

Location, location, location: surveying the intracellular real estate through proteomics in plants
A. Harvey MillarPlant Molecular Biology Group, School of Biomedical and Chemical Sciences, The University of Western Australia, Crawley, WA 6009, Australia. Corresponding author; email: hmillar@cyllene.uwa.edu.au
Functional Plant Biology 31(6) 563-582 https://doi.org/10.1071/FP04034
Submitted: 4 February 2004 Accepted: 16 March 2004 Published: 23 June 2004
Abstract
Knowledge of cellular compartmentation is critical to an understanding of many aspects of biological function in plant cells but it remains an under-emphasised concept in the use of and investment in plant functional genomic tools. The emerging effort in plant subcellular proteomics is discussed, and the current datasets that are available for a series of organelles and cellular membranes isolated from a range of plant species are noted. The benefit of knowing subcellular location in determining the role of proteins of unknown function is considered alongside the challenges faced in this endeavour. These include clear problems in dealing with contamination during the isolation of subcellular compartments, the meaningful integration of these datasets once completed to assemble a jigsaw of the cellular proteome as a whole, and the use of the wider literature in supplementing this proteomic discovery effort.
Keywords: mass spectrometry, membranes, organelles, plants, proteomics, subcellular fractionation.
Functional genomics and cell biology
Wide-scale genomic analysis of plants has greatly aided the scope and potential of discovering and exploiting novel capabilities in these organisms for the betterment of humans and the environment. The sequencing of the Arabidopsis genome (The Arabidopsis genome initiative 2000) as a model plant related to crops such as canola, and the rice genome (Goff et al. 2002) as a model grass related to the grain crops, have provided a foundation for this research activity. Clearly both models represent special value to Australia with its agricultural commitment to wheat, barley and oil seeds. However, many of the post-genomic experimental approaches applied today significantly neglect the cellular compartmentation of plant cells that fundamentally differentiates them from their bacterial, prokaryotic counterparts. That is, genomics, transcript analysis, metabolomics and much of proteomics fundamentally neglect a central tenant in cell biology — the separation of proteins and molecular functions by membrane barriers (Jung et al. 2000; Dreger 2003; Huber et al. 2003). Thus, while the current drive in proteomics to visualise more and more proteins in single cell homogenates is technically admirable, the approach neglects the cellular architecture that underlies the actual working environment of each gene product in a cell. Central metabolism and gas exchange, biosynthesis of high quality and high quantity products, and cellular signaling pathways in defense from the physical environment and invading pathogens, are all inextricably compartmented processes in plant cells. If we consider the cellular contents as the real estate market and proteins as the individual properties, then the three ‘ls’ of real estate, ‘location location location’, become a critical driver of a protein’s significance and function.
In mammals, the genomic revolution occurred on the backdrop of a relative wealth of biochemistry and cell fractionation studies undertaken for a century by a very large research community. There is a large array of reductionist studies that have localised gene products at the protein level to a large number of sub-cellular locations and many antibodies are available to track individual proteins or groups of proteins. In contrast, the availability of well annotated genomic information in plants; which has been comparable and often ahead of what is available for mammals; sits against a backdrop of a relatively small set of data on proteins and protein location. While some comparative genomics can be used to predict location by comparison to yeast and mammalian systems (Simpson and Pepperkok 2003), the divergence of plant genomic sequence means there are very large numbers of plant-specific proteins for which location cannot be sensibly predicted by cross-kingdom comparisons (The Arabidopsis genome initiative 2000).
Location as a key determinant of function
The products of the thousands of genes in plants are efficiently targeted to particular parts of the cell by elaborate targeting machinery. This uses targeting information within the amino acid sequence of the proteins (Emanuelsson and von Heijne 2001). Gene families of closely related products abound in plants, with over 50% of genes existing in families of at least two members (The Arabidopsis genome initiative 2000). There is much discussion about what level of redundancy exists within the coding regions of model plant genomes. Knockout studies in model plants target single genes and lack of phenotype is often explained by redundancy in gene families. However, while the protein products themselves appear to have redundant functions when tested in vitro, they can be non-redundant in vivo due to differences in cellular destination of the individual proteins in gene families. Often there is little idea of where to look for phenotype changes at the molecular level in these plant lines, because the location of the predicted protein product and its presence in time and space within the cell is not known at all, or is not known with accuracy. The subset of proteins found in a particular location is suited to this environment and facilitates the compartment’s function(s). Identifying these protein subsets is thus an initial step towards understanding cellular function as a whole, and provides a vital piece of the jigsaw puzzle in identifying the role of the many proteins currently designated as of unknown function in genome databases. We now need to work towards re-emphasising the compartmentation perspective and to integrating it into a functional genomic network in model plants as an essential tool for interpretation of genome function.
Targeting prediction tools in defining subcellular proteomes
Given these clear needs, several possible routes can be taken to place a cell biology perspective on plant genomic data. The simplest, cheapest and quickest, is to use bioinformatic targeting algorithms to predict where protein products will be located. An array of such programs exists including Psort [http://hypothesiscreator.net/iPSORT/ (validated 4 May 2004)], TargetP [http://www.cbs.dtu.dk/services/TargetP/ (validated 4 May 2004)], SubLoc [http://www.bioinfo.tsinghua.edu.cn/SubLoc/ (validated 4 May 2004)] and Predotar [http://www.inra.fr/Internet/Produits/Predotar/ (validated 4 May 2004)] reviewed by Emanuelsson and von Heijne (2001). Using a variety of these programs, proteins can be predicted to be localised to the nucleus, mitochondrion, plastid, peroxisome and endoplasmic reticulum (ER) based on primary sequence of the protein. A significant limitation of this approach is the lack of prediction capabilities to other membrane compartments such as the Golgi, vacuole and plasma membrane. Further, comparing the output of such programs across whole protein sets predicted from genome sequencing programs shows that such programs often disagree widely on the final location, leaving very small consensus sets that are generally predicted to be located in a given compartment (Richly et al. 2003; Heazlewood et al. 2004; Tanaka et al. 2004). A major reason for this inaccuracy is the lack of verified location data to train targeting algorithms in the first place (Emanuelsson 2002). In addition, when new experimental datasets are compared to the consensus prediction sets from the bioinformatic analyses, often the overlap between these sets is less than 50% (Sickmann et al. 2003; Heazlewood et al. 2004; Tanaka et al. 2004).
Experimentally defining subcellular proteomes
The clear alternative to these bioinformatic approaches is to undertake direct experimental analysis to locate proteins in cells. To do this one-by-one in the traditional manner adopted by mammalian researchers is clearly inappropriate today, and neglects to use the clear advantage we have in plants of having the genome sequence before knowing the location of its products. Post-genomic approaches to systematically locate proteins fall into two classes. Protein-centred approaches target individual products of unknown location to add to existing studies. While organelle-centred approaches would broadly identify proteins in a particular location to build a directory of protein location for both known and unknown proteins.
Genome sequence availability allows the synthesis of proteins with affinity or visual tags that allow transgene products to be located in vivo. This is a classical protein centred targeted approach. The use of green fluorescent protein (GFP) attached to proteins of interest is the best known of these technologies. High-throughput GFP screening of protein location on a genome scale is currently underway in model organisms (Huh et al. 2003) including model plants (Cutler et al. 2000; Escobar et al. 2003). However, as the amount of GFP location data increases, limitations of this approach are becoming apparent. By attaching a tag to a protein and expressing it from a non-physiological promoter, proteins can be targeted to non-physiological locations (Sickmann et al. 2003). Addition of large proteins like GFP or even small peptides such as a histidine (His) tag can result in non-physiological location because the native targeting information in the protein sequence is masked by the addition, or the tag itself has targeting capabilities which overcome the weak targeting of the protein (Zhou and Weiner 2001; Rial et al. 2002; Chew et al. 2003).
Alternatively, rather than introducing a transgene at all, the endogenous protein products can be identified in aliquots of compartments purified by subcellular fractionation studies (Fig. 1A ). This is now commonly referred to as ‘subcellular proteomics’ and it aims to define the protein set (or subproteome) of a particular location inside cells (Jung et al. 2000; Dreger 2003). This is a organelle-centred approach, building up subcellular protein sets which can ultimately provide a picture of the spatial location of proteins in cells (Fig. 1B ). Proteomics uses mass spectrometry and the genome sequence of an organism to link peptides identified in a sample back to protein sequences and the genes that encode them. The general principles and technologies have been well outlined by Jonsson (2001), and plant-specific issues previously discussed by Heazlewood and Millar (2003).
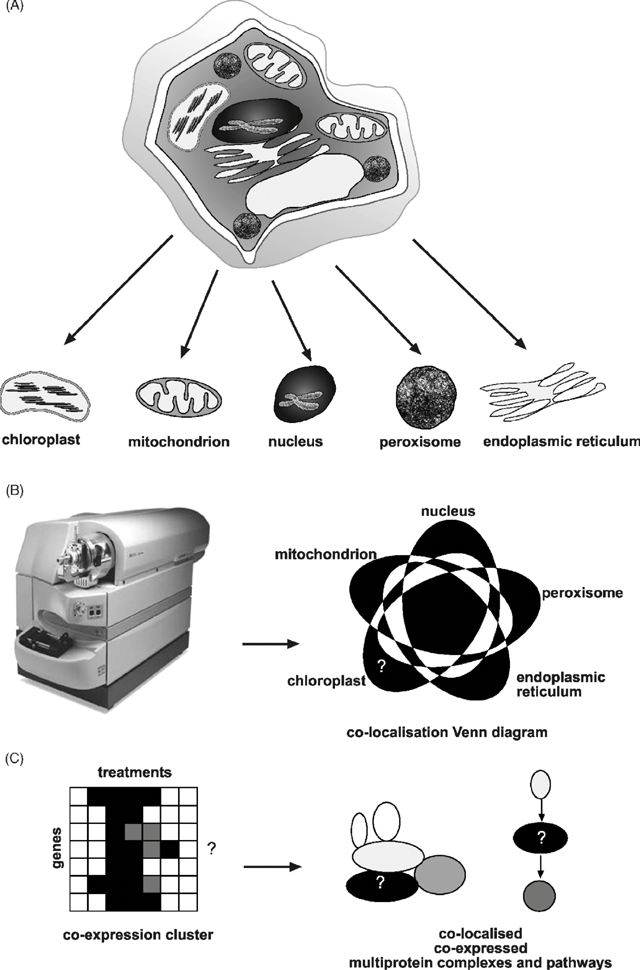
|
Subcellular proteomics in plants to date
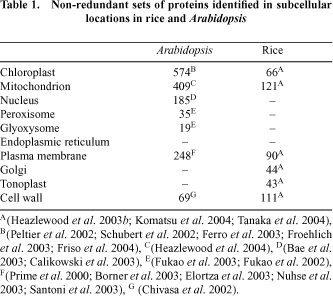
Sub-cellular proteomic discovery projects are typically undertaken by researchers with a long history of studying a particular function within a particular compartment of interest. For example researchers interested in photosynthesis analyse the chloroplast proteome to identify new proteins involved in this process. Such studies have been undertaken mainly in Arabidopsis and rice to date (Table 1). However, a variety of targeted studies in other plants have also appeared in the literature.
|
Plastids have been a favourite subcellular compartment for study in plants. This is no doubt because they are abundant, easy to purify and because chloroplasts house the reactions of photosynthesis in green tissues. However, it is also because plastids (present throughout plant tissues as proplastids, amyloplasts, etioplasts or chloroplasts) represent uniquely plant organelles with a variety of cellular functions. While targeted studies on particular proteins in chloroplasts have been undertaken for decades, the first major effort in defining its proteome by mass spectrometry was published in 2000, with a study in pea dedicated to the lumenal and peripheral thylakoid proteins (Peltier et al. 2000). This study identified 66 proteins; some 30% of which were previously unstudied in plants and showed decisively the value of subfractionation to yield both depth and biological location in proteome analysis. Subsequently, the focus has shifted to Arabidopsis, with a series of five reports providing in-depth analysis of the lumen (Peltier et al. 2002; Schubert et al. 2002), thylakoid membrane (Friso et al. 2004) and mixed envelope membranes (Ferro et al. 2003; Froehlich et al. 2003) of the chloroplast. In total, the Arabidopsis chloroplast set represents the localisation of the products from some 570 non-redundant genes. The proteome of amyloplasts has also been examined in wheat, identifying some 170 proteins (Andon et al. 2002).
Mitochondria have been next in line for subcellular proteomics. The relative ease of purifying these dense organelles from plant cells and their robustness to cell homogenisation has aided their analysis. A history of mitochondrial isolation for respiratory measurements and electron transport analysis in plants has provided assays for purity and integrity of these organelles. Again, while mitochondrial proteins have been isolated and displayed on gels for many years, two studies on Arabidopsis published in 2001 provided the foundation of this activity in the age of mass spectrometry based protein identification. Both studies identified some 50–100 proteins, giving a set of products from 92 non-redundant genes (Kruft et al. 2001; Millar et al. 2001). A variety of more targeted studies have since provided both techniques for further subdividing the mitochondrial proteome (Werhahn and Braun 2002; Herald et al. 2003; Millar and Heazlewood 2003) and detailed insights into the protein components of complexes I–V of the respiratory chain (Eubel et al. 2003; Heazlewood et al. 2003a , c ). More recently, a larger analysis using non-gel proteomic approaches based on liquid chromatography and tandem mass spectrometry has provided a set of over 400 non-redundant proteins from Arabidopsis mitochondrial samples (Heazlewood et al. 2004). In other plants, some 120–150 proteins have been identified in rice mitochondria (Heazlewood et al. 2003b ; Komatsu et al. 2004) and approximately 60 each in maize and pea mitochondria (Bardel et al. 2002; Hochholdinger et al. 2004). Several differential analyses of mitochondrial proteomes have been performed; in Arabidopsis during oxidative stress (Sweetlove et al. 2002), in maize comparing wild type and CMS lines (Hochholdinger et al. 2004) and in pea between different tissue types (Bardel et al. 2002).
The proteome of nuclei has received attention recently with two papers identifying a combined set of 185 non-redundant gene products from Arabidopsis (Bae et al. 2003; Calikowski et al. 2003). The study of Bae et al. (2003) further showed the changes in abundance of a set of over 50 of these proteins in response to cold treatment. A rice nuclei analysis is also underway, but is yet to be published (Komatsu et al. 2004). The peroxisome, has been less studied to date, with only two preliminary analyses identifying 30 proteins in greening Arabidopsis cotyledons (Fukao et al. 2002) and 19 proteins from this organelle in etiolated cotyledons (Fukao et al. 2003).
A series of studies has also identified proteins amongst the other intracellular membrane systems in plants. These included analysis of fairly crude fractions containing plasma membrane, Golgi and ER from Arabidopsis (Prime et al. 2000), and more purified plasma membrane fractions from Arabidopsis (Santoni et al. 1999) and an ER fraction from castor bean (Maltman et al. 2002). More focussed studies on glycosylphosphatidylinositol-anchored proteins (Borner et al. 2003; Elortza et al. 2003) and aquaporins (Santoni et al. 2003) and phosphoproteins (Nuhse et al. 2003) from plasma membranes in Arabidopsis have also been published. In total this represents a non-redundant set of 248 plasma membrane proteins. The systematic analysis of plasma membrane, Golgi and tonoplast in rice has yielded approximately 180 protein identifications (Komatsu et al. 2004). During the nitrogen fixing symbiosis between plants and Rhizobium bacteria, proliferation and budding of the plant plasma membrane occurs in specialised root nodule cells. Analysis of this peribacteroid membrane by proteomics has been undertaken in Lotus (Wienkoop and Saalbach 2003) and pea (Saalbach et al. 2002), revealing several proteins that may be important in transport of organic and inorganic ions between the plant and bacteria. However, the use of classical 2-dimensional gels for many of these studies still means that many hydrophobic protein classes await display and identification from these membrane systems.
A series of studies have also analysed the proteomes of the cell wall and of extracellular spaces. In Arabidopsis, rice and soybean, the cell wall proteome has been investigated resulting in the identification of 69, 111 and 4 proteins respectively (Chivasa et al. 2002; Mithoefer et al. 2002; Komatsu et al. 2004). Major constituents of the apoplastic proteome have been investigated in rice and Arabidopsis (Haslam et al. 2003) and in cowpea (Fecht-Christoffers et al. 2003). One study has also considered the protein content of the phloem in lupin (Hoffmann-Benning et al. 2002).
While at face value these studies represent an invaluable effort towards the goal of location determination, the details of their lists and the current state of accessing these data still leaves much to be desired. Three key problems exist which need to be tackled. These are noted below as the contamination, the integration and the wider literature problems.
The contamination problem
Probably the major and most pressing problem in subproteome analysis is the ever-increasing sensitivity of mass spectrometry. It is now capable of identifying the low level contaminants in compartment preparations that at one time were considered ‘pure’ or at least ‘pure enough’ for study. Thus contaminating proteins from other cellular locations are being erroneously allocated to particular subcellular structures. The first step in alleviating this problem is employing fractionation procedures to further improve purity in order to minimise cross-contaminants. A variety of density centrifugation techniques coupled to differential centrifugation sedimentations have traditionally been used to separate many of the organelles in plants. Increasingly these gradients need to be repeated and refined in order to more thoroughly reduce contamination during density band aspiration. The sensitivity of mass spectrometry also means that less and less material is required for final identification, and hence quality rather than quantity should be in the minds of researchers as they prepare subcellular fractions. However, often density and size either alone or together cannot reasonably be expected to cleanly separate membrane systems and organelle structures from each other. Techniques such as free flow electrophoresis separation of membranes and organelles on the basis of charge (Bardy et al. 1998), phase partitioning of membranes (Rochester et al. 1987; Faraday et al. 1996) or isolation on the basis of immuno-affinity (Burgess and Thompson 2002) are required. Combining these density, size, charge and affinity techniques will inevitably reduce the yield of subcellular fractions considerably, but may substantially increase purity for subproteome analysis.
Putative lists of contaminants between subproteomes are also complicated by the small but significant number of dual or multi-targeted proteins that are legitimately in more than one location in plant cells (Peeters and Small 2001). For example, we recently identified a range of ascorbate-glutathione cycle proteins in mitochondrial samples by subcellular proteomics (Heazlewood et al. 2004). These were generally considered to be chloroplast stromal proteins. Further analysis to determine if they were contaminants in mitochondrial fractions revealed they were dual-targeted to both compartments in plants (Chew et al. 2003). Also, the products of small gene families that encode major and minor isoforms or very similar proteins are generally considered to be co-located. However, when analysing the gene family for the chloroplast outer membrane receptor (TOC64) we found the major form is a chloroplast protein and a minor form is clearly a mitochondrial protein (Chew et al. 2004).
Another critical issue in solving the contamination problem is to remember that subproteomes are part of a whole cellular proteome. Hence, while defining the set of proteins in single locations is clearly the starting point, to fully appreciate contaminants we need to assemble all the subproteomes of a cell so that the primary and potentially secondary locations of proteins can be determined in a whole cell context. Currently these meta-datasets are rarely available, but increasing this integration will be vital in subproteome interpretation. In this context, a suite of new quantification techniques is being introduced, ostensibly for differential proteomics across treatments and development (Hamdan and Righetti 2002), however, several of these could also be adapted to quantify proteins between potentially contaminating structures in order to assess levels of contamination.
The data integration problem
As noted above, the integration of subproteome data will certainly have benefits for the contamination problem. However, integration also has scientific merit in its own right in understanding biology as it is played out in cellular locations. It is very difficult to see how this can be done properly by different researchers, using varying techniques and plant tissues, looking at their compartment of interest. Thus there is a significant need for a systematic analysis of sub-cellular proteomes in single model systems by the same techniques in a way that the raw data can be compared and queried to best define the primary location of each protein in the whole cell. In plants this has yet to be done fully, but is best exemplified to date by the work of Komatsu et al. (2004) as a part of the rice proteome database [http://gene64.dna.affrc.go.jp/RPD/ (validated 4 May 2004)]. A large series of subcellular locations is being investigated by these workers and a detailed database of subcellular location on gels, protein identifications and raw mass spectra has been developed. Further analysis of this dataset in rice is currently complicated by the still rather draft-like genome sequence. The open reading frames have yet to be fully annotated in rice, and only a preliminary non-redundant key for loci position and gene sequence has been released. In contrast, the Arabidopsis database provides a mature annotation, a primary key in the form of Arabidopsis genome initiative (AGI) numbers, and a wealth of integrated genomic resources for whole genome expression analysis and genetically manipulated plant lines [http://www.arabidopsis.org/ (validated 4 May 2004)]. However, an integrated subcellular proteomic dataset in Arabidopsis is not currently available. Several websites seek to highlight individual experimental or predicted subproteome sets in Arabidopsis, and some place these in a wider genomic context. The plastid proteome database at Cornell University, USA [http://cbsusrv01.tc.cornell.edu/users/ppdb/ (validated 4 May 2004)] provides data on experimental and predicted chloroplast proteins. Similarly, the Arabidopsis mitochondrial proteome project, Universität Hannover, Germany [http://www.gartenbau.uni-hannover.de/genetik/AMPP (validated 4 May 2004)] and our Arabidopsis Mitochondrial Protein Database, University of Western Australia, Australia [http://www.mitoz.bcs.uwa.edu.au (validated 4 May 2004)] provide such data for mitochondrial location. In addition, we have sought to add published experimental sets from other researchers into our database to build a broader subcellular location database for plastids, mitochondria, nuclei and peroxisomes [http://www.mitoz.bcs.uwa.edu.au (validated 4 May 2004)]. The Aramemnon database of membrane proteins, University of Cologne, Germany [http://aramemnon.botanik.uni-koeln.de/ (validated 4 May 2004)] seeks to classify and characterise membrane protein families and includes subcellular location predictions. The Max Plank Institute in Cologne, has released a dataset dedicated to the evolutionary diversification of mitochondrial and plastid proteomes [http://www.mpiz-koeln.mpg.de/~leister/ (validated 4 May 2004)]. Finally, valuable resources relating to protein–GFP fusions can be searched at the Carnegie Institute of Washington [http://deepgreen.stanford.edu (validated 4 May 2004)].
The wider literature problem
Proteomics is often accused of re-inventing the wheel in protein identification by only considering recent high-throughput protein location analyses, while a significant set of data that deals with protein locations in plants determined on a one-by-one basis already exists in the literature. This latter set was compiled using traditional and novel approaches in subcellular fractionation, activity assay, protein purification, immunological detection, protein microsequencing and targeting prediction. Combining complementary approaches in this manner often provides a strong argument for location. Incorporation of these data in assessing mass spectrometry analysis of subproteomes would be greatly beneficial. It can provide independently confirmed location and larger subproteomes including proteins that are too low in abundance for current mass spectrometry detection. Additionally, proteins that have physicochemical properties that prevent their display by typical proteomic techniques can be identified by these alternative approaches. However, building such datasets from the literature, even for Arabidopsis, is not easy, given that many of these studies were conducted before AGI numbers were introduced and still today only a small number of researchers use the AGI number in publications. Several bioinformatic attempts to build subcellular proteomes from the literature have been published; notably, Guo et al. (2004) have recently released DBSubLoc [http://www.bioinfo.tsinghua.edu.cn/dbsubloc.html (validated 4 May 2004)]. This is a database containing over 60 000 proteins from a range of organisms that are allocated to subcellular locations based on annotation in primary sequence databases, model organism genome projects and literature texts. For plants, nearly 5500 entries exist, which represent nearly 1400 non-redundant gene products that are allocated to one of eight subcellular locations. Building more species specific datasets of this type will be valuable to supplement current proteome projects.
How will location data actually help determine function of unknown proteins?
Scepticism about the value of location data alone in the apparently large jump from location to function may well turn out to have significant basis; however, its likely additive value is often overlooked. Initially it will clearly give a spatial home in the cell to a gene product (Fig. 1B ). For a significant number of gene products this will simply be confirmatory, especially for proteins widely considered to be present in a given location with long traditions of study of both molecular function and localisation. But for many other gene products, these location data may be the only piece of experimental information available apart from, for some, the suggestion of functional group assigned by comparative sequence analysis. In these cases, location is the first step towards defining function by providing a handle to enthuse focused researchers to further investigate the protein and a starting point for looking for a phenotype in genetically altered plant lines lacking or over-expressing the gene encoding it. Once larger datasets are available, then subproteomes can be built to reveal the array of protein in the particular subcellular fraction, which is effectively providing a roll call of the workforce in a location (Fig. 1B ). Coupled to large-scale expression analyses of these genes in response to treatments and development, provided through microarray datasets, these subproteomes can be grouped into co-expression clusters (Fig. 1C ). Tight co-location, co-expression sets form the basis of putative molecular machinery and biochemical pathways and as such, are indicators of cellular functionality. The grouping of unknown proteins together in this way provide discrete biological problems that can be probed with immunological and other protein–protein interaction techniques to link unknown-function proteins with known-function proteins.
Subproteomes are not static, they change through gene expression in different tissues, in the same tissue during development, and they even change through protein translocation from one compartment to another. Such translocation events have been shown to underlie a range of signaling events in eukaryotic cells. Defining subproteomes under different conditions and in different tissues can therefore also give a differential analysis of these translocations and a broader view of the potential proteomes in different subcellular compartments. For example, these analyses could reveal proteins that move from the cytosol to the nucleus, and also the changing lists of proteins that may exist in a given organelle across embryos, pollen, leaves and roots. Finally, as eukaryotic cells provide a breadth of genetic and functional divergence, subcellular proteomes will allow the comparison of these protein populations on a location basis between species. This can be used to cast light on evolutionary processes (Richly et al. 2003), but also represents a very practical tool for biotechnology and pharmacology that seek to move intervention treatments from model species to target species. In the pharmaceutical industry the value of rats, mice or monkeys as models for humans can be assessed not just on the basis of genetic similarity but also on the basis of similarity in molecular architecture at the level of protein location. This target location information is very valuable as most drugs operate on proteins and have restricted access and half-lives in different cellular compartments due to chemistry and degradation pathways. The agrochemical industry can also use this approach in a similar manner to assess model plant suitability in targeting disease tolerance and novel product formation in crops of interest.
Acknowledgments
AHM is an Australian Research Council QEII Research Fellow and also thanks the Australian Research Council for grants through the Discovery Program to fund this research.
Andon NL,
Hollingworth S,
Koller A,
Greenland AJ,
Yates JR, Haynes PA
(2002) Proteomic characterisation of wheat amyloplasts using identification of proteins by tandem mass spectrometry. Proteomics 2, 1156–1168.
| Crossref | GoogleScholarGoogle Scholar | PubMed |

Bae MS,
Cho EJ,
Choi EY, Park OK
(2003) Analysis of the Arabidopsis nuclear proteome and its response to cold stress. The Plant Journal 36, 652–663.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Bardel J,
Louwagie M,
Jaquinod M,
Jourdain A,
Luche S,
Rabilloud T,
Macherel D,
Garin J, Bourguignon J
(2002) A survey of the plant mitochondrial proteome in relation to development. Proteomics 2, 880–898.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Bardy N,
Carrasco A,
Galaud JP,
Pont-Lezica R, Canut H
(1998) Free-flow electrophoresis for fractionation of Arabidopsis thaliana membranes. Electrophoresis 19, 1145–1153.
| PubMed |
Borner GH,
Lilley KS,
Stevens TJ, Dupree P
(2003) Identification of glycosylphosphatidylinositol-anchored proteins in Arabidopsis. A proteomic and genomic analysis. Plant Physiology 132, 568–577.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Burgess RR, Thompson NE
(2002) Advances in gentle immunoaffinity chromatography. Current Opinion in Biotechnology 13, 304–308.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Calikowski TT,
Meulia T, Meier I
(2003) A proteomic study of the Arabidopsis nuclear matrix. Journal of Cellular Biochemistry 90, 361–378.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Chew O,
Whelan J, Millar AH
(2003) Molecular definition of the ascorbate–glutathione cycle in Arabidopsis mitochondria reveals dual targeting of antioxidant defenses in plants. Journal of Biological Chemistry 278, 46 869–46 877.
| Crossref | GoogleScholarGoogle Scholar |
Chew O,
Lister R,
Qbadou S,
Heazlewood JL,
Soll J,
Schleiff E,
Millar AH, Whelan J
(2004) A plant outer mitochondrial membrane protein with high amino acid sequence identity to a chloroplast protein import receptor. FEBS Letters 557, 109–114.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Chivasa S,
Ndimba BK,
Simon WJ,
Robertson D,
Yu XL,
Knox JP,
Bolwell P, Slabas AR
(2002) Proteomic analysis of the Arabidopsis thaliana cell wall. Electrophoresis 23, 1754–1765.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Cutler SR,
Ehrhardt DW,
Griffitts JS, Somerville CR
(2000) Random GFP::cDNA fusions enable visualisation of subcellular structures in cells of Arabidopsis at a high frequency. Proceedings of the National Academy of Sciences USA 97, 3718–3723.
| Crossref | GoogleScholarGoogle Scholar |
Dreger M
(2003) Subcellular proteomics. Mass Spectrometry Reviews 22, 27–56.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Elortza F,
Nuhse TS,
Foster LJ,
Stensballe A,
Peck SC, Jensen ON
(2003) Proteomic analysis of glycosylphosphatidylinositol–anchored membrane proteins. Molecular and Cellular Proteomics 2, 1261–1270.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Emanuelsson O
(2002) Predicting protein subcellular localisation from amino acid sequence information. Briefing in Bioinformatics 3, 361–376.
Emanuelsson O, von Heijne G
(2001) Prediction of organellar targeting signals. Biochimica et Biophysica Acta 1541, 114–119.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Escobar NM,
Haupt S,
Thow G,
Boevink P,
Chapman S, Oparka K
(2003) High-throughput viral expression of cDNA-green fluorescent protein fusions reveals novel subcellular addresses and identifies unique proteins that interact with plasmodesmata. The Plant Cell 15, 1507–1523.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Eubel H,
Jansch L, Braun HP
(2003) New insights into the respiratory chain of plant mitochondria. supercomplexes and a unique composition of complex II. Plant Physiology 133, 274–286.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Faraday CD,
Spanswick RM, Bisson MA
(1996) Plasma membrane isolation from freshwater and salt-tolerant species of Chara: antibody cross-reactions and phosphohydrolase activities. Journal of Experimental Botany 47, 589–594.
| PubMed |
Fecht-Christoffers MM,
Braun HP,
Lemaitre-Guillier C,
VanDorsselaer A, Horst WJ
(2003) Effect of manganese toxicity on the proteome of the leaf apoplast in cowpea. Plant Physiology 133, 1935–1946.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Ferro M,
Salvi D,
Brugiere S,
Miras S,
Kowalski S,
Louwagie M,
Garin J,
Joyard J, Rolland N
(2003) Proteomics of the chloroplast envelope membranes from Arabidopsis thaliana. Molecular and Cellular Proteomics 2, 325–345.
| PubMed |
Friso G,
Ytterberg AJ,
Giacomelli L,
Peltier JB,
Rudella A,
Sun Q, van Wijk KJ
(2004) In-depth analysis of the thylakoid membrane proteome of Arabidopsis thaliana chloroplasts; new proteins, functions and a plastid proteome database. The Plant Cell 16, 478–499.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Froehlich JE,
Wilkerson CG,
Ray K,
McAndrew RS,
Osteryoung KW,
Gage DA, Phinney BS
(2003) Proteomic study of the Arabidopsis thaliana chloroplastic envelope membrane utilising alternatives to traditional two-dimensional electrophoresis. Journal of Proteome Research 2, 413–425.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Fukao Y,
Hayashi M, Nishimura M
(2002) Proteomic analysis of leaf peroxisomal proteins in greening cotyledons of Arabidopsis thaliana. Plant and Cell Physiology 43, 689–696.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Fukao Y,
Hayashi M,
Hara-Nishimura I, Nishimura M
(2003) Novel glyoxysomal protein kinase, GPK1, identified by proteomic analysis of glyoxysomes in etiolated cotyledons of Arabidopsis thaliana. Plant and Cell Physiology 44, 1002–1012.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Goff SA,
Ricke D,
Lan T-H,
Presting G, Wang R, ,
et al
.
(2002) A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 296, 92–100.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Guo T,
Hua S,
Ji X, Sun Z
(2004) DBSubLoc: database of protein subcellular localisation. Nucleic Acids Research 32, D122–D124.
| Crossref | GoogleScholarGoogle Scholar |
Hamdan M, Righetti PG
(2002) Modern strategies for protein quantification in proteome analysis: advantages and limitations. Mass Spectrometry Reviews 21, 287–302.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Haslam RP,
Downie AL,
Raveton M,
Gallardo K,
Job D,
Pallett KE,
John P,
Parry MAJ, Coleman JOD
(2003) The assessment of enriched apoplastic extracts using proteomic approaches. The Annals of Applied Biology 143, 81–91.
Heazlewood JL, Millar AH
(2003) Integrated plant proteomics: putting the green genomes to work. Functional Plant Biology 30, 471–482.
| Crossref | GoogleScholarGoogle Scholar |
Heazlewood JL,
Howell KA, Millar AH
(2003a) Mitochondrial complex I form Arabidopsis and rice: orthologs of mammalian and funal components coupled with plant-specific subunits. Biochimica et Biophysica Acta 1604, 159–169.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Heazlewood JL,
Howell KA,
Whelan J, Millar AH
(2003b) Towards an analysis of the rice mitochondrial proteome. Plant Physiology 132, 230–242.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Heazlewood JL,
Whelan J, Millar AH
(2003c) The products of the mitochondrial orf25 and orfB genes are FO components in the plant F1FO ATP synthase. FEBS Letters 540, 201–205.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Heazlewood JL,
Tonti-Filippini JS,
Gout AM,
Day DA,
Whelan J, Millar AH
(2004) Experimental analysis of the Arabidopsis mitochondrial proteome highlights signalling and regulatory components, provides assessment of targeting prediction programs and points to plant specific mitochondrial proteins. The Plant Cell 16, 241–256.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Herald VL,
Heazlewood JL,
Day DA, Millar AH
(2003) Proteomic identification of divalent metal cation binding proteins in plant mitochondria. FEBS Letters 537, 96–100.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Hochholdinger F,
Guo L, Schnable PS
(2004) Cytoplasmic regulation of the accumulation of nuclear-encoded proteins in the mitochondrial proteome of maize. The Plant Journal 37, 199–208.
| PubMed |
Hoffmann-Benning S,
McIntosh DA,
Gage L,
Kende H, Zeevaart JAD
(2002) Comparison of peptides in the phloem sap of flowering and non-flowering Perilla and lupine plants using microbore HPLC followed by matrix-assisted laser desorption / ionisation time-of-flight mass spectrometry. Planta 216, 140–147.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Huber LA,
Pfaller K, Vietor I
(2003) Organelle proteomics: implications for subcellular fractionation in proteomics. Circulation Research 92, 962–968.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Huh WK,
Falvo JV,
Gerke LC,
Carroll AS,
Howson RW,
Weissman JS, O’Shea EK
(2003) Global analysis of protein localisation in budding yeast. Nature 425, 686–691.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Jonsson AP
(2001) Mass spectrometry for protein and peptide characterisation. Cellular and Molecular Life Sciences 58, 868–884.
| PubMed |
Jung E,
Heller M,
Sanchez JC, Hochstrasser DF
(2000) Proteomics meets cell biology: the establishment of subcellular proteomes. Electrophoresis 21, 3369–3377.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Komatsu S,
Kojima K,
Suzuki K,
Ozaki K, Higo K
(2004) Rice proteome database based on two-dimensional polyacrylamide gel electrophoresis: its status in 2003. Nucleic Acids Research 32, D388–D392.
| Crossref | GoogleScholarGoogle Scholar |
Kruft V,
Eubel H,
Jansch L,
Werhahn W, Braun HP
(2001) Proteomic approach to identify novel mitochondrial proteins in Arabidopsis. Plant Physiology 127, 1694–1710.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Maltman DJ,
Simon WJ,
Wheeler CH,
Dunn MJ,
Wait R, Slabas AR
(2002) Proteomic analysis of the endoplasmic reticulum from developing and germinating seed of castor (Ricinus communis). Electrophoresis 23, 626–639.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Millar AH, Heazlewood JL
(2003) Genomic and proteomic analysis of mitochondrial carrier proteins in Arabidopsis. Plant Physiology 131, 443–453.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Millar AH,
Sweetlove LJ,
Giege P, Leaver CJ
(2001) Analysis of the Arabidopsis mitochondrial proteome. Plant Physiology 127, 1711–1727.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Mithoefer A,
Mueller B,
Wanner G, Eichacker LA
(2002) Identification of defence-related cell wall proteins in Phytophthora
sojae-infected soybean roots by ESI–MS / MS. Molecular Plant Pathology 3, 163–166.
| Crossref | GoogleScholarGoogle Scholar |
Nuhse TS,
Stensballe A,
Jensen ON, Peck SC
(2003) Large-scale analysis of in vivo phosphorylated membrane proteins by immobilised metal ion affinity chromatography and mass spectrometry. Molecular and Cellular Proteomics 2, 1234–1243.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Peeters N, Small I
(2001) Dual targeting to mitochondria and chloroplasts. Biochimica et Biophysica Acta 1541, 54–63.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Peltier JB,
Friso G,
Kalume DE,
Roepstorff P,
Nilsson F,
Adamska I, van Wijk KJ
(2000) Proteomics of the chloroplast: systematic identification and targeting analysis of lumenal and peripheral thylakoid proteins. The Plant Cell 12, 319–341.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Peltier J-B,
Emanuelsson O,
Kalume DE,
Ytterberg J, Friso G ,
et al
.
(2002) Central functions of the lumenal and peripheral thylakoid proteome of Arabidopsis determined by experimentation and genome-wide prediction. The Plant Cell 14, 211–236.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Prime TA,
Sherrier DJ,
Mahon P,
Packman LC, Dupree P
(2000) A proteomic analysis of organelles from Arabidopsis thaliana. Electrophoresis 21, 3488–3499.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Rial DV,
Lombardo VA,
Ceccarelli EA, Ottado J
(2002) The import of ferredoxin-NADP+ reductase precursor into chloroplasts is modulated by the region between the transit peptide and the mature core of the protein. European Journal of Biochemistry 269, 5431–5439.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Richly E,
Chinnery PF, Leister D
(2003) Evolutionary diversification of mitochondrial proteomes: implications for human disease. Trends in Genetics 19, 356–362.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Rochester CP,
Kjellbom P,
Andersson B, Larsson C
(1987) Lipid composition of plasma membranes isolated from light-grown barley (Hordeum vulgare) leaves: identification of cerebroside as a major component. Archives of Biochemistry and Biophysics 255, 385–391.
| PubMed |
Saalbach G,
Erik P, Wienkoop S
(2002) Characterisation by proteomics of peribacteroid space and peribacteroid membrane preparations from pea (Pisum sativum) symbiosomes. Proteomics 2, 325–337.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Santoni V,
Doumas P,
Rouquie D,
Mansion M,
Rabilloud T, Rossignol M
(1999) Large scale characterisation of plant plasma membrane proteins. Biochimie 81, 655–661.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Santoni V,
Vinh J,
Pflieger D,
Sommerer N, Maurel C
(2003) A proteomic study reveals novel insights into the diversity of aquaporin forms expressed in the plasma membrane of plant roots. The Biochemical Journal 373, 289–296.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Schubert M,
Petersson UA,
Haas BJ,
Funk C,
Schroder WP, Kieselbach T
(2002) Proteome map of the chloroplast lumen of Arabidopsis thaliana. Journal of Biological Chemistry 277, 8354–8365.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Sickmann A,
Reinders J,
Wagner Y,
Joppich C, Zahedi R ,
et al
.
(2003) The proteome of Saccharomyces cerevisiae mitochondria. Proceedings of the National Academy of Sciences USA 100, 13207–13212.
| Crossref | GoogleScholarGoogle Scholar |
Simpson JC, Pepperkok R
(2003) Localising the proteome. Genome Biology 4, 240.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Sweetlove LJ,
Heazlewood JL,
Herald V,
Holtzapffel R,
Day DA,
Leaver CJ, Millar AH
(2002) The impact of oxidative stress on Arabidopsis mitochondria. The Plant Journal 32, 891–904.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Tanaka N,
Fujita M,
Handa H,
Murayama S, Uemura M ,
et al
.
(2004) Proteomics of the rice cell: systematic identification of the protein population in subcellular compartments. Molecular Genetics and Genomics In press ,
| Crossref | GoogleScholarGoogle Scholar |
The Arabidopsis genome initiative
(2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408, 796–815.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Werhahn W, Braun HP
(2002) Biochemical dissection of the mitochondrial proteome from Arabidopsis thaliana by three-dimensional gel electrophoresis. Electrophoresis 23, 640–646.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Wienkoop S, Saalbach G
(2003) Proteome analysis. Novel proteins identified at the peribacteroid membrane from Lotus japonicus root nodules. Plant Physiology 131, 1080–1090.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Zhou J, Weiner H
(2001) The N-terminal portion of mature aldehyde dehydrogenase affects protein folding and assembly. Protein Science 10, 1490–1497.
| Crossref | GoogleScholarGoogle Scholar | PubMed |