

A Biomimetic Synthetic Approach to the Frondosins*
Kevin K. W. Kuan A , Aylin M. C. Hirschvogel A and Jonathan H. George A BA Department of Chemistry, University of Adelaide, Adelaide, SA 5005, Australia.
B Corresponding author. Email: jonathan.george@adelaide.edu.au
Australian Journal of Chemistry 69(12) 1420-1423 https://doi.org/10.1071/CH16218
Submitted: 4 April 2016 Accepted: 2 June 2016 Published: 20 July 2016
Abstract
The frondosins are a family of five marine sponge-derived meroterpenoids. We propose that the 6–7 ring system common to each of the frondosins is biosynthesized via ring expansion of a 6–6 ring system. Compelling evidence in favour of this proposal was obtained in the form of a biomimetic synthesis of the frondosin 6–7 ring system, which features a highly stereo- and regio-selective ring expansion cascade reaction as the key step.
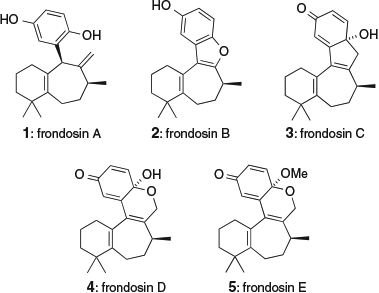
Frondosins A–E (1–5, Fig. 1) are a family of five meroterpenoid natural products first isolated from Dysidea frondosa in 1997.[1] The isolation of frondosin A and frondosin D from Euryspongia sp. was reported shortly afterwards.[2] The frondosins each possess a 6–7 carbocyclic framework that is either attached or fused to a hydroquinone, quinone, or benzofuran ring system. The unusual structures of the frondosins, combined with their reported inhibition of the binding of interleukin-8 to its receptor, has inspired significant attention from the synthetic community.[3] However, a general and divergent synthetic route to the whole frondosin family has remained elusive.
|
We aimed to use our proposal for the biosynthesis of the frondosins (Scheme 1) as the blueprint for a unified synthetic approach.[4] A similar proposal for frondosin biosynthesis has also been suggested by Pettus.[5] Although it has not yet been isolated as a natural product, we believe that hydroquinone 6 is a plausible intermediate in the biosynthesis of the frondosins. We have previously converted 6 into aureol, a marine sponge meroterpenoid closely related to the frondosins.[6] The bicyclic framework of 6 (with its characteristic Δ5,11-double bond) has also been formed by acid-catalyzed rearrangement of avarol[7] (via sequential 1,2-methyl and hydride shifts[8]) and by acid-catalyzed fragmentation of aureol.[9] Oxidation of hydroquinone 6 could form o-quinone methide 7, via either tautomerization of an intermediate quinone or direct oxidation at the benzylic C-10 position. We propose that the reactive o-quinone methide intermediate 7 could then undergo a highly stereo- and regio-selective ring expansion to form frondosin A (1). Further oxidation of 1 could form quinone 8, which could undergo epimerization at C-10 to form 10. The quinone carbonyl group of 10 could then participate in an intramolecular Paternò–Büchi reaction with the neighbouring Δ9,18-alkene to give a strained oxetane intermediate 11, which could fragment to give frondosin C (3). Alternatively, deprotonation of 8 at C-10 could form o-quinone methide 9, which could cyclize via a 6π-electrocyclic reaction to form 12. Oxidation of 12 could then give frondosins D and E (4 and 5) via the interception of an intermediate quinone cation with either H2O or MeOH respectively. Finally, frondosin B (2) could arise via intramolecular 1,6-conjugate addition of 4 followed by elimination of formaldehyde from 13 by a retro [4+2] cycloaddition.

|
The initial oxidation of frondosin A (1) to quinone 8 is likely to be a facile process and could occur spontaneously under aerobic oxidation conditions. As the later rearrangements to give frondosins B–E (2–5) could be predisposed to occur under non-enzymatic conditions, it is possible that these compounds are artefacts of the isolation process. In particular, the use of MeOH in both extraction and isolation procedures could explain the presence of the C-17 methoxy group of frondosin E. Nevertheless, we were attracted by the idea of using the unified proposal for the biosynthetic origin of the frondosins as outlined in Scheme 1 to inspire a divergent synthesis of all five family members.
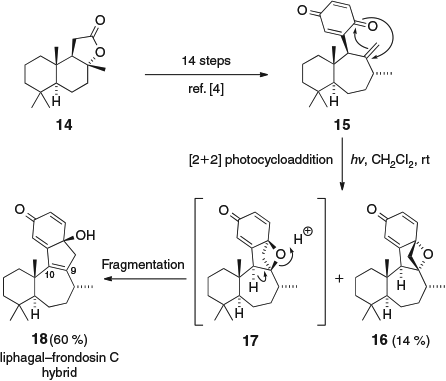
We have previously investigated our proposal for the biosynthetic origin of the frondosin C ring system via a model study (Scheme 2).[4] Quinone 15 was synthesized in 14 steps from (+)-sclareolide (14). Exposure of 15 to sunlight then formed the liphagal–frondosin C hybrid molecule 18 in 60 % yield, along with 14 % of the stable oxetane 16. We propose that intramolecular Paternò–Büchi reaction of 15 forms two diastereomeric oxetanes, 16 and 17. Oxetane 17 has an antiperiplanar relationship between the C–H bond at C-10 and the C–O bond at C-9, and therefore undergoes rapid fragmentation to give 18, whereas oxetane 16 is kinetically stable.
|
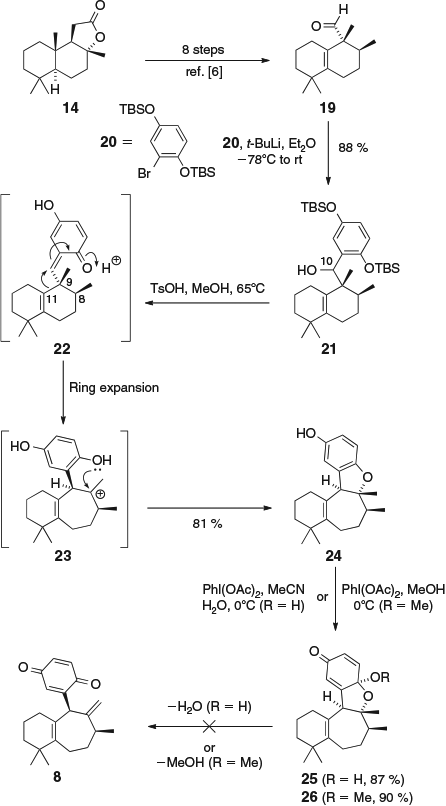
Our primary aim in the present investigation was to synthesize the 6–7 ring system of frondosin A via a ring-expansion cascade reaction of a 6–6 ring system. We have previously employed a similar strategy in two total syntheses of liphagal.[10] We soon realized that the major challenge in the synthesis of frondosin A via this strategy would be the selective installation of the exocyclic Δ9,18-alkene. The investigation began with synthesis of aldehyde 19 in eight steps from (+)-sclareolide (14) according to our previously published procedures[6] (Scheme 3). Nucleophilic addition of the aryllithium species generated from aryl bromide 20[11] and t-BuLi gave alcohol 21 as single diastereomer (although the configuration of the newly formed stereocentre at C-10 was not determined). Treatment of 21 with p-toluenesulfonic acid (TsOH) in MeOH then gave the ring-expanded frondosin analogue 24 in good yield and as a single diastereomer.[12] The structure of 24 was determined via 2D NMR studies (see the Supplementary Material for a full presentation of NMR data). We propose that this acid-catalyzed reaction proceeds via two-fold desilylation followed by dehydration to give o-quinone methide 22 and then ring expansion to give tertiary carbocation 23.[13] Alternatively, the reaction could be considered to involve ring expansion of a stabilized benzylic carbocation. The 1,2-shift is highly selective, with the vinylic C-9–C-11 bond migrating in preference to the C-8–C-9 bond. The cascade reaction is terminated by trapping of the tertiary carbocation 23 by the adjacent phenol to give 24, rather than deprotonation to form the exocylic alkene of frondosin A (1). However, given that we had efficiently synthesized the 6–7 ring system common to the frondosins, we sought methods to convert 24 into some of our natural product targets by fragmentation of the undesired dihydrobenzofuran ring. Our first strategy involved oxidative dearomatization of 24 using PhI(OAc)2 to give hemiacetal 25 (under aqueous conditions) or acetal 26 (with MeOH as solvent). We then attempted to fragment 25 and 26 to give quinone 8 under acid catalysis, base catalysis, and using dehydration reagents such as Martin’s sulfurane, but without success. This was unfortunate, as quinone 8 has previously been reduced to frondosin A, and it could also potentially be used to access frondosins B–E using our biosynthetic hypothesis outlined in Scheme 1.
|
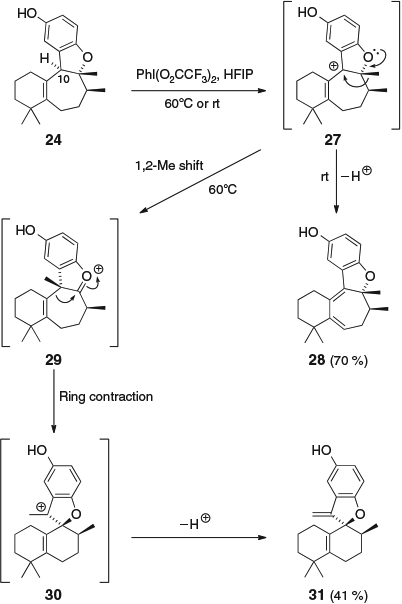
Oxidative dearomatization of 24 using PhI(O2CCF3)2 in hexafluoroisopropanol (HFIP) gave very different results (Scheme 4). When the oxidation was conducted at room temperature, diene 28 was formed, presumably via benzylic oxidation at C-10 to give carbocation 27 followed by deprotonation. Alternatively, heating the reaction at 60°C gave the ring-contracted isomer 31. The formation of 31 presumably proceeds via a 1,2-methyl shift of carbocation 27 to generate oxonium ion 29, followed by a ring contraction to give carbocation 30 and finally deprotonation.
|
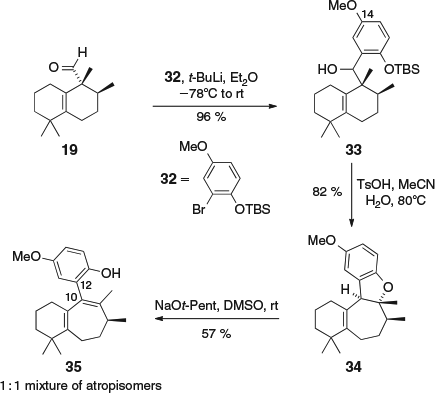
We also attempted the fragmentation of the dihydrobenzofuran ring of 24 under basic conditions. This reaction failed to proceed, perhaps owing to the presence of the unprotected phenol at C-14. We therefore synthesized 34 (a dihydrobenzofuran substrate with a methoxy group instead of a phenol at C-14) via the union of aldehyde 19 and aryl bromide 32[14] to give alcohol 33, followed by acid-catalyzed ring expansion (Scheme 5). Basic fragmentation of 34 with sodium pentoxide was possible, but we were unable to control the location of the newly formed alkene, with the undesired conjugated diene 35 formed as a 1 : 1 mixture of atropisomers (owing to restricted rotation about the C-10–C-12 bond). Attempts to oxidize 35 to a quinone, and then to potentially isomerize it to form the frondosin D and E ring system, were unsuccessful owing to its instability to even mild oxidants.
|
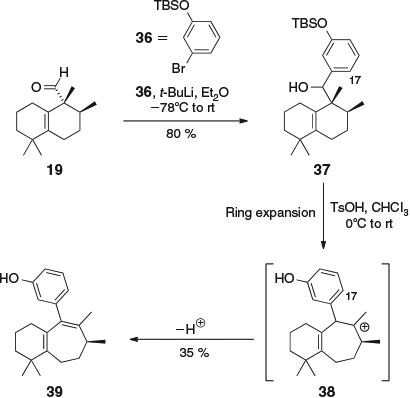
In a final study, we investigated the effect of removing the C-17 substituent altogether. Thus, aldehyde 19 was combined with aryl bromide 36[15] to give alcohol 37 (Scheme 6). Acid-catalyzed ring expansion of 37 presumably generated the tertiary carbocation 38, which lacked the C-17 phenol and therefore underwent deprotonation to give diene 39. The NMR spectra of 39 were very similar to those of 35, except for the absence of the doubling of signals due to atropisomerism.
|
In conclusion, we have used a biosynthetically inspired ring-expansion cascade reaction to construct the 6–7 ring system of frondosin A. However, the undesired formation of an additional dihydrobenzofuran ring, and its resistance towards selective fragmentation, have thwarted attempts to synthesize frondosin A itself.
Supplementary Material
Experimental procedures and full characterization data for all new compounds are available on the Journal’s website.
* The corresponding author, Jonathan H. George, was awarded the 2015 RACI Athel Beckwith Lectureship.
Acknowledgement
We thank the Australian Research Council for financial support in the form of a Discovery Project (DP160103393).
References
[1] A. D. Patil, A. J. Freyer, L. Killmer, P. Offen, B. Carte, A. J. Jurewicz, R. K. Johnson, Tetrahedron 1997, 53, 5047.| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXis1Wju7c%3D&md5=1ab085d67196790d75cee297f14d72caCAS |
[2] Y. F. Hallock, J. H. Cardellina, M. R. Boyd, Nat. Prod. Lett. 1998, 11, 153.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXhtVygtrs%3D&md5=649865a036cde4276b219473b9d3f62eCAS |
[3] (a) M. Inoue, A. J. Frontier, S. J. Danishefsky, Angew. Chem. Int. Ed. 2000, 39, 761.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXhsVGktb8%3D&md5=d0872e35405d9a9aeed2b486d813f23dCAS |
(b) M. Inoue, M. W. Carson, A. J. Frontier, S. J. Danishefsky, J. Am. Chem. Soc. 2001, 123, 1878.
| Crossref | GoogleScholarGoogle Scholar |
(c) C. C. Hughes, D. Trauner, Angew. Chem. Int. Ed. 2002, 41, 1569.
| Crossref | GoogleScholarGoogle Scholar |
(d) D. J. Kerr, A. C. Willis, B. L. Flynn, Org. Lett. 2004, 6, 457.
| Crossref | GoogleScholarGoogle Scholar |
(e) C. C. Hughes, D. Trauner, Tetrahedron 2004, 60, 9675.
| Crossref | GoogleScholarGoogle Scholar |
(f) I. Martinez, P. E. Alford, T. V. Ovaska, Org. Lett. 2005, 7, 1133.
| Crossref | GoogleScholarGoogle Scholar |
(g) X. Li, R. E. Kyne, T. V. Ovaska, Org. Lett. 2006, 8, 5153.
| Crossref | GoogleScholarGoogle Scholar |
(h) B. M. Trost, Y. Hu, D. B. Horne, J. Am. Chem. Soc. 2007, 129, 11781.
| Crossref | GoogleScholarGoogle Scholar |
(i) J. P. Olson, H. M. L. Davies, Org. Lett. 2008, 10, 573.
| Crossref | GoogleScholarGoogle Scholar |
(j) X. Li, A. Keon, J. A. Sullivan, T. V. Ovaska, Org. Lett. 2008, 10, 3287.
| Crossref | GoogleScholarGoogle Scholar |
(k) G. Mehta, N. S. Likhite, Tetrahedron Lett. 2008, 49, 7113.
| Crossref | GoogleScholarGoogle Scholar |
(l) T. V. Ovaska, J. A. Sullivan, S. I. Ovaska, J. B. Winegrad, J. D. Fair, Org. Lett. 2009, 11, 2715.
| Crossref | GoogleScholarGoogle Scholar |
(m) G. Mehta, N. S. Likhite, Tetrahedron Lett. 2009, 50, 5263.
| Crossref | GoogleScholarGoogle Scholar |
(n) K.-S. Masters, B. L. Flynn, Org. Biomol. Chem. 2010, 8, 1290.
| Crossref | GoogleScholarGoogle Scholar |
(o) M. Reiter, S. Torsell, S. Lee, D. W. C. MacMillan, Chem. Sci. 2010, 1, 37.
| Crossref | GoogleScholarGoogle Scholar |
(p) D. Garayalde, K. Kruger, C. Nevado, Angew. Chem. Int. Ed. 2011, 50, 911.
| Crossref | GoogleScholarGoogle Scholar |
(q) D. W. Lee, R. K. Pandey, S. Lindeman, W. A. Donaldson, Org. Biomol. Chem. 2011, 9, 7742.
| Crossref | GoogleScholarGoogle Scholar |
(r) J. Zhang, L. Li, Y. Wang, W. Wang, J. Xue, Y. Li, Org. Lett. 2012, 14, 4528.
| Crossref | GoogleScholarGoogle Scholar |
(s) D. R. Laplace, B. Verbraeken, K. Van Hecke, J. M. Winne, Chem. – Eur. J. 2014, 20, 253.
| Crossref | GoogleScholarGoogle Scholar |
(t) E. Z. Oblak, M. D. VanHeyst, J. Li, A. J. Wiemer, D. L. Wright, J. Am. Chem. Soc. 2014, 136, 4309.
| Crossref | GoogleScholarGoogle Scholar |
(u) M. D. VanHeyst, D. L. Wright, Eur. J. Org. Chem. 2015, 1387.
| Crossref | GoogleScholarGoogle Scholar |
[4] H. P. Pepper, K. K. W. Kuan, J. H. George, Org. Lett. 2012, 14, 1524.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XivVOhtbc%3D&md5=e1c7ac83530576a277a3c0db3e42f056CAS | 22360673PubMed |
[5] S. K. Jackson, K.-L. Wu, T. R. R. Pettus, in Biomimetic Organic Synthesis (Eds E. Poupon, B. Nay) 2011, Vol. 2, Ch. 20, pp. 723–749 (Wiley-VCH: Weinheim).
[6] K. K. W. Kuan, H. P. Pepper, W. M. Bloch, J. H. George, Org. Lett. 2012, 14, 4710.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xht12it7zF&md5=1f5f475ff3a1ae9dd31a795cc0e1fb48CAS |
[7] L. Minale, R. Riccio, G. Sodano, Tetrahedron Lett. 1974, 15, 3401.
| Crossref | GoogleScholarGoogle Scholar |
[8] J. H. George, M. McArdle, J. E. Baldwin, R. M. Adlington, Tetrahedron 2010, 66, 6321.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXptlaqsLk%3D&md5=d279c82290746a6a8e6d86efc6ea15f1CAS |
[9] P. Djura, D. B. Stierle, B. Sullivan, D. J. Faulkner, E. Arnold, J. Clardy, J. Org. Chem. 1980, 45, 1435.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL3cXhslOlur0%3D&md5=3ed173e6c1da81b2e5cb4e72c89f4ffbCAS |
[10] (a) J. H. George, J. E. Baldwin, R. M. Adlington, Org. Lett. 2010, 12, 2394.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXkvVahs7o%3D&md5=13d5e9753612462b99d9c9cf7805a419CAS | 20394432PubMed |
(b) A. W. Markwell-Heys, K. K. W. Kuan, J. H. George, Org. Lett. 2015, 17, 4228.
| Crossref | GoogleScholarGoogle Scholar |
[11] J. P. Willis, K. A. Z. Gogins, L. L. Miller, J. Org. Chem. 1981, 46, 3215.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL3MXks1Wlur4%3D&md5=9178ec694781ed621c0a082f6d271443CAS |
[12] For a related ring-expansion reaction, see: M. Zhou, H.-C. Geng, H.-B. Zhang, K. Dong, W.-G. Wang, X. Du, X.-N. Li, F. He, H.-B. Qin, Y. Li, J.-X. Pu, H.-D. Sun, Org. Lett. 2013, 15, 314.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhvV2gsL3O&md5=67446405b4f4b3c8a72831405fd08b1bCAS | 23265286PubMed |
[13] (a) For reviews of o-quinone methide chemistry, see: R. W. Van De Water, T. R. R. Pettus, Tetrahedron 2002, 58, 5367.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XkvVegtbg%3D&md5=3f97c39eba1e9f3904a074da117cfb9dCAS |
(b) T. P. Pathak, M. S. Sigman, J. Org. Chem. 2011, 76, 9210.
| Crossref | GoogleScholarGoogle Scholar |
(c) N. J. Willis, C. D. Bray, Chem. – Eur. J. 2012, 18, 9160.
| Crossref | GoogleScholarGoogle Scholar |
(d) W.-J. Bai, J. G. David, Z.-G. Feng, M. G. Weaver, K.-L. Wu, T. R. R. Pettus, Acc. Chem. Res. 2014, 47, 3655.
| Crossref | GoogleScholarGoogle Scholar |
[14] J. Bucher, T. Wurm, K. S. Nalivela, M. Rudolph, F. Rominger, A. S. K. Hashmi, Angew. Chem. Int. Ed. 2014, 53, 3854.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXjs1Kktbk%3D&md5=c438dad1f315dcf11bde648383aa54beCAS |
[15] R. N. Misra, B. R. Brown, W.-C. Han, D. N. Harris, A. Hedberg, M. L. Webb, S. E. Hall, J. Med. Chem. 1991, 34, 2882.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3MXltlGrsbo%3D&md5=ffe103d6d71e016102903d0837b7d014CAS | 1910091PubMed |