

Energetics of the Proton Transfer Pathway for Tyrosine D in Photosystem II
Keisuke Saito A B C , Naoki Sakashita A and Hiroshi Ishikita A BA Department of Applied Chemistry, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8654, Japan.
B Research Center for Advanced Science and Technology, The University of Tokyo, 4-6-1 Komaba, Meguro-ku, Tokyo 153-8904, Japan.
C Corresponding author. Email: ksaito@appchem.t.u-tokyo.ac.jp
Australian Journal of Chemistry 69(9) 991-998 https://doi.org/10.1071/CH16248
Submitted: 19 April 2016 Accepted: 29 May 2016 Published: 4 July 2016
Abstract
The proton transfer pathway for redox active tyrosine D (TyrD) in photosystem II is a hydrogen-bond network that involves D2-Arg180 and a series of water molecules. Using quantum mechanical/molecular mechanical calculations, the detailed properties of the energetics and structural geometries were investigated. The potential-energy profile of all hydrogen bonds along the proton transfer pathway indicates that the overall proton transfer from TyrD is energetically downhill. D2-Arg180 plays a key role in the proton transfer pathway, providing a driving force for proton transfer, maintaining the hydrogen-bond network structure, stabilising P680•+, and thus deprotonating TyrD-OH to TyrD-O•. A hydrophobic environment near TyrD enhances the electrostatic interactions between TyrD and redox active groups, e.g. P680 and the catalytic Mn4CaO5 cluster: the redox states of those groups are linked with the protonation state of TyrD, i.e. release of the proton from TyrD. Thus, the proton transfer pathway from TyrD may ultimately contribute to the conversion of S0 into S1 in the dark in order to stabilise the Mn4CaO5 cluster when the photocycle is interrupted in S0.
Introduction
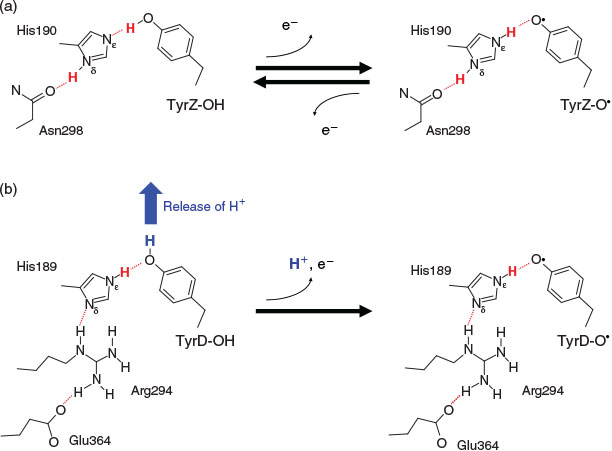
In photosystem II (PSII), the chlorophyll dimer P680 is composed of four chlorophyll a molecules, PD1, PD2, ChlD1, and ChlD2, and it absorbs light at a wavelength of 680 nm, leading to the formation of a range of charge-separated states with P680•+. These states are stabilized by electron transfer to the first quinone, QA, and by electron donation from a tyrosine residue, tyrosine Z (TyrZ, D1-Tyr161), to P680•+ (≈PD1•+). TyrZ then oxidizes the water-oxidizing Mn4CaO5 cluster. Oxidation of the Mn4CaO5 cluster occurs in the S0→S1→S2→S3→S0 transitions (as previously reviewed[1]). To serve as a redox active cofactor, TyrZ requires the hydrogen-bond partner D1-His190. Recently determined high-resolution PSII crystal structures[2] revealed that the phenolic O site of TyrZ forms a hydrogen bond with the Nϵ site of D1-His190 with an OTyrZ–NϵD1-His190 length of ~2.5 Å. The significantly shortened hydrogen-bond distance was not one of the most significant points about the crystal structure[2a] until Saito et al. demonstrated that the remarkably short hydrogen-bond distance can be quantum-chemically reproduced in the presence of the PSII protein environment.[3] The pKa values of TyrZ and D1-His190 are equal in the PSII protein environment,[3] leading to the formation of a significantly short, ‘single-well hydrogen bond’.[4] The proton is localized near the hydrogen-bond donor moiety in standard hydrogen bonds. In contrast, the proton is delocalized at the bottom of the nearly barrierless potential in single-well hydrogen bonds, which appears to be a theoretical basis for the so-called ‘proton-rocking mechanism’ of TyrZ (e.g.[3,5]), where the proton that belongs to the Nϵ site of D1-His190 is deprotonatable (note: this is energetically possible because the Nδ site donates a hydrogen bond to D1-Asn298 and is permanently protonated). Thus, the proton-rocking mechanism is possible only when the proton in the hydrogen bond can belong to both TyrZ and D1-His190 either (i) by having TyrZ and D1-His190 with similar pKa values in a single-well hydrogen bond, or (ii) by having the two cases, pKa(TyrZ) ≥ pKa(D1-His190) and pKa(TyrZ) ≤ pKa(D1-His190), depending on the TyrZ redox state: this means that the proton-rocking model requires the deprotonatable H+ at the Nϵ site of D1-His190 (Fig. 1a).
|
In PSII, another redox active tyrosine, tyrosine D (TyrD, D2-Tyr160), has D2-His189 as a hydrogen-bond partner. Because TyrD is geometrically isolated from the electron transfer pathway from the Mn4CaO5 cluster to P680 via TyrZ, it plays no obligatory role in enzyme function. It was generally thought that the phenolic proton of TyrD was most likely to transfer to the Nϵ site of D2-His189 (e.g.[6]), as is the case for the TyrZ–D2-His190 pair (summarized by Nakamura and Noguchi[7]). However, this appears to be impossible based on the geometry of the crystal structures. Ishikita and Knapp found that in contrast to D1-His190, the putative proton-rocking Nϵ site of D2-His189 cannot release the proton (unless D2-His189 becomes the doubly deprotonated anionic form), because the Nϵ site of D2-His189 is permanently protonated owing to the Nδ site of D2-His189 being hydrogen bonded by positively charged D2-Arg294 (Fig. 1b).[8]
In contrast, the permanently protonated Nϵ site of D2-His189 forces the -OH group of TyrD-OH to orient out of the TyrD–D2-His189 bond towards an external water molecule H2OD (Fig. 2) when TyrD-OH is formed.[9] H2OD has two binding positions, H2OD(prox) and H2OD(dist),[2] and it moves between TyrD and D2-Arg180 in response to changes in the TyrD oxidation state.[9] Using large-scale quantum mechanical/molecular mechanical (QM/MM) calculations and the crystal structure,[2a] Saito et al. demonstrated that in response to oxidation of TyrD-OH to TyrD-O•, the proton released from TyrD-OH is transferred along the proton transfer pathway involving D2-Arg180, D2-His61, and a series of water molecules (i.e. redox-coupled proton transfer).[9] Intriguingly, the corresponding pathway is also conserved on the active D1 side, involving TyrZ, water molecules adjacent to the Mn4CaO5 cluster, a Cl– ion interacting with D1-Asn181, and D1-Asp61;[9] this implies that in an ancestral homodimer, there was once an active Mn cluster in the cavity adjacent to TyrD.[10]
|
Notably, the proton transfer pathway from TyrD suggested by Saito et al. has been supported by recent FT-IR spectroscopy studies by Nakamura and Noguchi, where the release of the proton to the bulk was observed in response to the oxidation of TyrD-OH.[7] In the present study, we determined the detailed properties of energetics and structural geometries of the proton transfer pathway from TyrD.
Results and Discussion
Geometry and Energy Profile Along the Proton Transfer Pathway
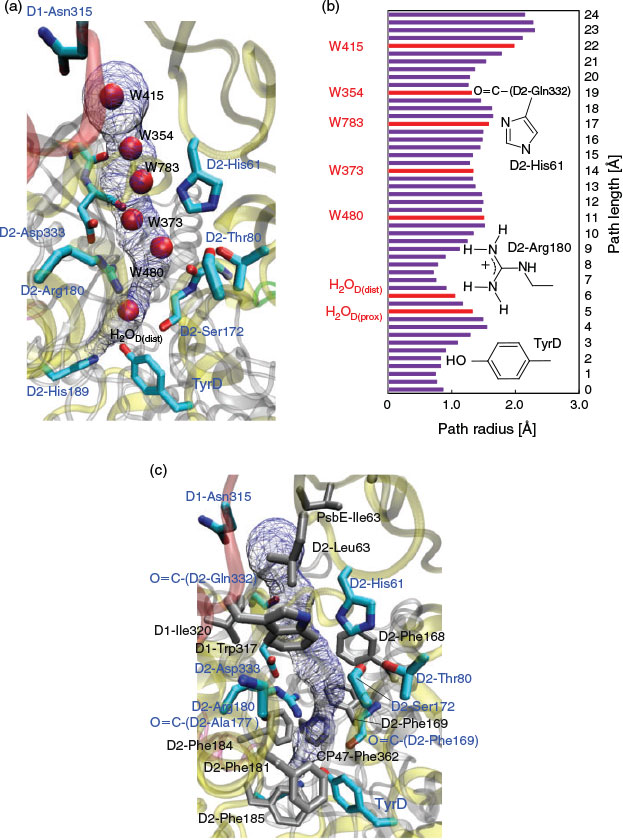
The proton transfer pathway from TyrD[9] is consistent with a hydrogen-bond network that has six water molecules conserved in the 1.9 Å[2a] and the free electron laser structures.[2b] The hydrogen-bond network is located along the cavity with a length of ~24 Å (Fig. 2a). H2OD, the initial proton acceptor from TyrD, is spatially isolated from the other five water molecules by the D2-Arg180 side chain (Fig. 2b).
Fig. 3a shows the potential-energy profile of all hydrogen bonds along the proton transfer pathway. The potential-energy profile indicates that overall proton transfer from TyrD is energetically downhill (Fig. 3a). The energy levels at the proton acceptor moieties along the pathway are consistent with those reported previously.[9] As proton transfer proceeds, the hydrogen-bond pattern of the entire proton transfer pathway is altered from the ‘pre-PT’ pattern to the ‘post-PT’ pattern (Fig. 4a). The downhill proton transfer steps from W480 to W373 (Fig. 4b) and from W373 to W783 (Fig. 4c) involve rearrangement of the hydrogen-bond network. The radical state was fully localized on TyrD-O• in the QM region (Fig. 3b).

|

|
Remarkably, the region near the TyrD (D2-Tyr160)/D2-His189 pair is hydrophobic because of the localization of several hydrophobic phenylalanine side chains near the TyrD/D2-His189 pair, e.g. D2-Phe181, D2-Phe184, D2-Phe185, and CP47-Phe362[8] (Figs 2c and 5), which destabilize the proton released from TyrD and serve as a driving force for proton transfer once TyrD-OH is oxidized. The proton transfer from D2-Arg180 to W373 is significantly downhill (Fig. 3a) because the acceptor W373 forms a hydrogen bond with negatively charged D2-Asp333 and provides a driving force for the proton transfer from the positively charged donor D2-Arg180 (Fig. 5).

|
Roles of D2-Arg180 in Proton Transfer from TyrD
It appears that D2-Arg180 is a key component that plays a role in releasing a proton from TyrD-OH and forming TyrD-O•, because (i) D2-Arg180 is the only titratable side chain involved in the proton conducting hydrogen-bond network via TyrD (Fig. 2) and (ii) in the D2-Arg180 mutants, the electron paramagnetic resonance (EPR) signals from TyrD (i.e. TyrD-O•) were small.[11] Considering the energy profile of the TyrD proton transfer presented here, the following roles of D2-Arg180 can be postulated:
D2-Arg180, Providing a Driving Force for the Proton Transfer
As described above, mutation of positively charged D2-Arg180 (i.e. loss of the positive charge) should result in a decrease in the driving force for the downhill proton transfer, specifically from D2-Arg180 to W373 (Fig. 3a), and loss of the TyrD-O• formation; this may explain why the D2-Arg180 mutation resulted in the decrease of the EPR signal from TyrD.[11]
D2-Arg180, Maintaining the Hydrogen-Bond Network Structure
D2-Arg180 is the only titratable side chain involved in the proton-conducting hydrogen-bond network from TyrD (Fig. 2). The removal of D2-Arg180 may result in the disorder of the hydrogen-bond network, leading to an increase in the activation energy for proton transfer (e.g.[12]).
D2-Arg180, Stabilizing P680•+ and Deprotonating TyrD-OH
D2-Arg180 also contributes to increasing the PD1•+ population.[13] Indeed, mutations at the D2-Arg180 residue have been shown to increase the charge recombination rate between QA•– and P680•+ (≈PD1•+), i.e. destabilize P680•+.[11] Rutherford et al. proposed that TyrD-O• formation was associated with localization of the highly oxidizing cation P680•+ to the chlorophyll nearest to TyrZ (i.e. PD1•+) for efficient electron transfer from the Mn4CaO5 cluster via TyrZ;[14c] this may be understood by considering the presence of D2-Arg180, which has an electrostatic link with both TyrD and P680•+. These observations may represent a functional link between the ‘formation of PD1•+’ and ‘release of the proton from TyrD-OH (i.e. formation of TyrD-O•)’ mediated by D2-Arg180.
When TyrD Uses the Proton Transfer Pathway: Electron Transfer via TyrD
A hydrophobic environment of TyrD in comparison with TyrZ (e.g.[2a,8,9,14c,15]) makes TyrD sensitive to the redox states of the other redox-active cofactors, e.g. P680 and the Mn4CaO5 cluster (e.g.[14c]). TyrD is involved in the following electron transfer processes (e.g.[14c]):
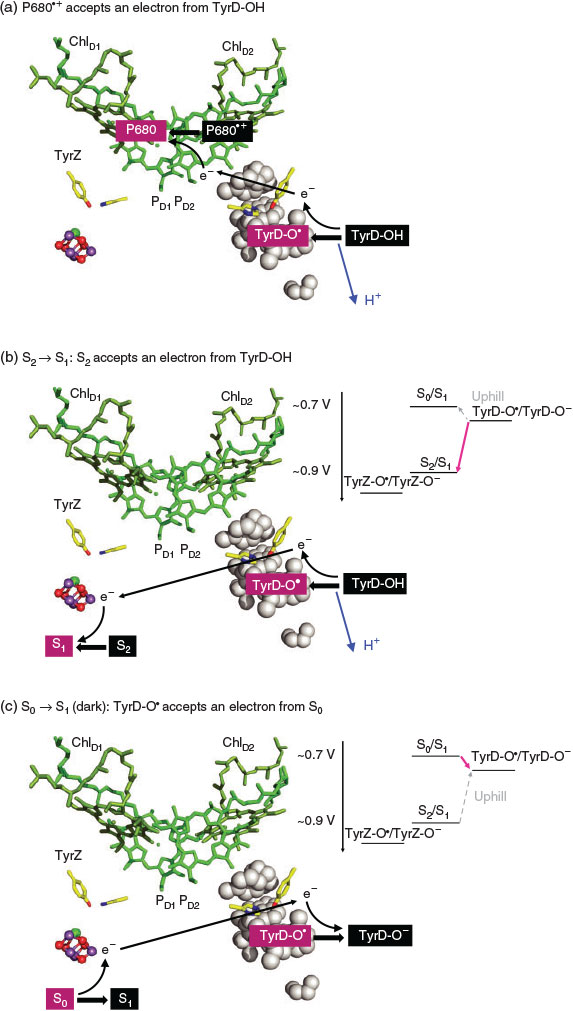
Electron Transfer from TyrD-OH to P680•+ (Fig. 6a)
|
The release of the proton from TyrD-OH was observed even in the Mn4CaO5-depleted PSII in response to the formation of P680•+ (e.g.[7,14]). TyrD-OH transfers the electron to highly oxidizing P680•+ and simultaneously releases the proton. This occurs on a time scale in the tens of milliseconds.[14b,16]
Electron Transfer from TyrD-OH to S2 (Fig. 6b)
EPR studies demonstrated that S2 can be reduced to S1 by TyrD-OH in the dark (1–2 s);[14c,17] this is possible because TyrD-O•/TyrD-O has a redox potential of ~0.7 V[8,18], which is lower than ~0.9 V for S1/S2.[18]
Electron Transfer from S0 to TyrD-O• (Fig. 6c)
Once formed, TyrD-O• is very stable for many hours.[14a,14c] Long dark adaptation of PSII resulted in the oxidation of S0 to S1 via electron transfer from S0 (S0/S1 ~0.7 V[18]) to TyrD-O•.[14c,17] In pea thylakoids, S0 disappears in ~20 min.[17a] In addition, TyrD oxidizes the over-reduced states of the Mn4CaO5 cluster, e.g. S–1 and S–2 (‘photoactivation’[19]).
Among the three cases, electron transfer from TyrD-OH to P680•+ [7,14] (Fig. 6a) or electron transfer from TyrD-OH to S2[14c,17] (Fig. 6b) requires the release of the proton from TyrD-OH along the proton transfer pathway, resulting in TyrD-O•. TyrD-O• is stable for many hours and can oxidize S0 to S1 in the dark, resulting in a 100 % S1 sample.[14a,14c] Since TyrD-O• is frequently present in PSII samples,[14a,14c] it is possible that the proton transfer pathway from TyrD[9] functions to facilitate the formation of TyrD-O•, which is ready for a conversion of S0 into S1 in the dark,[14c,17] presumably to stabilize the Mn4CaO5 cluster (e.g.[19]). All these redox roles of TyrD[14c] are possible because of the hydrophobic environment of TyrD,[2a,8,9,14c,15] where electrostatic interactions are pronounced.
Concluding Remarks
The proton transfer pathway that proceeds from TyrD towards the protein bulk surface, originally suggested in QM/MM studies[9] and further confirmed in recent FT-IR spectroscopy studies,[7] is present in all recent high-resolution crystal structures[2] (Table 1). The potential-energy profile of all hydrogen bonds along the proton transfer pathway indicates that the overall proton transfer from TyrD is energetically downhill (Fig. 3). D2-Arg180 plays a key role in the proton-conducting pathway, providing a driving force for proton transfer, maintaining the hydrogen-bond network structure as being the only titratable residue in the pathway (Figs. 2 and 4), stabilizing P680•+[11] (in particular by increasing the PD1•+ population over the PD2•+ population[13]), and thus deprotonating TyrD-OH to TyrD-O•.[7,14] Deprotonation of D2-Arg180 predominantly decreases the redox potential of TyrD but not that of TyrZ owing to the hydrophobic environment, which is formed by phenylalanine side chains near the TyrD/D2-His189 pair, e.g. D2-Phe181, D2-Phe184, D2-Phe185, and CP47-Phe362 as suggested previously.[8] The hydrophobic environment near TyrD also enhances the electrostatic interaction of TyrD with redox active cofactors, e.g. P680•+ (Fig. 6a) and the Mn4CaO5 cluster (Fig. 6b, c). In particular, the conversion of S0 into S1 in the dark (dark adaptation) is a redox role of TyrD-O•.[14c,17b] It can be postulated that the proton transfer pathway from TyrD ultimately contributes to the conversion of S0 into S1 in the dark to stabilize the Mn4CaO5 cluster when the photocycle is interrupted in S0.

|
Computational Procedures
The atomic coordinates of PSII were taken from the X-ray structure of the PSII monomer unit designated monomer A of the PSII complexes from Thermosynechococcus vulcanus at a 1.9 Å resolution (PDB code, 3ARC).[2a] Owing to the large system size of PSII, we considered residues and cofactors in subunits D1, D2, CP47, and CP43 only as the protein environment.[9] Hydrogen atoms were generated and energetically optimized using CHARMM,[20] whereas the positions of all non-hydrogen atoms were fixed and all titratable groups were kept in their standard protonation states. For the QM/MM calculations, we added additional counter ions to neutralize the entire system. Atomic partial charges of the amino acids were adopted from the all-atom CHARMM22[21] parameter set. The atomic charges of the cofactors were taken from our previous studies of PSII.[9]
We used the Qsite[22] program code as described in previous studies.[9] We employed the unrestricted density functional theory method with the B3LYP functional and LACVP**+ basis sets. The geometries were refined by constrained QM/MM optimization (see Supplementary Material). Specifically, the coordinates of the heavy atoms in the surrounding MM region were fixed to the original X-ray coordinates, whereas those of the hydrogen atoms in the MM region of the residues within 7.0 Å from the QM region were optimized using the OPLS2005 force field. All atomic coordinates in the QM region were fully relaxed (i.e. not fixed) in the QM/MM calculation. The QM region was defined as: D2-His61, TyrD, D2-Arg180, D2-His189, D2-Arg294, D2-Asp333, and water molecules that were within hydrogen-bond distance of these residues, namely, HOH-(chain D)-1(H2OD), -D354, -D364, -D366, -D373, -D390, -D480, and -D783. The potential-energy profile of the hydrogen bond was obtained as follows: first, we prepared for the QM/MM optimized geometry without constraints, and we used the resulting geometry as the initial geometry. The hydrogen atom was then moved from the hydrogen-bond donor atom (e.g. Odonor) to the acceptor atom (e.g. Oacceptor) by 0.05 Å, after which the geometry was optimized by constraining the Odonor–H and H–Oacceptor distances, and the energy of the resulting geometry was calculated. This procedure was repeated until the hydrogen atom reached the Oacceptor atom.
Supplementary Material
The QM/MM optimized geometries are available on the Journal’s website.
Acknowledgements
This research was supported by the JST PRESTO program (K.S.), JSPS KAKENHI (22740276 and 26800224 to K.S. and 15H00864, 26105012 and 26711008 to H.I.), Materials Integration for Engineering Polymers of Cross-Ministerial Strategic Innovation Promotion Program (SIP, H.I.), Tokyo Ohka Foundation for The Promotion of Science and Technology (H.I.), and Interdisciplinary Computational Science Program in CCS, University of Tsukuba. Theoretical calculations were partly performed using the Research Center for Computational Science, Okazaki, Japan.
References
[1] (a) H. Dau, I. Zaharieva, M. Haumann, Curr. Opin. Chem. Biol. 2012, 16, 3.| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xjt1Kjsbs%3D&md5=8d738a238ccd734da0822948379004f8CAS | 22387134PubMed |
(b) N. Cox, J. Messinger, Biochim. Biophys. Acta 2013, 1827, 1020.
| Crossref | GoogleScholarGoogle Scholar |
[2] (a) Y. Umena, K. Kawakami, J.-R. Shen, N. Kamiya, Nature 2011, 473, 55.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXkslCmtLg%3D&md5=abc7a38ea3183ce0a96d876da0716e61CAS | 21499260PubMed |
(b) M. Suga, F. Akita, K. Hirata, G. Ueno, H. Murakami, Y. Nakajima, T. Shimizu, K. Yamashita, M. Yamamoto, H. Ago, J.-R. Shen, Nature 2015, 517, 99.
| Crossref | GoogleScholarGoogle Scholar |
[3] K. Saito, J.-R. Shen, T. Ishida, H. Ishikita, Biochemistry 2011, 50, 9836.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXht1KktrrO&md5=52e20db6b95410e9cdab589b3938a22aCAS | 21972783PubMed |
[4] (a) A. Warshel, A. Papazyan, P. A. Kollman, Science 1995, 269, 102.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXmsl2isr4%3D&md5=ced73e9a477bd756c6b9ab07a165483aCAS | 7661987PubMed |
(b) C. L. Perrin, J. B. Nielson, Annu. Rev. Phys. Chem. 1997, 48, 511.
| Crossref | GoogleScholarGoogle Scholar |
[5] (a) R. Ahlbrink, M. Haumann, D. Cherepanov, O. Bogershausen, A. Mulkidjanian, W. Junge, Biochemistry 1998, 37, 1131.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXjtFOhtw%3D%3D&md5=282129d2a945d83d03c28483271c8e04CAS | 9454606PubMed |
(b) A.-M. A. Hays, I. R. Vassiliev, J. H. Golbeck, R. J. Debus, Biochemistry 1999, 38, 11851.
| Crossref | GoogleScholarGoogle Scholar |
(c) F. Rappaport, J. Lavergne, Biochim. Biophys. Acta 2001, 1503, 246.
| Crossref | GoogleScholarGoogle Scholar |
(d) S. Nakamura, R. Nagao, R. Takahashi, T. Noguchi, Biochemistry 2014, 53, 3131.
| Crossref | GoogleScholarGoogle Scholar |
[6] (a) C. Tommos, L. Davidsson, B. Svensson, C. Madsen, W. Vermaas, S. Styring, Biochemistry 1993, 32, 5436.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3sXitlSgtbc%3D&md5=ef38dbcd45b4be9838bd1bc917c5465eCAS | 8388721PubMed |
(b) X. S. Tang, D. A. Chisholm, G. C. Dismukes, G. W. Brudvig, B. A. Diner, Biochemistry 1993, 32, 13742.
| Crossref | GoogleScholarGoogle Scholar |
[7] S. Nakamura, T. Noguchi, Biochemistry 2015, 54, 5045.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXht1KkurfF&md5=8bbb13dd6c55e867037655f0fd635461CAS | 26241205PubMed |
[8] H. Ishikita, E. W. Knapp, Biophys. J. 2006, 90, 3886.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XltF2mtro%3D&md5=b57226a5c93d7043d2fd3f23855564d2CAS | 16513785PubMed |
[9] K. Saito, A. W. Rutherford, H. Ishikita, Proc. Natl. Acad. Sci. USA 2013, 110, 7690.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXptFWqsr0%3D&md5=971c0ed896199a7917841bba54eaa8f1CAS | 23599284PubMed |
[10] (a) A. W. Rutherford, W. Nitschke, in Origin and Evolution of Biological Energy Conversion (Ed. H. Baltscheffsky) 1996, pp. 143–176 (Wiley-VCH: New York, NY).
(b) A. W. Rutherford, P. Faller, Philos. Trans. R. Soc. Lond. B Biol. Sci. 2003, 358, 245.
| Crossref | GoogleScholarGoogle Scholar |
[11] P. Manna, R. LoBrutto, C. Eijckelhoff, J. P. Dekker, W. Vermaas, Eur. J. Biochem. 1998, 251, 142.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXnvV2ltQ%3D%3D&md5=159153a057579a36f3c0e77b37b88849CAS | 9492278PubMed |
[12] A. A. Stuchebrukhov, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys. 2009, 79, 031927.
| Crossref | GoogleScholarGoogle Scholar |
[13] K. Saito, T. Ishida, M. Sugiura, K. Kawakami, Y. Umena, N. Kamiya, J.-R. Shen, H Ishikita, J. Am. Chem. Soc. 2011, 133, 14379.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtVegt7rE&md5=a7a89c8386fc52c3608f8c156c1a3299CAS | 21805998PubMed |
[14] (a) G. T. Babcock, B. A. Barry, R. J. Debus, C. W. Hoganson, M. Atamian, L. McIntosh, I. Sithole, C. F. Yocum, Biochemistry 1989, 28, 9557.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3cXksVOn&md5=2cc2522bae8b437f6ba784a34999de3eCAS | 2692711PubMed |
(b) P. Faller, R. J. Debus, K. Brettel, M. Sugiura, A. W. Rutherford, A. Boussac, Proc. Natl. Acad. Sci. USA 2001, 98, 14368.
| Crossref | GoogleScholarGoogle Scholar |
(c) A. W. Rutherford, A. Boussac, P. Faller, Biochim. Biophys. Acta 2004, 1655, 222.
| Crossref | GoogleScholarGoogle Scholar |
(d) C. Berthomieu, R. Hienerwadel, Biochim. Biophys. Acta 2005, 1707, 51.
| Crossref | GoogleScholarGoogle Scholar |
[15] (a) S. V. Ruffle, D. Donnelly, T. L. Blundell, J. H. A. Nugent, Photosynth. Res. 1992, 34, 287.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3sXjs1SqtQ%3D%3D&md5=082949c28b28c4a4575095dcca6d9212CAS | 24408780PubMed |
(b) B. Svensson, C. Etchebest, P. Tuffery, P. van Kan, J. Smith, S. Styring, Biochemistry 1996, 35, 14486.
| Crossref | GoogleScholarGoogle Scholar |
[16] C. A. Buser, L. K. Thompson, B. A. Diner, G. W. Brudvig, Biochemistry 1990, 29, 8977.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3cXlsV2jtLY%3D&md5=f5f7d736e43eb8b28e783d719348dbf2CAS | 2176840PubMed |
[17] (a) W. F. J. Vermaas, G. Renger, G. Dohnt, Biochim. Biophys. Acta 1984, 764, 194.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2cXhtFOqu7Y%3D&md5=7cb1b2c90961233644971b31620a6ef7CAS |
(b) S. Styring, A. W. Rutherford, Biochemistry 1987, 26, 2401.
| Crossref | GoogleScholarGoogle Scholar |
[18] I. Vass, S. Styring, Biochemistry 1991, 30, 830.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3MXjvFertQ%3D%3D&md5=cfab17b4922320e2cb260d555735b1dfCAS | 1988070PubMed |
[19] J. Messinger, G. Renger, Biochemistry 1993, 32, 9379.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3sXltlGrsLk%3D&md5=3300fd7df31ecc41faf38a9d42e53a11CAS | 8369309PubMed |
[20] B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan, M. Karplus, J. Comput. Chem. 1983, 4, 187.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL3sXit1aiu7w%3D&md5=0c1d8a9547a3abf84bc32ec09d35c708CAS |
[21] A. D. MacKerell, D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiórkiewicz-Kuczera, D. Yin, M. Karplus, J. Phys. Chem. B 1998, 102, 3586.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXivVOlsb4%3D&md5=71b4a1e8e893cd039e356fa79522d1caCAS | 24889800PubMed |
[22] QSite, version 5.8 2012 (Schrödinger, LLC: New York, NY).
[23] M. Petrek, M. Otyepka, P. Banas, P. Kosinova, J. Koca, J. Damborsky, BMC Bioinformatics 2006, 7, 316.
| Crossref | GoogleScholarGoogle Scholar | 16792811PubMed |