

Isotopic composition of polyhalomethanes from marine macrophytes – systematic effects of the halogen substituents on isotopic composition
Enno Bahlmann A B C , Christian Stolle B , Ingo Weinberg B , Richard Seifert A , Detlef E. Schulz-Bull B and Walter Michaelis AA Department of Geosciences, University of Hamburg, Bundesstraße 55, D-20146 Hamburg, Germany.
B Leibniz Institute for Baltic Sea Research Warnemünde (IOW), Seestraße 15, D-18119 Rostock, Germany.
C Corresponding author. Email: enno.bahlmann@zmaw.de
Environmental Chemistry 12(4) 504-514 https://doi.org/10.1071/EN14210
Submitted: 2 October 2014 Accepted: 3 June 2015 Published: 20 July 2015
Environmental context. Once released to the atmosphere, halocarbons are involved in key chemical reactions. Stable carbon isotope measurements of halocarbons can provide valuable information on their sources and fate in the atmosphere. Here, we report δ13C values of 13 polyhalomethanes released from brown algae, which may provide a basis for inferring their sources and fate in future studies.
Abstract. Halocarbons are important vectors of reactive halogens to the atmosphere, where the latter participate in several key chemical processes. An improved understanding of the biogeochemical controls of the production–destruction equilibrium on halocarbons is of vital importance to address potential future changes in their fluxes to the atmosphere. Carbon stable isotope ratios of halocarbons could provide valuable additional information on their sources and fate that cannot be derived from mixing ratios alone. We determined the δ13C values of 13 polyhalomethanes from three brown algae species (Laminaria digitata, Fucus vesiculosus, Fucus serratus) and one seagrass species (Zostera noltii). The δ13C values were determined in laboratory incubations under variable environmental conditions of light, water levels (to simulate tidal events) and addition of hydrogen peroxide (H2O2). The δ13C values of the polyhalomethanes ranged from –42.2 ‰ (±3.5 s.d.) for CHCl3 to 6.9 ‰ (±4.5) for CHI2Br and showed a systematic effect of the halogen substituents that could empirically be described in terms of linear free energy relationships. We further observed an enrichment in the δ13C of the polyhalomethanes with decreasing polyhalomethane yield that is attributed to the competing formation of halogenated ketones. Though variable, the isotopic composition of polyhalomethanes may provide useful additional information to discriminate between marine polyhalomethane sources.
Additional keywords: brown algae, halocarbons, LFER, stable carbon isotopes.
Introduction
Once emitted into the atmosphere, halocarbons can release reactive halogen species, which participate in several key chemical processes. They affect the oxidation capacity of the troposphere and stratosphere by the destruction of ozone.[1–4] Additionally, they act as oxidants with either reactive chlorine or bromine species, thereby contributing to the degradation of hydrocarbons[5] or to the oxidation of elemental mercury[6] respectively. Some halocarbons, e.g. iodomethane (CH3I), are also known to contribute to aerosol formation in the marine boundary layer[7] and may influence atmospheric dimethyl sulphide (DMS) and nitrogen oxide cycles.[7]
Within the last few years, much progress has been made in quantifying the global emissions of various halocarbons. However, the current emissions estimates for these climate-relevant compounds remain fairly uncertain owing to the large spatial and temporal variability in observed halocarbon mixing ratios and fluxes. This is, in particular, true for short-lived halocarbons such as iodomethane (CH3I) and bromoform (tribromomethane, CHBr3),[8–14] with the largest uncertainties in the known sources being observed for coastal and near-shore emissions.[15] Despite the uncertainties in global halocarbon emission budgets, it is well accepted that in the marine realm, diverse autotrophic organisms contribute to halocarbon production. Marine macroalgae have long been recognised as potent sources of diverse halocarbons and their production has been studied in numerous laboratory and field experiments.[16–25] More recently, seagrass meadows have also been recognised as halocarbon sources.[26,27] Brown algae – particularly from the order Laminariales – produce a wide range of polyhalomethanes (PHMs).[16,22,23] Furthermore, production of monohalomethanes, e.g. CH3I,[18,24] and of several iodinated and brominated C2 to C4 monoalkylhalides[21,24] has been observed. The biogenic formation of PHMs proceeds by an enzyme-catalysed halogenation of organic substrates having an activated α-carbon atom, most likely β-diketones or β-ketoacids.[28] Halogenated heptanones were among the most abundant halometabolites found in the red algae Bonnemasiona hamifera. Based on this, 3-oxooctanoid acid has been suggested as a substrate for the enzymatic halogenation.[29,30] The halogenation occurs by haloperoxidase activity in the presence of H2O2, resulting in the formation of hypohalous acid as the halogenating agent. In the second step, the organic substrate undergoes stepwise electrophilic substitution followed by nucleophilic acyl substitution.[29–31] Halogenation of dissolved organic matter (DOM) in the water column, catalysed by haloperoxidase enzymes released into the water, may also be an important pathway.[32]
Once formed, halocarbons can be cleaved either by bacterial or chemical degradation such as hydrolysis or photolysis. Nucleophilic halide substitution, most likely with chloride, leads to the formation of new halocarbons, in the case of PHMs to mixed bromo-chloro and iodo-chloromethanes such as CH2BrCl, CHBr2Cl and CH2ClI.[33,34] Thus, the final release of halocarbons to the atmosphere is the result of complex production and decomposition processes.
A better understanding of the biogeochemical controls of the production–destruction equilibrium may substantially improve current emission estimates[10,12] and is of vital importance to address potential future changes. Carbon isotope signatures of individual halocarbons have been proposed as a valuable tool to distinguish between different sources, to obtain information on source and sink mechanisms and to refine the global budgets of CH3Br[35] and CH3Cl.[36–38] Tremendous progress has recently been made for the carbon isotopic analysis of dissolved halocarbons,[39,40] which now allows extension of this approach to short-lived halocarbons such as CH3I, CH2Br2 and CHBr3. Determination of the carbon isotope ratios of these compounds could provide valuable additional information about source–sink relationships that cannot be derived from the mixing ratios alone. Therefore, we determined the carbon isotope source signature of halocarbons produced by different marine macrophytes. The incubated species were three brown algae species (Laminaria digitata, Fucus vesiculosus, Fucus serratus) and one seagrass species (Zostera noltii). We here report carbon isotope signatures for 13 PHMs and discuss these data with respect to the underlying formation processes and assess their suitability for source assignment.
Methods
Three laboratory-based incubation experiments with different set-ups were performed. A first set of experiments with two brown algae species (Fucus vesiculosus, Fucus serratus) was conducted at the Institute of Baltic Seas Research (IOW) in Warnemünde in 2004. Algae were collected in September and October 2004 from the Baltic Sea near Todendorf (Germany, 53°57′57N, 10°55′43E). A second set of incubations with Zostera noltii was carried out at the Institute of Biogeochemistry and Marine Chemistry (IFBM) in Hamburg in 2010. The seagrass was collected from a dense seagrass meadow in Dagebüll (Germany, 54°54′12N; 8°41′52E) on 10 September 2010. A third set of incubations was conducted with two brown algae Laminaria digitata and F. vesiculosus in September 2013. These species were sampled at the shore of Helgoland island in the German Bight (54°11′17N, 7°53′11E).
For each incubation experiment, intact whole plants were sampled in the field and directly transported to the laboratory within 1 day. The seagrass and macroalgae were held in natural seawater collected at the sampling location until the beginning of the incubation experiments. The incubation experiments were generally conducted within 2 days of sampling.
IOW set-up
Details on the incubation and stable carbon isotope determination carried out at the IOW are given elsewhere.[39,40] Briefly, from each Fucus species, sections of the thallus including the central rib and air bladders (length up to 10 cm) were carefully cut using a scalpel, weighed (~200 g fresh weight, FW), and immediately placed in 5-L gas-tight Duran glass bottles. All incubations were performed in autoclaved sea water with no headspace. The sea water was purged for 7 to 8 days with nitrogen 5.0 (Westfalen, Muenster, Germany, purity >99.999 %) before incubations to remove halocarbons. Afterwards, the pH was adjusted to ~7. Three replicates of F. vesiculosus and four replicates of F. serratus were incubated at 15 °C for 24 h under light conditions with a photon density of 1000 to 1500 µmol m–2 s–1. Additionally, three replicates of each Fucus species were spiked with 3 mL of 30 % H2O2 (Merck, Darmstadt, Germany) in order to mimic enhanced oxidative stress. After the incubation, the water from the incubation vessels was filtered using glass-fibre filters (GFF, 140 mm, Whatman, Maidstone, UK) and directly transferred to a continuous-flow purge-and-trap system for halocarbon extraction. The halocarbons were enriched on Tenax TA (Sigma–Aldrich, Taufkirchen, Germany) at –35 °C, thermally desorbed and cryofocussed again before the injection. Measurements were performed by gas chromatography–combustion–isotope ratio mass spectrometry (GC-C-IRMS, trace GC with GCC III interface coupled to a MAT 253; all Thermo Electron, Bremen, Germany). An analytical standard solution containing 24 halogenated volatile organic compounds (HVOCs) was used for quantification. Standard injections of HVOCs with three to five replicates at different concentration levels revealed a precision of ≤20 %. The analytical precision of the isotopic determination of HVOCs ranged from <1 to 3 ‰ for carbon amounts from 0.5 to 20 ng.
IFBM set-up
The seagrass incubations were carried out with whole intact plants (~35 g FW) in a 1-L Duran glass bottle (Schott, Mainz, Germany) equipped with a three-port cap. One port was generally closed with a septum for injection of CO2 during the incubations. The other two ports were the inlet and outlet for the purge gas and the CO2 monitoring loop. The purge gas lines were equipped with ball valves that were opened during purging. The gas was introduced through the inlet through a stainless steel frit. The outlet was connected to a particle filter (Sartorius, Hoettingen, Germany, Teflon membrane filter, diameter, 45 mm; pore size, 0.2 µm) followed by a condenser kept at approximately –20 °C to reduce the water vapour pressure of the outgoing air. Prior to the seagrass incubation, 600–700 mL of seawater was filtered (GFF, Whatman) and purged for ~1 h with synthetic air in order to eliminate the VHOCs. During incubation, the CO2 concentration in the headspace (pCO2) was monitored in a closed loop at a flow rate of 60 mL min–1 with a CO2 analyser (Li-840, LI-COR Biosciences, Bad Homburg, Germany). At the end of each incubation cycle, the sample was purged with synthetic air (Westfalen) at a flow rate of 1 L min–1 for 30 min. The halocarbons produced were stripped from the water phase, preconcentrated in cryotraps and then transferred to adsorption tubes.[27] CO2 was injected with a gas-tight syringe when the concentration fell below 370 parts per million by volume (ppmv). The incubations were carried out at a constant temperature of 21 °C. Day–night cycles (10–14 h) were simulated using two 150-W metal lamps (OSram HQI TS, Muenchen, Germany) providing a photon flux of 1800 µmol m2 h–1 (±300 s.d.) with a solar spectrum between 300 and 700 nm. VHOCs were purged after 1–3 h of incubation in light and after 14 h of incubation in the dark. In total, 11 seagrass incubations and 5 sea water only incubations were carried out.
The macroalgae incubations were carried out with a similar set-up, applying ~200 g FW in a 5-L Duran glass bottle. To avoid degradation of very-short-lived iodine-containing PHMs,[33,34] the samples were continuously purged with synthetic air (Westfalen, Germany) doped with CO2 (Westfalen Germany), providing a pCO2 of 380 ppmv (±15) in the purge gas. The purge flow was set to 2 L min–1 and typical sampling times were 30 min, resulting in a sampling volume of 60 L. The sampling procedure was the same as described for the seagrass. In between samplings, the purge gas was vented into a fume hood. Various studies have reported high halocarbon production in sea water sampled in the vicinity of macroalgae, in particular of the order Laminariales.[17,22] In order to avoid these blank problems, the incubations were carried out with artificial sea water with a salinity of 30 PSU produced from commercially available sea salt. The water was purged prior the experiments for 24 h with ambient air (24 h, 2 L min–1) and VHOC-free synthetic air (4 h, 2 L min–1) to remove the VHOCs from the seawater solution and the pH was adjusted to 8.1 ± 0.1. Two incubations with L. digitata and one with F. serratus were conducted. First, we performed one long-term incubation of 30 h with L. digitata designated to assess the effect of radiation and tidal inundation on the production and isotopic composition of the halocarbons. Therefore, incubations were carried out in the presence of water under light (LW1, LW2) and dark (NLW) conditions as well as in the absence of water under light and dark conditions (LNW, NLNW). Prior to sampling, the system was allowed to equilibrate for at least 2 h under the respective incubation conditions. In total, 15 samples were taken during this experiment, with at least two replicates for each incubation condition. Further, three replicates from a second specimen of L. digitata and a specimen of F. vesiculosus were taken under light and no water conditions aiming to address the intra- and interspecies variability in the isotopic composition of the halocarbons.
The analytic procedure for the IFBM samples is based on those of Bahlmann et al.[41] for the isotopic determination of trace gases. A Scott TO EPA 15/17 standard (Air Liquide America Specialty Gases LLC, Plumsteadville, PA, USA) containing 32 halocarbons, among others, was used as a daily working standard. Analytes present in the standard were identified by comparison of their retention time and mass spectra and quantified against the Scott TOC EPA 15/17 standard. The overall measurement uncertainty was better than ±10 % Further compounds not present in the standard (CH2Br2, CH2ClI, CH2BrI, CH2I2, CHBr2I and CHBrI2) were identified by comparison of the mass spectra obtained with the National Institute of Standards mass spectral database version 2.0. These compounds were quantified on the IRMS through the CO2 intensities against CHBr3 as internal standard. The uncertainty of this procedure is ±15 % on the 1σ level.[41] Results are only reported for peaks that met the following quality criteria: (i) peak purity better than 90 %; (ii) peak separation better than 90 % valley. All carbon isotope ratios are reported in per mille relative to the Vienna Pee Dee Belemnite (VPDB) scale. The analytical precision of the carbon isotope ratio determination was from 0.3 to 2.6 ‰ on the 1σ level and the range varies depending on the amount of carbon. Linearity of the detector response has been shown for carbon amounts ranging from 0.1 to 240 ng.[41]
Results and discussion
In total, 24 compounds in sufficient amounts (>0.5 ng carbon) for carbon stable isotope determination were detected in the incubation experiments. In the following, only the results of the PHMs will be discussed to reduce the complexity of the data compiled. All polyhalomethane data from the incubation experiments are provided in the Supplementary material (Tables S1–S6).
Production rates
Apart from monohalomethanes (MHMs), the seagrass Zostera noltii surprisingly produced substantial amounts of CHBr3, accounting for ~40 % of total halocarbon production. To the best of our knowledge, no haloperoxidase activity has been reported from higher plants. Thus, the CHBr3 may originate from associated epiphytic microalgae. CHBr3 production rates ranged from 0.5 to 20.9 pmol g–1 FW h–1 (Table S5). On average, 8.5 times higher production rates were observed during light compared with dark incubations. Similar diel variations in PHM production have been reported from macroalgae incubations.[22,42] However, the source of these variations has not yet been fully clarified. It may either be related to diel variations in H2O2 concentration[42,43] or to diel variations in the PHM precursors. Some halocarbon production was observed in the filtered (0.7 µm) seagrass-free seawater controls. Total CHBr3 production rates ranged from 1.8 to 22.7 pmol h–1 (Table S5), equalling 0.09 to 0.67 pmol g–1 FW h–1 when normalised to the seagrass biomass, and thus contributed less than 10 % to the overall CHBr3 production in the seagrass incubations.
In contrast to the seagrass incubations, the IFBM macroalgae incubations were carried out with artificial sea water that was rigorously purged prior the experiments. The blank controls, taken before the addition of macroalgae, revealed negligible halocarbon production of less than 2.3 % of halocarbon production in the macroalgae incubation except for CH3Cl. In general, the macroalgae produced a broader spectrum of VHOCs, with PHMs accounting for 75 to 97 % of total halocarbon production. Halocarbon production rates of the macroalgae varied by almost four orders of magnitude. Laminaria digitata was the most productive species, with total halocarbon production rates ranging from 16.8 to 1355 pmol g–1 FW h–1 for L. digitata 1 and from 563 to 1270 pmol g–1 FW h–1 for L. digitata 2 (Table S3). The most abundant PHMs were CHBr3, CH2I2 and CH2Br2. Additionally, mixed bromo-chloromethanes (CH2BrCl, CHBrCl2 and CHBr2Cl) as well as several iodinated PHMs (CH2ClI, CH2BrI, CHBr2I and CHBrI2) were found in the L. digitata incubations. The production rates of bromine- and chlorine-containing PHMs were generally well correlated with each other (R2 > 0.8, P = 0.05). The iodine-containing PHMs showed a less pronounced correlation with each other (0.8 > R2 > 0.42, P = 0.05) and with the bromine- and chlorine-containing compounds (0.73 > R2 > 0.31, P = 0.05).
During the long incubation with L. digitata 1, we observed a decline in the production of PHMs, exemplified for CHBr3, CH2I2 and CH2Br2 in Fig. 1. From experiment LW1 carried out at the beginning of the incubation to LW2, which was carried out 16 h later, the PHM production declined by almost 90 %, although both incubations were performed under the same conditions (+light, +water). Consistently, the production rates determined for the second specimen of L. digitata (+light, –water) were in the same range as those at the beginning of the first incubation (LW1), but enhanced by 85 % relative to the production rates observed for specimen 1 under the same conditions 20 h after the start of the incubations (LNW). This strong temporal trend precludes any assessment of the incubation conditions on the production and release of the PHMs. The reason for this decline in production is not clear. It may be related to changes in the activity of bromoperoxidases, a decreasing production of PHM precursors, reduced oxidative stress, or the build-up of substrates that can be halogenated but compete with PHM formation. However, our results corroborate those of Leedham et al.,[25] who reported 3- to 10-fold higher production rates from 4-h incubations as compared with 24-h incubations.
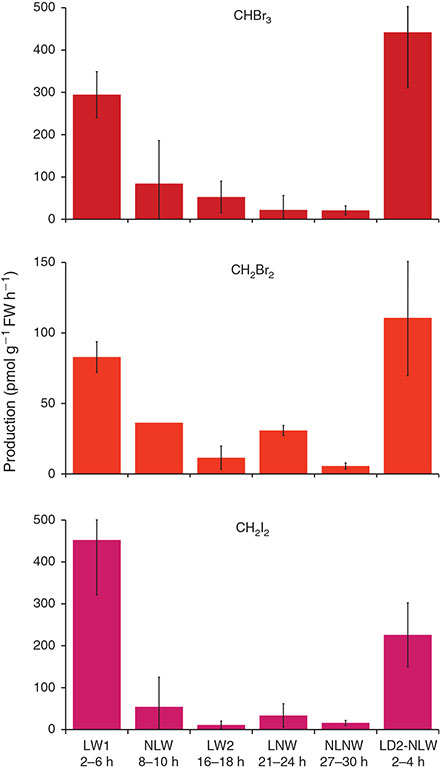
|
Total halocarbon production in the Fucus vesiculosus incubation at the IFBM was 15.6 pmol g–1 FW h–1 (±6.4), being less than 3 % of total halocarbon production by L. digitata. This is consistent with differences in halocarbon production previously reported for several Fucus and Laminaria species.[22,25] As for L. digitata, CHBr3 and CH2I2 were the most abundant compounds in the Fucus incubations, with average production rates of 7.2 and 5.9 pmol g–1 FW h–1 respectively. Other PHMs produced in sufficient amounts for carbon isotope determination were CH2Br2 and CH2Cl2. The 24-h incubations carried out at the IOW revealed a similar spectrum of halocarbons (Table S1). In addition to the above-mentioned compounds, mixed bromo-chlorocarbons were present in the incubations with H2O2. The halocarbon production of Fucus serratus was comparable with that of F. vesiculosus within a factor of 2.2. The total halocarbon production was below 1 pmol g–1 FW h–1 in the absence of H2O2 and increased to 8.0 pmol g–1 FW h–1 (F. vesiculosus) and 17.8 pmol g–1 FW h–1 (F. serratus) in the presence of H2O2. The highly elevated H2O2 levels in these experiments (~5 mmol L–1) may have fostered extracellular PHM production. However, Lin and Manley[32] reported a mean CH2Br2 : CHBr3 production ratio of 0.02. Another study[44] reported no CH2Br2 production from DOM for 28 DOM model components. In line with previous water chlorination studies, these results suggest dihalomethanes (DHMs) are a minor product of DOM halogenation. In our H2O2 experiments, CH2Br2 was the second most abundant PHM, with an average CH2Br2 : CHBr3 production ratio of 0.2 being typical for seaweeds. We thus conclude that the addition of H2O2 primarily triggered intracellular PHM formation. The total halocarbon production by F. vesiculosus in the IOW incubations in the absence of H2O2 is almost two orders of magnitude smaller than in the IFBM experiments. We can partly attribute this to the different incubation duration of 24 h (IOW) and 3 h (IFBM). Other factors that may account for the different production rates are the pH of the seawater (IFBM, 8.1; IOW, ~7), the sampling location and the sampling season. However, in summary, the production rates and their variability found here (Tables S1, S4, S5) fit in the range of previous studies,[25] with the IOW production rates being at the lower end and the IFBM production rates being in the middle to upper range.
Carbon isotope ratios
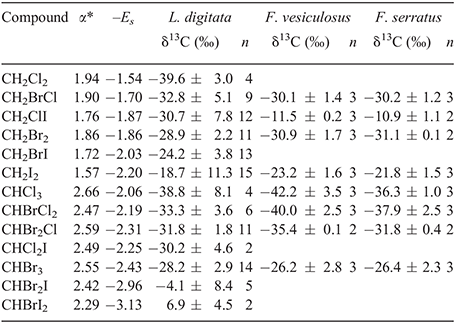
In the Z. noltii incubations, the δ13C of CHBr3 corrected for the seawater contribution ranged from –3.5 to –11.2 ‰, with a mean of 7.0 ‰ (±2.7) (Table S5). CHBr3 was depleted in 13C during the night when production was low and showed an enrichment in 13C over the day when production was elevated. In comparison with this, available field data from seagrass-dominated coastal sites suggest somewhat depleted δ13C values for CHBr3 from Z. noltii communities ranging from –8 to –18‰.[26,27] Substantially enriched δ13C values of –0.2 ‰ (±1.9) were observed for CHBr3 in the seawater controls performed along with the seagrass incubations (Table S5). In the Z. noltii incubations, the average stable carbon isotopic composition of the macroalgae-derived PHMs ranged from –38.7 ‰ (±3.5) (CH2Cl2) to 6.9 ‰ (±4.5) (CHBrI2) in the L. digitata incubations and from –34.9 ‰ (±4.1) (CH2Cl2) to –10.1 ‰ (CH2ClI) for F. vesiculosus in the IFBM incubations (Table S4) The δ13C of CHBr3 was –29.9 ‰ (±3.8) for L. digitata and –23.1 ‰ (±2.6) for F. vesiculosus. In the IOW incubations, δ13C values for F. vesiculosus ranged from –42.2 ‰ (±3.5) (CHCl3) to –11.5 ‰ (±0.2) (CH2ClI) and those for F. serrata ranged from –36.3 ‰ (±1.0) (CHCl3) to –10.9 ‰ (±0.5) (CH2ClI) (Table S2). These values were obtained during incubations in the presence of light and H2O2. In the incubations without additional H2O2, the carbon isotope ratios could only be determined for CHBr3 (average –15.8 ‰, ±2.8) and for CH2Br2 (average –19.1 ‰, ±0.9). Both were enriched in 13C by 10.5 and 11.4 ‰ compared with the incubation in the presence of H2O2.
PHMs can also be formed extracellularly from DOM.[32] This background may confound the isotopic composition of PHMs of macrophyte incubations in the present study. Indeed, the δ13C of CHBr3 in the seawater controls performed along with the seagrass incubations indicates an enrichment in 13C for the non-macrophyte-derived CHBr3. However, currently we cannot unambiguously attribute this enrichment to extracellular CHBr3 production. The seagrass data have been corrected for this background production. However, it is possible that the background production was higher in the presence of seagrass in comparison with the control experiments owing to higher levels of extracellular bromoperoxidases. We cannot fully rule out a background contribution for the macroalgae incubations because release of bromoperoxidases from the incubated macroalgae may have stimulated extracellular PHM production that is not captured by the procedural blanks. For the IFBM macroalgae experiments carried out in the absence of water, any PHM contribution from DOM is very unlikely. The maximum bromoform production rate from DOM reported by Lin was 0.48 pmol L–1 h–1,[32] equalling an extracellular bromoform production of ~2.5 pmol h–1 (60 pmol day–1) in 5-L incubations. These experiments were carried out with at elevated bromide concentrations and very high bromoperoxidase levels and thus provide an upper limit for extracellular bromoform production. During the IFBM incubations, total bromoform production in the presence of water ranged from 2400 to 49 200 pmol h–1. In the IOW experiments, total bromoform production ranged from 17 000 to 64 000 pmol in the presence of H2O2 and from 96 to 185 pmol in the absence of H2O2. Thus, except for the latter experiments, we can safely assume a negligible contribution (<4 %) from extracellular bromoform production and attribute the isotopic composition of the PHMs to the incubated macrophytes. The data from the IOW experiments in the absence of H2O2 have to be taken with care. Our data from the L. digitata incubation revealed no clear imprint of the incubation conditions on the isotopic composition of the PHMs. Thus, we cannot currently address how different environmental conditions may affect the isotopic composition of the PHMs.
The data rather suggest a tendency towards depletion in δ13C with increasing PHM production following a logarithmic trend, as shown for CHBr, CH2Br2 and CH2I2 in Fig. 2. For CHBr3 (R2 = 0.70, n = 34) and for CH2Br2 (R2 = 6.0, n = 29), this tendency was observed among all macroalgae incubations regardless of the incubation conditions. The δ13C values of CHBr3 from the seagrass incubations and the concurrent seawater controls do not fit this trend. They are more enriched in 13C and as mentioned before, they show a tendency towards more enriched values during daytime when production was higher. However, this tendency has to be taken with care because it may also result from unaccounted contributions of CHBr3 enriched in 13C from the sea water over the course of the day. Typical 13C values of bulk biomass are –11 ‰ (±2) for Z. noltii[45] and range from –15 to 20 ‰ for L. digitata.[46] Thus, differences in the substrates isotope composition may only partly account for the observed differences in the δ13C of CHBr3 from seagrass and macroalgae. Furthermore, the bulk δ13C values suggest an inverse apparent kinetic isotope effect (AKIE, enrichment in the product relative to the substrate) for CHBr3 in the seagrass incubations but a normal AKIE in the macroalgae incubations. The latter is further supported by the reversed relationship between the δ13C and the production rates. The opposite tendency in the seagrass incubations points to an inverse AKIE. Notably, the formation of chloroform on HOCl treatment of DOM model components at a typical seawater pH of 8 revealed a normal isotope effect for resorcinol, acetophenone, acetylacetone and 1,1,1-trichloropropanone, whereas phenol and 2,4,6-trichlorophenol showed an inverse isotope effect.[47] For phenol, this was confirmed in a follow-up study.[48] The latter study further revealed a normal carbon isotope effect for chloroform from propanone. In summary, these data suggest an opposed AKIE for ketone and phenolic moieties.[47] As outlined earlier, ketone moieties have been suggested as a substrate for the enzymatic halogenation in macroalgae. Seagrasses, however, produce lignins, making phenolic moieties likely PHM precursors in the seagrass incubations.[49] Thus, differences in the PHM precursors may further account for the different isotopic composition of CHBr3 from seagrass and macroalgae.
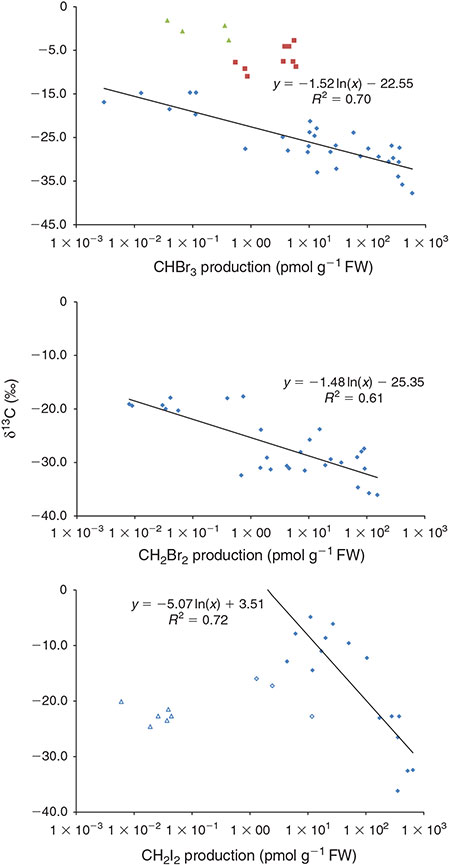
|
In contrast to CHBr3 and CH2Br2, the δ13C values of CH2I2 do not show a common trend among all macroalgae experiments but a significant correlation for L. digitata (R2 = 0.72, n = 16) Here, better fit is obtained using a linear regression (R2 = 0.83). F. vesiculosus shows a similar tendency but with overall lower production rates in the IFBM experiments. The differences between L. digitata and F. vesiculosus in the IFBM experiments may reflect the higher iodine accumulation in Laminariales as compared with Fucales.[50,51] With CHBr3 and CH2Br2 showing a common trend among all incubations, the differences between the IOW and IFBM experiments may also reflect differences in the availability or uptake of iodine.
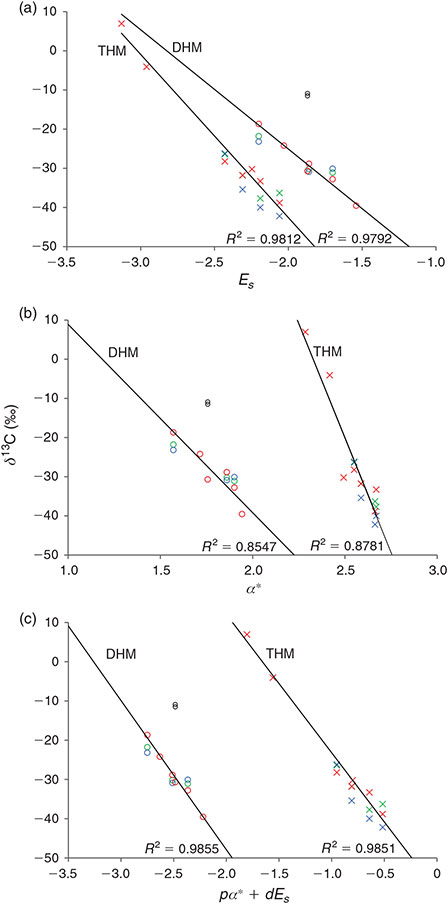
Our data set revealed a systematic imprint of the halogen substituents on the isotopic composition of the PHMs, i.e. enrichment in 13C from chloro- over bromo- to iodo-substituents (Table 1). The influence of substituents on the kinetics of chemical reactions can be quantitatively described by linear free-energy relationships (LFER).[52–54] Indeed, we found that the isotopic composition of the di- and trihalomethanes (THMs) from the L. digitata incubations are well correlated with Taft’s steric parameters (Es) (Fig. 3a) and polar parameter (α*) (Fig. 3b). The Taft equation is an empirical relation for aliphatic compounds separating the substituents’ effect on the rate constant of a reaction into a polar and steric component[55]:
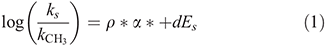
where log(ks/kCH3) is the ratio of the rate of the substituted reaction compared with the reference reaction (e.g. with CH3 as substituent), α* is the polar substituent constant, which describes field and inductive effects of the substituent, Es is the steric substituent constant, ρ* is the sensitivity factor for the reaction to polar effects and d (normally written as δ) is the sensitivity factor for the reaction to steric effects. Taft’s parameter for the chlorine- and bromine-containing compounds were taken from Glezer et al.[56] and those for the iodinated compounds were calculated as described therein.
|
|
The correlation of the δ13C values of the DHMs with Taft’s parameter are:
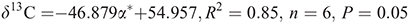
for the polar parameter and
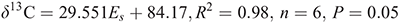
for Taft’s steric parameter. The respective correlations of the THMs are:
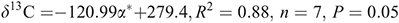
and
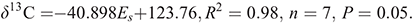
For the DHMs, the best fit to the combined Taft parameter (ρα* + dEs) is found for –0.31α* + Es (R2 = 0.99, n = 6, P = 0.05), and for the THMs, the best fit is obtained for 0.58α* + Es (R2 = 0.99 n = 7, P = 0.05) (Fig. 3c).
The observed trends in the isotopic composition of the polyhalomethanes have to be rationalised on the basis of the underlying reactions. The enzyme-catalysed halogenation involves several reaction steps and leads to a variety of products as shown in Fig. 4 for 3-oxooctanoid acid. A comprehensive explanation of the observed isotope discrimination among the PHMs would clearly require detailed information about the isotopic fractionation of each reaction step, but information on the intrinsic isotope fractionation factors for each reaction step is scarce. The substituents can in principle directly affect the intrinsic isotope fractionation of each reaction step. Further, in cases of branched reactions leading to more than one product, the isotopic composition of each product depends on the kinetic isotope effects of each reaction and the proportion of products.[57,58] Such an effect has been proposed to explain the isotope effects in the formation of chloroform from different precursors.[47] The correlation between the carbon isotope ratios of the PHMs and the Taft parameter suggests substituents effects on the relative rates being the decisive factor for the observed isotope discrimination among the PHMs rather than direct effects on the isotope fractionation. Thus, we can focus the discussion on the branching points of the reaction scheme that are the reactions of 2-halo-β-ketoacid and 1,1-dihalo-2-heptanone (Fig. 4). The 2-halo-β-ketoacid is either subject to further halogenation or decarboxylates to the respective 1-halo-2-heptanone. The decarboxylation of lactic acid is assigned with an intrinsic kinetic isotope effect (KIE) of 4.8 ‰ at the α-carbon, and for the decarboxylation of 2-benzoylpropionic acid, a 14C-KIE of 51 ‰ for the α-carbon has been reported.[59] Isotope effects in chemical reactions involving 14C are generally assumed to be 1.9 times larger than those involving 13C,[60,61] suggesting a 13C-KIE of ~27 ‰. We found no information on the carbon isotope effect of the enolisation, the rate-limiting step of the halogenation. However, for the bromination of 14C-labelled 4-nitro-4-methylstilbene, a 14C-KIE of 3.5 ‰ (±0.3) has been reported for the β-carbon,[62] suggesting a fairly small isotope effect for electrophilic halogenations. In any case, the isotope effect of the decarboxylation will induce an opposite isotope effect in the dihalogenated β-ketoacids, with the magnitude depending on the proportion of product. The rate-limiting step in the halogenation is the enolisation. The electron-withdrawing effect of the halogen substituent stabilises the enolate form and thus increases the relative rate of halogenation. The electron-withdrawing effect depends on the electronegativity of the substituents and can be described in terms of Taft’s polar parameter and increases in the order: I < Br < Cl, making the 2-chloro-β-ketoacid a more favourable substrate for halogenation relative to the 2-iodo-β-ketoacid. In addition, the reaction of the chlorinated substrate may be favoured for steric reasons.

|
As evident from the experiments of Beissner et al.,[30] the halogen substituent also promotes the decarboxylation. For the decarboxylation of trihaloacetic acids, a reverse substituent effect was observed, with triiodoacetic acids decarboxylating more readily than trifluoroacetic acid. The decarboxylation rate constants were found to correlate well with Taft’s steric parameter Es.[63] Notably, the hydrolysis rate constants of the THMs increasing from chloro- to iodo-substituents[64] also correlate with Taft’s steric parameter for the THMs. This may rather reflect the ability of the halogen substituents to stabilise the intermediate carbanion than be a steric effect. Thus, interpretation of the correlation in terms of steric effects has to be done with caution. Nevertheless, if such reversal of the substituents’ effect also holds true for the decarboxylation of the monohalogenated β-ketoacids, it would favour the decarboxylation of the iodine adduct relative to the chlorine adduct. In summary, we hypothesise that a chlorine substituent relative to an iodine substituent favours further halogenation, leading to the PHMs. However, an iodine substituent relative to a chlorine substituent favours decarboxylation over halogenation. As a consequence, the iodinated products are enriched in 13C relative to the chlorinated products (Table 1). The same principle applies to the reactions of 1,1-dihalo-2-heptanones. They may be further halogenated to the trihaloketones, yielding THMs, be hydrolysed to the respective DHMs or undergo further side reactions. Any isotope effect assigned to these further side reactions will concordantly affect the isotopic composition of the di- and trihalomethanes. The decisive reaction steps with respect to the isotope discrimination between the di- and trihalomethanes are thus hydrolysis and further halogenation. Again, the more electronegative chlorine substituents will enhance the halogenation relative to iodine substituents and as before, steric effects may also affect the relative reaction rates. The competing hydrolysis proceeds by the formation of an intermediate carbanion. We hypothesise that, in analogy to the THMs, iodine substituents can more readily stabilise the carbanion than chlorine substituents, and thus expect the 1,1-diiodo-2-heptanone to be hydrolysed more readily than the 1,1-dichloro-2-heptanone. The hydrolysis of 1,1,1-trichloropropanone is assigned a KIE of 14 ‰.[47] Any carbon isotope effect assigned to the hydrolysis will therefore tentatively lead to enrichment in the 13C of the THMs relative to the DHMs, with this enrichment depending on the ratio of the THMs to DHMs and the respective isotope effects. Our data from the L. digitata incubation show only low enrichment in the average isotopic composition of CHBr3 (–28.2 ‰) relative to CH2Br2 (–28.8 ‰). The product ratio of both is 5 : 1. For this case, we assume that the isotope effects of both reactions and the product yields level each other out, resulting in negligible isotope discrimination between both compounds. In line with this, a somewhat larger isotope discrimination but a closer product ratio of 2 : 1 is observed for chloroform (–37.0 ‰) and dichloromethane (–38.8 ‰). Much more pronounced isotopic discrimination occurs between the iodine-containing THMs and DHMs. CHBr2I (–4.1 ‰) and CHBrI2 (+6.9 ‰) are both strongly enriched relative to the dihalogenated products of their precursors CH2Br2 (–28.8 ‰), CH2BrI (–24.2 ‰) and CH2I2 (–18.6 ‰). In this case, the relative yield (compared with the dihalogenated products) is only 5 % for CHBr2I and less than 1 % for CHBrI2 and could thus easily account for the large isotope discrimination. We have to emphasise that the empiric correlations with Taft’s parameter found here depend on the specific conditions and thus cannot be generalised. Furthermore, different precursors having different apparent KIEs[47] may be involved in PHM formation. As outlined earlier, a generalised concept would clearly require detailed information about the precursors and the isotopic fractionation of each reaction step and is thus beyond the scope of the present work.
The iodine-containing PHMs are subject to rapid photolysis in aqueous solutions, with reported photolysis lifetimes ranging from 9.5 min for CH2I2 to 9 h for CH2ClI under mid-latitude summer conditions.[34] Any secondary reaction of the PHMs during the incubation should alter their isotopic composition and result in a deviation from the correlation with the Taft parameter. Surprisingly, a substantial deviation pointing towards secondary degradation is only observed in the δ13C values of CH2ClI (–11.5 and –10.9‰) obtained from F. vesiculosus and F. serratus in the 24-h incubation of the IOW, whereas the isotopic composition of CH2I2 having a photolysis lifetime of only 9.5 min fits well into the correlation with the Taft parameter. We thus can exclude any photochemical degradation altering the isotopic composition of the iodine-containing PHMs during these experiments. Instead, the presence of the two different halogens chlorine and iodine may facilitate nucleophilic reactions[33] of CH2ClI, leading to its partial degradation in the 24-h incubations.
In analogy to the isotope discrimination between the different PHMs, we can attribute the observed depletion in δ13C with increasing PHM production to the PHM yield relative to the halogenated products of the competing reaction pathways, in particular 1-halo-2-heptanone. A low product ratio of PHMs to 1-halo-2-heptanones is expected to result in fairly enriched δ13C of the PHMs. Conversely, a high product ratio will result in a relative 13C depletion of the PHMs. This may also explain the fairly loose correlation between the isotopic composition of the PHMs and their production rate because the latter is also influenced by other factors, such as overall enzyme activity. PHMs may also be formed from DOM in the presence of free extracellular bromoperoxidases.[32] Such a mechanism could account for the blank production in the filtered seawater controls in the seagrass experiments, though we cannot fully rule out other sources. In the study of Beissner et al.,[30] the product ratio was strongly pH-dependent. At pH 7.6, CHBr3 was the most abundant halogenated compound, accounting for ~60 % of the halogenated products. In contrast, at pH 8.0, which is close to the typical seawater pH, 1-bromo-2-heptanone was by far the main product (~90 %), whereas the relative CHBr3 yield was less than 5 %. This may explain the strong enrichment in 13C of CHBr3 (δ13C = +0.9 ‰, ±1.5) in the filtered seawater controls.
The δ13C values of CHBr3 ranged from –36.8 to 23.9 ‰ for L. digitata, –26.2 to –14.1 ‰ for F. vesiculosus and F. serratus, –11.2 to –3.5‰ for Z. noltii, and from –2.7 to 1.6 ‰ in the filtered sea water. In comparison with the incubation data, available field data from a seagrass-dominated coastal sites suggest somewhat depleted δ13C values for CHBr3 from Z. noltii communities between –8 and –18 ‰.[26,27] This difference may be related to contributions from other sources or reflect different haloform yields as described above. In any case, the isotopic source signature of seagrass-derived CHBr3 seems to be highly variable. THMs formed during water chlorination, the main anthropogenic THM source, have an isotopic source signature of –31.1 ‰ (±0.9),[65] overlapping with the source signature of L. digitata. Despite the large variability in the isotopic composition of PHMs at the species level, the isotopic source signature may still be useful to discriminate between different sources, when used carefully. An accompanying study in this issue[66] reports δ13C values of CHBr3 as a valuable tool to discriminate between phytoplankton and macroalgae sources. The mean δ13C value of CHBr3 was –12 ‰ (±4) from a diatom-dominated phytoplankton bloom, but –26 ‰ (±2) from a site strongly influenced by macroalgae. Thus, the latter values fit very well with our data derived from the macroalgae incubations. The δ13C of naturally produced CHCl3 in soil gas ranges from –22.8 to 26.2 ‰, resembling the isotopic composition of soil organic matter.[67] In contrast, industrially produced chloroform, presumably originating from the chlorination of methane, shows more depleted δ13C values ranging from –43.2 to –63.6 ‰.[67] CHCl3 formed on chlorination of Lake Zurich water showed a δ13C of –37 ‰,[47] being in the range reported here. Reported δ13C values of tropospheric CHCl3 mainly from marine-influenced sites are in the range of –37 ‰ (±5),[41] resembling the marine source signature. To this end, the available isotopic data for CHCl3 suggest that carbon isotope ratios can be used to discriminate between terrestrial, aquatic and industrial sources.
Conclusions
To the best of our knowledge, this is the first systematic study assessing species-specific carbon isotope signatures of naturally produced PHMs from marine macrophytes. Though variable, the isotopic composition of PHMs may provide useful additional information to discriminate between different PHM sources such as macroalgae, seagrass or abiotic formation from DOM. Further isotopic source signatures, in particular from phytoplankton sources, as well as more information on the temporal and spatial variability including isotopic information on the substrate undergoing halogenation are required to establish a robust isotopic data set that can be used for source assignment. Further, the effect of environmental conditions has to be elucidated in future studies.
We found a systematic effect of the halogen substituents on the isotopic composition of the PHMs that could be rationalised using LFER and is attributed to isotopic fractionation at the branching points of the enzyme-catalysed halogenation. On the same basis, we rationalised the observed trend towards more depleted δ13C values with increasing production rates to the PHM yield relative to other halogenated products. We are, currently, not aware of any previous study rationalising isotope effects on the basis of LFER. The dependence of the isotopic source signature on the halogen substituents allows secondary reactions of the PHMs to be addressed, as shown here for CH2ClI. Thus, carbon stable isotope analysis of dissolved PHMs may improve our understanding of their chemistry. The dependence of the isotopic composition of the PHMs, in particular of CHBr3, on the relative PHM yield may make stable carbon isotope analysis a valuable tool for monitoring changes in relative PHM yields that may arise from ocean acidification, as outlined before, but this requires further substantiation from systematic studies.
Acknowledgements
We thank Ralf Lendt, Sabine Beckmann, Tim Eckhardt and Alexander Ohnesorge for their invaluable help with the laboratory work and acknowledge the Bundesministerium für Bildung und Forschung (BMBF) for funding of this work in the frame of the SOPRAN project (grants 03F0611E, 03F0611B and 03F0662E).
References
[1] S. C. Wofsy, M. B. McElroy, Y. L. Yung, The chemistry of atmospheric bromine. Geophys. Res. Lett. 1975, 2, 215.| The chemistry of atmospheric bromine.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE28XhtFSrurw%3D&md5=a71da6ff29465f1dbf373a5362fdec98CAS |
[2] S. Solomon, R. R. Garcia, A. R. Ravishankara, On the role of iodine in ozone depletion. J. Geophys. Res. 1994, 99, 20491.
| On the role of iodine in ozone depletion.Crossref | GoogleScholarGoogle Scholar |
[3] W. T. Sturges, D. E. Oram, L. J. Carpenter, S. A. Penkett, A. Engel, Bromoform as a source of stratospheric bromine. Geophys. Res. Lett. 2000, 27, 2081.
| Bromoform as a source of stratospheric bromine.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXlslalt70%3D&md5=2d09400e295300baa825e406d3c29230CAS |
[4] R. von Glasow, Atmospheric chemistry: sun, sea and ozone destruction. Nature 2008, 453, 1195.
| Atmospheric chemistry: sun, sea and ozone destruction.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXnslGksrc%3D&md5=978a03ebd2e75b67f579d7be52b0dc9cCAS | 18580939PubMed |
[5] R. Sander, R. Vogt, G. W. Harris, P. J. Crutzen, Modelling the chemistry of ozone, halogen compounds, and hydrocarbons in the Arctic troposphere during spring. Tellus B Chem. Phys. Meterol. 1997, 49, 522.
| Modelling the chemistry of ozone, halogen compounds, and hydrocarbons in the Arctic troposphere during spring.Crossref | GoogleScholarGoogle Scholar |
[6] R. P. Mason, G. R. Sheu, Role of the ocean in the global mercury cycle. Global Biogeochem. Cycles 2002, 16, 1093.
| Role of the ocean in the global mercury cycle.Crossref | GoogleScholarGoogle Scholar |
[7] L. J. Carpenter, Iodine in the marine boundary layer. Chem. Rev. 2003, 103, 4953.
| Iodine in the marine boundary layer.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXnt1Kltbk%3D&md5=80f7191f08c71ef5800c9171e187eff1CAS | 14664638PubMed |
[8] B. Quack, D. W. R. Wallace, Air–sea flux of bromoform: controls, rates, and implications. Global Biogeochem. Cycles 2003, 17, 1023.
| Air–sea flux of bromoform: controls, rates, and implications.Crossref | GoogleScholarGoogle Scholar |
[9] J. H. Butler, D. B. King, J. M. Lobert, S. A. Montzka, S. A. Yvon-Lewis, B. D. Hall, N. J. Warwick, D. J. Mondeel, J. W. Aydin, Elkins, Oceanic distributions and emissions of short-lived halocarbons. Global Biogeochem. Cycles 2007, 21, GB1023.
| Elkins, Oceanic distributions and emissions of short-lived halocarbons.Crossref | GoogleScholarGoogle Scholar |
[10] L. J. Carpenter, C. E. Jones, R. M. Dunk, K. E. Hornsby, J. Woeltjen, Air–sea fluxes of biogenic bromine from the tropical and North Atlantic Ocean. Atmos. Chem. Phys. 2009, 9, 1805.
| Air–sea fluxes of biogenic bromine from the tropical and North Atlantic Ocean.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXntFKgs7o%3D&md5=02044f133528a6c9729b8f06e7ebf467CAS |
[11] C. J. Palmer, C. J. Reason, Relationships of surface bromoform concentrations with mixed-layer depth and salinity in the tropical oceans. Global Biogeochem. Cycles 2009, 23, GB2014.
| Relationships of surface bromoform concentrations with mixed-layer depth and salinity in the tropical oceans.Crossref | GoogleScholarGoogle Scholar |
[12] L. M. O’Brien, N. R. P. Harris, A. D. Robinson, B. Gostlow, N. Warwick, X. Yang, J. A. Pyle, Bromocarbons in the tropical marine boundary layer at the Cape Verde Observatory – measurements and modelling. Atmos. Chem. Phys. 2009, 9, 9083.
| Bromocarbons in the tropical marine boundary layer at the Cape Verde Observatory – measurements and modelling.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhs1Gjur4%3D&md5=f737240938b04c76bca017c93d386d9fCAS |
[13] Q. Liang, R. S. Stolarski, S. R. Kawa, J. E. Nielsen, A. R. Douglass, J. M. Rodriguez, D. R. Blake, E. L. Atlas, L. E. Ott, Finding the missing stratospheric Bry: a global modeling study of CHBr3 and CH2Br2. Atmos. Chem. Phys. 2010, 10, 2269.
| Finding the missing stratospheric Bry: a global modeling study of CHBr3 and CH2Br2.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXlslOqtLw%3D&md5=44fe3425fade90d1aafd042f92501863CAS |
[14] J. A. Pyle, M. J. Ashfold, N. R. P. Harris, A. D. Robinson, N. J. Warwick, G. D. Carver, B. Gostlow, L. M. O’Brien, A. J. Manning, S. M. Phang, S. E. Yong, K. P. Leong, E. H. Ung, S. Ong, Bromoform in the tropical boundary layer of the Maritime Continent during OP3. Atmos. Chem. Phys. 2011, 11, 529.
| Bromoform in the tropical boundary layer of the Maritime Continent during OP3.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXkvFClsbw%3D&md5=b83b452d0b8b1c2baed68606498f43caCAS |
[15] S. A. Montzka, S. Reimann, A. Engel, K. Krüger, S. O'Doherty, W. T. Sturges, D. R. Blake, M. Dorf, P. J. Fraser, L. Froidevaux, K. Jucks, K. Kreher, M. J. Kurylo, A. Mellouki, J. Miller, O. -J. Nielsen, V. L. Orkin, R. G. Prinn, R. Rhew, M. L. Santee, and D. P. Verdonik, Chapter 1. Ozone-depleting substances (ODSs) and related chemicals, in Scientific Assessment of Ozone Depletion: 2010, Global Ozone Research and Monitoring Project - Report 52 2011 (World Meteorological Organization: Geneva, Switzerland).
[16] P. M. Gschwend, J. K. MacFarlane, K. A. Newman, Volatile halogenated organic compounds released to seawater from temperate marine macroalgae. Science 1985, 227, 1033.
| Volatile halogenated organic compounds released to seawater from temperate marine macroalgae.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2MXhsFCnsrc%3D&md5=18174824e00a0911a3e756c90e0b947eCAS | 17794227PubMed |
[17] S. L. Manley, M. N. Dastoor, Methyl halide (CH3X) production from the giant kelp, Macrocystis, and estimates of global CH3X production by kelp. Limnol. Oceanogr. 1987, 32, 709.
| Methyl halide (CH3X) production from the giant kelp, Macrocystis, and estimates of global CH3X production by kelp.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2sXlslantbo%3D&md5=c697f0bea6c2de37dd68b33e05302cbbCAS |
[18] S. L. Manley, M. N. Dastoor, Methyl iodide (CH3I) production by kelp and associated microbes. Mar. Biol. 1988, 98, 477.
| Methyl iodide (CH3I) production by kelp and associated microbes.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL1cXlsVKgurk%3D&md5=6db7ec479be6b87771fc49b7a59c8fc0CAS |
[19] S. L. Manley, K. Goodwin, W. J. North, Laboratory production of bromoform, methylene bromide, and methyl iodide by macroalgae and distribution in near-shore southern California waters. Limnol. Oceanogr. 1992, 37, 1652.
| Laboratory production of bromoform, methylene bromide, and methyl iodide by macroalgae and distribution in near-shore southern California waters.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3sXlt1CmsLk%3D&md5=0178dc9446f6ffd5c45a812bc1c8a7abCAS |
[20] P. D. Nightingale, G. Marlin, P. S. Liss, Production of chloroform and other low-molecular-weight halocarbons by some species of macroalgae. Limnol. Oceanogr. 1995, 40, 680.
| Production of chloroform and other low-molecular-weight halocarbons by some species of macroalgae.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXos1GmsLs%3D&md5=2cb64aec84570c06e6f20d85c94479fdCAS |
[21] B. Giese, F. Laturnus, F. C. Adams, C. Wiencke, Release of volatile iodinated C1–C4 hydrocarbons by marine macroalgae from various climate zones. Environ. Sci. Technol. 1999, 33, 2432.
| Release of volatile iodinated C1–C4 hydrocarbons by marine macroalgae from various climate zones.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXjsFCqtr8%3D&md5=8a2a763149b29808130309ee8dd79166CAS |
[22] L. J. Carpenter, G. Malin, P. S. Liss, F. C. Küpper, Novel biogenic iodine-containing trihalomethanes and other short-lived halocarbons in the coastal east Atlantic. Global Biogeochem. Cycles 2000, 14, 1191.
| Novel biogenic iodine-containing trihalomethanes and other short-lived halocarbons in the coastal east Atlantic.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXktVGksA%3D%3D&md5=37912ff04f8080df94e590909ccbccb9CAS |
[23] F. Laturnus, B. Giese, C. Wiencke, F. C. Adams, Low-molecular-weight organoiodine and organobromine compounds released by polar macroalgae – the influence of abiotic factors. Fresenius J. Anal. Chem. 2000, 368, 297.
| Low-molecular-weight organoiodine and organobromine compounds released by polar macroalgae – the influence of abiotic factors.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXmtFGmurg%3D&md5=c4e557a6fc4f59c3a5007ce27a89e234CAS | 11220596PubMed |
[24] J. M. Baker, W. T. Sturges, J. Sugier, G. Sunnenberg, A. A. Lovett, C. E. Reeves, P. D. Nightingale, S. A. Penkett, Emissions of CH3Br, organochlorines, and organoiodines from temperate macroalgae. Chemosphere, Glob. Chang. Sci. 2001, 3, 93.
| Emissions of CH3Br, organochlorines, and organoiodines from temperate macroalgae.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXhtlGktbk%3D&md5=368d96054e64f9d048c4e1f0ab21502cCAS |
[25] E. C. Leedham, C. Hughes, F. S. L. Keng, S. M. Phang, G. Malin, W. T. Sturges, Emission of atmospherically significant halocarbons by naturally occurring and farmed tropical macroalgae. Biogeosciences 2013, 10, 3615.
| Emission of atmospherically significant halocarbons by naturally occurring and farmed tropical macroalgae.Crossref | GoogleScholarGoogle Scholar |
[26] I. Weinberg, E. Bahlmann, W. Michaelis, R. Seifert, Determination of fluxes and isotopic composition of halocarbons from seagrass meadows using a dynamic flux chamber. Atmos. Environ. 2013, 73, 34.
| Determination of fluxes and isotopic composition of halocarbons from seagrass meadows using a dynamic flux chamber.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXnsVyktr4%3D&md5=c72566fc4073bec8916d38258f26ff00CAS |
[27] I. Weinberg, E. Bahlmann, T. Eckhardt, W. Michaelis, R. Seifert, A halocarbon survey from a seagrass-dominated subtropical lagoon, Ria Formosa (Portugal): flux pattern and isotopic composition. Biogeosciences Discuss. 2014, 11, 10605.
| A halocarbon survey from a seagrass-dominated subtropical lagoon, Ria Formosa (Portugal): flux pattern and isotopic composition.Crossref | GoogleScholarGoogle Scholar |
[28] J. C. Morris, B. Baum, Precursors and Mechanisms of Haloform Formation in the Chlorination of Water Supplies, in Water Chlorination: Chemistry, Environmental Impact and Health Effects, Volume 2 (Eds R. L. Jolley, H. Gorchex and D. H. Hamilton Jr) 1978, pp. 29–48 (Ann Arbor Science Publishers: Ann Arbor, MI).
[29] R. Theiler, J. C. Cook, L. P. Hager, J. F. Siuda, Halohydrocarbon synthesis by bromoperoxidase. Science 1978, 202, 1094.
| Halohydrocarbon synthesis by bromoperoxidase.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE1MXot1yitg%3D%3D&md5=b276e0d79bde079a4b17ddedaf89441fCAS | 17777960PubMed |
[30] R. S. Beissner, W. J. Guilford, R. M. Coates, L. P. Hager, Synthesis of brominated heptanones and bromoform by a bromoperoxidase of marine origin. Biochemistry 1981, 20, 3724.
| Synthesis of brominated heptanones and bromoform by a bromoperoxidase of marine origin.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL3MXktlOqsLc%3D&md5=f0b678843f0fc4ded45d82c0ab3af643CAS | 7272274PubMed |
[31] S. Manley, Phytogenesis of halomethanes: a product of selection or a metabolic accident? Biogeochemistry 2002, 60, 163.
| Phytogenesis of halomethanes: a product of selection or a metabolic accident?Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XmtlGgtLw%3D&md5=9dbdb5c6112be59d3140c0295d609fefCAS |
[32] C. Y. Lin, S. L. Manley, Bromoform production from seawater treated with bromoperoxidase. Limnol. Oceanogr. 2012, 57, 1857.
| Bromoform production from seawater treated with bromoperoxidase.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXptlCgsg%3D%3D&md5=26baf1d0d724aa2e3c7b39440ce53eddCAS |
[33] K. Ballschmiter, Pattern and sources of naturally produced organohalogens in the marine environment: biogenic formation of organohalogens. Chemosphere 2003, 52, 313.
| Pattern and sources of naturally produced organohalogens in the marine environment: biogenic formation of organohalogens.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXjsVGnsLg%3D&md5=61e3cf9aaf5aa9aa8ec0b25ff4eefa02CAS | 12738255PubMed |
[34] C. E. Jones, L. J. Carpenter, Solar photolysis of CH2I2, CH2ICl, and CH2IBr in water, saltwater, and seawater. Environ. Sci. Technol. 2005, 39, 6130.
| Solar photolysis of CH2I2, CH2ICl, and CH2IBr in water, saltwater, and seawater.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXmtVWjurw%3D&md5=f37c93459f8e4c3faf302ed49ca019b6CAS | 16173573PubMed |
[35] S. E. McCauley, A. H. Goldstein, D. J. DePaolo, An isotopic approach for understanding the CH3Br budget of the atmosphere. Proc. Natl. Acad. Sci. USA 1999, 96, 10006.
| An isotopic approach for understanding the CH3Br budget of the atmosphere.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXlvFehu7c%3D&md5=327f55e78beeadc46d4922e0b7678aabCAS | 10468552PubMed |
[36] D. B. Harper, J. T. G. Hamilton, V. Ducrocq, J. T. Kennedy, A. Downey, R. M. Kalin, The distinctive isotopic signature of plant-derived chloromethane: possible application in constraining the atmospheric chloromethane budget. Chemosphere 2003, 52, 433.
| The distinctive isotopic signature of plant-derived chloromethane: possible application in constraining the atmospheric chloromethane budget.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXjsVGnsbk%3D&md5=d276cbe2f03eaedf78043d00ad18fc2bCAS | 12738266PubMed |
[37] F. Keppler, D. B. Harper, T. Röckmann, R. M. Moore, J. T. G. Hamilton, New insight into the atmospheric chloromethane budget gained using stable carbon isotope ratios. Atmos. Chem. Phys. 2005, 5, 2403.
| New insight into the atmospheric chloromethane budget gained using stable carbon isotope ratios.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXht1Kgs7rM&md5=69b9b7a8984e8a5964cd3a15e1365070CAS |
[38] T. Saito, Y. Yokouchi, Stable carbon isotope ratio of methyl chloride emitted from glasshouse-grown tropical plants and its implication for the global methyl chloride budget. Geophys. Res. Lett. 2008, 35, L08807.
| Stable carbon isotope ratio of methyl chloride emitted from glasshouse-grown tropical plants and its implication for the global methyl chloride budget.Crossref | GoogleScholarGoogle Scholar |
[39] N. R. Auer, B. U. Manzke, D. E. Schulz-Bull, Development of a purge-and-trap continuous flow system for the stable carbon isotope analysis of volatile halogenated organic compounds in water. J. Chromatogr. A 2006, 1131, 24.
| Development of a purge-and-trap continuous flow system for the stable carbon isotope analysis of volatile halogenated organic compounds in water.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XhtVGjtbjM&md5=84abc41d533351d046b83e14a04522f9CAS | 16899248PubMed |
[40] N. R. Auer, Stable carbon isotope analysis of volatile halogenated organic compounds (VHOCs) in the marine environment 2005, Ph.D. thesis, University of Rostock, Rostock, Germany.
[41] E. Bahlmann, I. Weinberg, R. Seifert, C. Tubbesing, W. Michaelis, A high-volume sampling system for isotope determination of volatile halocarbons and hydrocarbons. Atmos. Meas. Tech. 2011, 4, 2073.
| A high-volume sampling system for isotope determination of volatile halocarbons and hydrocarbons.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XisVGmu7Y%3D&md5=2281cd7f1b4fa5d2e1639ae72e2999f4CAS |
[42] A. Ekdahl, M. Pedersén, K. Abrahamsson, A study of the diurnal variation of biogenic volatile halocarbons. Mar. Chem. 1998, 63, 1.
| A study of the diurnal variation of biogenic volatile halocarbons.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXnslKitLY%3D&md5=76ee28e7953758315dedce32df191193CAS |
[43] K. Abrahamsson, K.-S. Choo, M. Pedersén, G. Johansson, P. Snoeijs, Effects of temperature on the production of hydrogen peroxide and volatile halocarbons by brackish-water algae. Phytochemistry 2003, 64, 725.
| Effects of temperature on the production of hydrogen peroxide and volatile halocarbons by brackish-water algae.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXnt1yitb0%3D&md5=a9cfd7a731a99f6d89b245acc1e024ffCAS | 13679095PubMed |
[44] Y. Liu, D. C. O. Thornton, T. S. Bianchi, W. A. Arnold, M. R. Shields, J. Chen, S. A. Yvon-Lewis, Dissolved organic matter composition drives the marine production of brominated very short-lived substances. Environ. Sci. Technol. 2015, 49, 3366.
| Dissolved organic matter composition drives the marine production of brominated very short-lived substances.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXjsFCgs7s%3D&md5=3c15952b051431f6cd57b12e445947faCAS | 25723123PubMed |
[45] H. Kennedy, J. Beggins, C. M. Duarte, J. W. Fourqurean, M. Holmer, N. Marbà, J. J. Middelburg, Seagrass sediments as a global carbon sink: isotopic constraints. Global Biogeochem. Cycles 2010, 24, GB4026.
| Seagrass sediments as a global carbon sink: isotopic constraints.Crossref | GoogleScholarGoogle Scholar |
[46] G. Schaal, P. Riera, C. Leroux, Trophic significance of the kelp Laminaria digitata (Lamour.) for the associated food web: a between-sites comparison. Estuar. Coast. Shelf Sci. 2009, 85, 565.
| Trophic significance of the kelp Laminaria digitata (Lamour.) for the associated food web: a between-sites comparison.Crossref | GoogleScholarGoogle Scholar |
[47] W. A. Arnold, J. Bolotin, U. v. Gunten, T. B. Hofstetter, Evaluation of functional groups responsible for chloroform formation during water chlorination using compound-specific isotope analysis. Environ. Sci. Technol. 2008, 42, 7778.
| Evaluation of functional groups responsible for chloroform formation during water chlorination using compound-specific isotope analysis.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXotlOmsr8%3D&md5=9fc068deb884d868f2e9722406f1e366CAS | 19031860PubMed |
[48] F. Breider, D. Hunkeler, Mechanistic insights into the formation of chloroform from natural organic matter using stable carbon isotope analysis. Geochim. Cosmochim. Acta 2014, 125, 85.
| Mechanistic insights into the formation of chloroform from natural organic matter using stable carbon isotope analysis.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXitVWju7fE&md5=3c9f10eacec4a44fd3781c1978d11c4bCAS |
[49] V. A. Klap, M. A. Hemminga, J. J. Boon, Retention of lignin in seagrasses: angiosperms that returned to the sea. Mar. Ecol. Prog. Ser. 2000, 194, 1.
| Retention of lignin in seagrasses: angiosperms that returned to the sea.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXjvVWitbc%3D&md5=bd2776a5ea15afd611375461c279ac24CAS |
[50] W. A. P. Black, Seasonal variation in chemical composition of some of the littoral seaweeds common to Scotland. Part II. Fucus serratus, Fucus vesiculosus, Fucus spiralis and Pelvetia canaliculata. J. Soc. Chem. Ind. 1949, 68, 183.
| Seasonal variation in chemical composition of some of the littoral seaweeds common to Scotland. Part II. Fucus serratus, Fucus vesiculosus, Fucus spiralis and Pelvetia canaliculata.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaH1MXks1yjtA%3D%3D&md5=56d9bdab0742baec4e3796ac89cfbbe6CAS |
[51] W. A. P. Black, The seasonal variation in chemical constitution of some of the sub-littoral seaweeds common to Scotland. Part II. Laminaria digitata. J. Soc. Chem. Ind. 1948, 67, 169.
| The seasonal variation in chemical constitution of some of the sub-littoral seaweeds common to Scotland. Part II. Laminaria digitata.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaH1MXptlU%3D&md5=0550df6e077b11f0243eddd9fa48cd19CAS |
[52] R. W. Taft, Separation of Polar, Steric and Resonance Effects in Reactivity 1956 (Wiley: New York).
[53] C. Hansch, A. Leo, R. W. Taft, A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 1991, 91, 165.
| A survey of Hammett substituent constants and resonance and field parameters.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3MXhs1ehsLo%3D&md5=5c1f3bbe1f238860ce5c0b31a0ce6598CAS |
[54] R. P. Schwarzenbach, P. M. Gschweno, D. M. Imboden, Environmental Organic Chemistry 1993 (Wiley: New York).
[55] R. W. Taft, Linear free-energy relationships from rates of esterification and hydrolysis of aliphatic and ortho-substituted benzoate esters. J. Am. Chem. Soc. 1952, 74, 2729.
| Linear free-energy relationships from rates of esterification and hydrolysis of aliphatic and ortho-substituted benzoate esters.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaG38Xls1CnsQ%3D%3D&md5=043aa67f7633721ee18779ec60be7a56CAS |
[56] V. Glezer, B. Harris, N. Tal, B. Iosefzon, O. Lev, Hydrolysis of haloacetonitriles: linear free-energy relationships, kinetics and products. Water Res. 1999, 33, 1938.
| Hydrolysis of haloacetonitriles: linear free-energy relationships, kinetics and products.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXivFKmurc%3D&md5=e09d20e1912a5df4a8da7819f420badaCAS |
[57] M. DeNiro, S. Epstein, Mechanism of carbon isotope fractionation associated with lipid synthesis. Science 1977, 197, 261.
| Mechanism of carbon isotope fractionation associated with lipid synthesis.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE2sXkvVWjsLo%3D&md5=f174d3ea2b273dda5247612c2da57164CAS | 327543PubMed |
[58] M. H. O’Leary, Carbon isotope fractionation in plants. Phytochemistry 1981, 20, 553.
| Carbon isotope fractionation in plants.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL3MXltFWlsLY%3D&md5=ba2d6bb060f3d17ce2497bbd4d9c569aCAS |
[59] E. M. Hodnett, R. L. Rowton, 14C-isotope effects in the decarboxylation of 2-benzoylpropionic acid, in Radioisotopes in the Physical Science and Industry 1962, pp. 225–234 (International Atomic Energy Agency: Vienna, Austria).
[60] P. E. Yankwich, A. L. Promislow, R. F. Nystrom, 14C and 13C intramolecular isotope effects in the decarboxylation of liquid malonic acid at 140.5°. J. Am. Chem. Soc. 1954, 76, 5893.
| 14C and 13C intramolecular isotope effects in the decarboxylation of liquid malonic acid at 140.5°.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaG2MXltF2gtA%3D%3D&md5=42be48c1c1efd1f4860fc03ac685f1c8CAS |
[61] M. J. Stern, P. C. Vogel, Relative 14C–13C kinetic isotope effects. J. Chem. Phys. 1971, 55, 2007.
| Relative 14C–13C kinetic isotope effects.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE3MXltVWltLY%3D&md5=fc3fe698dca93c38f3af83abaacf123fCAS |
[62] P. B. Kokil, A. Fry, Isotope effects and mechanism in the bromination of alpha- and beta-carbon-14 labeled 4-nitro-4′-methylstilbenes. Tetrahedron Lett. 1986, 27, 5051.
| Isotope effects and mechanism in the bromination of alpha- and beta-carbon-14 labeled 4-nitro-4′-methylstilbenes.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2sXkslOjtrs%3D&md5=a7b85dfca5882ab5f7da444500485cc7CAS |
[63] X. Zhang, R. A. Minear, Decomposition of trihaloacetic acids and formation of the corresponding trihalomethanes in drinking water. Water Res. 2002, 36, 3665.
| Decomposition of trihaloacetic acids and formation of the corresponding trihalomethanes in drinking water.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XlvFOmu7c%3D&md5=bf246ec32b29c27688a7c5443d292766CAS | 12230213PubMed |
[64] J. Hine, N. W. Burske, M. Hine, P. B. Langford, The relative rates of formation of carbanions by haloforms. J. Am. Chem. Soc. 1957, 79, 1406.
| The relative rates of formation of carbanions by haloforms.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaG2sXls1CgtQ%3D%3D&md5=496e304501f650808a25a8432d60b350CAS |
[65] B. A. Bergamaschi, M. S. Fram, R. Fujii, G. R. Aiken, C. Kendall, S. R. Silva, in Proceedings of the Technical Meeting, Charleston South Carolina 1999 (Charleston).
[66] B. A. Bergamaschi, M. S. Fram, R. Fujii, G. R. Aiken, C. Kendall, S. R. Silva, The carbon isotopic composition of trihalomethanes formed from chemically distinct dissolved organic carbon isolates from the Sacramento–San Joaquin River Delta, California, USA, in US Geological Survey Toxic Substances Hydrology Program – Proceedings of the Technical Meeting, Volume 2 – Contamination of Hydrologic Systems and Related Ecosystems, Water-Resources Investigation Report 99-4018BA, 8–12 March 1999, Charleston, SC, USA 1999 (US Geological Survey: Charleston, SC). Available at http://toxics.usgs.gov/pubs/wri99-4018/Volume2/sectionA/2212_Bergamaschl/pdf/2212_Berga.pdf [Verified 10 July 2015].
[67] A. Orlikowska, C. Stolle, F. Pollehne, K. Juergens, D. E. Schulz-Bull, Dynamics of halocarbons in coastal surface waters during short term mesocosm experiments. Environ. Chem. 2015, 12, 515.
| Dynamics of halocarbons in coastal surface waters during short term mesocosm experiments.Crossref | GoogleScholarGoogle Scholar |
[68] D. Hunkeler, T. Laier, F. Breider, O. S. Jacobsen, Demonstrating a natural origin of chloroform in groundwater using stable carbon isotopes. Environ. Sci. Technol. 2012, 46, 6096.
| Demonstrating a natural origin of chloroform in groundwater using stable carbon isotopes.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XmsVWgu7k%3D&md5=c54e6f036fa7b07f4bf8bbbebc805cb5CAS | 22554551PubMed |