

The Evolution of New Catalytic Mechanisms for Xenobiotic Hydrolysis in Bacterial Metalloenzymes*
Elena Sugrue A , Carol J. Hartley B , Colin Scott B and Colin J. Jackson A CA Research School of Chemistry, Australian National University, Canberra, ACT 2601, Australia.
B CSIRO Land and Water, Canberra, ACT 2600, Australia.
C Corresponding author. Email: colin.jackson@anu.edu.au

Elena Sugrue is a Ph.D. scholar at the Research School of Chemistry, Australian National University (ANU). She completed her Bachelor of Science degree at the University of Canterbury, receiving first class honours before taking up doctoral studies at the ANU in 2013. Her research interests include investigating the chemical basis of biological function and how chemical and biophysical features can change over evolutionary trajectories. |

Dr Carol Hartley is a research scientist and leader of the Biocatalysis and Synthetic Biology research team within the Commonwealth Scientific and Industrial Research Organisation (CSIRO) in Canberra, Australia. She obtained a Ph.D. in microbiology from Rhodes University, South Africa, before joining the CSIRO and has a strong interest in biocatalysis and the use of enzymes to advance biotechnology and synthetic biology. |

Dr Colin Scott obtained his Ph.D. in molecular microbiology from the University of Sheffield in the UK in 2000 before taking up a post-doctoral fellowship with the Commonwealth Scientific and Industrial Research Organisation (CSIRO). He currently leads the Biotechnology and Synthetic Biology Group at the CSIRO. He has strong interests in enzyme evolution, biocatalysis, microbial physiology, and synthetic biology. |

Associate Professor Colin Jackson has held research positions at the Commonwealth Scientific and Industrial Research Organisation (CSIRO) and was a visiting research fellow at the Weizmann Institute of Science (Rehovot, Israel) and a Marie Curie Research Fellow at the Institut de Biologie Structurale (Grenoble, France), before his appointment at the Research School of Chemistry at the Australian National University. He is an ARC Future Fellow and has been awarded an ARC DECRA, a Human Frontiers in Science Young Investigator Award, an AIPS Tall Poppy Award, ACT Tall Poppy of the Year, the 2015 ACT Scientist of the Year Award, and the 2015 RACI Rennie Memorial Medal. |
Australian Journal of Chemistry 69(12) 1383-1395 https://doi.org/10.1071/CH16426
Submitted: 21 July 2016 Accepted: 6 September 2016 Published: 23 September 2016
Abstract
An increasing number of bacterial metalloenzymes have been shown to catalyse the breakdown of xenobiotics in the environment, while others exhibit a variety of promiscuous xenobiotic-degrading activities. Several different evolutionary processes have allowed these enzymes to gain or enhance xenobiotic-degrading activity. In this review, we have surveyed the range of xenobiotic-degrading metalloenzymes, and discuss the molecular and catalytic basis for the development of new activities. We also highlight how our increased understanding of the natural evolution of xenobiotic-degrading metalloenzymes can be been applied to laboratory enzyme design.
Introduction
Almost half of all proteins are predicted to associate with a metal ion to function, harnessing the reactivity of metal ions for catalysis or using them to stabilise protein structure to allow them to carry out essential functions.[1–3] A metal ion cofactor enhances the catalytic diversity provided by the 20 canonical amino acids, as the redox states, coordination preferences, and Lewis acidities of metal ions can facilitate novel reactions. In accordance with this, enzyme activity and reactivity can be tuned through the use of different metal ions, scaffold-imposed ligand coordination, and redox states.[4,5] In this review, we will primarily discuss the metal-dependent hydrolysis of xenobiotic compounds (Table 1) where metal ions can act as strong Lewis acids at neutral pH to generate catalytic nucleophiles, polarise substrates for nucleophilic attack, and stabilise charges in the transition state.[6–10] We will also discuss how metal ions allow reactions with diverse anthropogenic (synthetic) chemicals to proceed through different mechanisms (Table 2).

|

|
There is great diversity in both the nature of metal ion coordination and associated functions among contemporary metalloenzymes.[11–13] For example, even enzymes that break down the same substrates, such as glycerophosphodiesterase (GpdQ) from Enterobacter aerogenes, and phosphotriesterases (PTE) (Pseudomonas diminuta Pd-PTE, Sphingobium sp. TCM1 Sb-PTE, and Agrobacterium radiobacter Ar-PTE) all break down the pesticide paraoxon, but have different protein folds and metal ion binding sites (Table 2).[6,7,14,15] Xenobiotic hydrolysing enzymes have frequently been used to simplify the study of metalloenzyme evolution, as they represent a known evolutionary starting point: the introduction of the xenobiotic chemical into the environment.[16–19] Bacteria in environments into which synthetic herbicide and pesticide compounds have been introduced have frequently evolved complex systems to break them down, as there is a strong selective pressure to have additional energy sources for survival, particularly in nutrient-lacking soil.[20] Numerous studies have characterised diverse bacterial species that can use different xenobiotics as the sole carbon source.[21–24] Often, consortia of bacteria are required for the complete breakdown of the compound, as the breakdown products are frequently more toxic than the unmodified xenobiotic.[17,25,26] The most intensively studied example of this is the well-known evolution of bacteria to break down newly developed synthetic antibiotics. The evolution can be rapid as there is extremely strong selective pressure for bacteria in environments exposed to these xenobiotics that directly target bacterial survival.[27–29] This provides another advantage to studying xenobiotic-degrading enzymes as model systems for molecular evolution: they are frequently under intense selective pressure, which reduces the amount of neutral variation between sequences and results in functional changes often occurring in short timeframes. For instance, the triazine herbicides were introduced to the environment in the 1950s and bacteria capable of breaking these compounds down were discovered less than 50 years later.[30–32] Thus, recently evolved xenobiotic-degrading enzymes provide excellent model systems to investigate the molecular mechanisms of enzyme evolution, as the directionality of the evolutionary processes is known, the selective pressure is easily identified, the evolved protein can often be traced back to a known ancestral enzyme, and the selective pressure is frequently intense enough that new functions are established over short time periods.[9,10,33,34]
As metalloenzymes represent such a large proportion of the proteome,[1–3] investigating the diverse means by which they catalyse reactions with similar substrates, or how they converge on similar reactions using alternative protein scaffolds, coordination, or metal ions is an important area of research for the fundamental understanding of biological (and inorganic) catalysis. The study of xenobiotic-degrading metalloenzymes provides valuable information beyond an improved understanding of catalysis; by carefully following the evolutionary process, these enzymes allow us to better understand fundamental aspects of molecular evolution and how the reactivity of metal ions in enzymes can be tuned through evolution. The aim of the present review is to summarise a selection of the molecular mechanisms by which different metalloenzymes have evolved to break down anthropogenic chemicals. The subsequent sections each introduce and discuss molecular mechanisms that contribute and promote the divergence of metalloenzymes, to facilitate new catalytic mechanisms for xenobiotic hydrolysis. The importance of metal ion promiscuity, modification of active site residues, changes to metal ion coordination, stabilisation of the unbound state of metalloenzymes, and variability in loop regions in the evolution of new catalytic activity are all discussed. This information is useful for applied work on the specific enzymes discussed, for artificial enzyme design, and to improve our fundamental understanding of metalloenzymes.
Metal Ion Promiscuity and Metalloenzyme Evolution
The coordination of a metal ion to a protein is mediated by many factors: the properties and preferred coordination geometries of the various metal ions, the metal ion ligands available at the binding site, the bioavailability of the respective metal ions in the cell, and the inherent flexibility of protein scaffolds.[35–38] There is great variation in the amino acid side chains used that coordinate to metal ions.[39,40] Generally, comparatively ‘hard’ metal ions like manganese (according to the hard–soft acid–base scale) will coordinate to oxygen atoms in aspartate, glutamate, asparagine, and glutamine or on the carbon backbone,[39,41] whereas more ‘borderline’ metal ions like zinc and cobalt often coordinate to the nitrogen atoms of histidine or sulfur-containing cysteine.[39] The first-shell residues that directly coordinate a metal ion in the active site are stabilised and optimised by second-shell residues and the entire enzyme scaffold. The importance of these remote residues has been highlighted by the observation that mutations in these remote regions can alter the configuration, or conformational stability, of the binding site, with drastic impact on the metal ion specificity, affinity, stability, and function of the enzyme.[39,42–45] The significance of outer residues is demonstrated in the differing metal ion specificities and reaction mechanisms observed between the diesterase Rv0805 from Mycobacterium tuberculosis and the enzyme GpdQ from Enterobacter aerogenes despite these having identical first-shell coordination of the respective metal ions.[46] This also demonstrates the intrinsic challenge associated with the generation of biomimetics that match the catalytic properties of their biological representatives.[47] Owing to this apparent optimised structural specificity, it has often been assumed that metalloenzymes are specific for the metal ion they are bound to when purified.[48,49] However, a growing number of metalloenzymes have been shown to have extensive flexibility in metal ion incorporation, where several different metal ions can be incorporated in metalloenzyme active sites without sacrificing stability or abolishing activity.[50–52] In fact, the incorporation of alternative metal ions can often enhance promiscuous activities, as observed in the metallo-β-lactamase (MβL) superfamily.[53] In a physiologically relevant context and environment, metal ions are present at variable concentrations. This is one of the reasons why bacterial cells have evolved such complex systems to attempt to maintain metal ion homeostasis and overcome the challenges associated with specifically incorporating different metal ions into different proteins.[38,50,54,55] The metal ion homeostasis mechanisms used and resultant bioavailability may differ between different bacterial species,[56–58] which is important to consider, as many of the genes encoding xenobiotic-hydrolysing proteins are transposable.[59–62] Therefore, there is potential for different bacterial environments to influence the ratio of metal isoforms produced, which could promote the evolution of novel metal preferences. Overall, alternatively metalated enzyme isoforms are a predicted aspect of bacterial cells that is likely to impact both the survival and evolvability of the host system.
Flexibility in metal ion cofactor preference allows catalysis to be tuned to the properties of the metal ion and enables catalysis to occur in more diverse environments, which is particularly advantageous for xenobiotic-degrading enzymes essential for survival. A frequent example of metal ion promiscuity is observed in hydrolases that can incorporate iron or zinc. These metal ions have different Lewis acidities and pKA values, which enables an adjustment of catalysis and the optimum pH for activity on differential metal ion incorporation. In binuclear hydrolases like PTE derived from Agrobacterium radiobacter and the previously mentioned glycerophosphodiesterase (GpdQ) (Table 2), which catalyse the hydrolysis of PTE pesticides, both Zn2+–Zn2+ and mixed Fe2+–Zn2+ binding sites are commonly observed.[15,63,64] In PTEs, the more buried Mα site generally displays a higher affinity for iron, whereas the lower-affinity Mβ site generally favours zinc binding, depending on the environmental concentration.[15] This flexibility in metal ion binding enables the utilisation of both metal ion properties: iron as a stronger Lewis acid better suited to generate a nucleophile at neutral pH,[15,65] and zinc better suited for substrate binding with less of a preference for a symmetrical coordination sphere.[15]
Beyond the benefits of tuning metal ion preference for the catalysis of xenobiotics, the ability of a metalloenzyme to function with both Zn–Zn and Fe–Zn active sites could increase bacterial survival in different conditions. For example, IMP-1 from Pseudomonas aeruginosa, a β-lactamase involved in antibiotic resistance,[66] was previously thought to exist only in Zn–Zn form, potentially owing to experimental artefacts; however, recent analysis revealed that a heterobinuclear Fe–Zn complex is produced in the presence of even small amounts of iron.[67] This could create an evolutionary advantage in environments with limited zinc bioavailability and varying pH ranges, as this enzyme is essential for organism fitness. Zinc is abundant in the environment, but it is restricted to very low bioavailable concentrations in the cell;[50] however, in varying pH and redox environments, redox-sensitive iron is more likely to exhibit changes in bioavailability.[68,69] Therefore, the ability to use both iron and zinc is of evolutionary advantage in environments that can exhibit variation in relative bioavailability of both metal ions in response to different factors. A similar conclusion has been drawn for the organophosphate-degrading enzyme GpdQ, where its promiscuity for different metal ions and substrates means it can easily adapt to changes in environmental conditions, increasing the odds of survival of its host cell.[70] Finally, the alternative metal isoforms of PTEs appear to utilise different mechanisms for hydrolysis.[71] Zn–Zn PTE isoforms appear to utilise a bridging hydroxide nucleophile (Fig. 1), whereas the Fe–Zn and Co–Co isoforms generally lead to the production of a bridging product that is only consistent with a terminally coordinated hydroxyl nucleophile (Fig. 1).[71–73] The mechanistic flexibility afforded by the use of alternative metal ions, without the need to alter the protein scaffold, is an advantageous trait for the evolution of xenobiotic-degrading enzymes in different environments.
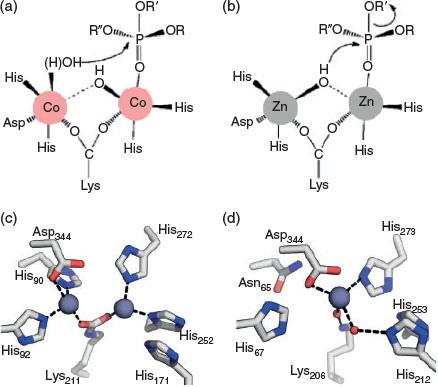
|
The extent of metalloenzyme cofactor promiscuity is best represented by the numerous bacterial zinc hydrolases that can catalyse reactions with comparatively poorly bioavailable metal ions such as cobalt, nickel, cadmium, and manganese.[74,75] In different metallohydrolase scaffolds, it has been observed that a cobalt isoform will often have enhanced activity compared with the zinc isoform. For example, cobalt-bound PTE has a kcat/Km value (turnover number/substrate concentration at half maximum velocity, a second-order rate constant that is commonly used as a measure of catalytic specificity)[76] that is an order of magnitude higher with the organophosphate insecticide paraoxon compared with the untreated PTE isoform.[15] This has also been seen in cobalt-bound molinate hydrolase derived from Gulosibacter molinativorax ON4T (MolA) where the kcat/Km with its herbicide substrate molinate is an order of magnitude higher when compared with the activity of the more likely native zinc form.[10] It is thought that cobalt might be more efficient than zinc with ‘softer’ substrates, according to the hard–soft acid–base scale, because cobalt can reduce non-productive binding owing to an improved affinity for the substrate.[41,77] The similar coordination ligand preferences, pKA values, and atomic radii of cobalt and zinc also mean that they are almost always easily interchanged in zinc-dependent enzymes.[78] Cobalt-, cadmium-, manganese-, and nickel-treated PTE can retain activity with paraoxon, despite the octahedral coordination spheres that these metal ions preferentially adopt, compared with the tetrahedral or trigonal bipyramidal coordination of zinc ions, the native ion of PTE.[74,79–81] This flexibility in metal ion incorporation appears to occur more frequently when the metal binding site is optimised for fewer interactions, as water molecules can be utilised to replace missing ligands or fulfil additional coordination positions.[70,80] However, when iron is replaced by zinc in Geobacillus kaustophilus lactonase and in GpdQ, the resultant enzymes are inactive.[63,82] A non-optimal active site arrangement, or the lack of a stronger Lewis acid may prevent promiscuity in these cases; however, cobalt may still be incorporated due to the previously mentioned ease with which cobalt replaces zinc. The flexibility of enzyme scaffolds to incorporate metal ions that are many orders of magnitude less frequent in the cell and thus unlikely to be the native metal illustrates the inherent promiscuity of metal ion preferences in metalloenzymes. This promiscuity may have been utilised in the evolution of contemporary enzymes that preferentially bind and catalyse reactions with different metal ions, specialising for different environmental niches.[83–86] To summarise, metal ion promiscuity is a property that can increase the survivorship of bacteria through facilitating catalysis in different conditions, in addition to increasing the evolvability of an enzyme towards novel xenobiotic functions by enabling the recruitment of different catalytic properties.
Active Site Changes and Activity Divergence
Although function-changing mutations are often located in loop regions, changes to catalytic residues within the active site can occur to facilitate the hydrolysis of xenobiotic compounds.[34] A clear example of a short evolutionary trajectory to generate a novel xenobiotic hydrolase is seen in the natural evolution of atrazine chlorohydrolase (AtzA, Pseudomonas sp. strain ADP) from the iron hydrolase melamine deaminase (TriA, Pseudomonas sp. strain NRRL B-12227).[34,87,88] Only nine amino acids differ between these two enzymes, yet they cause a complete divergence of activity from a deaminase (kcat/Km = 20810 M–1 s–1), TriA, with residual dechlorinase activity (kcat/Km = 60 M–1 s–1), to a dechlorinase, AtzA (14600 M–1 s–1), with no detectable deaminase activity.[34] To understand the evolutionary divergence between TriA and AtzA, we must understand the different leaving group requirements of the respective substrates (Fig. 2). The herbicide atrazine, which is hydrolysed by AtzA, has an electron-withdrawing Cl– leaving group that is stable as an anion (pKa –7), whereas the melamine substrate, which is hydrolysed by TriA, has an electron-donating NH2– leaving group that will require protonation for the reaction to proceed (pKa 34).[34] These distinct leaving group requirements demand differences in the catalytic dyad used in leaving group stabilisation. TriA has an ionisable cysteine residue that can donate a proton to the NH2– leaving group, which is stabilised by an adjacent aspartate that can act as a general acid. In contrast, the developing negative charge on the Cl– leaving group of atrazine is stabilised by a serine hydroxyl in the transition state, but no proton transfer is necessary, and the serine residue is stabilised by an adjacent asparagine residue (Fig. 2). Changing the cysteine and aspartate observed in TriA to the serine and asparagine observed in AtzA completely changes the activity and reactivity of the enzyme to match that of AtzA, via the direct modification of active site residues. The evolution of AtzA from TriA is uncommon, as many more mutations are usually observed in natural evolutionary pathways of enzymes towards a novel function, owing to both neutral drift and also the accumulation of stabilising mutations to compensate for the frequently destabilising effect of mutations.[89–91]
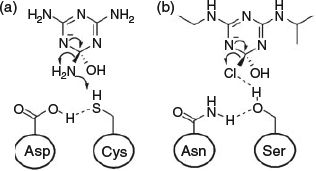
|
Metal Ion Rearrangements Can Initiate Catalytic Divergence
As previously described, the coordination of metal ions involves both directly ligating residues and the entire enzyme scaffold to support this interaction. Therefore, when classifying metalloenzyme superfamilies, metal ion coordination motifs are often used, as they are crucial to the structure and a useful metric to separate large groups of enzymes into functional classes.[11–13,92] Consequently, it is expected that changes in metal ion binding motifs-classified enzyme superfamilies are frequently associated with the evolution of new function. This has been shown to be the case with the molinate (MolA) and phenylurea (PuhB, Mycobacterium brisbanense JK1) hydrolases: these herbicide-degrading enzymes belong to the amidohydrolase superfamily and exhibit unusual metal ion coordination modes (Table 2, Fig. 1).[10,93] The classic members of this family, which have evolved for ancient functions, such as dipeptidases, have binuclear active sites, commonly with tetrahedral or trigonal bipyramidal coordination geometries.[94–96] PuhB and MolA bear the closest structural similarity to binuclear carboxypeptidase enzymes but have lost one of the metal ion binding sites.[10] Using phylogenetic, structural, and kinetic analysis, it was shown that, in PuhB and MolA, the loss of the second metal binding site in the evolution of these herbicide-degrading enzymes facilitated the development of a completely different catalytic mechanism.[10] Loss of the second metal binding site through a conserved mutation prevents the post-translational carboxylation of a lysine residue, as two metal ions are required to stabilise the carboxylate group in binuclear enzymes in this superfamily (Fig. 1).[95,97] When the lysine residue is no longer carboxylated, it is free to coordinate to a water molecule that has an essential role protonating the electron-donating leaving groups of the xenobiotic substrates (Table 1). This protonation is not essential for the dipeptide substrates of the binuclear carboxypeptidase ancestors. Thus, in the evolutionary trajectory of the molinate and diuron hydrolases, rearrangement of the metal ion binding site facilitated a change in mechanism to enable the catalysis of herbicide compounds with different catalytic requirements.
Alternative binding modes of catalytic metal ions are known to occur during catalysis and on substrate binding, and to be associated with promiscuous activity.[7,98–100] The flexibility of metal ion coordination in a B1 MβL (Bacillus cereus), through the variable positioning of metal ion-coordinating ligands on flexible loop regions and the use of coordination state-flexible zinc ions, is thought to be crucial for its catalysis of diverse substrates and antibiotics.[100] Rapid-freeze–quench double–electron electron resonance spectroscopy was used to analyse loop movements in the New Delhi metallo-β-lactamase (NDM, Klebsiella pneumoniae).[101] It was shown that loop movements occur during catalysis, and it has previously been shown that there are metal–metal distance changes between resting, intermediate-bound, and product-bound states; it seems possible that these two factors are linked.[102–104] It is thought that the flexibility in coordination states observed in MβL enables hydrolysis with different compounds, but at the cost of efficient hydrolysis.[100] The promiscuous reactions that different MβLs catalyse generally have lower kcat, where it is hypothesised that promiscuous substrates may not be well positioned relative to the nucleophilic hydroxide for catalysis.[105] Flexibility in metal ion coordination in MβLs enables catalysis of diverse compounds, albeit at a reduced efficiency, but this promiscuity would improve survival of host organisms challenged with new xenobiotic substrates, and provide a starting point for improvement over generations.
In the lactonase PON1, which can be derived from different sources,[106,107] the catalytic calcium ion is most frequently coordinated by three asparagine residues, a glutamate residue, and an aspartate residue.[99] However, a secondary low-metal-ion-occupancy binding mode is also possible, where the asparagine coordinating ligands coordinate to a water molecule absent in the most common binding mode, and exhibit different orientations.[99] When PON1 was subjected to directed evolution to enhance its promiscuous paraoxonase activity, the variants had higher metal ion occupancy in the alternative metal binding site.[99,108] This alternative binding site, 1.8 Å away from the native site, is postulated to be the initiator of divergence in activity between lactonase and paraoxonase enzymes. The mutation of a histidine residue in the directed evolution of PON1 is thought to change the calcium ion binding position, optimising the ligating residues for paraoxon hydrolysis. The catalytic role that the histidine residue has in activating the nucleophilic water molecule for attack of the original lactone substrates is also prevented.[99,108] It seems likely that the switching between native lactonase hydrolysis and promiscuous organophosphate hydrolysis in PON1 is mediated by use of alternative binding modes.[99] Alternative binding modes are also observed when metalloenzymes are bound to different substrates;[98,109,110] optimising the best metal ion binding mode to accommodate a new xenobiotic substrate may be an evolutionary strategy used in the evolution of metalloenzymes.
Metal ion coordination changes were also observed in the directed evolution of a xenobiotic-hydrolysing PTE towards arylesterase function, where a novel metal site was found after the last round of evolution.[111] Prior to the final round of evolution, metal coordination was the same as observed in wild-type PTE, whereas the enzyme from the eighteenth round exhibited an ensemble of metal coordination modes. One conformer exhibits the same metal coordination sphere as in the wild type, with two metal ions bridged by a carboxylated lysine. However, an alternative conformation has only one metal ion positioned in a novel site not occupied in the wild-type binding mode. Changes to the distance between the two metal ions in PTE have also been observed in directed evolution experiments; the 0.5-Å increase in distance is optimal for the hydrolysis of paraoxon.[112] Conformational dynamics through long-range interactions are a potential explanation for this change in metal ion positioning,[113] which may also explain the increase in possible metal ion coordination modes in the round 18 PTE variant.[111] Changes in the conformational dynamics of metalloenzymes along their evolutionary trajectories may improve their evolvability through increasing the number of potential metal coordination modes.
Reorganisation of metal ions in the active site, or solidifying alternative binding modes and associated promiscuous activities both appear to be important mechanisms of evolution towards xenobiotic hydrolysis in metalloenzymes. The ability to repurpose existing catalytic infrastructure for new or improved functionality is an efficient strategy from an evolutionary standpoint, and it may explain the many overlapping catalytic activities observed in metalloenzyme superfamilies.[105]
Instability of Apo-Enzyme States Influences the Evolvability of Metalloenzymes
In addition to their catalytic roles within metalloenzymes, metal ions also often contribute to the stability and the correct folding of metalloenzymes.[114–117] Metal ions have the potential to associate at various stages of the folding pathway: they can associate with the unfolded protein, with partially folded intermediate states, or with the folded apo-form of the enzyme.[118–121] It has been shown that the apo-form of a metalloenzyme is more unstable than the bound form,[122,123] where the addition of native metal ions increases stability.[75,124] For example, the addition of cobalt or zinc to carbonic anhydrase refolds the molten globule state; it has been proposed that metal ions may associate with a folding intermediate to prevent aggregation pathways for this enzyme.[119,125] Furthermore, different metal ions have been shown to contribute differently to the stability of bovine carbonic anhydrase, highlighting the specificity with which metal ions contribute to stability and folding pathways.[126] The involvement of metal ions in protein folding makes the modelling of these processes more complex, as there can be cooperation between the coordination landscape and the folding landscape.[127,128] The binding of a charged metal ion can influence folding dynamics via the deprotonation of amino acids, polarisation of the microenvironment and resultant changes to electrostatics and coordination networks, and coupling between metal binding and the global motions the polypeptide.[127] The diverse contributions of metal ion binding to protein folding indicate that modifications to the scaffold that influence these contributions could also change the folding landscape, and this may impact the evolvability of metalloenzymes.
In the directed evolution of xenobiotic-degrading metalloenzymes towards improved catalysis, it has been observed that improvement of apoenzyme stability also results in an increase in soluble expression.[114,116] In bacteria, there is a trade-off between the advantage of having unbound metal ions available to bind to proteins and the toxicity of excess metal ions.[50,129] Therefore, the stabilisation of the apo-form of folding intermediates, or the folded apo-protein itself could enable more metalloenzymes to accumulate in low metal ion concentrations, which would be an evolutionary advantage for the host system. In the directed evolution of triazine hydrolase (TrzN, Arthrobacter aurescens) towards enhanced hydrolysis of triazine-based herbicides, mutations that stabilised the apo-form of TrzN but had no effect on the holo-form were identified.[114] The mutations had a structural impact on TrzN; the more stabilised TrzN variant (G3) exhibited a novel loop conformation in the apo-form, where loop two collapses into the active site cavity.[114] This alternative loop conformation was not observed in any TrzN structures that had complete metal ion occupation in the active site.[114] The recruitment of an alternative loop conformation to perform this role in the folding pathway of TrzN has a sound basis, as cavity-filling mutations are known to stabilise protein structures.[130–132] Stabilising the apo-form of TrzN resulted in 300-fold improvement in soluble expression; this indicates the bottleneck that instable folding intermediates can form towards soluble protein expression. In the directed evolution of a PTE towards enhanced solubility, it was observed that the evolved variant with the highest expression (S5) actually had lower thermostability, and a poorer affinity for zinc.[116] However, despite these disadvantages, it was determined that S5 can maintain its apo-form, which allows its accumulation, whereas the wild-type PTE cannot.[116] The instability of the apo-forms of metalloenzymes is likely to act as a bottleneck towards their high expression intracellularly. This is important to consider in the evolution of xenobiotic-hydrolysing enzymes, as mutations that will impact on the correct folding of the metalloenzyme, interfering with the intertwined coordination, and folding landscapes would not be favoured, restricting evolutionary paths.
Loop Rearrangements and Flexibility Promote Novel Function
In many metal ion-dependent enzyme superfamilies, recently diverged members will exhibit superimposable core protein folds and metal ion binding sites, despite reasonably low levels (<30 %) of amino acid sequence identity to other members.[133–135] However, loop regions are often highly variable, even between the most similar groups within superfamilies.[11,136] Loop insertions and deletions and concentrated sequence changes in loops are observed within enzyme superfamilies, where conformational sampling by loops and direct interactions between the loop and the rest of the structure can be modified.[137] Several articles describe loop modifications as being one of the most powerful evolutionary tools towards new enzymatic function, as the core fold, which is essential for the structural integrity of the enzyme, and the active site, which provides the catalytic ‘power’, remain fairly invariant, but substrate recognition and interactions can drastically change.[33,138–140] As loop regions are generally not tethered to the enzyme by extensive intra-protein amino acid interactions, they can adopt a greater range of conformations and mutations are less likely to perturb the protein fold.[141] There is often an accumulation of mutations in the loop regions in xenobiotic-degrading enzymes correlated with the new function. This has been observed extensively in the natural, designed, and directed evolution towards enhanced organophosphate hydrolysis in proteins from several folds.[33,111,112,142–146]
In the evolution of PTE from the amidohydrolase superfamily, its latent lactonase activity and the promiscuity of PTE-like lactonases (PLLs) from different sources with organophosphate compounds indicated a likely evolutionary trajectory of PTE from PLLs, despite low sequence similarity (<30 %).[147–149] When analysing the sequence differences between PTEs and PLLs, the majority of the insertions, deletions and amino acid substitutions are located in the loop regions (Fig. 3).[33] Remodelling an active-site loop in Pseudomonas diminuta PTE to resemble that of the equivalent Sulfolobus solfataricus PLL loop, combined with an essential epistatic mutation, resulted in quorum-sensing homoserine lactonase activity that was not previously detectable.[33] Further work has identified how essential the flexibility of the loops in PLL is to facilitate the promiscuous PTE activity.[144,149] In accordance with this, it has also been identified that loop conformations and flexibility are tuned through both directed and natural evolution.[142,145] Conformational substates occur throughout an enzyme-catalysed reaction, as product binding and release may require a more ‘open’ substate, and the chemical reaction itself will often require a ‘closed’ preorganised substate, as is observed in PTE.[145] Improving the transitions between these substates, optimising the conformational landscape, can result in an improved kcat and therefore through evolution, there are often concentrated sequence changes associated with regions that influence the dynamics of these areas.[111,112,145]
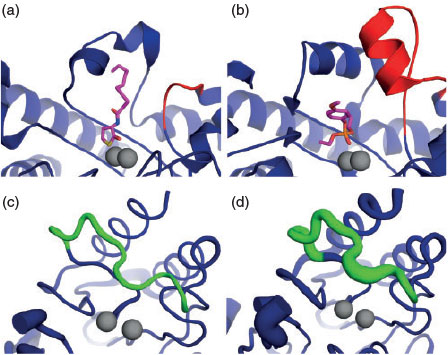
|
Active site loop flexibility has also been identified as contributing to the concerning substrate promiscuity of different MβLs towards almost all β-lactam antibiotics.[100,150,151] Computational modelling of a loop located above the active site of an MβL showed that this loop can interact with three different antibiotics in distinct conformations, allowing the enzyme to bind to each antibiotic with significant affinity.[151] The importance of active-site loop plasticity was also identified the directed evolution of the Bacillus cereus MβL, BcII, to improve activity with a reasonably poor substrate, cephalexin,[152] where it was recognised that increased active-site loop flexibility contributed to improved catalysis (1.97 × 105 M–1 s–1 kcat/Km, compared with the initial 2.65 × 104 M–1 s–1) (Fig. 3).[152,153] In summary, the frequent association of active-site loop modification with the evolution of xenobiotic hydrolysis indicates the importance of loop flexibility as a tuneable factor in the evolution of metalloenzymes.
Engineering Metalloenzymes for Novel Functions Using Lessons from Natural Evolution
Extensive fundamental work has examined how metalloenzymes function and evolve new activity and this basic knowledge has been applied to the artificial design of metalloenzymes with novel functions.[154,155] Two aspects are often separately optimised in metalloenzyme engineering, the inorganic complex and coordinating residues best suited for the desired reaction, and the entire enzyme scaffold to confer enantioselectivity and protect unstable intermediates (Fig. 4).[156–162] As previously described, in natural evolution, the use of alternative metal ions can greatly change the specific activity of a metalloenzyme with different substrates. As the use of bioavailable metal ions is not a concern in the artificial design of metalloenzymes, many unique chemical reactions can take place through the use of precious metal ions instead, opening up the potential to harness the vast catalytic scope of transition metals in the design of novel enzymatic catalysts.

|
There are essentially two routes available for engineering novel metalloenzyme function: engineering an existing metalloenzyme towards novel function and de novo creation of an artificial metalloenzyme with novel function; both of these draw on current understanding about the natural evolution of metalloenzymes. Creation of the first ever cofactor-independent reductase is an example of the first approach. Rhodium, used commonly in chemical synthesis as a complex catalyst for hydrogenation reactions, was incorporated into carbonic anhydrase.[163,164] As an analogous example, but using the second approach, Pecoraro and colleagues successfully developed novel carbonic anhydrase mimics by incorporation of metal-binding centres into de novo designed protein folds, demonstrating highly efficient catalysts with both a supercoiled coil structure (SCCS) and a single-stranded antiparallel three helix bundle (a3DH3).[165,166] Drawing inspiration from natural di-metal proteins, and applying a reductionist approach also resulted in the creation of the de novo engineered ‘Due Ferri’ proteins, such as DF1, a four-helical bundle di-iron phenol oxidase enzyme.[167] As another example of de novo design, the use of a covalently linked di-rhodium catalyst within an optimised prolyl oligopeptidase resulted in an artificial metalloenzyme able to catalyse an olefin cyclopropanation.[168] A similar result was achieved in the artificial design of a copper binding site in a thermostable protein scaffold. Knowledge of both the requirements of ligating residues in the protein scaffold and the potential of copper to catalyse Diels–Alder reactions were used to create this novel metalloenzyme.[169,170] Information taken from nature, of the ligating residues required for certain metal ions and the benefits of using the correct metal ion for the desired reaction have all been applied in the design of these artificial enzymes. However, without optimising the enzyme scaffold further, the turnover rates of these artificial metalloenzymes are inherently limited.
There is great variability in the methods used and the level of optimisation performed on protein scaffolds in artificial metalloenzyme design. If the scaffold is not extensively optimised, the affinity of the metal ion complex for the protein and the ability of the protein to shield intermediates from solvent and prevent side reactions can still lead to reasonable turnover rates.[51,171] However, extensive rational design, computational enzyme design, and rounds of directed evolution are routinely employed to improve the substrate and metal ion affinity and overall turnover rate in artificial metalloenzymes.[172–175] Just as in natural metalloenzyme evolution towards xenobiotic hydrolysis, structural optimisation will lead to improved specificity and turnover rates. To improve the activity of a transfer hydrogenase, it was computationally optimised.[173] A mutation on a loop region creates a new hydrogen bond, rigidifying a loop that is likely to help orientate the bulky iridium cofactor into a more stable orientation for improved catalysis. The importance of having a cofactor in a stable conformation for enhanced catalysis was also shown for a rhodium-containing catalyst that catalyses the polymerisation of phenylacetylene using molecular dynamic simulations.[175] The rationally designed variant with the highest trans selectivity was observed to have the most defined orientation within the binding cavity in the simulations. Residues that make up the second coordination sphere are known to be important for tuning the specificity of naturally evolved metalloenzymes; this has been incorporated in the rational improvement of an artificial transfer hydrogenase where two mutations in the second coordination sphere are thought to improve interactions with the dialkyl ketone substrates of this artificial enzyme.[176] In the design and improvement of artificial metalloenzymes, there is extensive overlap in the molecular mechanisms observed compared with those seen in the natural evolution of metalloenzymes. Although there have been great strides made in the design strategies to match the activity of natural enzymes, there is still very limited diversity in the successful cases when compared with nature.
Conclusions
In the molecular evolution of xenobiotic-degrading metalloenzymes, several different mechanisms have enabled the tuning of catalysis towards xenobiotic hydrolysis (Fig. 5). Factors that enhance the evolvability of the respective enzymes have been utilised to promote novel function, such as flexibility in metal ion incorporation, loop conformational sampling, and the use of alternative metal ion binding modes. More drastic changes to active site residues, direct metal coordinating residues, and the insertion or deletion and extensive modification of loop regions have been shown to significantly change activity along evolutionary trajectories to xenobiotic hydrolysis. Analysing the different molecular mechanisms by which metalloenzymes have evolved to break down xenobiotic compounds is important for the fundamental understanding of enzyme catalysis and evolution. The most comprehensive way to test this understanding is to artificially design metalloenzymes with desired functions; more work needs to be done to routinely artificially design enzyme catalysts that can match the specificity and efficiency of naturally evolved xenobiotic-hydrolysing metalloenzymes.

|
References
[1] K. J. Waldron, J. C. Rutherford, D. Ford, N. J. Robinson, Nature 2009, 460, 823.| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXpvFCruro%3D&md5=2d049972c1512bf28680582b00ec62d1CAS | 19675642PubMed |
[2] A. Cvetkovic, A. L. Menon, M. P. Thorgersen, J. W. Scott, F. L. Poole, F. E. Jenney, W. A. Lancaster, J. L. Praissman, S. Shanmukh, B. J. Vaccaro, Nature 2010, 466, 779.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXovFCns7s%3D&md5=7e2bfe616497ea933c96f826dfc0cfa9CAS | 20639861PubMed |
[3] A. J. Thomson, H. B. Gray, Curr. Opin. Chem. Biol. 1998, 2, 155.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXjs1Olt70%3D&md5=0193b6198d38341a4657c9e93d825b37CAS | 9667942PubMed |
[4] Y. Lu, S. M. Berry, T. D. Pfister, Chem. Rev. 2001, 101, 3047.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXntlGntbk%3D&md5=9c2a94f860ae4cc4fdbc3e600083e848CAS | 11710062PubMed |
[5] J. Reedijk, E. Bouwman, Bioinorganic Catalysis 1999 (CRC Press: Boca Raton, FL).
[6] A. N. Bigley, F. M. Raushel, Biochim. Biophys. Acta, Proteins Proteomics 2013, 1834, 443.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XnslOjur4%3D&md5=73e4981a22e6fcbbf6b6782a13864738CAS |
[7] K. S. Hadler, E. A. Tanifum, S. H.-C. Yip, N. Mitic, L. W. Guddat, C. J. Jackson, L. R. Gahan, K. Nguyen, P. D. Carr, D. L. Ollis, J. Am. Chem. Soc. 2008, 130, 14129.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXht1Sjt7vE&md5=a9a5fdd6aeedccf93030709bbdec2a12CAS | 18831553PubMed |
[8] H. Feng, J. Ding, D. Zhu, X. Liu, X. Xu, Y. Zhang, S. Zang, D.-C. Wang, W. Liu, J. Am. Chem. Soc. 2014, 136, 14694.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXhs1CnsrbK&md5=1770942ca36136de16f613820b511ab7CAS | 25268575PubMed |
[9] J. L. Seffernick, E. Reynolds, A. A. Fedorov, E. Fedorov, S. C. Almo, M. J. Sadowsky, L. P. Wackett, J. Biol. Chem. 2010, 285, 30606.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXht1SlsrrE&md5=2d7db9c8b7bef75710b425f4cbe664a9CAS | 20659898PubMed |
[10] E. Sugrue, N. J. Fraser, D. H. Hopkins, P. D. Carr, J. L. Khurana, J. G. Oakeshott, C. Scott, C. J. Jackson, Appl. Environ. Microbiol. 2015, 81, 2612.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXkvV2htLw%3D&md5=144c2961a9327da21fe7a0ee9d6df45bCAS | 25636851PubMed |
[11] C. M. Seibert, F. M. Raushel, Biochemistry 2005, 44, 6383.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXjtVOjsrk%3D&md5=d8d8145afd302ad9d4b46046e1bb7d69CAS | 15850372PubMed |
[12] C. Bebrone, Biochem. Pharmacol. 2007, 74, 1686.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhtlWrt7vE&md5=24cbfab5b8f33ccf29cc4d78428876e3CAS | 17597585PubMed |
[13] G. Schenk, N. Mitic, L. R. Gahan, D. L. Ollis, R. P. McGeary, L. W. Guddat, Acc. Chem. Res. 2012, 45, 1593.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XosFemtL4%3D&md5=7f6fd4cdae3568c9f165bedbaf66608bCAS | 22698580PubMed |
[14] M. F. Mabanglo, D. F. Xiang, A. N. Bigley, F. M. Raushel, Biochemistry 2016, 55, 3963.
| 1:CAS:528:DC%2BC28XhtVOisL3N&md5=c79f6698c92ac3a9919b536ffb1fb2b0CAS | 27353520PubMed |
[15] C. J. Jackson, P. D. Carr, H.-K. Kim, J.-W. Liu, P. Herrald, N. Mitić, G. Schenk, C. A. Smith, D. L. Ollis, Biochem. J. 2006, 397, 501.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XmvFaku78%3D&md5=25ce699e66e53a285094eeeb1878bf3fCAS | 16686603PubMed |
[16] L. J. Krutz, D. L. Shaner, C. Accinelli, R. M. Zablotowicz, W. B. Henry, J. Environ. Qual. 2008, 37, 848.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXmtlahtrc%3D&md5=8dac8fa77e4148629e9fb16ccb23a3f2CAS | 18453406PubMed |
[17] L. Barreiros, B. Nogales, C. M. Manaia, A. C. S. Ferreira, D. H. Pieper, M. A. Reis, O. C. Nunes, Environ. Microbiol. 2003, 5, 944.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXos1Cltb8%3D&md5=41b2c2a10b94764fc568749b068cb0dfCAS | 14510848PubMed |
[18] I. Horne, T. D. Sutherland, R. L. Harcourt, R. J. Russell, J. G. Oakeshott, Appl. Environ. Microbiol. 2002, 68, 3371.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38Xlt1Snt7g%3D&md5=188c7a4a1683ad308032eaafe520a60eCAS | 12089017PubMed |
[19] J. E. Cullington, A. Walker, Soil Biol. Biochem. 1999, 31, 677.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXjs1Citr8%3D&md5=c08e86dcf4c92ab8ca90b09112be21fcCAS |
[20] J. L. Seffernick, L. P. Wackett, Biochemistry 2001, 40, 12747.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXntF2itb8%3D&md5=60d6f1d8dad0616b9d8ade282681f6d2CAS | 11669610PubMed |
[21] B. K. Singh, A. Walker, J. A. W. Morgan, D. J. Wright, Appl. Environ. Microbiol. 2004, 70, 4855.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXms1eksr0%3D&md5=bd5ef653d9879f7c212efa6643a82399CAS | 15294824PubMed |
[22] L. Smith-Grenier, A. Adkins, Can. J. Microbiol. 1996, 42, 221.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK28XhvVGjtr4%3D&md5=5dd4c7ed998eaf2782aa782c36247ad6CAS |
[23] V. Tett, A. Willetts, H. Lappin-Scott, FEMS Microbiol. Ecol. 1994, 14, 191.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2cXlt1OnsLo%3D&md5=1555dbd19f4251aab19463822361aa08CAS |
[24] C. Yanze-Kontchou, N. Gschwind, Appl. Environ. Microbiol. 1994, 60, 4297.
| 1:CAS:528:DyaK2MXitlygur8%3D&md5=f59dc940b2d90c5ca088ded0daae7a0eCAS | 7811069PubMed |
[25] W. Dejonghe, E. Berteloot, J. Goris, N. Boon, K. Crul, S. Maertens, M. Höfte, P. De Vos, W. Verstraete, E. M. Top, Appl. Environ. Microbiol. 2003, 69, 1532.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXitlCls7s%3D&md5=f59905bfbd694f78054d8c018b4bd24dCAS | 12620840PubMed |
[26] C. van Ginkel, Biodegradation 1996, 7, 151.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK28XjtlSgtLs%3D&md5=9910c1563ba5a30ca280229a8728b84eCAS | 8882807PubMed |
[27] R. Cantón, T. M. Coque, Curr. Opin. Microbiol. 2006, 9, 466.
| Crossref | GoogleScholarGoogle Scholar | 16942899PubMed |
[28] M. Watanabe, S. Iyobe, M. Inoue, S. Mitsuhashi, Antimicrob. Agents Chemother. 1991, 35, 147.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3MXlvVOrtg%3D%3D&md5=808d074800f2f1a51e98d6802fd2f1b5CAS | 1901695PubMed |
[29] E. Meyer, P. Gastmeier, M. Deja, F. Schwab, Int. J. Med. Microbiol. 2013, 303, 388.
| Crossref | GoogleScholarGoogle Scholar | 23727396PubMed |
[30] H. M. LeBaron, J. Mc Farland, O. Burnside, The Triazine Herbicides 2011 (Elsevier: Amsterdam).
[31] M. Wenk, T. Baumgartner, J. Dobovšek, T. Fuchs, J. Kucsera, J. Zopfi, G. Stucki, Appl. Microbiol. Biotechnol. 1998, 49, 624.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXkt1Klt7g%3D&md5=9f08d3763bfbf71877b953383f83ef35CAS | 9650261PubMed |
[32] C. Scott, C. J. Jackson, C. W. Coppin, R. G. Mourant, M. E. Hilton, T. D. Sutherland, R. J. Russell, J. G. Oakeshott, Appl. Environ. Microbiol. 2009, 75, 2184.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXksFWltb4%3D&md5=3324b2e9dce3ae718749f913b9fddc71CAS | 19201959PubMed |
[33] L. Afriat-Jurnou, C. J. Jackson, D. S. Tawfik, Biochemistry 2012, 51, 6047.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhtVGhs77I&md5=79762eb93c2c5ef86939ff022d18d5e1CAS | 22809311PubMed |
[34] S. Noor, M. C. Taylor, R. J. Russell, L. S. Jermiin, C. J. Jackson, J. G. Oakeshott, C. Scott, PLoS One 2012, 7, e39822.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XpvVCnsrc%3D&md5=270b3da1e37a9a0f4bbcd6f97dda7d46CAS | 22768133PubMed |
[35] R. H. Holm, P. Kennepohl, E. I. Solomon, Chem. Rev. 1996, 96, 2239.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK28Xmt1Gnu7o%3D&md5=34767a75ec553f1cdf97860c47e33da9CAS | 11848828PubMed |
[36] D. S. Auld, in Zinc Biochemistry, Physiology, and Homeostasis (Ed. W. Maret) 2001, pp. 85–127 (Springer: Dordrecht).
[37] Z. Ma, F. E. Jacobsen, D. P. Giedroc, Chem. Rev. 2009, 109, 4644.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtF2qtLzN&md5=36dd37208627157a7d42ecf6553f846bCAS | 19788177PubMed |
[38] K. J. Waldron, N. J. Robinson, Nat. Rev. Microbiol. 2009, 7, 25.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhsFWitrfI&md5=975887e75703543cd4ec1a3642615448CAS | 19079350PubMed |
[39] T. Dudev, Y.-l. Lin, M. Dudev, C. Lim, J. Am. Chem. Soc. 2003, 125, 3168.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXht1Sgt7w%3D&md5=889d545871e8c877fd637edcb4bb95d7CAS | 12617685PubMed |
[40] R. Jernigan, G. Raghunathan, I. Bahar, Curr. Opin. Struct. Biol. 1994, 4, 256.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2cXmslCnurc%3D&md5=b512fd630cc431619b08bf0e73672490CAS |
[41] R. G. Pearson, J. Am. Chem. Soc. 1963, 85, 3533.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaF2cXksV0%3D&md5=e5e42dec9101100828b79122edf9d296CAS |
[42] J.-L. Foo, C. J. Jackson, P. D. Carr, H.-K. Kim, G. Schenk, L. R. Gahan, D. L. Ollis, Biochem. J. 2010, 429, 313.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXotVajsL0%3D&md5=22d8996bcd985ca366ebb70d074be274CAS | 20459397PubMed |
[43] R. Levy, V. Sobolev, M. Edelman, Hum. Mutat. 2011, 32, 1309.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtlagurvK&md5=28850e50031dc50b632b3b6d8093e5f9CAS | 21898656PubMed |
[44] J. D. Cox, J. A. Hunt, K. M. Compher, C. A. Fierke, D. W. Christianson, Biochemistry 2000, 39, 13687.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXntFWjt7s%3D&md5=c984cadf84afcf7949cc89999e2098b5CAS | 11076507PubMed |
[45] N. Mitic, K. S. Hadler, L. R. Gahan, A. C. Hengge, G. Schenk, J. Am. Chem. Soc. 2010, 132, 7049.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXlsVeju7w%3D&md5=198f9665857db95011b174d5f367f608CAS | 20433174PubMed |
[46] M. M. Pedroso, J. A. Larrabee, F. Ely, S. E. Gwee, N. Mitić, D. L. Ollis, L. R. Gahan, G. Schenk, Chem. – Eur. J. 2016, 22, 999.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXitVWru73N&md5=ef4634e55cf7177d9eda4b055a20bacaCAS | 26662456PubMed |
[47] S. J. Smith, A. Casellato, K. S. Hadler, N. Mitić, M. J. Riley, A. J. Bortoluzzi, B. Szpoganicz, G. Schenk, A. Neves, L. R. Gahan, J. Biol. Inorg. Chem. 2007, 12, 1207.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhtFKrsb3K&md5=e972752b368632b08a972bdde7e15ef9CAS | 17701232PubMed |
[48] N. Laraki, N. Franceschini, G. M. Rossolini, P. Santucci, C. Meunier, E. De Pauw, G. Amicosante, J. M. Frère, M. Galleni, Antimicrob. Agents Chemother. 1999, 43, 902.
| 1:CAS:528:DyaK1MXisVSnsrw%3D&md5=4e74815db9bedea6e7f3dff6541548ffCAS | 10103197PubMed |
[49] A. D. Cameron, M. Ridderström, B. Olin, B. Mannervik, Structure 1999, 7, 1067.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXmtlartL0%3D&md5=d673acf34a66f500a6b8f549dff2aff4CAS | 10508780PubMed |
[50] C. E. Outten, T. V. O’Halloran, Science 2001, 292, 2488.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXkvFWlsrk%3D&md5=0b73521ffb31cb8ba9849806c0fed6baCAS | 11397910PubMed |
[51] A. Fernández‐Gacio, A. Codina, J. Fastrez, O. Riant, P. Soumillion, ChemBioChem 2006, 7, 1013.
| Crossref | GoogleScholarGoogle Scholar | 16688707PubMed |
[52] G. F. da Silva, L.-J. Ming, J. Am. Chem. Soc. 2005, 127, 16380.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXhtFOisLzE&md5=113bb6f9662c921bfa1197c19bbbeabfCAS | 16305209PubMed |
[53] F. Baier, J. Chen, M. Solomonson, N. C. Strynadka, N. Tokuriki, ACS Chem. Biol. 2015, 10, 1684.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXmtV2ltLY%3D&md5=1c275a93160cb56c3af9c9a46f04591fCAS | 25856271PubMed |
[54] S. C. Andrews, A. K. Robinson, F. Rodríguez-Quiñones, FEMS Microbiol. Rev. 2003, 27, 215.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXksVOlt70%3D&md5=0d2373c09f7b81912b3f8c5b3f6e2333CAS | 12829269PubMed |
[55] N. S. Jakubovics, H. F. Jenkinson, Microbiology 2001, 147, 1709.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXlsVClur8%3D&md5=90ab0c96e36bad11d9ddd18030284714CAS | 11429449PubMed |
[56] H. Reyes-Caballero, G. C. Campanello, D. P. Giedroc, Biophys. Chem. 2011, 156, 103.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXmtFeju7w%3D&md5=5807b32a1bf6e8313cf098a483433ac4CAS | 21511390PubMed |
[57] I. J. Schalk, M. Hannauer, A. Braud, Environ. Microbiol. 2011, 13, 2844.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhs1emtbbF&md5=d7e36f44ac13088126621462dc8f7ce9CAS | 21883800PubMed |
[58] B. Montanini, D. Blaudez, S. Jeandroz, D. Sanders, M. Chalot, BMC Genomics 2007, 8, 107.
| Crossref | GoogleScholarGoogle Scholar | 17448255PubMed |
[59] R. J. Russell, C. Scott, C. J. Jackson, R. Pandey, G. Pandey, M. C. Taylor, C. W. Coppin, J. W. Liu, J. G. Oakeshott, Evol. Appl. 2011, 4, 225.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XltFOru7o%3D&md5=78579a24edfb0643d9574e8161661ee6CAS | 25567970PubMed |
[60] B. K. Singh, Nat. Rev. Microbiol. 2009, 7, 156.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhsFajtbbO&md5=abf754397ab20fbc5773967028cd122cCAS | 19098922PubMed |
[61] I. Horne, X. Qiu, R. J. Russell, J. G. Oakeshott, FEMS Microbiol. Lett. 2003, 222, 1.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXjvV2lu7w%3D&md5=8df6374df32d6c5ec4b3c0457d4b8c8aCAS | 12757939PubMed |
[62] D. Siddavattam, S. Khajamohiddin, B. Manavathi, S. B. Pakala, M. Merrick, Appl. Environ. Microbiol. 2003, 69, 2533.
| Crossref | GoogleScholarGoogle Scholar | 12732518PubMed |
[63] C. J. Jackson, K. S. Hadler, P. D. Carr, A. J. Oakley, S. Yip, G. Schenk, D. L. Ollis, Acta Crystallogr. Sect. F: Struct. Biol. Cryst. Commun. 2008, 64, 681.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXptlOqsrY%3D&md5=c7b28e5edca1e789bf43b1ec7c65f748CAS |
[64] C. J. Jackson, P. D. Carr, J.-W. Liu, S. J. Watt, J. L. Beck, D. L. Ollis, J. Mol. Biol. 2007, 367, 1047.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXjtFKisbk%3D&md5=f216497e262eb468732b6b88ef941facCAS | 17306828PubMed |
[65] D. Dempsey, D. Perrin, Buffers for pH and Metal Ion Control 1974 (Chapman and Hall: London).
[66] S. Kouda, M. Ohara, M. Onodera, Y. Fujiue, M. Sasaki, T. Kohara, S. Kashiyama, S. Hayashida, T. Harino, T. Tsuji, J. Antimicrob. Chemother. 2009, 64, dkp142.
[67] T. J. Carruthers, P. D. Carr, C. T. Loh, C. J. Jackson, G. Otting, Angew. Chem. Int. Ed. 2014, 53, 14269.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXitVGlsrzE&md5=043f11e040216dfa1aebf37ead289205CAS |
[68] W. Maret, J. Nutr. 2000, 130, 1455S.
| 1:CAS:528:DC%2BD3cXivFKms7o%3D&md5=34de04cab5a3eac9694cf0e7a3df50cfCAS | 10801959PubMed |
[69] P. Hinsinger, Plant Soil 2001, 237, 173.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XovVWlsQ%3D%3D&md5=99a7d5937d05b2142b7de9b3ada6ca8fCAS |
[70] L. J. Daumann, B. Y. McCarthy, K. S. Hadler, T. P. Murray, L. R. Gahan, J. A. Larrabee, D. L. Ollis, G. Schenk, Biochim. Biophys. Acta, Proteins Proteomics 2013, 1834, 425.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xjt1GrsL8%3D&md5=ece24a16448bd944fb79f5f40303b209CAS |
[71] F. Ely, K. S. Hadler, L. R. Gahan, L. W. Guddat, D. L. Ollis, G. Schenk, Biochem. J. 2010, 432, 565.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsVOkt7rP&md5=2d148263560bfe3679e82c62e5975dddCAS | 20868365PubMed |
[72] C. J. Jackson, J.-L. Foo, H.-K. Kim, P. D. Carr, J.-W. Liu, G. Salem, D. L. Ollis, J. Mol. Biol. 2008, 375, 1189.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXlsFOmsA%3D%3D&md5=9e600194d0707232bf9bdb176481c036CAS | 18082180PubMed |
[73] C. Jackson, H.-K. Kim, P. D. Carr, J.-W. Liu, D. L. Ollis, Biochim. Biophys. Acta, Proteins Proteomics 2005, 1752, 56.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXptVWnu78%3D&md5=ba29259cdeb062da068472c5b8c15f36CAS |
[74] G. A. Omburo, J. M. Kuo, L. S. Mullins, F. Raushel, J. Biol. Chem. 1992, 267, 13278.
| 1:CAS:528:DyaK38XltVOht7s%3D&md5=a1960b9ac66c685b6a17749f4611817aCAS | 1320014PubMed |
[75] D. Rochu, N. Viguie, F. Renault, D. Crouzier, M.-T. Froment, P. Masson, Biochem. J. 2004, 380, 627.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXltVahurc%3D&md5=625ac6f0b127f34f8634d00568435d3cCAS | 15018612PubMed |
[76] R. Eisenthal, M. J. Danson, D. W. Hough, Trends Biotechnol. 2007, 25, 247.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXlsFGrtrY%3D&md5=4bb95a9957066c1b8fa8fd28ed65eefeCAS | 17433847PubMed |
[77] C. J. Jackson, J.-W. Liu, M. L. Coote, D. L. Ollis, Org. Biomol. Chem. 2005, 3, 4343.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXht1OisrfP&md5=6bbb9d2163920a140515303dd35e49a2CAS | 16327895PubMed |
[78] L. Sillén, A. Martell, Stability Constants of Metal Ion Complexes: Supplement 1. Special Publication No. 25 1971 (Royal Society of Chemistry: London).
[79] D. W. Christianson, Prog. Biophys. Mol. Biol. 1997, 67, 217.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXjvFOiuw%3D%3D&md5=239ad59e376c8ab6e408420a67414149CAS | 9446936PubMed |
[80] M. M. Benning, H. Shim, F. M. Raushel, H. M. Holden, Biochemistry 2001, 40, 2712.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXosVGjtQ%3D%3D&md5=c3db3433199eb4c04ba5814b9c799ff1CAS | 11258882PubMed |
[81] L. R. Rulíšek, J. Vondrášek, J. Inorg. Biochem. 1998, 71, 115.
| Crossref | GoogleScholarGoogle Scholar |
[82] B. Xue, J. Y. Chow, A. Baldansuren, L. L. Yap, Y. H. Gan, S. A. Dikanov, R. C. Robinson, W. S. Yew, Biochemistry 2013, 52, 2359.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXjsVymu7k%3D&md5=b964f0f38d940b249bb54d49362ae1d6CAS | 23461395PubMed |
[83] A. Espart, M. Marín, S. Gil-Moreno, O. Palacios, F. Amaro, A. Martín-González, J. C. Gutiérrez, M. Capdevila, S. Atrian, Int. J. Biol. Sci. 2015, 11, 456.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXlsFShs7k%3D&md5=8e2475dcb688e478da7ab17d7610de2bCAS | 25798065PubMed |
[84] B. C. Tripp, C. B. Bell, F. Cruz, C. Krebs, J. G. Ferry, J. Biol. Chem. 2004, 279, 6683.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXht1Cltb0%3D&md5=0281d22087ce26e6b0a8fb65650acb10CAS | 14662760PubMed |
[85] M. M. He, S. L. Clugston, J. F. Honek, B. W. Matthews, Biochemistry 2000, 39, 8719.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXksVagtbw%3D&md5=ac8a8038d23a7f94c7d1bb4e6c798416CAS | 10913283PubMed |
[86] Ò. Palacios, A. Pagani, S. Pérez-Rafael, M. Egg, M. Höckner, A. Brandstätter, M. Capdevila, S. Atrian, R. Dallinger, BMC Biol. 2011, 9, 4.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsFWitr8%3D&md5=c717268b2fa3bbae73cde7542774ca09CAS | 21255385PubMed |
[87] H. Renata, Z. J. Wang, F. H. Arnold, Angew. Chem. Int. Ed. 2015, 54, 3351.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXjslCiur4%3D&md5=f479d8835db265587e3438566df646b2CAS |
[88] J. L. Seffernick, M. L. de Souza, M. J. Sadowsky, L. P. Wackett, J. Bacteriol. 2001, 183, 2405.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXis1arsbs%3D&md5=a2c43a049cf3f13faefc3ab83c3b9b17CAS | 11274097PubMed |
[89] S. Bershtein, K. Goldin, D. S. Tawfik, J. Mol. Biol. 2008, 379, 1029.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXmsFKqtrc%3D&md5=3049092308f80308e397a550b5a83601CAS | 18495157PubMed |
[90] N. Tokuriki, F. Stricher, L. Serrano, D. S. Tawfik, PLOS Comput. Biol. 2008, 4, e1000002.
| Crossref | GoogleScholarGoogle Scholar | 18463696PubMed |
[91] J. D. Bloom, F. H. Arnold, Proc. Natl. Acad. Sci. USA 2009, 106, 9995.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXotFKnsb8%3D&md5=9af75d9bb67b5e7ed2ac6316404966e3CAS | 19528653PubMed |
[92] A. I. Karsisiotis, C. Damblon, G. C. Roberts, Metallomics 2014, 6, 1181.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXhtVKjsLrN&md5=2d12c00ee016052584f3fa5de7f93a4cCAS | 24696003PubMed |
[93] J. L. Khurana, C. J. Jackson, C. Scott, G. Pandey, I. Horne, R. J. Russell, A. Herlt, C. J. Easton, J. G. Oakeshott, Biochem. J. 2009, 418, 431.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhs1Onsrc%3D&md5=28b4c06b99cdd44ef3e7f5c603ee0336CAS | 19000034PubMed |
[94] D. F. Xiang, Y. Patskovsky, C. Xu, A. J. Meyer, J. M. Sauder, S. K. Burley, S. C. Almo, F. M. Raushel, Biochemistry 2009, 48, 3730.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXkt1Omt7w%3D&md5=510e26d790e3f47b3f32210932d6c037CAS | 19281183PubMed |
[95] D. F. Xiang, Y. Patskovsky, C. Xu, A. A. Fedorov, E. V. Fedorov, A. A. Sisco, J. M. Sauder, S. K. Burley, S. C. Almo, F. M. Raushel, Biochemistry 2010, 49, 6791.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXovFelsbw%3D&md5=07b28e35226cbb35af19f75e16220060CAS | 20604542PubMed |
[96] D. F. Xiang, C. Xu, D. Kumaran, A. C. Brown, J. M. Sauder, S. K. Burley, S. Swaminathan, F. M. Raushel, Biochemistry 2009, 48, 4567.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXlt12mt7w%3D&md5=6f3d3248b7ca2dc9881bdeff655f3769CAS | 19358546PubMed |
[97] H. Shim, F. M. Raushel, Biochemistry 2000, 39, 7357.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXjsFGqu7w%3D&md5=b4fa470e9e70c0bf48f2daaeb3bd1463CAS | 10858282PubMed |
[98] A. Lavie, K. N. Allen, G. A. Petsko, D. Ringe, Biochemistry 1994, 33, 5469.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2cXjtFyhtLc%3D&md5=200f79b2bcf09357c74ebc069c193364CAS | 8180169PubMed |
[99] M. Ben-David, G. Wieczorek, M. Elias, I. Silman, J. L. Sussman, D. S. Tawfik, J. Mol. Biol. 2013, 425, 1028.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXitVGmtbY%3D&md5=255ec2ecca29c431d9247d40df6843c0CAS | 23318950PubMed |
[100] J. M. González, A. Buschiazzo, A. J. Vila, Biochemistry 2010, 49, 7930.
| Crossref | GoogleScholarGoogle Scholar | 20677753PubMed |
[101] M. Aitha, A. J. Moller, I. D. Sahu, M. Horitani, D. L. Tierney, M. W. Crowder, J. Inorg. Biochem. 2016, 156, 35.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXitVyqtLbI&md5=8739146e99ec1b6f9b20bb048f200601CAS | 26717260PubMed |
[102] R. M. Breece, Z. Hu, B. Bennett, M. W. Crowder, D. L. Tierney, J. Am. Chem. Soc. 2009, 131, 11642.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXptlGnsr8%3D&md5=9238435255739d9a49f3fc8fd2b2ddbcCAS | 19653676PubMed |
[103] H. Zhang, Q. Hao, FASEB J. 2011, 25, 2574.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXpvFersrk%3D&md5=f980918fc0e68dfded42cbeb2efdef67CAS | 21507902PubMed |
[104] A. Costello, G. Periyannan, K.-W. Yang, M. W. Crowder, D. L. Tierney, J. Biol. Inorg. Chem. 2006, 11, 351.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XjtFamtrw%3D&md5=6f7f800a49708886476c9a9b28dd0229CAS | 16489411PubMed |
[105] F. Baier, N. Tokuriki, J. Mol. Biol. 2014, 426, 2442.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXns1Crsbw%3D&md5=4068388e0964e93e3216e7c40b9feaa8CAS | 24769192PubMed |
[106] A. Aharoni, L. Gaidukov, S. Yagur, L. Toker, I. Silman, D. S. Tawfik, Proc. Natl. Acad. Sci. USA 2004, 101, 482.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXmsFGgsA%3D%3D&md5=b90ac61c247bf570f7e17d161e06bf98CAS | 14695884PubMed |
[107] R. Shamir, C. Hartman, R. Karry, E. Pavlotzky, R. Eliakim, J. Lachter, A. Suissa, M. Aviram, Free Radic. Biol. Med. 2005, 39, 336.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXlvVWksrs%3D&md5=103ee204f558da1e133a5fed2f139fe5CAS | 15993332PubMed |
[108] M. Goldsmith, Y. Ashani, Y. Simo, M. Ben-David, H. Leader, I. Silman, J. L. Sussman, D. S. Tawfik, Chem. Biol. 2012, 19, 456.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XlvFygtrg%3D&md5=74459747acf58a0cfce7322217e965fdCAS | 22520752PubMed |
[109] M. Sendovski, M. Kanteev, V. S. Ben-Yosef, N. Adir, A. Fishman, J. Mol. Biol. 2011, 405, 227.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsF2itrvL&md5=c1122594c098d2631be25c74ded291edCAS | 21040728PubMed |
[110] H. Yoshida, M. Yamaji, T. Ishii, K. Izumori, S. Kamitori, FEBS J. 2010, 277, 1045.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhvV2jsbo%3D&md5=5e87b007a53679b4314ec183538febbcCAS | 20088877PubMed |
[111] N. Tokuriki, C. J. Jackson, L. Afriat-Jurnou, K. T. Wyganowski, R. Tang, D. S. Tawfik, Nat. Commun. 2012, 3, 1257.
| Crossref | GoogleScholarGoogle Scholar | 23212386PubMed |
[112] M. Kaltenbach, C. J. Jackson, E. C. Campbell, F. Hollfelder, N. Tokuriki, eLife 2015, 4, e06492.
| Crossref | GoogleScholarGoogle Scholar |
[113] C. M. Miton, N. Tokuriki, Protein Sci. 2016, 25, 1260.
| 1:CAS:528:DC%2BC28Xht1Ontr0%3D&md5=bcb0aec8ac989a80608628a1c47ab5c5CAS | 26757214PubMed |
[114] C. J. Jackson, C. W. Coppin, P. D. Carr, A. Aleksandrov, M. Wilding, E. Sugrue, J. Ubels, M. Paks, J. Newman, T. S. Peat, Appl. Environ. Microbiol. 2014, 80, 4003.
| Crossref | GoogleScholarGoogle Scholar | 24771025PubMed |
[115] K. E. R. Duncan, M. J. Stillman, J. Inorg. Biochem. 2006, 100, 2101.
| Crossref | GoogleScholarGoogle Scholar |
[116] C. Roodveldt, D. Tawfik, Protein Eng. Des. Sel. 2005, 18, 51.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXivFWhtrs%3D&md5=87f76abee18e0b187b18e14778382078CAS | 15790580PubMed |
[117] L. Perezgasga, L. Sánchez-Sánchez, S. Aguila, R. Vazquez-Duhalt, Appl. Biochem. Biotechnol. 2012, 166, 1236.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XisFCqu7s%3D&md5=0d7160b5445d952245f38cb04b3ef7a9CAS | 22249853PubMed |
[118] C. J. Wilson, D. Apiyo, P. Wittung-Stafshede, Q. Rev. Biophys. 2004, 37, 285.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXhtVGjsL%2FE&md5=1959ec2963e9af07f373d4da66339287CAS | 16194296PubMed |
[119] D. Andersson, P. Hammarström, U. Carlsson, Biochemistry 2001, 40, 2653.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXosVGjsA%3D%3D&md5=6e9a286def99f88751017ccae120981fCAS | 11258876PubMed |
[120] J. W. Liu, K. S. Hadler, G. Schenk, D. Ollis, FEBS J. 2007, 274, 4742.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhtVyksLrI&md5=e11636cb8ad6b4a911e4243f7d4b491dCAS | 17714507PubMed |
[121] A. E. Proudfoot, L. Goffin, M. A. Payton, T. N. Wells, A. R. Bernard, Biochem. J. 1996, 318, 437.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK28XlvFSmtb4%3D&md5=1feb4f353abdabee9181905250cdc4daCAS | 8809030PubMed |
[122] W. Niu, Q. Shu, Z. Chen, S. Mathews, E. Di Cera, C. Frieden, J. Phys. Chem. B 2010, 114, 16156.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtFSlsr%2FK&md5=05ca855933300ed8b7fb220065587262CAS | 20815357PubMed |
[123] K.-P. Wong, L. M. Hamlin, Arch. Biochem. Biophys. 1975, 170, 12.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE2MXlt1yhsr8%3D&md5=475c94a1e3b26fbac53677a0328656a2CAS | 240319PubMed |
[124] X. L. Xu, J. X. Chen, L. Y. Zhang, X. H. Liu, W. Q. Liu, Q. L. Liu, Biopolymers 2006, 82, 167.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XkvFSmsrs%3D&md5=a62dca30a31f6f5eb7ae5a38bc1fdff7CAS | 16475157PubMed |
[125] K. L. Gudiksen, A. R. Urbach, I. Gitlin, J. Yang, J. A. Vazquez, C. E. Costello, G. M. Whitesides, Anal. Chem. 2004, 76, 7151.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXptlygs7s%3D&md5=2d9a253b0018ae11199b13928eadfe94CAS | 15595855PubMed |
[126] G. P. Lisi, R. P. Hughes, D. E. Wilcox, J. Biol. Inorg. Chem. 2016, 21, 659.
| 1:CAS:528:DC%2BC28XhtVOnsrbP&md5=71a045083f4c93cd7493ecc2f66fe839CAS | 27350155PubMed |
[127] W. Li, J. Wang, J. Zhang, W. Wang, Curr. Opin. Struct. Biol. 2015, 30, 25.
| Crossref | GoogleScholarGoogle Scholar | 25523438PubMed |
[128] W. Li, W. Wang, S. Takada, Proc. Natl. Acad. Sci. USA 2014, 111, 10550.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXhtFCnsrfM&md5=a5dd2cef16ef31b77ccaad9b2c074d0cCAS | 25002491PubMed |
[129] D. Wang, C. A. Fierke, Metallomics 2013, 5, 372.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXltVWqtrg%3D&md5=c92202a1f34c27f3988922cdc36e71efCAS | 23446818PubMed |
[130] M. Saito, H. Kono, H. Morii, H. Uedaira, T. H. Tahirov, K. Ogata, A. Sarai, J. Phys. Chem. B 2000, 104, 3705.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXhslKisLk%3D&md5=7e74d2006d6f517a40bd28d9afdccd1bCAS |
[131] T. Sengupta, Y. Tsutsui, P. L. Wintrode, Biochemistry 2009, 48, 8233.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXpsFaktb8%3D&md5=75328248e275f555990c5639d5600e90CAS | 19624115PubMed |
[132] K. Ishikawa, H. Nakamura, K. Morikawa, S. Kanaya, Biochemistry 1993, 32, 6171.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3sXks1Knu7w%3D&md5=51cc1234b8c6f9c74c5b2848b4aa8d01CAS | 8390295PubMed |
[133] J. A. Gerlt, P. C. Babbitt, Annu. Rev. Biochem. 2001, 70, 209.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXlsVeht70%3D&md5=c32ee46deb9929effc3654a94dbdf7b1CAS | 11395407PubMed |
[134] P. He, G. R. Moran, J. Inorg. Biochem. 2011, 105, 1259.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtFymtrbK&md5=51032bece40548269ad339b4014cbabaCAS | 21820381PubMed |
[135] T. T. Nguyen, S. Brown, A. A. Fedorov, E. V. Fedorov, P. C. Babbitt, S. C. Almo, F. M. Raushel, Biochemistry 2008, 47, 1194.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXitFartw%3D%3D&md5=1378768bf83031f6174fc940e4d354e5CAS | 18171028PubMed |
[136] D. J. Wichelecki, B. M. Balthazor, A. C. Chau, M. W. Vetting, A. A. Fedorov, E. V. Fedorov, T. Lukk, Y. V. Patskovsky, M. B. Stead, B. S. Hillerich, Biochemistry 2014, 53, 2722.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXlsFymsbk%3D&md5=737c705ee822b3d9242ca612df026942CAS | 24697546PubMed |
[137] A. Aharoni, L. Gaidukov, O. Khersonsky, S. M. Gould, C. Roodveldt, D. S. Tawfik, Nat. Genet. 2005, 37, 73.
| 1:CAS:528:DC%2BD2cXhtFChsL%2FE&md5=863dece69975a356747c8b50be6af2f7CAS | 15568024PubMed |
[138] D. S. Tawfik, Science 2006, 311, 475.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XpslegtQ%3D%3D&md5=e0b664a832ccb3ff0b4614ae772ff9dbCAS | 16439649PubMed |
[139] H.-S. Park, S.-H. Nam, J. K. Lee, C. N. Yoon, B. Mannervik, S. J. Benkovic, H.-S. Kim, Science 2006, 311, 535.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XmvVymtQ%3D%3D&md5=db4c37eab4487c7be2681c9aad8203cdCAS | 16439663PubMed |
[140] A. Ochoa-Leyva, F. Barona-Gómez, G. Saab-Rincón, K. Verdel-Aranda, F. Sánchez, X. Soberón, J. Mol. Biol. 2011, 411, 143.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXpt1Oqurc%3D&md5=3345097e5e9aa5ef30137a89a4728fbfCAS | 21635898PubMed |
[141] E. Dellus-Gur, A. Toth-Petroczy, M. Elias, D. S. Tawfik, J. Mol. Biol. 2013, 425, 2609.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXmtV2rsbs%3D&md5=c5a5971ba6c823bbd194d7cb1cc3de97CAS | 23542341PubMed |
[142] H. Yang, P. D. Carr, S. Y. McLoughlin, J.-W. Liu, I. Horne, X. Qiu, C. Jeffries, R. Russell, J. G. Oakeshott, D. Ollis, Protein Eng. 2003, 16, 135.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXislektrs%3D&md5=b4c0be1bce937cd984e680732651a8acCAS | 12676982PubMed |
[143] J. Hiblot, G. Gotthard, E. Chabriere, M. Elias, Sci. Rep. 2012, 2, 779.
| 23139857PubMed |
[144] J. Hiblot, G. Gotthard, M. Elias, E. Chabriere, PLoS One 2013, 8, e75272.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXhsFeku73E&md5=f7a492ed98b00d966c25f4f6565eb65eCAS | 24086491PubMed |
[145] C. J. Jackson, J.-L. Foo, N. Tokuriki, L. Afriat, P. D. Carr, H.-K. Kim, G. Schenk, D. S. Tawfik, D. Ollis, Proc. Natl. Acad. Sci. USA 2009, 106, 21631.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXltlKjsw%3D%3D&md5=5af6eff6432c9241c98b4096f8c6453aCAS | 19966226PubMed |
[146] M. M. Meier, C. Rajendran, C. Malisi, N. G. Fox, C. Xu, S. Schlee, D. P. Barondeau, B. Höcker, R. Sterner, F. M. Raushel, J. Am. Chem. Soc. 2013, 135, 11670.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXhtVKqtLvL&md5=2ad4f7e67e2db9fef9abbaec0c202805CAS | 23837603PubMed |
[147] L. Afriat, C. Roodveldt, G. Manco, D. S. Tawfik, Biochemistry 2006, 45, 13677.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XhtFChu7nP&md5=755e254be9199ffd8d482e81623658cfCAS | 17105187PubMed |
[148] J. Hiblot, G. Gotthard, E. Chabriere, M. Elias, PLoS One 2012, 7, e47028.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhsFOkurnE&md5=76f4e6c20cb49c7988149d51996da314CAS | 23071703PubMed |
[149] J. Hiblot, J. Bzdrenga, C. Champion, E. Chabriere, M. Elias, Sci. Rep. 2015, 5, 8372.
| 25670483PubMed |
[150] M. W. Crowder, J. Spencer, A. J. Vila, Acc. Chem. Res. 2006, 39, 721.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28Xmsl2ksLY%3D&md5=e24d23d008bdd7f71b2e8f8610674224CAS | 17042472PubMed |
[151] C. E. Valdez, M. Sparta, A. N. Alexandrova, J. Chem. Theory Comput. 2013, 9, 730.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhsFClsLjE&md5=9c28b9a666415baa98bf278e7f99428bCAS | 26589068PubMed |
[152] P. E. Tomatis, S. M. Fabiane, F. Simona, P. Carloni, B. J. Sutton, A. J. Vila, Proc. Natl. Acad. Sci. USA 2008, 105, 20605.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXks1GntA%3D%3D&md5=a22ed8bff4176fc355f3966631186d82CAS | 19098096PubMed |
[153] P. E. Tomatis, R. M. Rasia, L. Segovia, A. J. Vila, Proc. Natl. Acad. Sci. USA 2005, 102, 13761.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXhtVOqsb3J&md5=d0601d8c566dda7368c4d02678297754CAS | 16172409PubMed |
[154] M. Dürrenberger, T. R. Ward, Curr. Opin. Chem. Biol. 2014, 19, 99.
| Crossref | GoogleScholarGoogle Scholar | 24608081PubMed |
[155] Y. Lu, N. Yeung, N. Sieracki, N. M. Marshall, Nature 2009, 460, 855.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXpvFCkt7g%3D&md5=93ebc590dae03307c42bc45b000bcf39CAS | 19675646PubMed |
[156] P. J. Deuss, R. den Heeten, W. Laan, P. C. Kamer, Chem. – Eur. J. 2011, 17, 4680.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXksVehtLY%3D&md5=be8be78e5fa183627319b04cb6214efdCAS | 21480401PubMed |
[157] C. Esmieu, M. V. Cherrier, P. Amara, E. Girgenti, C. Marchi‐Delapierre, F. Oddon, M. Iannello, A. Jorge‐Robin, C. Cavazza, S. Ménage, Angew. Chem. Int. Ed. 2013, 52, 3922.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXjtVaqurg%3D&md5=f9674af668e8a33395201d37cc4ecd2dCAS |
[158] M. Creus, T. R. Ward, Org. Biomol. Chem. 2007, 5, 1835.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXmtVyltLw%3D&md5=55fa5d7776305827903588e56d84b91bCAS | 17551630PubMed |
[159] Y. Lu, Curr. Opin. Chem. Biol. 2005, 9, 118.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXivFSmu70%3D&md5=63b831d8b89997b6e291bac55fa808f6CAS | 15811795PubMed |
[160] T. Himiyama, D. F. Sauer, A. Onoda, T. P. Spaniol, J. Okuda, T. Hayashi, J. Inorg. Biochem. 2016, 12, 1314.
[161] A. Pordea, M. Creus, J. Panek, C. Duboc, D. Mathis, M. Novic, T. R. Ward, J. Am. Chem. Soc. 2008, 130, 8085.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXmsVelsL0%3D&md5=5dadaa324622279d6930f623198442a4CAS | 18507383PubMed |
[162] I. N. Ugwumba, K. Ozawa, Z.-Q. Xu, F. Ely, J.-L. Foo, A. J. Herlt, C. Coppin, S. Brown, M. C. Taylor, D. L. Ollis, J. Am. Chem. Soc. 2011, 133, 326.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsFKqurzF&md5=87649860f921151e749ec4b30080475eCAS | 21162578PubMed |
[163] D. A. Evans, M. M. Morrissey, J. Am. Chem. Soc. 1984, 106, 3866.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2cXkslWrs7g%3D&md5=da90c4597e0f79f37f8a3fc9ddf29d30CAS |
[164] Q. Jing, K. Okrasa, R. J. Kazlauskas, Chem. – Eur. J. 2009, 15, 1370.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhslCqsLY%3D&md5=bfb53d39a162ca77e1c216c6a652586dCAS | 19115310PubMed |
[165] J. S. Plegaria, V. L. Pecoraro, Methods Mol. Biol. 2016, 1414, 187.
| Crossref | GoogleScholarGoogle Scholar | 27094292PubMed |
[166] V. M. Cangelosi, A. Deb, J. E. Penner-Hahn, V. L. Pecoraro, Angew. Chem. Int. Ed. 2014, 53, 7900.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXhtVShurnI&md5=83b3a9c26d837f2e73e659e734cd214fCAS |
[167] M. Faiella, C. Andreozzi, R. T. M. de Rosales, V. Pavone, O. Maglio, F. Nastri, W. F. DeGrado, A. Lombardi, Nat. Chem. Biol. 2009, 5, 882.
| 1:CAS:528:DC%2BD1MXhsVWlsbrN&md5=613b31b7e6d435269c3a4451b197c2bbCAS | 19915535PubMed |
[168] P. Srivastava, H. Yang, K. Ellis-Guardiola, J. C. Lewis, Nat. Commun. 2015, 6, 7789.
| 1:CAS:528:DC%2BC2MXhtlWhurrF&md5=8c7c6d03dcb34fd8ebd77b4047b11e6fCAS | 26206238PubMed |
[169] D. A. Evans, D. M. Barnes, J. S. Johnson, T. Lectka, P. von Matt, S. J. Miller, J. A. Murry, R. D. Norcross, E. A. Shaughnessy, K. R. Campos, J. Am. Chem. Soc. 1999, 121, 7582.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXkvVynur8%3D&md5=ab380a187ba18794497e16a66e14fe3eCAS |
[170] J. Podtetenieff, A. Taglieber, E. Bill, E. J. Reijerse, M. T. Reetz, Angew. Chem. 2010, 122, 5277.
| Crossref | GoogleScholarGoogle Scholar |
[171] K. Okrasa, R. J. Kazlauskas, Chem. – Eur. J. 2006, 12, 1587.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XhvFKksbw%3D&md5=0360273fec5538039e247fc3cdda503bCAS | 16416502PubMed |
[172] C. S. Mocny, V. L. Pecoraro, Acc. Chem. Res. 2015, 48, 2388.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXht1Oqs7%2FE&md5=44f97a7d99602ae3fca44dc54791b206CAS | 26237119PubMed |
[173] T. Heinisch, M. Pellizzoni, M. Dürrenberger, C. E. Tinberg, V. Köhler, J. Klehr, D. Häussinger, D. Baker, T. R. Ward, J. Am. Chem. Soc. 2015, 137, 10414.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXht1GmsrrM&md5=b3b9bd84e74a4847c6021d9e79bb7fd0CAS | 26226626PubMed |
[174] V. Köhler, J. Mao, T. Heinisch, A. Pordea, A. Sardo, Y. M. Wilson, L. Knörr, M. Creus, J. C. Prost, T. Schirmer, Angew. Chem. Int. Ed. 2011, 50, 10863.
| Crossref | GoogleScholarGoogle Scholar |
[175] K. Fukumoto, A. Onoda, E. Mizohata, M. Bocola, T. Inoue, U. Schwaneberg, T. Hayashi, ChemCatChem 2014, 6, 1229.
| 1:CAS:528:DC%2BC2cXisFSrsbY%3D&md5=b6e8f31a269efd3db74643107052ccb5CAS |
[176] M. Creus, A. Pordea, T. Rossel, A. Sardo, C. Letondor, A. Ivanova, I. LeTrong, R. E. Stenkamp, T. R. Ward, Angew. Chem. Int. Ed. 2008, 47, 1400.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXisVylsro%3D&md5=93851363c3af6bf07b95f00610de572bCAS |
[177] N. Shapir, C. Pedersen, O. Gil, L. Strong, J. Seffernick, M. J. Sadowsky, L. P. Wackett, J. Bacteriol. 2006, 188, 5859.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XosVehtLs%3D&md5=a348670076bb1a4030c22e3b3aae116eCAS | 16885454PubMed |
[178] L. Lauretti, M. L. Riccio, A. Mazzariol, G. Cornaglia, G. Amicosante, R. Fontana, G. M. Rossolini, Antimicrob. Agents Chemother. 1999, 43, 1584.
| 1:CAS:528:DyaK1MXktlCks74%3D&md5=e631a282672bc9afa41225ed60864794CAS | 10390207PubMed |
[179] D. Yong, M. A. Toleman, C. G. Giske, H. S. Cho, K. Sundman, K. Lee, T. R. Walsh, Antimicrob. Agents Chemother. 2009, 53, 5046.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsF2gs7jK&md5=4547d420e408fc1d9576e754a9b70d4fCAS | 19770275PubMed |
[180] M.-R. Meini, L. I. Llarrull, A. J. Vila, Antibiotics 2014, 3, 285.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXht1GjtLjP&md5=ae7c429d51ab89952ef738ac6104f327CAS | 25364574PubMed |
* Colin Jackson is the winner of the 2015 RACI Rennie Memorial Medal.