

Redox-State Dependent Spectroscopic Properties of Porous Organic Polymers Containing Furan, Thiophene, and Selenophene*
Carol Hua A , Stone Woo A , Aditya Rawal B , Floriana Tuna C , James M. Hook B , David Collison C and Deanna M. D’Alessandro A DA School of Chemistry, The University of Sydney, Sydney, NSW 2006, Australia.
B NMR Facility, Mark Wainwright Analytical Centre, University of New South Wales, Sydney, NSW 2052, Australia.
C School of Chemistry, University of Manchester, Manchester M13 9PL, UK.
D Corresponding author. Email: deanna.dalessandro@sydney.edu.au
Australian Journal of Chemistry 70(11) 1227-1234 https://doi.org/10.1071/CH17335
Submitted: 15 June 2017 Accepted: 27 September 2017 Published: 25 October 2017
Abstract
A series of electroactive triarylamine porous organic polymers (POPs) with furan, thiophene, and selenophene (POP-O, POP-S, and POP-Se) linkers have been synthesised and their electronic and spectroscopic properties investigated as a function of redox state. Solid state NMR provided insight into the structural features of the POPs, while in situ solid state Vis-NIR and electron paramagnetic resonance spectroelectrochemistry showed that the distinct redox states in POP-S could be reversibly accessed. The development of redox-active porous organic polymers with heterocyclic linkers affords their potential application as stimuli responsive materials in gas storage, catalysis, and as electrochromic materials.
Introduction
Porous organic polymers (POPs) comprise only lightweight elements with covalent linkages, and typically have high porosities and low densities.[1] They are particularly versatile as the intrinsic properties of the polymer, such as conductivity, pore size, or catalytic activity, can be tuned by varying the monomer components used.[1] The high chemical stabilities resulting from the linking covalent bonds have made them ideal candidates for applications in gas storage,[2,3] separation,[4] and catalysis, among others.[5–8]
Of particular interest is the inclusion of electroactive components into POPs to yield materials capable of switching between different redox states.[9–11] Due to the different spectroscopic, host–guest, and conductive properties afforded by the different states, redox-active POPs may have applications in organic electronics, in electroswing adsorption, and as electrochromic materials. The high degree of tunability afforded by the inclusion of different linker substituents allows the desired properties of the material to be engineered on the molecular scale.
Triarylamines are redox-active groups capable of a one electron oxidation to form a radical cation state. The efficiency of triarylamines as hole conductors in organic polymers and films has been previously demonstrated where the degree of electron transfer is able to be tuned by variation of the linker between triarylamine cores.[9,10,12–14] Heterocycles such as thiophenes have been demonstrated to provide efficient electron transfer between ‘holes’ in a system, with high charge carrier mobility in addition to good thermal and photo stabilities.[15,16] Systematic variation in the heterocycle used, from furan through to thiophene and selenophene, is hypothesised to result in increasingly electronically delocalised materials by virtue of the more diffuse orbitals of the heteroatoms further down the periodic table.[17]
In this work, we have synthesised six new electroactive porous organic polymers with furan, thiophene, and selenophene linkers in combination with the redox-active triarylamine core (POP-O, POP-S, and POP-Se) and redox-inactive benzene core (POP-O1, POP-S1, and POP-Se1). The porosities of the polymers have been investigated using gas adsorption measurements with N2 and H2 while the spectroscopic features of the polymers in each of their redox states have been determined using both in situ spectroelectrochemical and ex situ chemical oxidation approaches. The interplay of the electronic and spectroscopic properties in these organic materials is of particular interest with regards to the design of future electrochromic and conductive systems.
Results and Discussion
Synthesis and Characterisation
The organic polymers were synthesised via a Sonogashira–Hagihara cross coupling reaction with tris(4-ethynylphenyl)amine (POP-O, POP-S, and POP-Se) or 1,3,5-tris(4-ethynylphenyl)benzene cores (POP-O1, POP-S1, and POP-Se1) and 2,5-dibromofuran, 2,5-dibromothiophene, or 2,5-dibromoselenophene linkers (Scheme 1). The polymers were amorphous and were obtained as either yellow (POP-O1, POP-S1, POP-Se1) or red solids (POP-S, POP-O, POP-Se). The neutral polymers displayed relatively high thermal stabilities with decomposition occurring above 350°C. The initial weight loss below 100°C can be attributed to loss of methanol from the pores of the polymer (Fig. S1, Supplementary Material).

|
The successful formation and characterisation of the polymers was verified by the solid state NMR 13C cross polarisation (CP) magic-angle spinning (MAS) and 13C CP non-quaternary suppression (NQS) spectra (Fig. 1). For all polymers, the successful coupling of the linker to the core was indicated by the appearance of two peaks in the region of 80–95 ppm which can be assigned to the quaternary alkynyl carbons formed. The appearance of two peaks is due to the chemical inequivalence of the two carbon positions. The resonance at 140 ppm in the 13C CP NQS spectra of the furan-containing polymers, POP-O and POP-O1, can be assigned to the quaternary carbons present on the furan ring. In the thiophene and selenophene polymers this peak (along with the alkynyl carbon peaks) was increasingly shifted upfield due to the greater electron density present on the sulfur and selenium atoms when compared with oxygen. The greater rigidity of the benzene core also appears to lead to better defined quaternary carbon peaks between 110 and 135 ppm when compared with the triarylamine core. The broadness of the peaks in the spectra was due to the amorphous nature of these materials.

|
Electrochemical and Spectroscopic Properties of the Neutral and Oxidised POPs
The cyclic voltammogram of POP-O displays one broad oxidation peak at ~0.75 V versus ferrocene (Fc/Fc+) (Fig. S2a, Supplementary Material) due to overlapping contributions from oxidation of the triarylamine core to its radical cation and oxidation of the furan ring.[15,18,19] This corresponds well to the electrochemical data previously reported for the 2,5-bis[4-(N,N′-di(4-methoxyphenyl)amino)phenylethynyl]furan monomer system where the first three oxidation peaks observed were ascribed to oxidation of the triarylamine core and furan linker.[14] The cyclic voltammograms for POP-S and POP-Se are similar to that of POP-O with one broad oxidation peak at ~0.5 V versus Fc/Fc+ (Fig. S2b,c; Supplementary Material), assigned to oxidation of the triarylamine core to its radical cation state in addition to oxidation of the thiophene or selenophene rings. The cyclic voltammograms for POP-O1, POP-S1, and POP-Se1 exhibit a very broad oxidation peak attributed to oxidation of the respective heterocyclic linker (Fig. S3, Supplementary Material). As expected, the oxidation peak was not as prominent as in the voltammograms of POP-O, POP-S, and POP-Se due to the absence of the triarylamine core.
In POP-O, a broad band centred at ~12500 cm−1 corresponds to the formation of the triarylamine radical cation (Fig. 2a) and indicates that a significant amount of oxidation has already occurred in the as synthesised material, such that the polymer exists in a partially oxidised state.[20] The presence of a radical species in the as synthesised material was verified by electron paramagnetic resonance (EPR) spectroscopy for POP-O which exhibited a strong EPR signal at g 2.005 (Fig. S5, Supplementary Material). The band at ~20000 cm−1 is attributed to a charge transfer interaction from the triarylamine core to the furan bridge and is consistent with previous reports for monomeric systems.[14] The broad overlapping bands at > 22500 cm−1 are assigned to the localised aromatic π → π* transitions of the triarylamine core and furan linker. The spectra of POP-S and POP-Se display a strong charge transfer band from the triarylamine core to the thiophene or selenophene bridge at ~21000 cm−1, while bands > 22500 cm−1 are assigned to localised aromatic π → π* transitions of the triarylamine core and thiophene linker.[19] No EPR signals were observed for the neutral POP-S or POP-Se polymers (Fig. S5, Supplementary Material), which is supported by the lack of bands in the NIR region of the solid state UV/vis/NIR spectrum.
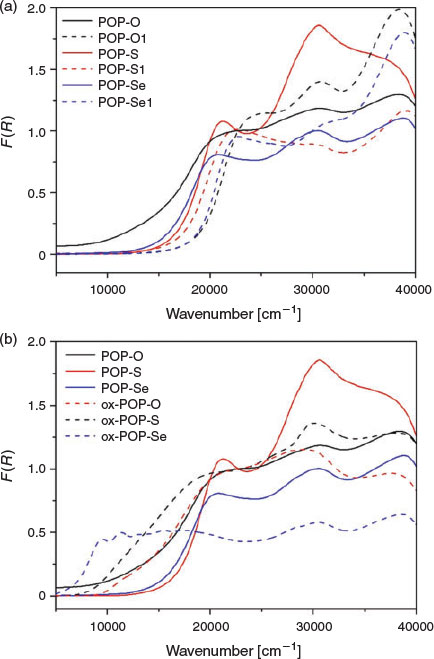
|
The UV/vis/NIR spectra of POP-O1, POP-S1, and POP-Se1 displayed bands at energies above 20000 cm−1 attributed to π → π* transitions of the aromatic moieties in 1,3,5-tris(4-ethynylphenyl)benzene (Fig. 2). The lowest energy band for POP-O1 (24300 cm−1), POP-S1 (21800 cm−1), and POP-Se1 (22790 cm−1) was assigned to the intramolecular charge-transfer transition from the HOMO to the LUMO (or π → π* transition) of the heterocycle linker. The transition was at higher energy for POP-O1 than POP-S1, which was consistent with prior observations that the HOMO–LUMO gap is smaller for thiophene than for furan bridged linkers.[19,21,22]
The polymers POP-O, POP-S, and POP-Se were oxidised by the addition of a saturated solution of NOBF4 in CH3CN to generate the oxidised polymers, ox-POP-O, ox-POP-S, and ox-POP-Se respectively. In all cases, the solid changed colour from red to dark brown/black. Oxidation of the triphenylamine units has been hypothesised to enable better hole conduction due to a more electron-poor π-conjugated system. In the UV/vis/NIR spectra of all the oxidised polymers the band located at ~12500 cm−1 was assigned to the D0 to D1 transition of the radical cation state of the triarylamine core (Fig. 2). The presence of the radical was verified by a strong EPR signal at g 2.005 for ox-POP-O and ox-POP-S and g 2.001 for ox-POP-Se (Fig. S5, Supplementary Material). A red shift in the band due to the π → π* transition of the triarylamine core at ~20000 cm−1 in the UV/vis/NIR spectra was observed for the oxidised polymers when compared with their neutral analogues, which is in accordance with previously reported triarylamine systems.[12]
The increasing overlap of the bands at ~12500 and ~20000 cm−1 in the oxidised polymers as the heteroatom is varied from oxygen to sulfur and selenium results in increasingly delocalised and dispersed orbitals. The increased orbital overlap has important implications for electron transfer as this indicates that electrons can be transferred from the excited state of the triarylamine radical cation to the heterocyclic bridge enabling effective conduction along the polymer network.
In situ Vis/NIR and EPR Spectroelectrochemistry of the POPs
To verify the observations made in the ex situ chemical oxidation experiments, in situ vis/NIR and EPR spectroelectrochemical experiments were additionally performed on the polymer networks. In the solid-state vis/NIR spectroelectrochemical experiment on POP-O, two bands at 11500 and 15550 cm−1 appeared upon application of a positive potential (Fig. 3a). The band at 11500 cm−1 can be attributed to formation of the triarylamine radical cation while the band at 15550 cm−1 was due to a charge transfer process between the triarylamine core and furan bridge. A colour change from red to black was observed during the spectroelectrochemical process (Fig. 3b). The processes were not reversible, which was consistent with the irreversibility of the anodic processes in the electrochemical data obtained for POP-O (Fig. S2a, Supplementary Material). The appearance of bands at 11500 and 15550 cm−1 during the in situ measurement corresponds well to the spectra obtained for the ex situ chemically oxidised ox-POP-O.
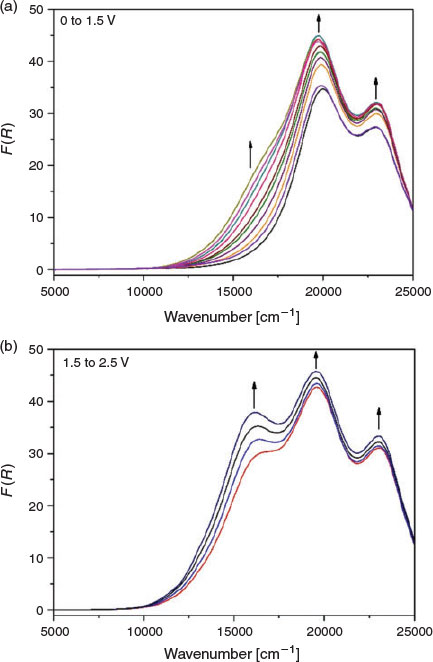
|
In POP-S and POP-Se, very broad bands in the NIR region appeared as a positive potential was applied (Fig. 4 and Fig. S4, Supplementary Material). This low energy transition was assigned to an intervalence charge transfer (IVCT) band potentially indicating electronic communication between adjacent triarylamine cores through the thiophene or selenophene linker. The formation of a shoulder at ~17500 cm−1 (Fig. 4 and Supplementary Material) in POP-S and POP-Se was assigned to a charge transfer process between the triarylamine core and the thiophene or selenophene bridge.[20] Upon oxidation of the triarylamine core to create a ‘hole’ in the system, an increase in electron transfer between triarylamine centres through the heterocyclic linker manifests as an increase in the intensity of the band at 23000 cm−1. The formation of a broad band in the NIR region corresponds to the UV/vis/NIR spectra of the chemically oxidised ox-POP-S and ox-POP-Se where all bands were red shifted upon oxidation.

|
Given the distinct differences in the EPR spectrum of POP-S when compared with that of ox-POP-S (Fig. S5, Supplementary Material), we chose to study the reversibility of the different redox states in POP-S by EPR spectroelectrochemistry. A small EPR signal for the polymer in the neutral state indicated that a small quantity of the triarylamine core exists in the oxidised state before application of a potential. As a positive potential of 1.7 V was applied to the polymer, the signal increased (Fig. 5) to yield an anisotropic EPR signal that was simulated as an axial system with g┴ 2.0068 and g|| 2.0045 (Fig. S6, Supplementary Material). A signal decrease was observed as the applied potential was returned to 0 V (Fig. 5). Despite the potential being held at 0 V for an extended period of time, a small EPR signal was still observed which was similar to that of the original polymer. The polymer was subsequently oxidised and reduced a further two times to assess the reversibility of the system (Figs S6 and S7, Supplementary Material). The increase in signal intensity of the radical with each subsequent oxidation over the same time period suggested structural changes which allowed the polymer to be oxidised with greater ease (Fig. S7, Supplementary Material). We hypothesise that the pores and interlinked networks of the polymer have potentially been altered to allow greater diffusion of counter-ions into the structure. The peak intensities decreased to those of the starting material indicating that the original neutral state was able to be fully regenerated after each oxidation cycle.

|
Porosity of the Organic Polymers
Gas adsorption experiments with N2 and H2 at 77 K were conducted to determine the porosity of the polymers. All the neutral polymers except POP-Se, which was non-porous to N2, displayed similar uptakes of nitrogen and similar Brunauer–Emmett–Teller (BET) surface areas (POP-O: 497, POP-O1: 513, POP-S: 520, POP-S1: 540, and POP-Se1: 396 m2 g−1) (Fig. 6a). A significant hysteresis was observed in the N2 isotherms at 77 K and may be indicative of flexibility in the polymer networks. The porosities of the polymers decreased markedly upon oxidation to essentially non-porous systems with BET values close to 0. This could be attributed either to collapse of the polymeric networks or occlusion of the pore space by the counter-ion in the oxidised material. The H2 uptakes of POP-O, POP-O1, POP-S, and POP-S1 were all comparable with a moderate H2 adsorption of ~5 mmol g−1 at 1100 mbar (Fig. 6b). The selenophene containing polymers, POP-Se and POP-Se1, exhibited a smaller H2 uptake of 2–3 mmol g−1 at 1100 mbar despite POP-Se being non-porous to N2 which may suggest the presence of ultramicropores.

|
Conclusion
A series of POPs with furan, thiophene, and selenophene linkers combined with the redox-active triarylamine core have been synthesised and their properties characterised as a function of redox state through in situ spectroelectrochemical and ex situ chemical oxidation experiments. The spectroscopic and electronic properties of the polymers changed upon oxidation to the triarylamine radical cation resulting in a darkening in colour corresponding to greater electronic delocalisation and orbital overlap, particularly for POP-S and POP-Se. The robust and reversible redox cycling of the materials, as demonstrated by POP-S, demonstrate the versatility of these electroactive polymers for potential use in electroswing adsorption and electrochromic applications.
Experimental
General Considerations
All chemicals and solvents were used as obtained and without further purification. Tris(p-iodophenyl)amine,[23] 2,5-dibromofuran,[24] 2,5-dibromoselenophene,[25] and Pd(PPh3)4[26] were synthesised according to literature procedures. Tris(p-ethynylphenyl)amine was synthesised according to a modification of the reported literature procedure.[27] Acetonitrile and triethylamine were dried over CaH2 and DMF was dried over activated CaSO4. All other reagents and solvents used were obtained commercially and used without further purification. Microanalyses were carried out at the Chemical Analysis Facility – Elemental Analysis Service in the Department of Chemistry and Biomolecular Science at Macquarie University, Australia.
POP-O, POP-S, and POP-Se
Tris(4-ethynylphenyl)amine (200 mg, 0.630 mmol), the heterocylic linker (2,5-dibromofuran, 2,5-dibromothiophene, or 2,5-dibromoselenophene) (0.504 mmol), and Pd(PPh3)4 (5.80 mg, 0.00504 mmol) were dissolved in a mixture of DMF (5.0 mL) and triethylamine (5.0 mL) to yield a dark reaction mixture that was stirred at room temperature for 5 min before the addition of copper(i) iodide (1.92 mg, 0.0101 mmol). This was then heated under nitrogen at 90°C for 3 days upon which an orange red solid was obtained. The reaction mixture was filtered and the solid washed consecutively with DMF, chloroform, methanol, water, methanol, and acetone before being air-dried. The solid was then washed via Soxhlet extraction with methanol overnight before being dried under vacuum (250 mg).
POP-O
Yield 250 mg. λmax (solid state)/cm−1 12400, 20250, 30060, 38840. Found: C 83.92, H 4.15, N 4.21. C60H30N2O·3.6CH4O requires C 83.92, H 4.92, N 3.08 %.
POP-S
Yield 268 mg. λmax (solid state)/cm−1 21120, 30270, 38540. Found: C 78.84, H 3.91, N 4.54. C60H30N2S·5.72H2O requires C 78.84, H 4.57, N 3.06 %.
POP-Se
Yield 271 mg. λmax (solid state)/cm−1 20670, 30310, 38880. Found: C 74.09, H 3.78, N 3.18. C60H30N2Se·7.25CH4O requires C 74.09, H 5.46, N 2.57 %.
POP-O1, POP-S1, and POP-Se1
1,3,5-Tris(4-ethynylphenyl)benzene (100 mg, 0.264 mmol), the heterocylic linker (2,5-dibromofuran, 2,5-dibromothiothene, or 2,5-dibromoselenophene) (0.211 mmol), and Pd(PPh3)4 (2.40 mg, 0.00211 mmol) were dissolved in a mixture of DMF (3.0 mL) and triethylamine (3.0 mL) and stirred under nitrogen at room temperature for 5 min before the addition of copper(i) iodide (0.800 mg, 0.00422 mmol). The brown solution was heated at 90°C for 3 days to obtain a yellow solid. The reaction mixture was filtered and the solid washed consecutively with DMF, chloroform, methanol, water, methanol, and acetone before being air-dried. The solid was then washed with methanol via Soxhlet extraction overnight before being dried under vacuum.
POP-O1
Yield 70 mg. λmax (solid state)/cm−1 24270, 30750, 38420. Found: C 86.47, H 3.86, N 0.36. C62H31O3·0.22DMF·1.68H2O requires C 86.47, H 4.16, N 0.36 %.
POP-S1
Yield 109 mg. λmax (solid state)/cm−1 21830, 30340, 39190. Found: C 85.55, H 3.99. C62H31S3 requires C 85.39, H 3.58 %.
POP-Se1
Yield 45.4 mg. λmax (solid state)/cm−1 22790, 31480, 38880. Found: C 76.85, H 3.65. C62H31Se3 requires C 76.53, H 3.08 %.
Oxidation of Polymers using NOBF4
The POP (20 mg) was suspended in dry and distilled acetonitrile (2 mL) and the mixture thoroughly degassed. A solution of nitrosonium tetrafluoroborate (10 mg, 0.0860 mmol) in acetonitrile (2 mL) was added slowly dropwise with vigorous bubbling of nitrogen to yield an immediate colour change from yellow to dark brown. The mixture was left to stir at room temperature with vigorous bubbling of nitrogen for 30 min, before the solid was filtered off and dried.
ox-POP-O
λmax (solid state)/cm−1 19630, 29740, 38690. Found: C 65.66, H 4.05, N 9.39. C60H30N2O·8.48DMF·1.71BF4 requires C 65.66, H 5.76, N 9.39 %.
ox-POP-S
λmax (solid state)/cm−1 20680, 29250, 38260. Found: C 67.67, H 3.23, N 5.24. C60H30N2S·2.48DMF·2.36BF4 requires C 67.67, H 3.23, N 2.63 %.
ox-POP-Se
λmax (solid state)/cm−1 9480, 16620, 30310, 39000. Found: C 53.84, H 3.58, N 5.35. C60H30N2Se·4.18DMF·5.24BF4 requires C 53.84, H 3.69, N 5.35 %.
Physical Measurements and Characterisation
Solid State NMR
The 13C CPMAS solid state NMR experiments were carried out on a wide-bore Bruker Biospin Avance III solids-300 MHz spectrometer operating at a frequency of 75 MHz for the 13C nucleus. Approximately 80 mg of sample was placed into 4 mm zirconia rotors fitted with Kel-f caps and spun in a double resonance H-X probehead at 8 kHz MAS (magic angle spinning). The 13C and 1H 90° radio frequency pulse lengths were optimised to 3.5 μs each. The 13C spectra were acquired with 1 ms cross polarisation contact time with a total suppression of spinning sidebands (TOSS) scheme, followed by 1H decoupling at 75 kHz field strength using spinal-64 decoupling. The 13C NQS (non-quaternary carbon suppression) spectra were recorded by turning off the 1H decoupling for 40 μs during the TOSS period. For sufficient signal-to-noise, ~1 k transients were acquired for each sample with recycle delays of 3.0 s in between to ensure sufficient relaxation of the 1H nuclei. The spectra were obtained at room temperature. The 13C chemical shifts were referenced to the glycine CO peak at 176 ppm.
Infrared Spectroscopy (DRIFTS)
FT-IR spectroscopy was performed on samples in a KBr matrix over the range 4000–400 cm−1 on a Bruker Tensor 27 FT-IR spectrometer with a resolution of 4 cm−1.
Thermal Gravimetric Analysis (TGA)
TGA was performed under a flow of nitrogen (0.1 L min−1) on a TA Instruments Hi-Res Thermogravimetric Analyser from 25–700°C at 1°C min−1.
Solid State Electrochemistry
Solid state electrochemical measurements were performed using a Bioanalytical Systems Electrochemical Analyser. Argon was bubbled through solutions of 0.1 M [(n-C4H9)4N]PF6 dissolved in distilled CH3CN. The cyclic voltammograms were recorded using a glassy carbon working electrode (1.5 mm diameter), a platinum wire auxiliary electrode, and an Ag/Ag+ wire quasi reference electrode. The sample was mounted on the glassy carbon working electrode by dipping the electrode into a paste made of the powder sample in CH3CN. Ferrocene was added as an internal standard upon completion of each experiment. All potentials are quoted in V versus Fc+/Fc.
Solid State UV/Vis/NIR Spectroscopy
UV/vis/NIR spectra were obtained on the samples at room temperature using a CARY5000 Spectrophotometer equipped with a Harrick Praying Mantis accessory over the wavenumber range 5000–40000 cm−1. BaSO4 was used for the baseline. Spectra are reported as the Kubelka–Munk transform, where F(R) = (1 – R)2/2R (R is the diffuse reflectance of the sample as compared with BaSO4).
Solid State Spectroelectrochemistry (Vis/NIR)
In the solid state, the diffuse reflectance spectra of the electrogenerated species were collected in situ in a 0.1 M [(n-C4H9)4N]PF6/CH3CN electrolyte over the range of 5000–25000 cm−1 using a Harrick Omni Diff Probe attachment and a custom built solid state spectroelectrochemical cell.[28] The cell consisted of a Pt wire counter electrode and a Ag/Ag+ quasi-reference electrode. The solid sample was immobilised onto a 0.1 mm thick indium-tin-oxide coated quartz slide (which acted as the working electrode) using a thin strip of Teflon tape. The applied potential (from −2.0 to 2.0 V) was controlled using an eDAQ potentiostat. Continuous scans of the sample were taken and the potential increased gradually until a change in the spectrum was observed.
Electron Paramagnetic Resonance (EPR)
EPR spectroscopy (Bruker EMX-X band) was performed at room temperature on freshly synthesised materials (~12 mg). The microwave frequency was between 9.75 and 9.85 GHz for all measurements and the microwave power below that required for saturation.
Solid State Spectroelectrochemistry (EPR)
The procedure and cell set up used were those previously described.[29] A three-electrode assembly based on simple narrow wires (A–M Systems) as electrodes where Teflon coated platinum and silver wires were used for the working and quasi-reference electrodes respectively, and a naked platinum wire as the counter electrode. The sample of interest was wrapped in a small piece of platinum mesh (~5 mm × 3 mm) lengthwise and the exposed end of the working electrode carefully wrapped in a spiral shape around the platinum mesh. A small piece of platinum mesh (~5 mm × 4 mm) was rolled up lengthwise and attached in a similar fashion to the counter electrode to ensure that the surface area of the counter electrode was larger than that of the working electrode. The working electrode was positioned lowest such that the redox product of interest was generated at the bottom of the tube and was well separated from the counter electrode. The electrodes were soldered to a narrow three-core microphone wire. The cell used was made by flame sealing the tip of a glass pipette. The potential was controlled with a portable μAutolab II potentiostat and the EPR spectra obtained using an EMX Micro X-band EPR spectrometer with 1.0 T electromagnet.
Gas Adsorption
Gas adsorption measurements over the 0–1 bar range were conducted using a Micromeritics Accelerated Surface Area and Porosity (ASAP) 2020 or 3-Flex instrument. Approximately 50–100 mg of the polymer sample was degassed under vacuum at 70°C for ~14 h. Nitrogen and hydrogen adsorption isotherms were measured at 77 K where the temperature was controlled by use of a liquid nitrogen bath. The surface area was determined using the BET method using the ASAP2020 V4.01 or 3-Flex Microactive software.
Supplementary Material
Details of the TGA, electrochemistry, EPR of all polymers, solid state Vis/NIR spectroelectrochemistry of POP-Se and solid state EPR spectroelectrochemistry of POP-S are available on the Journal’s website.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgements
The authors gratefully acknowledge the support of the Australian Research Council, the Australian Institute of Nanoscale Science and Technology at the University of Sydney, and the EPSRC UK National Electron Paramagnetic Resonance Service at the University of Manchester. The authors thank Dr Marcello Solomon for the collection of several TGA measurements.
References
[1] Y. Xu, S. Jin, H. Xu, A. Nagai, D. Jiang, Chem. Soc. Rev. 2013, 42, 8012.| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXhsVyrtbjL&md5=fc0ad34b6dbe7f4bf58a75cf0fd122a4CAS |
[2] H. A. Patel, S. Hyun Je, J. Park, D. P. Chen, Y. Jung, C. T. Yavuz, A. Coskun, Nat. Commun. 2013, 4, 1357.
| Crossref | GoogleScholarGoogle Scholar |
[3] W. Wang, M. Zhou, D. Yuan, J. Mater. Chem. A 2017, 5, 1334.
| Crossref | GoogleScholarGoogle Scholar |
[4] O. K. Farha, A. M. Spokoyny, B. G. Hauser, Y.-S. Bae, S. E. Brown, R. Q. Snurr, C. A. Mirkin, J. T. Hupp, Chem. Mater. 2009, 21, 3033.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXnvFWrs70%3D&md5=76b316a91ca83e8c0406a34acbc0b7a7CAS |
[5] Y. Zhang, S. N. Riduan, Chem. Soc. Rev. 2012, 41, 2083.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XivFWlsb8%3D&md5=65e60667472b9dc5d92d6019ec02ebebCAS |
[6] P. Kaur, J. T. Hupp, S. T. Nguyen, ACS Catal. 2011, 1, 819.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXnsVSjsbw%3D&md5=d01fe65dc82c7244678164417c00d604CAS |
[7] J.-X. Jiang, C. Wang, A. Laybourn, T. Hasell, R. Clowes, Y. Z. Khimyak, J. Xiao, S. J. Higgins, D. J. Adams, A. I. Cooper, Angew. Chem. Int. Ed. 2011, 50, 1072.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtFKntrs%3D&md5=96f924f6144d3f81e94930c1a5cf0aadCAS |
[8] K. Zhang, D. Kopetzki, P. H. Seeberger, M. Antonietti, F. Vilela, Angew. Chem. Int. Ed. 2013, 52, 1432.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhvVKgu7fK&md5=e173c85a4b6bfb18d8117c0a853c8472CAS |
[9] C. Hua, A. Rawal, T. B. Faust, P. D. Southon, R. Babarao, J. M. Hook, D. M. D’Alessandro, J. Mater. Chem. A 2014, 2, 12466.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXhtVKmu73K&md5=73fc23ad9730053a8f1b987a9c410519CAS |
[10] C. Hua, B. Chan, A. Rawal, F. Tuna, D. Collison, J. M. Hook, D. M. D’Alessandro, J. Mater. Chem. C 2016, 4, 2535.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC28XjtlKqu78%3D&md5=b470106a0605afcb5aa70e9c30019c5eCAS |
[11] G. Das, T. Prakasam, S. Nuryyeva, D. S. Han, A. Abdel-Wahab, J.-C. Olsen, K. Polychronopoulou, C. Platas-Iglesias, F. Ravaux, M. Jouiad, A. Trabolsi, J. Mater. Chem. A 2016, 4, 15361.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC28XhsVKisLzM&md5=99d5ef566c35be2a30cbbcc58903e205CAS |
[12] S. Amthor, B. Noller, C. Lambert, Chem. Phys. 2005, 316, 141.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXpvVGqtrc%3D&md5=47a94f9a4465e7bc174eb505c1922002CAS |
[13] M. h. Chahma, J. B. Gilroy, R. G. Hicks, J. Mater. Chem. 2007, 17, 4768.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXht12hsL3F&md5=f424329be6e2666986c716c65e80dd64CAS |
[14] G. Nöll, M. Avola, M. Lynch, J. Daub, J. Phys. Chem. C 2007, 111, 3197.
| Crossref | GoogleScholarGoogle Scholar |
[15] K. Idzik, J. Sołoducho, M. Łapkowski, S. Golba, Electrochim. Acta 2008, 53, 5665.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXlsV2lt7c%3D&md5=d5c5b46d0b006a301c004a142e0c85d9CAS |
[16] X. Qian, Z.-Q. Zhu, H.-X. Sun, F. Ren, P. Mu, W. Liang, L. Chen, A. Li, ACS Appl. Mater. Interfaces 2016, 8, 21063.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC28Xht1equ7nJ&md5=b918eda75209764bb47a628b6179b2e4CAS |
[17] P.-F. Li, T. B. Schon, D. S. Seferos, Angew. Chem. Int. Ed. 2015, 54, 9361.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXhtVensbvF&md5=1c50492497bdf1f538dbd0d0121ca5bfCAS |
[18] M. J. Plater, T. Jackson, Tetrahedron 2003, 59, 4687.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXksVegsb4%3D&md5=e645f0c1f9f82f65b0e89315a301a913CAS |
[19] K. Yamamoto, M. Higuchi, K. Uchida, Y. Kojima, Macromolecules 2002, 35, 5782.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XksVKksb4%3D&md5=ea41ddfb719d9ad43d7f1466fb680dbaCAS |
[20] V. Lukeš, P. Rapta, K. Haubner, M. Rosenkranz, H. Hartmann, L. Dunsch, J. Phys. Chem. A 2013, 117, 6702.
| Crossref | GoogleScholarGoogle Scholar |
[21] C.-J. Chen, Y.-C. Hu, G.-S. Liou, Polym. Chem. 2013, 4, 4162.
| 1:CAS:528:DC%2BC3sXhtVCqtbnI&md5=a1ddd8e6ed99a9b9df4d720199ce76eaCAS |
[22] P. Surawatanawong, A. K. Wójcik, S. Kiatisevi, J. Photochem. Photobiol. Chem. 2013, 253, 62.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXit1ensbs%3D&md5=467b0d09c6b24dc6897b357131787ec0CAS |
[23] Y. Jiang, Y. Wang, J. Yang, J. Hua, B. Wang, S. Qian, H. Tian, J. Polym. Sci. Part A: Polym. Chem. 2011, 49, 1830.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjt1Ghsrg%3D&md5=b01504cf52f2b6d141cfaa185c227723CAS |
[24] C. Ramalingan, I.-S. Lee, Y.-W. Kwak, Chem. Pharm. Bull. 2009, 57, 591.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXpvVylsb8%3D&md5=01122e9042a9279c6987a51ceaa69aa8CAS |
[25] Y.-J. Hwang, T. Earmme, S. Subramaniyan, S. A. Jenekhe, Chem. Commun. 2014, 50, 10801.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXht1Khu7bF&md5=a9f9883273465f2157fbe043dd106f6fCAS |
[26] D. R. Coulson, L. C. Satek, S. O. Grim, in Inorganic Syntheses (Ed. F. A. Cotton) 1972, pp. 121–124 (John Wiley & Sons, Inc.: Hoboken, NJ).
[27] N. Niamnont, N. Kimpitak, K. Wongravee, P. Rashatasakhon, K. K. Baldridge, J. S. Siegel, M. Sukwattanasinitt, Chem. Commun. 2013, 49, 780.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhvVykt7fK&md5=2107c496ffd32a5a0f0e3c80e2a29813CAS |
[28] P. M. Usov, C. Fabian, D. M. D’Alessandro, Chem. Commun. 2012, 48, 3945.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XksVOlurs%3D&md5=ea3cabda65ac3b37afcd0106371c29afCAS |
[29] P. R. Murray, D. Collison, S. Daff, N. Austin, R. Edge, B. W. Flynn, L. Jack, F. Leroux, E. J. L. McInnes, A. F. Murray, D. Sells, T. Stevenson, J. Wolowska, L. J. Yellowlees, J. Magn. Reson. 2011, 213, 206.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtl2gs7jM&md5=7a13ee310bbaa9df2c7d3279c55b34e1CAS |
* Dr Carol Hua is the recipient of the 2016 RACI Cornforth Medal and Associate Professor Deanna D’Alessandro is the recipient of the 2017 Australian Academy of Science Le Fèvre Memorial Prize.