

The Palladium-Catalysed Intramolecular Alder-ene (IMAE) Reactions of Certain Heteroatom-Linked 1,6-Enynes: The Formation of Hexahydro-Indoles and -Benzofurans*
Antony L. Crisp A , Jiwen Li A , Ping Lan A , Jeremy Nugent A , Eliška Matoušová B and Martin G. Banwell A CA Research School of Chemistry, Institute of Advanced Studies, The Australian National University, Canberra, ACT 2601, Australia.
B Department of Organic Chemistry, Faculty of Science, Charles University in Prague, Hlavova 8, 128 43 Praha 2, Czech Republic.
C Corresponding author. Email: Martin.Banwell@anu.edu.au
Australian Journal of Chemistry 68(8) 1183-1189 https://doi.org/10.1071/CH15340
Submitted: 10 June 2015 Accepted: 15 June 2015 Published: 16 July 2015
Abstract
A short review of the literature on palladium-catalysed intramolecular Alder-ene reactions of C-, N-, and O-linked 1,6-enynes is provided with a particular focus on the use of the latter two processes in the authors’ laboratories for the purposes of constructing various alkaloids.
Introduction
Palladium-catalysed processes are amongst the most important reactions in organic chemistry today.[1] Particularly notable examples include the Tsuji–Trost and Mizoroki–Heck reactions as well as the suite of related cross-coupling processes bearing names such as Sonogashira, Stille, Kumada, Suzuki–Miyaura, and Negishi.[2,3] Of course, the range of palladium-catalysed processes extends well beyond these, with this metal being used (often ‘suspended’ on an inert solid support such as charcoal) to effect the addition of dihydrogen to unsaturated organic compounds.[4] Indeed, these venerable and completely atom-economical processes,[5] in which all of the constituent atoms of the reacting partners end up in the product(s), represent some of the most important industrial processes known and the commercial value of them can be measured in the billions of dollars.[6] A perhaps less well-known type of process that can be catalysed by various palladium-based species are cycloisomerisations wherein polyunsaturated, open-chain or semi-cyclic species are converted, through intramolecular sigma-bond-forming processes, into cyclic or even polycyclic products.[7] These processes are not only completely atom-economical ones but they often also proceed with exquisite levels of regio- and/or stereo-chemical control. Furthermore, and as is often the case with other palladium-catalysed reactions, changing the nature of the ligands co-ordinating to the metal can have a profound influence not only on the type of product formed but also on the enantioselectivities that might be observed when potentially desymmetrising transformations are involved.[7] The primary focus of this short review is on one such cycloisomerisation process, namely the palladium-catalysed intramolecular Alder-ene (IMAE) reaction[8] of 1,6-enynes.
This has been described as the archetypal cycloisomerisation reaction.[7d] Two distinct variants are possible depending upon whether the atom-chain linking the reacting centres of unsaturation is an all-carbon unit or one incorporating heteroatoms such as nitrogen or oxygen. In the latter cases the products of reaction are substituted pyrrolidines and tetrahydrofurans, respectively.
The Alder-ene-Based Cycloisomerisation Reactions of ‘All Carbon’ 1,6-Enynes and Related Systems
While thermally-promoted variants of the title process have been known for well over half a century,[7d,8] a metal-catalysed form that proceeded efficiently and under mild conditions was only discovered in 1985 by Trost and Lautens and co-workers.[9] So, for example, these researchers established (Scheme 1) that treatment of the 1,6-enyne 1 (itself prepared via a Tsuji–Trost reaction) with 5 mol-% of the PdII species (Ph3P)2Pd(OAc)2 in hot d6-benzene affords the bicyclic diene 2 in 85 % yield.[9a]
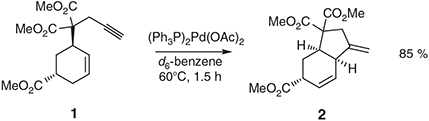
|
Many useful variants of this type of process, including asymmetric ones, followed thereafter,[10] as did the discovery and application of homologous processes employing 1,7-enynes as substrates.[11] Interestingly, in those substrates lacking a hydrogen-bearing substituent at the ‘outer’ allylic position or where there is branching at C3, the isomerisation reaction often yields 1,3-dienes. This is exemplified by the conversion of the 1,6-enyne 3 into the isomer 4 (Scheme 2) when N,N-bis-(benzylidene)ethylenediamine (BBEDA) is used as ligand.[9d] Elegant applications of such processes in natural products synthesis abound.[7,10–13]
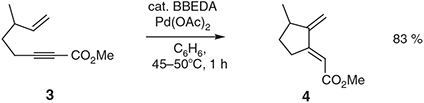
|
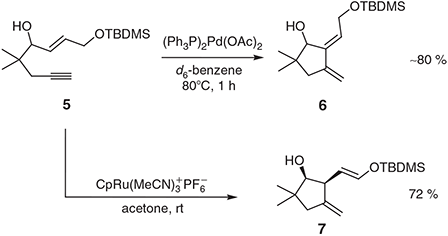
Since the original discovery of Trost and Lautens,[9] a range of other metal catalysts has been shown to effect the cycloisomerisations of 1,6- and 1,7-enynes and often in ways that are complementary to the processes observed using palladium. So, for example, when 1,6-enyne 5 (Scheme 3) is treated under ‘conventional’ conditions with (Ph3P)2Pd(OAc)2 then the 1,3-diene 6 is formed[12] while reaction of the same substrate with the cationic ruthenium catalyst CpRu(MeCN)3+PF6– affords the isomeric 1,4-diene 7 in a completely regio- and stereo-selective manner.[14] The latter conversion is notable for the efficient and selective formation of a silylenol ether in the presence a free hydroxyl group and stands as testimony to the mild nature of the reactions involved.
|
A new reaction pathway is observed when a quaternary carbon centre is introduced at the propargylic (C5) position of the substrate. For example, compound 8 is transformed into isomer 9 (40 %) (Chart 1) upon treatment under the same reaction conditions as employed for the conversion 5 → 7. A complete change in the mechanism of the cycloisomerisation is occurring in such instances with a C–H insertion pathway now being followed.[14b]

|
Stereochemically divergent outcomes have been observed in the ruthenium- and palladium-catalysed cycloisomerisations of cyclohexene-derived 1,7-enynes leading to decalin-containing products.[15] Extensions of such Ru-catalysed processes to the synthesis of certain alkaloid frameworks have been reported recently.[16]
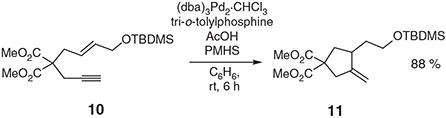
Reductive cycloisomerisations of 1,6-enynes catalysed by either palladium or rhodium have been reported by Trost and Rise[17] and Jang and Kirsche,[18] respectively. In the first case (involving palladium) either triethylsilane or polymethylhydrosiloxane (PMHS) serves as the reducing agent, while dihydrogen is employed for the same purpose in the second (i.e. when rhodium is the catalyst). The conversion 10 → 11 (Scheme 4) is illustrative of the types of transformations that can be achieved by such means. Recently, the ‘Trost variant’ of this process has been used to construct the A-ring of various daphnane congeners.[19]
|
Cp2Ti(CO)2[20] and certain iron(0)-ate complexes[21] have also been shown to effect the cycloisomerisation of a range of 1,6-enynes to the corresponding cyclic 1,4-dienes in toluene at temperatures between 80 and 105°C. A novel nickel-chromium catalyst system supported on an insoluble phosphinylated polymer has also been described.[22]
The Alder-ene-Based Cycloisomerisation Reactions of Heteroatom-Linked 1,6-Enynes and Related Systems
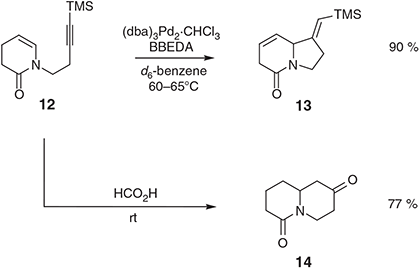
Substrates in which heteroatom linkers connect the reacting olefinic and acetylenic residues of 1,6-enynes can also be engaged in palladium-catalysed cycloisomerisation reactions[7] and, as illustrated below, the products of such processes have served as precursors to a range of natural products. One of the earliest examples of the title process was reported in 1992 by Trost and Pedregal[23] who demonstrated that the alkynyl N-acyl enamine 12 (Scheme 5) is converted into the isomeric indolizidine 13 (90 %) on exposure to 2.5 mol-% (dba)3Pd2·CHCl3 and 10 % BBEDA in d6-benzene at temperatures between 60 and 65°C. In stark contrast, when the same substrate is exposed to formic acid at room temperature (rt) it undergoes cationic cyclisation to generate the quinolizidine 14 in 77 % yield.
|
Naturally enough, reports detailing attempts to effect these types of conversions in an enantioselective manner soon followed with enantiomeric excesses of >99 % being observed in certain cases.[24,25] Related palladium(ii)-catalysed IMAE reactions have been described in which accompanying acetoxy group transfer is observed as highlighted by the enantioselective conversion 15 → 16 (Scheme 6).[26,27]
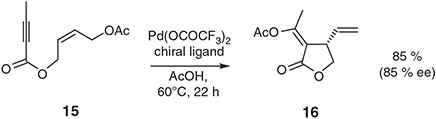
|
Tandem IMAE cross-coupling reactions involving N- and O-linked 1,6-enynes and aryl halides have been reported[28,29] as have oxidative variants wherein diarylation of the cyclisation product is achieved.[29] An example of the former process is shown in Scheme 7. Thus, the N-linked system 17 is engaged in an IMAE reaction and the palladated product of this process is intercepted in a cross-coupling reaction with bromoarene 18 such that the (presumably cis-ring fused) polycyclic product 19 can be obtained in 66 % yield.[28]
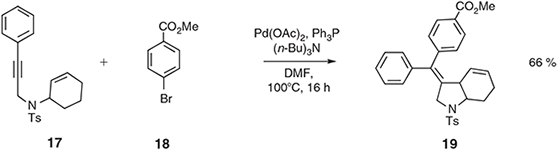
|
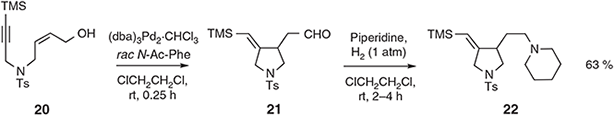
When the olefinic component of the substrate is part of a terminal allylic alcohol then the enolic residue in the product tautomerises to the corresponding aldehyde that can be engaged, in situ, in reductive amination reactions. So, for example, treatment of sulfonamide 20 under the conditions shown in Scheme 8 leads to the pyrrolidine aldehyde 21 (96 %) that on exposure, in the same reaction vessel, to a range of secondary-amines in the presence of dihydrogen affords the expected tertiary-amines such as 22.[30]
|
As was the case with the corresponding all carbon-linked 1,6-enynes detailed above, when the heteroatom-linked systems lack allylic hydrogens at the ‘outer’ position then 1,3-dienes (rather than 1,4-dienes) are formed in the cycloisomerisation process. The conversion 23 → 24 (Scheme 9) exemplifies matters and the products of such processes (e.g. 24) can participate in successive Diels–Alder cycloaddition and allylboration reactions with dienophiles and aldehydes, respectively.[31]
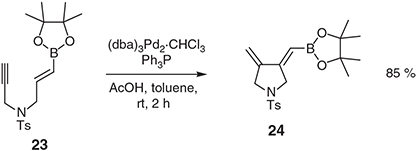
|
Anderson and co-workers have recently shown[32] that in certain heteroatom linked 1,6-enynes where a sulfonamide nitrogen is attached to the internal carbon of the alkyne residue (i.e. the substrate contains an ynamide residue) then 1,3-dienes incorporating an enamine unit are formed exclusively when a combination of Pd(OAc)2 and BBEDA is used to effect the cycloisomerisation process. In some instances, ruthenium-based catalysts can be used to effect the same conversions.[32]
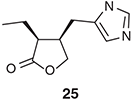
Rhodium(i)-based catalysts can also effect the IMAE reactions of heteroatom linked 1,6-enynes.[33–38] These can proceed in a highly enantioselective manner when appropriate chiral ligands are deployed. Zhang and co-workers have used such protocols in developing an elegant synthesis[34] of the butyrolactone-containing natural product (+)-pilocarpine (25) (Chart 2).
|
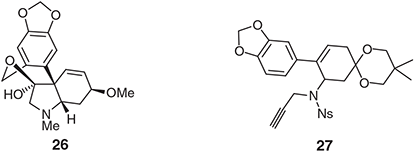
In 2010 we reported[39] the outcomes of our initial studies on the palladium-catalysed IMAE reactions of N-linked 1,6-enynes as a means for constructing the C3a-arylhexahydroindole substructure associated with the Amaryllidaceae alkaloid tazettine (26). While we rapidly established a method for preparing an enyne, 27, that seemed suitable for participation in the desired cycloisomerisation reaction, this process was overwhelmed by a competing coupling of the terminal alkyne residues of two separate substrate molecules to afford a 1,3-enyne-containing heterodimer (Chart 3).
|
In order to avoid such an event, a substrate incorporating a ‘capping’ methyl group was prepared by the pathway shown in Scheme 10. Thus, commercially available diallyl ketone (28) was converted into the ketal 29 under standard conditions and this was, in turn, subjected to a ring-closing metathesis (RCM) using Grubbs’ second-generation catalyst to afford cyclopentene 30. Addition of dibromocarbene, generated under phase-transfer conditions using triethylbenzylammonium chloride (TEBAC) as catalyst, to compound 30 then afforded adduct 31 that could be engaged in a silver cyanate promoted electrocyclic ring-opening reaction. The π-allyl cation so-formed was trapped as the corresponding isocyanate that was itself treated, in situ, with t-butanol and thereby affording the Boc-protected aminocyclohexene 32. This was converted into the corresponding nosyl-based sulfonamide, 33, in a straightforward manner. Compound 33 participated in a Suzuki–Miyaura cross-coupling reaction with the relevant arylboronic acid to afford the arylated cyclohexene 34 that was N-propargylated using 1-bromobut-2-yne in the presence of sodium hydride to give the ‘capped’ substrate 35 required for the IMAE reaction. In the event, when compound 35 was treated with Pd(OAc)2 and BBEDA an essentially quantitative yield of the desired C3a-arylhexahydroindole 36 was obtained. Interestingly, and in keeping with the observations made by Trost and Jebaratnam,[40] this particular ligand/palladium(ii) catalyst combination has proven to be the most effective one we have encountered so far.

|
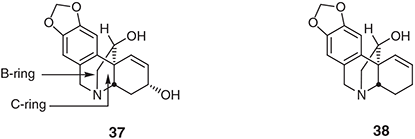
Using protocols related to those detailed immediately above we have been able to complete a total synthesis of the racemic modifications of the crinine alkaloid hamayne (37)[41] and the structure assigned to the related natural product haemultine (38) (Chart 4).[42] In each instance the exocyclic olefin associated with the product of the relevant IMAE reaction was manipulated in order to introduce the C-ring hydroxyl group with the B-ring being established by subjecting a late-stage derivative of the product of the IMAE process to a Pictet–Spengler reaction.
|
Very recently we have completed total syntheses of both the (+)- and (–)- forms of tazettine [viz. (+)- and (–)-26] using the same types of protocols as shown in Scheme 10 and utilising commercially available chiral amines for resolving the products of the desymmetrising electrocyclic ring opening of 6,6-dibromobicyclo[3.1.0]hexane (viz. the non-oxygenated congener of compound 31) (M. G. Banwell, P. Lan, A. C. Willis, unpubl. data).
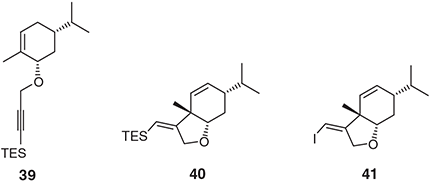
Encouraged by our successes in constructing hexahydroindoles using the palladium-catalysed IMAE reactions of N-linked 1,6-enynes, we sought to establish if the analogous oxygen heterocycles (viz. hexahydrobenzofurans) could be assembled by analogous means. In an extensive and just completed survey (J. Nugent, E. Matoušová, J. Li, M. G. Banwell, A. C. Willis, unpubl. data) that included an exploration of alternative and synthetically more useful alkyne capping groups, we prepared, inter alia, triethylsilyl (TES)-containing substrates such as 39 (Chart 5) and established that on exposure to Pd(OAc)2 and BBEDA in benzene under microwave irradiation conditions this affords the expected heterocycle, 40, in 80 % yield (the analogous conversion of the substrate lacking the TES group proceeds in just 16 % yield under the same conditions) (Chart 5). Furthermore, on treatment with N-iodosuccinimide in acetonitrile at 60°C alkenylsilane 40 undergoes an ipso-substitution reaction to give, in 82 % yield, the corresponding iodide 41 that is itself capable of engaging in a range of metal catalysed cross-coupling processes. Acyclic substrates also participate in the IMAE reaction under analogous conditions giving highly substituted furans as exemplified by the conversion 42 → 43 (the illustrated product was obtained in 74 % yield as a single diastereoisomer of yet-to-be determined configuration) (Chart 6). Spirocyclic furans are available by related means. Interestingly, various attempts to effect the same conversions using a range of seemingly relevant ruthenium and rhodium based systems were unsuccessful.
|

|
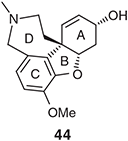
Our continuing interest in the natural product galanthamine (44) (Chart 7), a hexahydrobenzofuran-containing alkaloid used clinically in the treatment of the early stages of Alzheimer’s disease,[43] prompted us to consider its assembly using the title protocols.
|
In the event,[44] we were able to effect the IMAE reaction of the readily accessible O-linked 1,6-enyne 45 (Scheme 11) under our now standard conditions and thereby generate the bicyclic compound 46 in 71 % yield. On treatment with (Ph3P)4Pd and the non-nucleophilic base DBU, the elements of acetic acid were lost from compound 46 and the semi-cyclic and electron-rich diene 47 thereby obtained in 85 % yield. Compound 47 readily participated in a completely regioselective Diels–Alder reaction with propynal and the initially formed adduct was aromatised by treatment with manganese oxide and thus forming the benzaldehyde 48 in 61 % yield. On exposure to m-chloroperbenzoic acid (m-CPBA) compound 48 participated in a Dakin oxidation reaction and the product formate ester was immediately hydrolysed with potassium carbonate in methanol to give phenol 49 in 69 % yield over the two steps involved. O-Methylation of compound 49 under standard conditions then afforded the anisole 50 in quantitative yield. The construction of the seven-membered D-ring of galanthamine was straightforward and involved the two step conversion of sulfonamide 50 into formamide 51 (83 %) that was engaged in a modified Bischler–Napieralski type cyclodehydration reaction to give, after a reductive ‘workup’ using sodium triacetoxy(hydrido)borate and subsequent treatment with aqueous sodium bicarbonate, (±)-narwedine 52 (44 %), an established precursor to both (+)- and (–)-galanthamine.

|
While this synthesis of galanthamine is not the shortest one reported it is nevertheless notable for several reasons. It is the first synthesis in which the aromatic C-ring has been constructed de novo. Perhaps more significantly, especially in terms of the general theme of this article, the Pd(OAc)2-catalysed IMAE reaction that results in the formation of compound 46 proceeds effectively within a multi-functional substrate 45. It also allows for the construction of the quaternary carbon centre of galanthamine and establishes an allylic acetate moiety that only engages in Tsuji–Trost type chemistry on exposure to a palladium(0)-species. In other words, the IMAE and Tsuji–Trost reactions involved here are effectively ‘orthogonal’ processes.
Conclusions and Future Prospects
The palladium(ii)-catalysed IMAE reactions of N- and O-linked 1,6-enynes offer extraordinary capacities for the construction, under relatively mild and highly chemoselective conditions, of complex hexahydro-indoles and -benzofurans, respectively. The opportunities for the application of such processes to the synthesis of natural products and various analogues seem almost boundless. An important area of future endeavour will be the identification of enantioselective variants of broad scope and that can be conducted in operationally simple ways.
Acknowledgements
We thank the Australian Research Council for funding, the China Scholarship Council of the People’s Republic of China for awarding a stipend to PL and the Australian Government for the provision of an Australian Postgraduate Award to JN.
References
[1] R. Bates, Organic Synthesis Using Transition Metals, 2nd edn 2012 (John Wiley and Sons: Chichester).[2] See p. 1078 in: J. Clayden, N. Greeves, S. Warren, Organic Chemistry, 2nd edn 2012 (Oxford University Press: Oxford).
[3] S. Hadlington, Chem. World 2010, 11, 40.
[4] (a) See pp. 1–34 in: H. O. House, Modern Synthetic Reactions, 2nd edn 1972 (W. A. Benjamin Inc.: Menlo Park, CA).
(b) H.-U. Blaser, C. Malan, B. Pugin, F. Spindler, H. Steiner, M. Studer, Adv. Synth. Catal. 2003, 345, 103.
| Crossref | GoogleScholarGoogle Scholar |
(c) J. G. de Vries, C. J. Elsevier, Handbook of Homogeneous Hydrogenation 2007 (Wiley-VCH: Weinheim).
(d) S. Nishimura, Handbook of Heterogeneous Catalytic Hydrogenation for Organic Synthesis 2001 (John Wiley and Sons: New York, NY).
[5] B. M. Trost, Angew. Chem. Int. Ed. Engl. 1995, 34, 259.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXjvFCisL0%3D&md5=396cd2d6baa59b700b5932d782de779eCAS |
[6] The development of metal-free hydrogenation systems is an emerging area that is likely to assume considerable significance in due course. For a recent review see: L. J. Hounjet, D. W. Stephan, Org. Process Res. Dev. 2014, 18, 385.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXhslygsbg%3D&md5=86de24d2fe6167bca3d488b1bbfb6ef0CAS |
[7] (a) For reviews of certain relevant aspects of this rapidly expanding topic see: (a) B. M. Trost, Acc. Chem. Res. 1990, 23, 34.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3cXpslOgtA%3D%3D&md5=03cbccf3cf44e3359e4ab8d995370e36CAS |
(b) V. Michelet, P. Y. Toullec, J.-P. Genet, Angew. Chem. Int. Ed. 2008, 47, 4268.
| Crossref | GoogleScholarGoogle Scholar |
(c) A. Marinetti, H. Jullien, A. Voituriez, Chem. Soc. Rev. 2012, 41, 4884.
| Crossref | GoogleScholarGoogle Scholar |
(d) I. D. G. Watson, F. D. Toste, Chem. Sci. 2012, 3, 2899.
| Crossref | GoogleScholarGoogle Scholar |
[8] (a) D. F. Taber, Intramolecular Diels-Alder and Alder-ene Reactions 1984 (Springer Verlag: Berlin).
(b) K. Mikami, M. Shimizu, Chem. Rev. 1992, 92, 1021.
| Crossref | GoogleScholarGoogle Scholar |
[9] (a) B. M. Trost, M. Lautens, J. Am. Chem. Soc. 1985, 107, 1781.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2MXhtlWmt7c%3D&md5=eea0d6be240f26b01fc8eb975f295b5eCAS |
(b) B. M. Trost, M. Lautens, Tetrahedron Lett. 1985, 26, 4887.
| Crossref | GoogleScholarGoogle Scholar |
(c) B. M. Trost, M. Lautens, C. Chan, D. J. Jebaratnam, T. Mueller, J. Am. Chem. Soc. 1991, 113, 636.
| Crossref | GoogleScholarGoogle Scholar |
(d) B. M. Trost, G. J. Tanoury, M. Lautens, C. Chan, D. T. MacPherson, J. Am. Chem. Soc. 1994, 116, 4255.
| Crossref | GoogleScholarGoogle Scholar |
[10] B. M. Trost, B. A. Czeskis, Tetrahedron Lett. 1994, 35, 211.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2cXjt1akurc%3D&md5=9cb7e148c5cbec1e75503bdb41e9d88dCAS |
[11] B. M. Trost, Y. Li, J. Am. Chem. Soc. 1996, 118, 6625.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK28XjvVSisLc%3D&md5=9fb961de01cda48ab62fee242a055ab6CAS |
[12] B. M. Trost, J. Y. L. Chung, J. Am. Chem. Soc. 1985, 107, 4586.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2MXlsVahsrc%3D&md5=0028aa8720fc47708137532c807e434cCAS |
[13] B. M. Trost, P. A. Hipskind, J. Y. L. Chung, C. Chan, Angew. Chem. Int. Ed. Engl. 1989, 28, 1502.
| Crossref | GoogleScholarGoogle Scholar |
[14] (a) B. M. Trost, F. Dean Toste, J. Am. Chem. Soc. 2000, 122, 714.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXjsFWqtA%3D%3D&md5=1fc60e402a6caa33d4ef1df77ab87820CAS |
(b) B. M. Trost, F. Dean Toste, J. Am. Chem. Soc. 2002, 124, 5025.
| Crossref | GoogleScholarGoogle Scholar |
[15] B. M. Trost, A. C. Gutierrez, E. M. Ferreira, J. Am. Chem. Soc. 2010, 132, 9206.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXnsFSqsbw%3D&md5=ebf6494ae9efdce381702208d2d2b5d0CAS | 20545356PubMed |
[16] H. Cheng, F.-H. Zeng, D. Ma, M.-L. Jiang, L. Xu, F.-P. Wang, Org. Lett. 2014, 16, 2299.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXls1Crtb8%3D&md5=27a7e8e4361e6931ae46c6d47b12c8e0CAS | 24701960PubMed |
[17] B. M. Trost, F. Rise, J. Am. Chem. Soc. 1987, 109, 3161.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2sXltFGhtL4%3D&md5=73896c32cb2e854c810279a1a79de75aCAS |
[18] H.-Y. Jang, M. J. Kirsche, J. Am. Chem. Soc. 2004, 126, 7875.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXksVGqtr4%3D&md5=0e7077b44e090f40f59a1ba64aa98f53CAS | 15212535PubMed |
[19] P. A. Wender, N. Buschmann, N. B. Cardin, L. R. Jones, C. Kan, J.-M. Kee, J. A. Kowalski, K. E. Longcore, Nat. Chem. 2011, 3, 615.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXnslSltbs%3D&md5=16579c48088a66569c02ba186f0fe3c1CAS | 21778981PubMed |
[20] S. J. Sturla, N. M. Kablaoui, S. L. Buchwald, J. Am. Chem. Soc. 1999, 121, 1976.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXhtFagt7g%3D&md5=c5ee7e20da09ad2a14724402ba13202fCAS |
[21] A. Fürstner, R. Martin, K. Majima, J. Am. Chem. Soc. 2005, 127, 12236.
| Crossref | GoogleScholarGoogle Scholar | 16131197PubMed |
[22] B. M. Trost, J. M. Tour, J. Am. Chem. Soc. 1987, 109, 5268.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2sXltFGhu7c%3D&md5=7b750607e804dd350c0c05288ec8a296CAS |
[23] B. M. Trost, C. Pedregal, J. Am. Chem. Soc. 1992, 114, 7292.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK38XltFWnsLY%3D&md5=480cfac1092377ef57a8d460e8f3fdf2CAS |
[24] A. Goeke, M. Sawamura, R. Kuwano, Y. Ito, Angew. Chem. Int. Ed. Engl. 1996, 35, 662.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK28XitFWgtbY%3D&md5=6e7d800173158683c6bafbc7f54142ebCAS |
[25] M. Hatano, M. Terada, K. Mikami, Angew. Chem. Int. Ed. 2001, 40, 249.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXltlyksA%3D%3D&md5=3aa62827a5d39c49719e551cc858dea2CAS |
[26] Q. Zhang, X. Lu, X. Han, J. Org. Chem. 2001, 66, 7676.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXnslaqs7o%3D&md5=d432fdf1f9320b95365ccb86f48363f8CAS | 11701020PubMed |
[27] C. Muthiah, M. A. Arai, T. Shinihara, T. Arai, S. Takizawa, H. Sasai, Tetrahedron Lett. 2003, 44, 5201.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXks1Wht7w%3D&md5=ff2b45daee6e079533f71240210340bcCAS |
[28] T.-j. Meng, Y.-m. Hu, Y.-j. Sun, T. Zhu, S. Wang, Tetrahedron 2010, 66, 8648.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtlSmtb7J&md5=a393100b48f4eefe81b65e576641f70dCAS |
[29] M. Jiang, T. Jiang, J.-E. Bäckvall, Org. Lett. 2012, 14, 3538.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XovFaqsrg%3D&md5=9b9718b36d79559cc09e2bbea99610bbCAS | 22716930PubMed |
[30] C. J. Kressierer, T. J. J. Müller, Org. Lett. 2005, 7, 2237.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXjvVSksrY%3D&md5=318a5a8fc6338344e1fbf569bc034107CAS | 15901178PubMed |
[31] A. Hercouet, F. Berée, C. H. Lin, L. Toupet, B. Carboni, Org. Lett. 2007, 9, 1717.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXjvFSrtrs%3D&md5=fcefcc5a3465c32ff051c9af7a725cceCAS | 17408278PubMed |
[32] P. R. Walker, C. D. Campbell, A. Suleman, G. Carr, E. A. Anderson, Angew. Chem. Int. Ed. 2013, 52, 9139.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXhtFSgtrfJ&md5=ac7b8afddc79c8deacee98f778e96ea9CAS |
[33] P. Cao, B. Wang, X. Zhang, J. Am. Chem. Soc. 2000, 122, 6490.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXktFOgsr4%3D&md5=df95e947db6be491d8222b321806050eCAS |
[34] A. Lei, M. He, X. Zhang, J. Am. Chem. Soc. 2002, 124, 8198.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38Xks1aqsbY%3D&md5=7f594aaac2bdddde803dd0a186a29bf1CAS | 12105894PubMed |
[35] K. Mikami, Y. Yusa, M. Hatano, K. Wakabayashi, K. Aikawa, Chem. Commun. 2004, 98.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXpvFSisw%3D%3D&md5=cd79e3f8c6d14ca86a253b0788631ed8CAS |
[36] X. Tong, D. Li, Z. Zhang, X. Zhang, J. Am. Chem. Soc. 2004, 126, 7601.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXktlWhtbk%3D&md5=f1d66a7f686783a20e4e13b3ef97052aCAS | 15198608PubMed |
[37] K. Mikami, Y. Yusa, M. Hatano, K. Wayabayashi, K. Aikawa, Tetrahedron 2004, 60, 4475.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXjsVOltbg%3D&md5=840ab0eec913f1d995265bb5a08e22eaCAS |
[38] R. Okamoto, E. Okazaki, K. Noguchi, K. Tanaka, Org. Lett. 2011, 13, 4894.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtVOns7vN&md5=49ca609a832b1f2df264bf9222753c6fCAS | 21866888PubMed |
[39] A. L. Lehmann, A. C. Willis, M. G. Banwell, Aust. J. Chem. 2010, 63, 1665.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsFaqsbvL&md5=1b0247576166c7b86a42bdf605981b6bCAS |
[40] B. M. Trost, D. J. Jebaratnam, Tetrahedron Lett. 1987, 28, 1611.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL1cXhtFaktLw%3D&md5=1323bf849fa39ce3545a6caba9a8622dCAS |
[41] L. Petit, M. G. Banwell, A. C. Willis, Org. Lett. 2011, 13, 5800.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXht1Oju7fO&md5=f5c92030c38b9dc65f59e745559b483aCAS | 21970722PubMed |
[42] N. Gao, X. Ma, L. Petit, B. D. Schwartz, M. G. Banwell, A. C. Willis, I. A. Cade, A. D. Rae, Aust. J. Chem. 2013, 66, 30.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXhtVylsb4%3D&md5=21746d667993a29bfa5e6cc84c10dd20CAS |
[43] M. G. Banwell, J. Buckler, C. J. Jackson, P. Lan, X. Ma, E. Matoušová, J. Nugent, in Strategies and Tactics in Organic Synthesis (Ed. M. Harmata) 2015 (Academic Press: Oxford), in press.
[44] J. Nugent, E. Matoušová, M. G. Banwell, Eur. J. Org. Chem. 2015, 2015, 3771.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXnsFCnt7g%3D&md5=254a777dd644a98efd8b49ac8863df7bCAS |
* The corresponding author, Martin G. Banwell, is the winner of the 2014 RACI H. G. Smith Memorial Medal.