides and exposing them to cells to determine the degree of cell attachment.[59] A 576-member polymer microarray was incubated with suspensions of three pathogenic bacteria (Pseudomonas aeruginosa (PA), Staphylococcus aureus (SA), and uropathogenic Escherichia coli (UPEC)) and the attached bacteria were counted.[59] We used these data to construct linear and nonlinear models for cellular adhesion as a function of polymer surface chemistry for each pathogen type.[60] As Fig. 5 shows, we could make accurate quantitative predictions of the degree of bacterial adhesion for the three common pathogens, and could determine which aspects of the surface chemistry favoured low attachment. For P. aeruginosa and S. aureus it was found that cyclic hydrocarbon groups, tertiary butyl groups, and aliphatic groups on the meth/acrylate polymer were required for low bacterial attachment.

|
Culture of pluripotent cells such as induced pluripotent stem cells is a major research focus in regenerative medicine. Present methods to culture them and expand their populations rely on animal-derived products now increasingly under scrutiny. There is an urgent need for chemically defined, serum-free, feeder-free synthetic substrates and media to support robust self-renewal of pluripotent cells. Changes in cellular properties such as adhesion, morphology, motility, gene expression, and differentiation are influenced by chemistry, wettability, topography, and elastic modulus of the surface on which they are cultured. Using a 496-member polymer library, Yang et al. determined the degree of attachment of human embryonic stem cell embryoid bodies.[61] We modelled the relationship between surface chemistry and the degree of embryoid body attachment. Nonlinear models accounted for 80 % of the variance in the data, made quantitative predictions of the degree of attachment of embryoid bodies to new polymers, and elucidated the relationship between surface chemistry and cell attachment.[29]
Design of New Drugs Using Tripeptide Motifs
Nature tends to reuse motifs and structural components such as helices, sheets, and barrels in proteins. The concept that specific recognition events occur between amino acids in endogenous ligands and their native receptors forms a fundamental tenet of structural biology and drug design. While many of these recognition events are complex, given the economy of Nature it is interesting to speculate on what is the minimal information-bearing structure in proteins and peptides.
We reviewed the literature and established that this minimal information unit may be a tripeptide motif.[39] Some tripeptides have been mimicked by small molecules, while many more potentially important motifs have yet to be discovered and used for drug design. The idea that three contiguous amino acids is biologically important is consistent with Reynolds et al. and Neduva and Russell’s suggestion that 25 heavy atoms (the average number in a tripeptide) provides optimal ligand affinity.[62,63] Hann et al. also showed that in drug-like libraries and the World Drug Index, the heavy atom profile peaks at 25 heavy atoms.[64] As many drugs mimic biological signals, the over-representation of drugs this size is consistent with peptide motifs around three amino acids in length having biological relevance. Table 1 summarises some of the most relevant and biologically important motifs.

|
An example of the application of the tripeptide motif concept to drug discovery is HAV, an N-cadherin antagonist. Cadherins are calcium-dependent glycoproteins that play a central biological role, particularly in cell adhesion, morphogenesis, neurogenesis, and many other important functions. A key recognition sequence, HAV (His-Ala-Val), in cadherin was first identified by Byers et al.[65] Burden-Gully et al. have very recently reported small molecule mimics (Fig. 6) that show moderate cadherin antagonist activities.[66]

|
We applied this tripeptide motif concept to the design of potent agonists and antagonists of a key cytokine receptor, and antimicrobial agents operating via a novel mode of action.
Thrombopoietin (TPO) is an important cytokine that directs the differentiation of stem cells towards platelet production. Its receptor C-Mpl, is also implicated in a range of myeloproliferative diseases such as leukaemias. Cwirla et al.[67] conducted phage display experiments to identify families of peptides that bound to the C-Mpl receptor. We identified a key binding motif, RQW, in these peptide libraries and used this to develop potent small molecule and peptide agonists and antagonists of the TPO receptor.[22,23] The agonists were capable of potently stimulating platelet counts in animals, and also increased megakaryocyte ploidy. Peptide TPO mimetics could replace native TPO in the expansion of CD34+CD38– primary cells up to 8 days. These mimetics also promoted megakaryocyte development (CD41a+) up to 8 days primary cell culture. The design of agonists also allowed us to develop the first C-Mpl antagonists that are being developed as anticancer treatments.
The problem of bacterial resistance to drugs has become severe and new antibiotics that function via novel mechanisms, preferably those that are very difficult to acquire resistance to, is essential. Using a deep sequence similarity search, Dalrymple et al. identified a conserved tripeptide motif in the bacterial replisome, a viable target for the development of new antibiotics.[68] The SLF and DLF peptide motifs were highly conserved in the binding site of the β2 sliding clamp, an essential component of bacterial replicative machinery. These clamps are homodimeric ring-shaped molecules that encircle DNA and facilitate the operation of the polymerase. They interact with many proteins involved in bacterial DNA replication and repair, and drugs that interfere with this binding site will disrupt DNA synthesis. We developed a three-dimensional pharmacophore for this peptide motif based on a novel analysis of the structures in the protein crystallographic database. The 3D structures of the peptide motifs, DLF and SLF, were surprisingly well conserved in Nature, and provided a search query that allowed large chemical libraries to be searched for small molecules mimicking that 3D motif. This led to the identification of compounds that, on further development, inhibited β2 interactions at low micromolar concentrations and showed broad-spectrum antibiotic activity. Subsequent crystal structure determinations of the complex containing one of these inhibitors (Fig. 7) validated the concept of conservation of the 3D conformation of this tripeptide motif.[40] The candidate drugs arising from this research provided the intellectual property for the establishment of the company, Betabiotics.
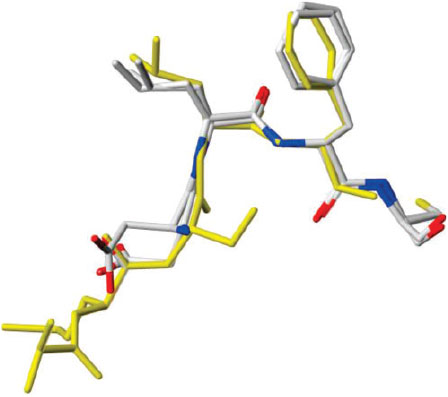
|
Where to next?
Given the rapid increase in computing power, the increased rate of automation of drug discovery and materials science, and a greater appreciation of the size of chemistry space, the prospects for computational molecular design are very bright. Areas of future research focus and application are likely to include:
-
Rational design of novel drug delivery vehicles based on self-assembly principles.
-
Design of evolutionary algorithms that allow molecular leads to be evolved towards desired properties, searching very large chemistry spaces more efficiently.[69]
-
Significantly more reliable methods for docking molecules into proteins and for scoring these interactions.
-
Improved pharmacokinetic, toxicity, and metabolism prediction methods, potentially using machine-learning models based on large datasets.[70–72]
-
The leveraging of computational tools from drug discovery to new areas of science – biomaterials, regenerative medicine, and tissue engineering.[5]
-
Increasingly robust and autonomous machine learning methods for modelling very large datasets and the application of these models to drug and materials discovery and optimisation.
-
Consensus tools that can integrate information from disparate sources for molecular design.
-
Understanding and modelling drug, target, and disease networks, exploiting multi-target drugs, and the use of the concepts of complex systems science to understand diseases.[73,74]
-
Design of small molecules to reprogram somatic and stem cells, and to selectively kill cancer stem cells.[21]
Clearly science and technology have moved on very rapidly since Adrien Albert’s day, but he would be very pleased to see his area of selective toxicity flourishing in the twenty-first century.
Acknowledgements
I gratefully acknowledge receipt of the Adrien Albert award from the Royal Australian Chemical Institute. Financial support from the CSIRO Advanced Materials Platform, a Newton Turner award for Exceptional Senior Scientists, and a Monash University-Nottingham travelling fellowship are also gratefully acknowledged. This work involved very substantial contributions of my past and present research team and CSIRO collaborators (Tu Le, Frank Burden, Vidana Epa, Anna Tarasova, David Haylock, Julianne Halley, Xavier Mulet, Charlotte Conn, Brian Dalrymple, Gene Wijffels, Kathleen Turner, Adam Meyer, Jacinta White, Jess Andrade), visiting scientists (Johnny Gasteiger, Maryam Salahinejad), and collaborators at the University of Nottingham (Andrew Hook, Jing Yang, Paul Williams, Morgan Alexander, Adam Celiz), MIT (Ying Mei, Robert Langer and Daniel Anderson), Shandong University (Bing Yan), Imperial College London (Molly Stevens, Eileen Gentleman, Helen Autefage), and Asymmetrex (James Sherley).
References
[1] A. Albert, Nature 1950, 165, 12.| Crossref | GoogleScholarGoogle Scholar | 1:STN:280:DyaG3c%2FhvVGgug%3D%3D&md5=20742bc7e0682c997991e4a43608b073CAS | 15408906PubMed |
[2] A. Albert, Selective Toxicity, with Special Reference to Chemotherapy 1951 (Methuen: London).
[3] A. Albert, Nature 1958, 182, 421.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaG1MXhvVymug%3D%3D&md5=afe2fe77ee30676d1a56c6a3d2ef9bceCAS | 13577867PubMed |
[4] A. Albert, Selective Toxicity, 2nd edn 1960 (Methuen: London).
[5] T. Le, V. C. Epa, F. R. Burden, D. A. Winkler, Chem. Rev. 2012, 112, 2889.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XpsVyisA%3D%3D&md5=b1864c63d05b44766ceb05223091d140CAS | 22251444PubMed |
[6] B. K. Shoichet, Nat. Chem. 2013, 5, 9.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhvVCktbzK&md5=a7db0a7a08da5d734160da72156e7b4dCAS | 23247169PubMed |
[7] F. R. Burden, B. S. Rosewarne, D. A. Winkler, Chemom. Intell. Lab. Syst. 1997, 38, 127.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXmsl2gurs%3D&md5=87f6df59e2e41da14cc69c86aa73f3aeCAS |
[8] D. Winkler, F. Burden, J. Halley, Drugs Future 2007, 32, 26.
[9] F. R. Burden, M. G. Ford, D. C. Whitley, D. A. Winkler, J. Chem. Inf. Comput. Sci. 2000, 40, 1423.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXnsFOitLo%3D&md5=da23f507e1080e08060875586736009aCAS | 11128101PubMed |
[10] F. R. Burden, M. J. Polley, D. A. Winkler, J. Chem. Inf. Model. 2009, 49, 710.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhslyjsbw%3D&md5=141e7a9c057b6cc50b2cbae575b88eddCAS | 19434903PubMed |
[11] F. R. Burden, D. A. Winkler, J. Med. Chem. 1999, 42, 3183.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXksFGhsro%3D&md5=5bd5a36d4ebde2ba3254abf6a7aad61fCAS | 10447964PubMed |
[12] D. A. Winkler, Mol. Biotechnol. 2004, 27, 139.
| Crossref | GoogleScholarGoogle Scholar | 15208456PubMed |
[13] D. A. Winkler, F. R. Burden, Mol. Simul. 2000, 24, 243.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXnvF2jtLc%3D&md5=c51d4abc1cfc8a51130b5a1a8d708ee0CAS |
[14] F. R. Burden, D. A. Winkler, QSAR Comb. Sci. 2009, 28, 1092.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXht1yrsr7K&md5=7f3ccbc97a63b4223ef9f0d3b9e0bbc0CAS |
[15] F. R. Burden, D. A. Winkler, QSAR Comb. Sci. 2009, 28, 645.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXptFeit74%3D&md5=c26d73ddee271601db240b65d7028089CAS |
[16] R. J. Hill, L. D. Graham, K. A. Turner, L. Howell, D. Tohidi-Esfahani, R. Fernley, J. Grusovin, B. Ren, P. Pilling, L. Lu, T. Phan, G. O. Lovrecz, M. Pollard, A. Pawlak-Skrzecz, V. A. Streltsov, T. S. Peat, D. A. Winkler, M. C. Lawrence, Adv. Insect Physiol. 2012, 43, 299.
| Crossref | GoogleScholarGoogle Scholar |
[17] W. Birru, R. T. Fernley, L. D. Graham, J. Grusovin, R. J. Hill, A. Hofmann, L. Howell, P. J. James, K. E. Jarvis, W. M. Johnson, D. A. Jones, C. Leitner, A. J. Liepa, G. O. Lovrecz, L. Lu, R. H. Nearn, B. J. O’Driscoll, T. Phan, M. Pollard, K. A. Turner, D. A. Winkler, Bioorg. Med. Chem. 2010, 18, 5647.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXptlGhurs%3D&md5=935e6a0adb8e77a7e0351bd8097aa343CAS | 20619664PubMed |
[18] A. Ali, T. M. Altamore, M. Bliese, P. Fisara, A. J. Liepa, A. G. Meyer, O. Nguyen, R. M. Sargent, D. G. Sawutz, D. A. Winkler, K. N. Winzenberg, A. Ziebell, Bioorg. Med. Chem. Lett. 2008, 18, 252.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXis1Squw%3D%3D&md5=6c87cea6889f506cf3d21aa7d64219c0CAS | 18006308PubMed |
[19] A. Ali, M. Bliese, J. A. M. Rasmussen, R. M. Sargent, S. Saubern, D. G. Sawutz, J. S. Wilkie, D. A. Winkler, K. N. Winzenberg, R. C. J. Woodgate, Bioorg. Med. Chem. Lett. 2007, 17, 993.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXht12qsbY%3D&md5=190d0cd3c6669b7cb689caf4d23ba622CAS | 17150358PubMed |
[20] J. A. Carmichael, M. C. Lawrence, L. D. Graham, P. A. Pilling, V. C. Epa, L. Noyce, G. Lovrecz, D. A. Winkler, A. Pawlak-Skrzecz, R. E. Eaton, G. N. Hannan, R. J. Hill, J. Biol. Chem. 2005, 280, 22258.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXksl2htrY%3D&md5=90d8f2939d84ae6b967e881f98ffefd2CAS | 15809296PubMed |
[21] F. Ismail, D. A. Winkler, ChemMedChem 2014, 9, 885.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXms1ajs7Y%3D&md5=ded7a40259a47e71505a1ce6cc2ca4eaCAS | 24760779PubMed |
[22] A. Tarasova, D. N. Haylock, L. Meagher, C. L. Be, J. White, S. K. Nilsson, J. Andrade, K. Cartledge, D. A. Winkler, ChemMedChem 2013, 8, 763.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXkslWrt7g%3D&md5=4f57d5c5a5eaa1594d1ac4efb93cd171CAS | 23554275PubMed |
[23] A. Tarasova, D. A. Winkler, ChemMedChem 2009, 4, 2002.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsFWrs7zI&md5=2261460ece8dd7d9fbb602d4a3c32006CAS | 19810084PubMed |
[24] A. Tarasova, D. Haylock, D. Winkler, Cytokine Growth Factor Rev. 2011, 22, 231.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsV2rsrbE&md5=1d853420880382454e30afd7a448cf35CAS | 21975328PubMed |
[25] D. A. Winkler, F. R. Burden, Mol. Biosyst. 2012, 8, 913.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XitVSmsbo%3D&md5=97746da100e1e72dc75f1c9f2da45125CAS | 22282302PubMed |
[26] J. D. Halley, K. Smith-Miles, D. A. Winkler, T. Kalkan, S. Huang, A. Smith, Stem Cell Res. 2012, 8, 324.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xht1Ont74%3D&md5=3344445614b96663a14ce8dc4676de5bCAS | 22169460PubMed |
[27] J. D. Halley, D. A. Winkler, F. R. Burden, Stem Cell Res. 2008, 1, 157.
| Crossref | GoogleScholarGoogle Scholar | 19383397PubMed |
[28] O. E. Hutt, S. Saubern, D. A. Winkler, Bioorg. Med. Chem. 2011, 19, 5903.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtFyntbvF&md5=f1276c7dec941c5105664ccc0d2b869eCAS | 21889349PubMed |
[29] V. C. Epa, J. Yang, Y. Mei, A. L. Hook, R. Langer, D. G. Anderson, M. C. Davies, M. R. Alexander, D. A. Winkler, J. Mater. Chem. 2012, 22, 20902.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhtlGqsLvP&md5=026d6a6243834d7725deb91ddbe646aaCAS | 24092955PubMed |
[30] T. C. Le, X. Mulet, F. R. Burden, D. A. Winkler, Mol. Pharmaceutics 2013, 10, 1368.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXjsVymt7Y%3D&md5=318f28e42f59c2f6ed948f18f8cf3fa3CAS |
[31] S. Marchesan, L. Waddington, C. D. Easton, D. A. Winkler, L. Goodall, J. Forsythe, P. G. Hartley, Nanoscale 2012, 4, 6752.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhsV2qsrbO&md5=320186073d3da9a20047696ead2e9fc1CAS | 22955637PubMed |
[32] A. D. Celiz, J. G. W. Smith, R. Langer, D. G. Anderson, D. A. Winkler, D. A. Barrett, M. C. Davies, L. E. Young, C. Denning, M. R. Alexander, Nat. Mater. 2014, 13, 570.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXot1ylsLY%3D&md5=c700c37ca4cc7b7364846b42cd9f0e25CAS | 24845996PubMed |
[33] H. Autefage, E. Gentleman, E. Littmann, M. A. B. Hedegaard, T. Von Erlach, M. O’Donnell, F. R. Burden, D. A. Winkler, M. M. Stevens, Proc. Natl. Acad. Sci. USA 2015, 112, 4280.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXkvFaqurc%3D&md5=9d4023ed2db29d94ce022683800d5ec6CAS | 25831522PubMed |
[34] W. B. Wan, L. L. Li, Z. B. Zhao, H. Hu, X. J. Hao, D. A. Winkler, L. C. Xi, T. C. Hughes, J. S. Qiu, Adv. Funct. Mater. 2014, 24, 4915.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXmslamsr8%3D&md5=1438bc92de059aac0f83224ce2cc291aCAS |
[35] See pp. 85–96 in V. C. Epa, D. A. Winkler, L. Tran, Computational Nanotoxicology: Adverse Effects of Engineered Nanoparticles 2012 (London: Academic Press).
[36] V. C. Epa, F. R. Burden, C. Tassa, R. Weissleder, S. Shaw, D. A. Winkler, Nano Lett. 2012, 12, 5808.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhsVOhsr7L&md5=66cb6dfc2493a754a8a3471293a2736fCAS | 23039907PubMed |
[37] D. A. Winkler, F. R. Burden, B. Yan, R. Weissleder, C. Tassa, S. Shaw, V. C. Epa, SAR QSAR Environ. Res. 2014, 25, 161.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXktlCksbg%3D&md5=75859e0dc153e97c12dd3ec3de5bf438CAS | 24625316PubMed |
[38] D. A. Winkler, E. Mombelli, A. Pietroiusti, L. Tran, A. Worth, B. Fadeel, M. J. McCall, Toxicology 2013, 313, 15.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhvVWit73J&md5=ec63dd66849c8b09e3b65c4d77ea27eaCAS | 23165187PubMed |
[39] P. Ung, D. A. Winkler, J. Med. Chem. 2011, 54, 1111.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXht1Sltbw%3D&md5=1c210586dfabad1adc7eb0fee5018769CAS | 21275407PubMed |
[40] G. Wijffels, W. M. Johnson, A. J. Oakley, K. Turner, V. C. Epa, S. J. Briscoe, M. Polley, A. J. Liepa, A. Hofmann, J. Buchardt, C. Christensen, P. Prosselkov, B. P. Dalrymple, P. F. Alewood, P. A. Jennings, N. E. Dixon, D. A. Winkler, J. Med. Chem. 2011, 54, 4831.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXnt1agsb4%3D&md5=cced8060d56680869c0cf2e131105aebCAS | 21604761PubMed |
[41] C. Hansch, T. Fujita, J. Am. Chem. Soc. 1964, 86, 1616.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaF2cXptF2gsQ%3D%3D&md5=744a7bb7eb0666d0d1295b06506e0d12CAS |
[42] C. Hansch, P. P. Maloney, T. Fujita, Nature 1962, 194, 178.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaF38XktFaltLg%3D&md5=af0d3ac6cb6635f9f889f133a1abbfedCAS |
[43] C. Hansch, T. Fujita, ACS Symp. Ser. 1995, 606, 1.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXps1ehsLY%3D&md5=86868de9bb685faa1a8777dfa4022e84CAS |
[44] F. R. Burden, D. A. Winkler, J. Chem. Inf. Comput. Sci. 1999, 39, 236.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXmsFWksLY%3D&md5=ae4515eebed23fbf2a114b3ec369cad6CAS |
[45] M. A. T. Figueiredo, IEEE Trans. Pattern Anal. Mach. Intell. 2003, 25, 1150.
| Crossref | GoogleScholarGoogle Scholar |
[46] D. Winkler, Drug Discov. Today 2001, 6, 1198.
| Crossref | GoogleScholarGoogle Scholar | 11722868PubMed |
[47] F. R. Burden, D. A. Winkler, in Artificial Neural Networks: Methods and Applications (Ed. D. Livingston) 2009, Methods in Molecular Biology Vol. 458, 25–44 (Humana Press: Totowa, NJ).
[48] H. T. Kiiveri, BMC Bioinf. 2008, 9, 195.
| Crossref | GoogleScholarGoogle Scholar |
[49] M. J. Polley, D. A. Winkler, F. R. Burden, J. Med. Chem. 2004, 47, 6230.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXptVyqsro%3D&md5=15b1005c1dc0a3cbfe3099d2c729191eCAS | 15566293PubMed |
[50] M. Salahinejad, T. C. Le, D. A. Winkler, Mol. Pharmaceutics 2013, 10, 2757.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXotlOgs7s%3D&md5=ba0ee1504b9f78e10787ecfb7bce25d5CAS |
[51] T. C. Le, M. Ballard, P. Casey, M. S. Liu, D. A. Winkler, Mol. Inf. 2015, 34, 18.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXhvVGku7zI&md5=8b3f800888817d174d9a385fda7ed40eCAS |
[52] P. J. Meunier, C. Roux, E. Seeman, S. Ortolani, J. E. Badurski, T. D. Spector, J. Cannata, A. Balogh, E.-M. Lemmel, S. Pors-Nielsen, R. Rizzoli, H. K. Genant, J.-Y. Reginster, N. Engl. J. Med. 2004, 350, 459.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXos1Snsw%3D%3D&md5=3026171a1b9dfcf7a7922fd09a710a87CAS | 14749454PubMed |
[53] J. Y. Reginster, E. Seeman, M. C. De Vernejoul, S. Adami, J. Compston, C. Phenekos, J. P. Devogelaer, M. Diaz Curiel, A. Sawicki, S. Goemaere, O. H. Sorensen, D. Felsenberg, P. J. Meunier, J. Clin. Endocrinol. Metab. 2005, 90, 2816.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXkt1Wgt78%3D&md5=ffb4861c3869cec85c9e710eb4bc8e9aCAS | 15728210PubMed |
[54] Y. H. Huh, M. Noh, F. R. Burden, J. C. Chen, D. A. Winkler, J. L. Sherley, Stem Cell Res. 2015, 14, 144.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXovF2lsw%3D%3D&md5=f1fa5f8ad7d7083c5b88ce912f6ed317CAS | 25636161PubMed |
[55] Y. H. Huh, J. L. Sherley, Stem Cells 2011, 29, 1620.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsV2itbzI&md5=6f2ecc76390fc0df43bff6536165aaceCAS | 21905168PubMed |
[56] X. Mulet, D. F. Kennedy, C. E. Conn, A. Hawley, C. J. Drummond, Int. J. Pharm. 2010, 395, 290.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXosF2it7Y%3D&md5=41f9a940cb7a7ea409d500eb40edc908CAS | 20580796PubMed |
[57] T. C. Le, C. E. Conn, F. R. Burden, D. A. Winkler, Cryst. Growth Des. 2013, 13, 1267.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXhsVemt78%3D&md5=d79cbcd6e8ca9fc570d4896bd9c63074CAS |
[58] T. C. Le, C. E. Conn, F. R. Burden, D. A. Winkler, Cryst. Growth Des. 2013, 13, 3126.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXotFyitLg%3D&md5=cdbb653a66b38fcc2af5b3363d0ae639CAS |
[59] A. L. Hook, C. Y. Chang, J. Yang, J. Luckett, A. Cockayne, S. Atkinson, Y. Mei, R. Bayston, D. J. Irvine, R. Langer, D. G. Anderson, P. Williams, M. C. Davies, M. R. Alexander, Nat. Biotechnol. 2012, 30, 868.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhtFOgsbzI&md5=88a62f1cefc755990459b7c473406719CAS | 22885723PubMed |
[60] V. C. Epa, A. L. Hook, C. Chang, J. Yang, R. Langer, D. G. Anderson, P. Williams, M. C. Davies, M. R. Alexander, D. A. Winkler, Adv. Funct. Mater. 2014, 24, 2085.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXhvVOhsLnM&md5=40b1c571f5c06de952b03a573a8204d1CAS |
[61] J. Yang, Y. Mei, A. L. Hook, M. Taylor, A. J. Urquhart, S. R. Bogatyrev, R. Langer, D. G. Anderson, M. C. Davies, M. R. Alexander, Biomaterials 2010, 31, 8827.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXht1egsLzF&md5=35171e13bba7a9297ec37ecc814bd2f0CAS | 20832108PubMed |
[62] V. Neduva, R. B. Russell, Nucleic Acids Res. 2006, 34, W350.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28Xps1yitLs%3D&md5=197a8ea50025a435901305380e8a1e4eCAS | 16845024PubMed |
[63] C. H. Reynolds, S. D. Bembenek, B. A. Tounge, Bioorg. Med. Chem. Lett. 2007, 17, 4258.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXnsVKntbo%3D&md5=3c799866e7e71f1f7899d7feb3afc7e5CAS | 17532632PubMed |
[64] M. M. Hann, A. R. Leach, G. Harper, J. Chem. Inf. Comput. Sci. 2001, 41, 856.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXivFynt7w%3D&md5=18b7e9da72e50540f7d55e8fc87078b3CAS | 11410068PubMed |
[65] S. Byers, E. Amaya, S. Munro, O. Blaschuk, Dev. Biol. 1992, 152, 411.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK38XltVCnsrg%3D&md5=238e46fd01cd10b8a154911cbfd1f24fCAS | 1322849PubMed |
[66] S. M. Burden-Gulley, T. J. Gates, S. E. L. Craig, S. F. Lou, S. A. Oblander, S. Howell, M. Gupta, S. M. Brady-Kalnay, Peptides 2009, 30, 2380.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVKntLvI&md5=a471e25b6801a67764ff1032c125b162CAS | 19765627PubMed |
[67] S. E. Cwirla, P. Balasubramanian, D. J. Duffin, C. R. Wagstrom, C. M. Gates, S. C. Singer, A. M. Davis, R. L. Tansik, L. C. Mattheakis, C. M. Boytos, P. J. Schatz, D. P. Baccanari, N. C. Wrighton, R. W. Barrett, W. J. Dower, Science 1997, 276, 1696.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXjvV2mtrw%3D&md5=765f9350eb72e2d32978a8da7c62e59aCAS | 9180079PubMed |
[68] B. P. Dalrymple, K. Kongsuwan, G. Wijffels, N. E. Dixon, P. A. Jennings, Proc. Natl. Acad. Sci. USA 2001, 98, 11627.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXnt12jsL4%3D&md5=48d6ac6c95c694b1370958a17fd2eb7dCAS | 11573000PubMed |
[69] R. V. Devi, S. S. Sathya, M. S. Coumar, Appl. Soft Comput. 2015, 27, 543.
| Crossref | GoogleScholarGoogle Scholar |
[70] M. J. Sorich, R. A. McKinnon, J. O. Miners, D. A. Winkler, P. A. Smith, J. Med. Chem. 2004, 47, 5311.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXnslejurs%3D&md5=ae6c5768d2f8c056602a49492e4df262CAS | 15456275PubMed |
[71] M. J. Sorich, J. O. Miners, R. A. McKinnon, D. A. Winkler, F. R. Burden, P. A. Smith, J. Chem. Inf. Comput. Sci. 2003, 43, 2019.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXnt1Ohsbs%3D&md5=1adbfed5d565ac0b8615c5f848c88bb8CAS | 14632453PubMed |
[72] M. J. Sorich, P. A. Smith, D. A. Winkler, F. R. Burden, R. A. McKinnon, J. O. Miners, Drug Metab. Rev. 2003, 35, 167.
[73] A. L. Hopkins, Nat. Chem. Biol. 2008, 4, 682.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXht1Kgs7fK&md5=c2b1283e4791c6205ec672b2af72844fCAS | 18936753PubMed |
[74] M. A. Yildirim, K. I. Goh, M. E. Cusick, A. L. Barabasi, M. Vidal, Nat. Biotechnol. 2007, 25, 1119.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhtFagt7rO&md5=914b83e382df20d749fa0cefc88807c3CAS | 17921997PubMed |
† Prof. Adrien Albert (1907–1989) was widely considered the ‘father of selective toxicity’ in Australia. He championed the idea that chemical compounds could be developed that were toxic to a pathogen or disease state but were much less so to normal tissues. He received his BSc (H1) and the University Medal from the University of Sydney in 1932. He completed his PhD in 1937 and a DSc in 1947 at the University of London. He was a Lecturer at the University of Sydney (1938–1947), during which time he advised the Medical Directorate, Australian Army (1942–1947). After the war, he worked at the Wellcome Research Institute in London (1947–1948), then returned to Australia as the Foundation Chair of Medical Chemistry, John Curtin School of Medical Research at the Australian National University in 1948. Apart from establishing the Department of Medical Chemistry, he was also elected a Fellow of the Australian Academy of Science, and authored a seminal textbook Selective Toxicity: The Physico-Chemical Basis of Therapy, published by Chapman and Hall in 1951 and extensively reprinted. In 1981 he won the Bristol-Myers Squibb Smissman Award and became Patron of the Royal Australian Chemical Institute (RACI) Division of Medicinal Chemistry. He also received the Officer of the Order of Australia honour for contributions to medical research. After his death, the Adrien Albert Laboratory of Medicinal Chemistry was established at the University of Sydney in 1989. The Royal Society of Chemistry created the Adrien Albert Lectureship in his honour and the Stony Brook University established the Albert lectures. Adrien was awarded a posthumous honorary DSc by the University of Sydney. The Adrien Albert Award was presented for outstanding research in medicinal chemistry by the Medicinal Chemistry and Chemical Biology Division of RACI at the RACI National Congress in Adelaide in 2014.
‡ ‘Only two things are infinite, the universe and human stupidity, and I’m not sure about the former.’ — Albert Einstein