

Chemistry of the Synthetic Strigolactone Mimic GR24*,**
Liam J. Bromhead A , Jason Smith B and Christopher S. P. McErlean A CA School of Chemistry, University of Sydney, Sydney, NSW 2006, Australia.
B School of Physical Sciences (Chemistry), University of Tasmania, Hobart, Tas. 7001, Australia.
C Corresponding author. Email: christopher.mcerlean@sydney.edu.au
Australian Journal of Chemistry 68(8) 1221-1227 https://doi.org/10.1071/CH15298
Submitted: 25 May 2015 Accepted: 11 June 2015 Published: 9 July 2015
Abstract
This research paper describes a second-generation, enantioselective synthesis of (+)-GR24. This new strategy circumvents the need for ozonolysis, which may not be suitable for reactions performed on scale. This flexible new approach is then used to synthesise a novel bromo-GR24 analogue. Finally, the optimal solvent for making GR24 stock solutions is identified.
Introduction
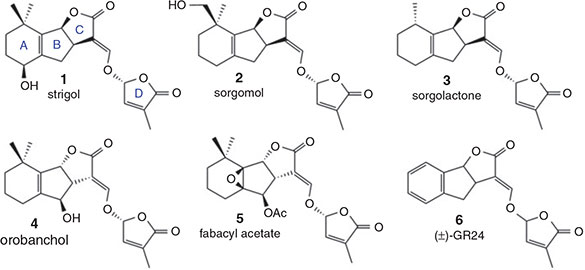
The identification of strigolactones as seed-germinating agents, phytohormones, and mediators of critical plant–fungi interactions, has resulted in an increasing number of research projects seeking to uncover new modes of biological activity and in planta targets.[1–7] While the number of naturally occurring strigolactones is now over 20 (Fig. 1), the extremely low natural abundance these compounds has necessitated the application of the aromatic A-ring analogue (±)-GR24 (6) in most plant-based assays. First reported in 1981,[8] (±)-GR24 has been the target of several syntheses but is still commonly employed as a racemic mixture of diastereomers.[9–13] The importance of employing single enantiomers has been underscored by the recent discovery that GR24 isomers with the absolute stereochemistry corresponding to natural (+)-strigol (1) and (–)-orobanchol (4), exert different biological effects.[14] As such, we recently reported a concise enantioselective route to any stereoisomer of GR24.[15] In this article, we report a second-generation synthetic strategy to GR24 stereoisomers with improved scalability. In addition, we demonstrate that this second-generation route may be applied to the generation of GR24-based molecular probes. Finally, we examine the stability of stock solutions of GR24 in various solvents to help guide future storage and application considerations.
|
Results and Discussion
Synthetic Studies
Our first-generation synthesis of (+)-GR24 (7) and (–)-epi-GR24 (8) began from the economical starting material, indene (9) (Scheme 1).[15] However, this approach involved an ozonolytic cleavage of the indene bond. The operation of ozonolysis on large scales always involves a certain amount of trepidation due the highly reactive nature of the intermediate molozonides and ozonides. While the application of flow-technology may alleviate the associated safety concerns,[16,17] the Wittig olefination to produce 10 was not highly chemoselective. Therefore, we elected to modify a procedure described by Cignarella and co-workers[18] to generate the indanone 12, which could subsequently undergo the stereo-defining Noyori asymmetric transfer hydrogenation.[19]
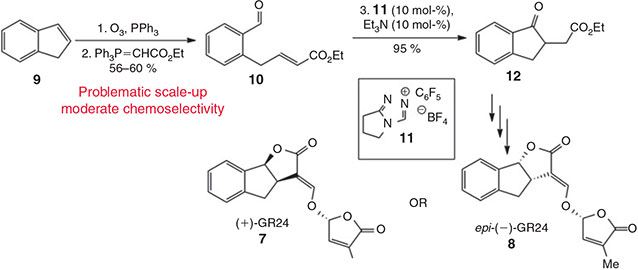
|
As shown in Scheme 2, 3-benzoylpropionic acid (13) was subjected to an aldol reaction with formaldehyde followed by in situ lactonisation to give compound 14.[18] Careful control of pH was critical for achieving a satisfactory yield for this transformation as excessive acid favoured a competing elimination reaction. Treatment of 14 with sulfuric acid effected the desired intramolecular Friedel–Crafts acylation, which was followed by Fischer esterification to give the desired indanone 12. Although this approach requires one further synthetic operation than our first-generation approach, the ready availability of all materials and reagents, and the circumvention of explosive reaction intermediates, meant that this procedure can be conducted on relatively large scales (up to 50 g batches). Application of our previously reported procedures enabled the gram-scale synthesis of (+)-GR24 (7). When conducted on a 10 g scale, the amount of RuTsDPEN catalyst employed for the dynamic kinetic asymmetric Noyori transfer hydrogenation could be conveniently lowered to 1 mol-% without any detriment to the yield or enantioselectivity of the reaction. In this instance we generated (+)-GR24 (7) with the stereochemistry corresponding to (+)-strigol (1), but the process is suitable for the generation of any GR24 stereoisomer.

|
Accessing GR24-Based Molecular Probes
Current research in the plant sciences is focussed on elucidating the in planta targets for strigolactone molecules to elucidate the origin of their biological activity. To meet this new challenge, fluorescent GR24 analogues are required. Previous efforts at generating fluorescently labelled strigolactone analogues have concentrated on substituting the A/B ring system with an indole or isoindole unit.[20–23] We felt that a more flexible strategy would be to append a fluorophore directly to the GR24 scaffold, allowing for tailored molecules for individual assays. We therefore targeted 6-Br-(+)-GR24 (18) as a proof of concept.
As depicted in Scheme 3, 4-bromoacetophenone (19) was subjected to an aldol–dehydration sequence to give 20. Chemoselective reduction gave 3-(4-bromobenzoyl)propionic acid (21) which could be employed in the previously described second-generation strategy. Aldol reaction with formaldehyde and ring closure gave compound 22. Intramolecular Friedel–Crafts acylation was complicated by a proto-debromination reaction, but conducting the reaction at 80°C gave an acceptable yield of the indanone 23. Esterification to compound 24 was straightforward and set the scene for the stereochemistry-defining reaction. Our concerns about competing reaction of the aryl bromide proved groundless, and compound 24 underwent smooth Noyori dynamic kinetic asymmetric transfer hydrogenation to give the lactone 25. The recrystallised compound displayed an enantiomeric excess of >99 % (Fig. 2). Formylation and attachment of the D-ring gave (+)-6-Br-GR24 (18) as a single enantiomer alongside (+)-6-Br-epi-GR24 (26).

|

|
|
|
Stability Studies
The quantity of (±)-GR24 (6) required to elicit biological responses in plant-based assays is usually so small that practical considerations dictate the generation of a stock solution of known concentration, followed by the administration of precise aliquots. Such stock solutions are generally refrigerated to avoid compound decomposition, but we became aware that the efficacy of some stock solutions appeared to deteriorate over time. We decided that an understanding of the relative stability of GR24 in various solvents would therefore be useful, as this potential instability is not understood by the users of the material. In addition, because of the pressure differences occurring with continually cooling and re-warming the stock solutions, contamination by atmospheric moisture is highly likely. Therefore, the effect of a water co-solvent was also investigated.
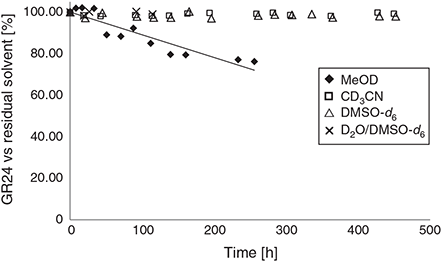
A sample of freshly prepared (±)-GR24 (6) was dissolved in a series of deuterated solvents and 1H NMR spectra were recorded after defined time intervals at room temperature (Fig. 3). The deterioration of the material was quantified by monitoring the integration ratio for a signal corresponding to (±)-GR24 (6) relative to residual non-deuterated solvent. It was immediately apparent that acetonitrile and DMSO were suitable solvents for the generation of stock solutions. Even after 18 days in these solvents, no deterioration of the sample could be detected by 1H NMR analysis. Furthermore, the inclusion of 30 % water into the DMSO solvent (v/v) resulted in no deterioration after more than 100 h. In contrast, methanol is not a suitable solvent for GR24 stock solutions.
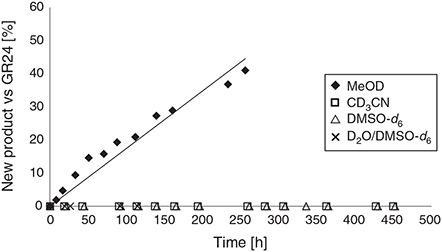
While quantifying the amount of deterioration by using the residual solvent as an internal standard is reliable for high boiling solvents, the volatility of methanol required a secondary measurement. Therefore, a signal corresponding to (±)-GR24 (6) was also compared with a new signal that appeared over the time-course of the experiment (Fig. 4). A line of best fit suggests that stock solutions of (±)-GR24 (6) in methanol have a half-life of ~12 days.
Conclusion
We report a synthetic strategy that enables the multigram-scale synthesis of (+)-GR24 (7), and that is flexible enough to allow for the synthesis of 6-Br-(+)-GR24 (18) as a single enantiomer. We anticipate that this latter molecule will allow ready access to a variety of fluorescently labelled GR24 analogues. Furthermore, we report the outcomes of GR24 stability studies that point to DMSO as a suitable solvent for the generation of stock solutions. The synthesis of the 5-Br, 7-Br, and 8-Br-(+)-GR24 analogues, as well as the attachment of several fluorophores is underway in our laboratory.
Experimental
General Experimental
Reagent grade dichloromethane and triethylamine were freshly distilled over calcium hydride. Tetrahydrofuran and methanol were collected using an Innovative Technology Inc. PureSolv solvent purification system. All other solvents and reagents were used as received from commercial sources. Melting points were determined using a Stanford Research Systems Optimelt automated melting point system and are uncorrected. Infrared spectra were acquired neat on a Bruker Alpha-E ATR spectrometer. 1H and 13C NMR spectra were recorded on a Bruker AVANCE DPX300 (1H frequency 300 MHz, 13C frequency 75 MHz) or Bruker AVANCE DPX500 (1H frequency 500 MHz, 13C frequency 125 MHz). 1H chemical shifts are expressed as parts per million (ppm) with residual chloroform (δ 7.26) or tetramethylsilane (δ 0.00) as reference and are reported as chemical shift (δH), relative integral, multiplicity (s = singlet, br = broad, d = doublet, t = triplet, dd = doublet of doublets, dt = doublet of triplets, m = multiplet), and coupling constants (J) reported in Hz. 13C NMR chemical shifts are expressed as parts per million (ppm) with residual chloroform (δ 77.16) as internal reference and are reported as chemical shift (δC) and multiplicity (assigned from DEPT experiments). NMR assignments were made on the basis of HSQC, HMBC, NOESY, and COSY 2D experiments. High resolution mass spectra were recorded on a Bruker ApexII Fourier Transform Ion Cyclotron Resonance mass spectrometer with a 7.0 T magnet, fitted with an off-axis Analytical electrospray source. Column chromatography was performed using Grace Davidson 40–63 μm (230–400 mesh) silica gel using distilled solvents. Analytical thin-layer chromatography was performed using preconditioned plates (Merck TLC silica gel 60 F254 on aluminium) and visualised using UV light (254 and 365 nm) and ethanolic anisaldehyde.
4-Benzoyldihydrofuran-2(3H)-one (14)[24]
A solution of aqueous sodium hydroxide (0.5 M, 620 mL) was added to β-benzoylpropionic acid (50.0 g, 280 mmol). Formalin (37 %, 23 mL) was added and the solution stirred for 1 h. Aqueous hydrochloric acid (32 %, 40 mL) was added and the solution stirred overnight. The liquid was decanted and the solid product was dissolved in dichloromethane (400 mL). The aqueous solution was extracted with dichloromethane (2 × 200 mL). The combined organic extracts were then washed with brine, dried over Na2SO4, and the solvent was removed under vacuum to give the title compound (41.0 g, 77 %, determined by NMR analysis) as a tan solid contaminated with an alkene by-product. Mp 57–60°C, lit.[24] 63–65°C. νmax/cm–1 2980, 1773, 1681, 1596, 1580, 1478, 1449, 1415, 1359, 1286, 1231, 1177, 1028, 1005. δH (300 MHz, CDCl3) 7.90–7.82 (2H, m), 7.60–7.53 (1H, m), 7.49–7.40 (2H, m), 4.57–4.28 (3H, m), 2.95–2.68 (2H, m). δC (75 MHz, CDCl3) 196.5 (C), 175.4 (C), 134.9 (C), 134.3 (CH), 129.1 (2 CH), 128.5 (2 CH), 69.0 (CH2), 42.1 (CH), 30.8 (CH2).
Ethyl 2-(1-Oxo-2,3-dihydro-1H-inden-2-yl)acetate (12)
In two separate batches, lactone (14) (11.08 g, 58.25 mmol) was taken up in concentrated sulfuric acid (98 %, 40 mL) and heated at 100°C for 10 min. The solution was poured into a separating funnel containing ice water. The solution was extracted with dichloromethane (3 × 200 mL) and the organic extracts were washed with brine, dried over Na2SO4, and the solvent removed under vacuum to give an orange solid (5.83 g). The batches were combined to give (9.60 g, 50.5 mmol) of the acid which was taken up in ethanol (100 mL) and acetyl chloride (5.38 mL, 75.7 mmol) was added. The reaction mixture was stirred overnight. Most of the solvent was removed under vacuum, and then the residue was quenched with saturated aqueous sodium hydrogen carbonate (100 mL). The aqueous layer was extracted with ethyl acetate (3 × 100 mL). The combined organic extracts were washed with brine, dried over Na2SO4, and the solvent was removed under vacuum to give a brown residue. The residue was purified by column chromatography, eluting with 10 % ethyl acetate in hexanes, to give the title compound 12 (10.98 g, 53 % over 2 steps) as a yellow oil. νmax/cm–1 2983, 1708, 1609, 1466, 1373, 1294, 1279, 1217, 1174, 1029. δH (300 MHz, CDCl3) 7.77 (1H, d, J 7.5), 7.59 (1H, dd, J 7.8, 7.2), 7.45 (1H, d, J 7.8), 7.37 (1H, dd, J 7.5, 7.2), 4.13 (2H, q, J 7.2), 3.45 (1H, dd, J 7.8, 17.0), 2.95 (3H, m), 2.62 (1H, dd, J 8.1, 16.2), 1.21 (3H, t, J 7.2). δC (75 MHz, CDCl3) 206.9 (C), 172.1 (C), 153.4 (C), 136.5 (C), 135.0 (CH), 127.6 (CH), 126.6 (CH), 124.1 (CH), 60.9 (CH2), 43.7 (CH), 35.4 (CH2), 33.1 (CH2), 14.3 (CH3). m/z (HRMS ESI) 241.08317; [C13H14O3 + Na]+ requires 241.08352.
(3aR,8bS)-3,3a,4,8b-Tetrahydro-2H-indeno[1,2-b]furan-2-one (15)
Dichloro(p-cymene)ruthenium (ii) dimer (140 mg, 0.23 mmol) and (1S,2S)-(+)-N-p-tosyl-1,2-diphenylethylenediamine (168 mg, 0.46 mmol) were taken up in isopropyl alcohol (5 mL). Triethylamine (130 μL, 920 μmol) was added and the reaction mixture stirred at 80°C for 1.5 h. The solvent was removed under vacuum to give (S,S)-RuTsDPEN which was used directly in the following step. Formic acid (8.64 mL) and Hünig’s base (15.96 mL) were mixed at 0°C. After 15 min, a solution of indanone 12 (10 g, 46 mmol) in dichloromethane (15 mL) was added via cannula, followed by a solution of the freshly prepared (S,S)-RuTsDPEN in dichloromethane (5 mL). The dichloromethane was removed under nitrogen flow and the mixture stirred overnight at 40°C. Saturated aqueous sodium hydrogen carbonate (40 mL) was added. The aqueous phase was extracted with ethyl acetate (3 × 70 mL). The combined organic extracts were then washed with brine, dried over Na2SO4, and the solvent was removed under vacuum. The residue was taken up in toluene (25 mL), pyridinium para-toluenesulfonate (500 mg, 2.00 mmol) was added, and the reaction mixture heated at 80°C. The solvent was removed under vacuum and the residue purified by column chromatography, eluting with 20 % ethyl acetate in hexanes, to give the title compound (6.89 g, 86 %) as colourless crystals, which were recrystallised from hexanes and ethyl acetate. Mp 93°C. [α]D20 +110°C (c 0.985, CHCl3), lit –107°C (c 0.4, CHCl3). νmax/cm–1 2954, 1753, 1610, 1450, 1349, 1250, 1145, 990. δH (300 MHz, CDCl3) 7.40 (1H, d, J 7.2), 7.24 (3H, m), 5.81 (1H, d, J 6.9), 3.26 (2H, m), 2.82 (2H, m), 2.32 (1H, dd, J 5.1, 18.1). δC (75 MHz, CDCl3) 176.9 (C), 142.5 (C), 138.8 (C), 130.0 (CH), 127.6 (CH), 126.4 (CH), 125.3 (CH), 87.7 (CH), 37.9 (CH2), 37.3 (CH), 35.7 (CH2). m/z (HRMS ESI) 197.05728; [C11H10O2 + Na]+ requires 197.05730.
(E)-4-(4-Bromophenyl)-4-oxobut-2-enoic Acid (20)
4′-Bromoacetophenone (30.0 g, 150 mmol) and glyoxylic acid monohydrate (13.9 g, 151 mmol) were stirred at 95°C under reduced pressure (34 mbar) for 3 h. The reaction mixture was cooled, dissolved in aqueous potassium carbonate (5 %, 600 mL), and extracted with ethyl acetate (2 × 200 mL). The aqueous layer was acidified with aqueous hydrochloric acid (10 %, 600 mL) and extracted with ethyl acetate (2 × 400 mL). The combined organic extracts were washed with brine, dried over Na2SO4, and the solvent removed under vacuum to give an orange solid. The solid was taken up in glacial acetic acid (30 mL), concentrated hydrochloric acid (32 %, 3 mL) was added, and the reaction mixture heated under reflux for 4 h. The acetic acid was removed under vacuum. The residue was taken up in ethyl acetate (300 mL) and washed with brine (3 × 300 mL). The organic extract was dried over Na2SO4 and the solvent removed under vacuum to give the title compound (21.25 g, 55 %) as a yellow solid. Mp 150 °C, lit.[25] 159–160°C. νmax/cm–1 3093 (br), 1713, 1689, 1666, 1633, 1585, 1568, 1421, 1398, 1296, 1282, 1193, 1071. δH (400 MHz, DMSO-d6) 13.20 (1H, br s), 7.99–7.95 (2H, m), 7.85 (1 H, d, J 15.6), 7.80–7.77 (2 H, m), 6.68 (1 H, d, J 15.6). δC (100 MHz, DMSO-d6) 188.8 (C), 166.3 (C), 135.9 (CH), 135.2 (C), 133.3 (CH), 132.1 (2 × CH), 130.8 (2 × CH), 128.3 (C). m/z (ESI–) 253 and 255 ([M – H]–, 100 and 81 %).
4-(4-Bromophenyl)-4-oxobutanoic Acid (21)
Zinc dust (10.9 g, 166 mmol) was added to a stirred solution of 20 (37.5 g, 147 mmol) in acetic acid (210 mL) and water (75 mL) over 1 h. The mixture was stirred for a further 3 h, the reaction mixture was then filtered, and the solid was taken up in ethyl acetate (300 mL). This solution was washed with brine (4 × 500 mL), dried over Na2SO4, and the solvent was removed under vacuum to give the title compound (20.7 g, 55 %). Mp 139°C, lit.[26] 146°C. νmax/cm–1 3044 (br), 1698, 1673, 1585, 1568, 1446, 1410, 1335, 1280, 1200, 1074. δH (400 MHz, CDCl3) 12.00 (1H, br s), 7.91 (2H, dapp, J 8.6), 7.74 (2H, dapp, J 8.5), 3.23 (2H, t, J 6.26), 2.57 (2H, t, J 6.24). δC (100 MHz, CDCl3) 198.3 (C), 174.2 (C), 135.9 (C), 132.3 (2 CH), 130.4 (2 CH), 127.7 (C), 33.6 (CH2), 28.3 (CH2). m/z (ESI–) 255 and 257 ([M – H]–, 86 and 83 %), 271 (100).
4-(4-Bromobenzoyl)dihydrofuran-2(3H)-one (22)
Aqueous sodium hydroxide (0.5 M, 170 mL) was added to 4′-bromobenzoyl propionic acid (21) (20.0 g, 77.8 mmol). Formalin (37 %, 6.37 mL) was added and the solution was stirred for 3 h. Aqueous hydrochloric acid (32 %, 20 mL) was added and the solution stirred overnight. The liquid was decanted and the solid product was dissolved in dichloromethane (300 mL). The previously decanted aqueous solution was washed twice with dichloromethane (100 mL). The combined organic extracts were then washed with brine, dried over Na2SO4, and the solvent removed under vacuum to give a brown oil. This was taken up in ethyl acetate (150 mL) and washed with aqueous potassium carbonate solution (5 %, 2 × 200 mL). The organic layer was then washed with brine, dried over Na2SO4, and the solvent removed under vacuum to give the title compound (12.6 g, 60 %) as a brown solid. Mp 90°C, lit.[24] 98–100°C. νmax/cm–1 2975, 1768, 1679, 1583, 1568, 1484, 1397, 1377, 1354, 1281, 1217, 1173, 1070, 1029, 1003. δH (400 MHz, CDCl3) 7.81–7.78 (2H, m), 7.69–7.65 (2H, m), 4.63–4.58 (1H, m), 4.83–4.39 (1H, m), 4.40–4.31 (1H, m), 2.96 (1H, dd, J 17.78, 7.22), 2.81 (1H, dd, J 17.78, 9.42). δC (100 MHz, CDCl3) 195.5 (C), 175.2 (C), 133.6 (C), 132.5 (2 CH), 130.0 (2 CH), 129.6 (C), 68.8 (CH2), 42.1 (CH), 30.8 (CH2). m/z (HRMS ESI) 268.98056, 270.97860; [C11H9BrO3 + H]+ requires 268.98078, 270.97874.
2-(5-Bromo-1-oxo-2,3-dihydro-1H-inden-2-yl)acetic Acid (23)
Compound 22 (4.70 g, 17.5 mmol) was taken up in concentrated sulfuric acid (98 %, 10 mL) and heated at 80°C for 3 h. The solution was transferred to a separating funnel containing ice water, using dichloromethane and water. The solution was extracted with dichloromethane (4 × 100 mL). The combined organic layers were washed with brine, dried over Na2SO4, and the solvent was removed under vacuum. The residue was taken up in ethyl acetate and washed with aqueous potassium carbonate solution (10 %, 2 × 50 mL). The combined aqueous extracts were acidified with concentrated hydrochloric acid and extracted with ethyl acetate (3 × 50 mL). The combined organic extracts were washed with brine, dried over Na2SO4, and the solvent was removed under vacuum to give the title compound (2.01 g, 43 %). νmax/cm–1 3065 (br), 1708, 1698, 1598, 1573, 1436, 1394, 1255, 1201. δH (400 MHz, CDCl3) 7.65 (1H, s), 7.62 (1H, d, J 8.24), 7.53 (1H, d, J 8.04), 3.45 (1H, dd, J 17.2, 7.76), 3.05–2.97 (2H, m), 2.90 (1H, dd, J 17.22, 4.38), 2.69 (1H, dd, J 17.9, 9.06). δC (100 MHz, CDCl3) 205.3 (C), 176.2 (C), 154.7 (C), 135.0 (C), 131.3 (CH), 130.5 (C), 129.8 (CH), 125.3 (CH), 43.4 (CH), 24.6 (CH2), 32.6 (CH2). m/z (HRMS ESI) 290.96274, 292.96096; [C11H9BrO3 + Na]+ requires 290.96273, 292.96068.
Ethyl 2-(5-Bromo-1-oxo-2,3-dihydro-1H-inden-2-yl)acetate (24)
Compound 23 (4.21 g, 15.6 mmol) was taken up in ethanol (25 mL) and acetyl chloride (1.7 mL, 23 mmol) was added. The reaction mixture was stirred overnight. The majority of the solvent was removed under vacuum and the residue was then quenched with saturated aqueous sodium hydrogen carbonate (10 mL). The aqueous layer was extracted with ethyl acetate (3 × 30 mL). The combined organic extracts were washed with brine, dried over Na2SO4, and the solvent removed under vacuum to give a brown residue. The residue was purified by column chromatography, eluting with 5 % ethyl acetate in hexanes, to give the title compound (3.13 g, 67 %) as a yellow solid. νmax/cm–1 2980, 1710, 1595, 1574, 1413, 1373, 1317, 1266, 1218, 1197, 1175. δH (400 MHz. CDCl3) 7.65–7.59 (2 H, m), 7.51 (1 H, dtapp, J 8.08, 0.75), 4.13 (2 H, q, J 7.13), 3.43 (1 H, dd, J 17.08, 7.8), 3.03–2.85 (3 H, m), 2.67 (1 H, dd, J 16.64, 8.04), 1.21 (3 H, t, J 7.16). δC (100 MHz, CDCl3) 205.4 (C), 171.7 (C), 154.8 (C), 135.3 (C), 131.1 (CH), 130.2 (C), 129.8 (CH), 125.1 (CH), 60.8 (CH2), 43.5 (CH), 35.0 (CH2), 32.6 (CH2), 14.1 (CH3). m/z (HRMS ESI) 318.99376, 320.99174; [C13H13BrO3 + Na]+ requires 318.99403, 320.99198.
(3aR,8bS)-6-Bromo-3,3a,4,8b-tetrahydro-2H-indeno[1,2-b]furan-2-one (25)
Dichloro(p-cymene)ruthenium (ii) dimer (62 mg, 0.10 mmol) and (1S,2S)-(+)-N-p-tosyl-1,2-diphenylethylenediamine (74 mg, 0.20 mmol) were taken up in isopropyl alcohol (3 mL). Triethylamine (56 μL, 0.40 mmol) was added and the reaction mixture stirred at 80°C for 1.5 h. The solvent was removed under vacuum to give (S,S)-RuTsDPEN which was used directly. Formic acid (0.95 mL) and Hünig’s base (1.76 mL) were mixed at 0°C. After 15 min, a solution of 24 (1.50 g, 5.00 mmol) in dichloromethane (5 mL) was added via cannula, followed by a solution of freshly prepared (S,S)-RuTsDPEN in dichloromethane (2 mL). The dichloromethane was removed under nitrogen flow and the mixture was stirred overnight. Saturated aqueous sodium hydrogen carbonate (15 mL) was added. The aqueous phase was extracted with ethyl acetate (3 × 20 mL). The combined organic extracts were then washed with brine, dried over Na2SO4, and the solvent was removed under vacuum. The residue was purified by column chromatography, eluting with 10 % ethyl acetate in hexanes followed by 30 % ethyl acetate in hexanes, to give a colourless solid. The solid product was taken up in toluene (10 mL), pyridinium para-toluenesulfonate (125 mg, 0.5 mmol) was added, and the reaction mixture heated at 80°C overnight. The solution was diluted with ethyl acetate (20 mL), washed with water (20 mL), brine (20 mL), and was then dried over Na2SO4. The solvent was removed under vacuum to give the title compound (1.14 g, 89 %) as colourless crystals, which were recrystallised from hexanes and ethyl acetate. Mp 156–157°C. [α]D20 +60.4°C (c 0.50, CHCl3). νmax/cm–1 2958, 1768, 1598, 1578, 1474, 1444, 1412, 1323, 1196, 1159. δH (400 MHz, CDCl3) 7.44–7.40 (2 H, m), 7.35–7.32 (1 H, m), 5.81 (1 H, d, J 7.08), 3.42–3.33 (1 H, m), 3.30 (1 H, dd, J 16.48, 8.56), 2.94–2.83 (2 H, m), 2.37 (1 H, dd, J 18.12, 5.52). δC (100 MHz, CDCl3) 176.3 (C), 144.8 (C), 137.9 (C), 130.9 (CH), 128.6 (CH), 127.8 (CH), 124.3 (C), 86.7 (CH), 37.8 (CH2), 37.6 (CH), 35.5 (CH2). m/z (HRMS ESI) 274.96758, 276.96554; [C11H9BrO2 + Na]+ requires 274.96781, 276.96577.
(+)-6-Bromo-GR24 (18) and (+)-Epi-6-bromo-GR24 (26)
To a solution of the compound 25 (400 mg, 1.58 mmol) in methyl formate (10 mL) at 0°C, was added potassium tert-butoxide (1.06 g, 9.50 mmol). The reaction mixture was stirred for 3 h and then quenched with hydrochloric acid (1.0 M, 15 mL). The aqueous phase was extracted with ethyl acetate (3 × 20 mL). The combined organic extracts were then washed with brine, dried over Na2SO4, and the solvent removed under vacuum. The colourless solid residue and potassium carbonate (330 mg, 2.37 mmol) were taken up in DMF (5 mL) and a solution of the bromobutenolide 16 (420 mg, 2.37 mmol) in DMF (2 mL) was added via cannula at 0°C. The solution was stirred overnight. The reaction was quenched with saturated aqueous ammonium chloride (10 mL). The reaction mixture was diluted with ethyl acetate (50 mL) and washed with water (3 × 50 mL). The organic extract was then washed with brine, dried over Na2SO4, and the solvent removed under vacuum. The residue was purified by column chromatography, eluting with 80 % diethyl ether in hexanes to give 18 (120 mg 19 %) as colourless crystals. Mp 184–186°C. [α]D20 +403°C (c 0.275, CHCl3). νmax /cm–1 2920, 1782, 1749, 1680, 1598, 1473, 1348, 1323, 1183, 1089, 1019. δH (400 MHz, CDCl3) 7.49 (1 H, d, J 2.5), 7.42 (1 H, d, J 8.0), 7.39 (1 H, s), 7.37 (1 H, d, J 8.0), 6.96 (1 H, tapp, J 1.5), 6.18 (1 H, tapp, J 1.3), 5.88 (1 H, d, J 7.8), 3.99–3.93 (1 H, m), 3.41 (1 H, dd, J 17.1, 9.2), 3.10 (1 H, dd, J 17.1, 2.8), 2.05 (3 H, tapp, J 1.5). δC (100 MHz, CDCl3) 170.9 (C), 170.0 (C), 151.2 (CH), 144.8 (C), 140.7 (CH), 137.9 (C), 136.2 (C), 130.9 (CH), 128.3 (CH), 127.9 (CH), 124.3 (C), 112.7 (C), 100.5 (CH), 84.9 (CH), 39.1 (CH), 37.0 (CH2), 10.8 (CH3). m/z (HRMS ESI) 398.98347, 400.98146; [C17H13BrO5 + Na]+ requires 398.98386, 400.98181.
Compound 26 (120 mg 19 %) was obtained as colourless crystals. Mp 156–157°C. [α]D20 +233°C (c 0.28, CHCl3). νmax/cm–1 2926, 1783, 1748, 1680, 1598, 1475, 1451, 1348, 1322, 1182, 1088, 1018. δH (400 MHz, CDCl3) 7.47 (1 H, d, J 2.5), 7.41 (1 H, d, J 8.0), 7.39 (1 H, s), 7.35 (1 H, d, J 8.0), 6.94 (1 H, tapp, J 1.5), 6.17 (1 H, tapp, J 1.2), 5.87 (1 H, d, J 7.9), 3.97–3.91 (1 H, m), 3.39 (1 H, dd, J 17.0, 9.2), 3.08 (1 H, dd, J 17.1, 3.1), 2.04 (3 H, tapp, J 1.4). δC (100 MHz, CDCl3) 170.8 (C), 170.0 (C), 151.2 (CH), 144.9 (C), 140.8 (CH), 137.9 (C), 136.2 (C), 130.9 (CH), 128.5 (CH), 127.8 (CH), 124.4 (C), 113.0 (C), 100.6 (CH), 85.0 (CH), 39.1 (CH), 37.2 (CH2), 10.8 (CH3). m/z (HRMS ESI) 398.98358, 400.98156; [C17H13BrO5 + Na]+ requires 398.98386, 400.98181.
Supplementary Material
1H and 13C NMR spectra for all synthesised compounds are available on the Journal’s website.
References
[1] M. Vurro, K. Yoneyama, Pest Manag. Sci. 2012, 68, 664.| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XitVWmsrY%3D&md5=372a77007fbb33d67f76ad109a1f5541CAS | 22323399PubMed |
[2] K. Yoneyama, X. Xie, K. Yoneyama, Phytopathology 2011, 101, S239.
[3] K. Yoneyama, X. Xie, K. Yoneyama, Phytopathology 2011, 101, S233.
[4] K. Ueno, M. Fujiwara, S. Nomura, M. Mizutani, M. Sasaki, H. Takikawa, Y. Sugimoto, J. Agric. Food Chem. 2011, 59, 9226.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtVGjtbfJ&md5=e875c2423e24eb1609c2729eb879d11eCAS | 21819156PubMed |
[5] X. N. Xie, K. Yoneyama, K. Yoneyama, Annu. Rev. Phytopathol. 2010, 48, 93.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXht1Wgt77K&md5=084ca6585085130cf7800ec946947a7aCAS |
[6] M. Sasaki, J. Pestic. Sci. 2009, 34, 315.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhs1aks7rJ&md5=478de498440c232a45f731a04c19bc18CAS |
[7] K. Akiyama, K. Matsuzaki, H. Hayashi, Nature 2005, 435, 824.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXkvVGgsL4%3D&md5=1b0b45eca068d9513a90c8aba439dfe8CAS | 15944706PubMed |
[8] A. W. Johnson, G. Gowda, A. Hassanali, J. Knox, S. Monaco, Z. Razavi, G. Rosebery, J. Chem. Soc., Perkin Trans. 1 1981, 1734.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL3MXkvFOnurw%3D&md5=0f23e1627de8424b631bd8f2f2f027c3CAS |
[9] H. Malik, F. Rutjes, B. Zwanenburg, Tetrahedron 2010, 66, 7198.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXps1Cmtbs%3D&md5=e149847e182c464a5f31a75da1139acbCAS |
[10] E. M. Mangnus, J. Agric. Food Chem. 1992, 40, 1230.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK38XksVyiu78%3D&md5=50175677429b9374eb941c4416a5effbCAS |
[11] J. W. J. F. Thuring, G. H. L. Nefkens, B. Zwanenburg, J. Agric. Food Chem. 1997, 45, 2278.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXjsVSlsrw%3D&md5=38b83f234a1b217bf693600d61190eccCAS |
[12] A. Reizelman, B. Zwanenburg, Eur. J. Org. Chem. 2002, 810.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XitVWiu70%3D&md5=754a525cab4fa51d1751fedbaa401d1bCAS |
[13] M. Lachia, P. M. J. Jung, A. De Mesmaeker, Tetrahedron Lett. 2012, 53, 4514.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XpvVSktL4%3D&md5=eb5f1a26921e9e1841cc44ebcff1ae4fCAS |
[14] A. Scaffidi, M. T. Waters, Y. K. Sun, B. W. Skelton, K. W. Dixon, E. L. Ghisalberti, G. R. Flematti, S. M. Smith, Plant Physiol. 2014, 165, 1221.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXhtFOqsbvE&md5=ce01e0161c4ec79479599f2184cf1e68CAS | 24808100PubMed |
[15] L. J. Bromhead, J. Visser, C. S. P. McErlean, J. Org. Chem. 2014, 79, 1516.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXotVKnsg%3D%3D&md5=15c3017f89f2d051b4545a472c79a459CAS | 24422520PubMed |
[16] M. D. Roydhouse, A. Ghaini, A. Constantinou, A. Cantu-Perez, W. B. Motherwell, A. Gavriilidis, Org. Process Res. Dev. 2011, 15, 989.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXosFyksro%3D&md5=231aae3ee51cfde7ae70f3ee1f05a566CAS |
[17] M. D. Roydhouse, W. B. Motherwell, A. Constantinou, A. Gavriilidis, R. Wheeler, K. Down, I. Campbell, RSC Adv. 2013, 3, 5076.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXktFSntLo%3D&md5=c16a6b1688d3b5be904a9819e2bab013CAS |
[18] G. Cignarella, G. Grella, M. M. Curzu, Synthesis 1980, 825.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL3MXotlylsg%3D%3D&md5=32b7b0ceb7cfbfc6bb381a619139271aCAS |
[19] A. Fujii, S. Hashiguchi, N. Uematsu, T. Ikariya, R. Noyori, J. Am. Chem. Soc. 1996, 118, 2521.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK28Xht1Wnu78%3D&md5=61fa76745e1f9b172802001d85f342fcCAS |
[20] H. Goossens, T. S. A. Heugebaert, B. Dereli, M. Van Overtveldt, O. Karahan, I. Dogan, M. Waroquier, V. Van Speybroeck, V. Aviyente, S. Catak, C. V. Stevens, Eur. J. Org. Chem. 2015, 1211.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXhtFOiu74%3D&md5=26d1eb3a779273a317c0c8746871a9e7CAS |
[21] A. Rasmussen, T. Heugebaert, C. Matthys, R. Van Deun, F.-D. Boyer, S. Goormachtig, C. Stevens, D. Geelen, Mol. Plant 2013, 6, 100.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXhtFCnurk%3D&md5=7f58b9ec1029218e5f4ded91790d6defCAS | 23024210PubMed |
[22] C. Prandi, E. G. Occhiato, S. Tabasso, P. Bonfante, M. Novero, D. Scarpi, M. E. Bova, I. Miletto, Eur. J. Org. Chem. 2011, 3781.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXotVCmsbk%3D&md5=78239978bef9d801b1f4e375d392fbb9CAS |
[23] C. Bhattacharya, P. Bonfante, A. Deagostino, Y. Kapulnik, P. Larini, E. G. Occhiato, C. Prandi, P. Venturello, Org. Biomol. Chem. 2009, 7, 3413.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXpsl2itrY%3D&md5=439eae956b75a85d8cb98e00a118f045CAS | 19675895PubMed |
[24] G. Cignarella, G. Grella, M. M. Curzu, Synthesis 1980, 1980, 825.
| Crossref | GoogleScholarGoogle Scholar |
[25] D. Papa, U.S. Patent 2562208 1951.
[26] K. Mogilaiah, N. Vasudeva Reddy, G. Randheer Reddy, Synth. Commun. 2003, 33, 3109.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXmt1KgtrY%3D&md5=e1fe4cb499294de3bb6f4f17da9b7c47CAS |
* The corresponding author, Christopher S. P. McErlean, was awarded the 2014 RACI Athel Beckwith Lectureship.
** In memory of Professor Michael McKenzie, 1968–2015.