

Functional genomics to study stress responses in crop legumes: progress and prospects
Himabindu Kudapa A , Abirami Ramalingam A B , Swapna Nayakoti A , Xiaoping Chen C , Wei-Jian Zhuang D , Xuanqiang Liang C , Guenter Kahl E F , David Edwards G and Rajeev K. Varshney A C H IA International Crops Research Institute for the Semiarid Tropics (ICRISAT), Patancheru 502324, India.
B Swinburne University of Technology, PO Box 218, John St, Hawthorn, Vic. 3122, Australia.
C Crops Research Institute, Guangdong Academy of Agricultural Sciences, Guangzhou, China.
D Fujian Provincial Key Laboratory of Plant Molecular and Cell Biology, Fujian Agriculture and Forestry University, Fuzhou 350002, PR China.
E Goethe University Frankfurt am Main, Institute for Molecular BioSciences, Max-von-Laue-Str. 9, Frankfurt am Main 60438, Germany.
F GenXPro GmbH, Frankfurt Biotechnology Innovation Center (FIZ), Altenhöferallee3, Frankfurt am Main 60438, Germany.
G School of Agriculture and Food Sciences, University of Queensland, Brisbane, St Lucia, Qld 4072, Australia.
H School of Plant Biology, The University of Western Australia, 35 Stirling Highway, Crawley, Perth, WA 6009, Australia.
I Corresponding author. Email: r.k.varshney@cgiar.org
This paper originates from a presentation at the ‘VI International Conference on Legume Genetics and Genomics (ICLGG)’ Hyderabad, India, 2–7 October 2012.
Functional Plant Biology 40(12) 1221-1233 https://doi.org/10.1071/FP13191
Submitted: 25 June 2013 Accepted: 22 August 2013 Published: 7 October 2013
Journal Compilation © CSIRO Publishing 2013 Open Access CC BY-NC-ND
Abstract
Legumes are important food crops worldwide, contributing to more than 33% of human dietary protein. The production of crop legumes is frequently impacted by abiotic and biotic stresses. It is therefore important to identify genes conferring resistance to biotic stresses and tolerance to abiotic stresses that can be used to both understand molecular mechanisms of plant response to the environment and to accelerate crop improvement. Recent advances in genomics offer a range of approaches such as the sequencing of genomes and transcriptomes, gene expression microarray as well as RNA-seq based gene expression profiling, and map-based cloning for the identification and isolation of biotic and abiotic stress-responsive genes in several crop legumes. These candidate stress associated genes should provide insights into the molecular mechanisms of stress tolerance and ultimately help to develop legume varieties with improved stress tolerance and productivity under adverse conditions. This review provides an overview on recent advances in the functional genomics of crop legumes that includes the discovery as well as validation of candidate genes.
Additional keywords: abiotic and biotic stresses, expression profiling, stress tolerance, transcriptomics.
Introduction
Fabaceae, the legume family, comprising more than 650 genera and 20 000 species, is the third largest family of higher plants and the second most important family among crop plants after Poaceae (the grass family). Legumes account for ~27% of crop production in agriculture worldwide based on area harvested and total production (Graham and Vance 2003). Crop legumes complement cereals, the primary source of carbohydrates in the human diet, in terms of amino acid composition, and provide around one-third (20–40%) of all dietary protein (Zhu et al. 2005). Legumes produce secondary metabolic compounds that can protect the plant against pathogens and pests. The economic importance of legumes and some of their salient biological features including symbiotic nitrogen fixation, the prevention of erosion, suppression of weeds and adding organic matter to the soil, provide ample justification for a significant investment in genomics based improvement of this important crop family.
Some legumes constitute an important component of the human diet in developing countries; include soybean (Glycine max), peanut or groundnut (Arachis hypogaea), chickpea (Cicer arietinum), cowpea (Vigna unguiculata), common bean (Phaseolus vulgaris), pigeonpea (Cajanus cajan), pea (Pisum sativum), lentil (Lens culinaris), faba bean (Vicia faba), mungbean (Vigna radiata) and lupin (Lupinus luteus). Despite having an important role in food security, the majority of these legume crops demonstrate low productivity due to biotic (e.g. bacteria, fungi, nematodes, viruses and insects) and abiotic (e.g. drought, salinity, heat and waterlogging) stresses. For example, in peanut and chickpea, drought is an important abiotic stress constrain and major biotic stresses include anthracnose, angular leaf spot, bean rust, bacterial blight in common bean, Ascochyta blight and Fusarium wilt in chickpea. Thus, it is necessary to enhance our understanding of specific aspects of defence/stress responses to improve crop productivity. Towards this aim, emerging genomics technology can be applied to interrogate the basis of stress response and identify candidate genes or key loci controlling stress tolerance or resistance. Subsequently these genes can be used in genetic modification or molecular breeding programs to develop improved varieties with enhanced resistance/tolerance to stress.
Due to their small genome sizes, their simple genetic system and amenability to forward and reverse genetic analyses, two legume species, namely Medicago (Medicago truncatula) and Lotus (Lotus japonicus) have been used for extensive molecular studies in the past two decades (Handberg and Stougaard 1992; Cook 1999). Recently, the legume community has adopted next generation sequencing (NGS) and high-throughput genotyping technologies to undertake functional genomics studies in the crop legumes. As a result, a vast amount of genomic resources have been developed that enable isolation and characterisation of key genes involved in legume stress response. Once candidate genes are identified, it is important to validate their function before their application in crop improvement strategies (Valliyodan and Nguyen 2006). The successful application of biotechnological tools to alleviate the biotic/abiotic constraints of crop legumes will require both biological knowledge of the target species and the underlying mechanisms of crop stress response.
In view of the above considerations, this article summarises and presents a critical appraisal of the development/availability of genomic resources and their use for the identification, isolation and validation of candidate genes conferring resistance/tolerance to biotic/abiotic stress. Finally, an overview has been presented on the integration of various functional genomics approaches towards the genetic improvement of leguminous crops.
Gene discovery through sequencing of transcriptomes and genomes
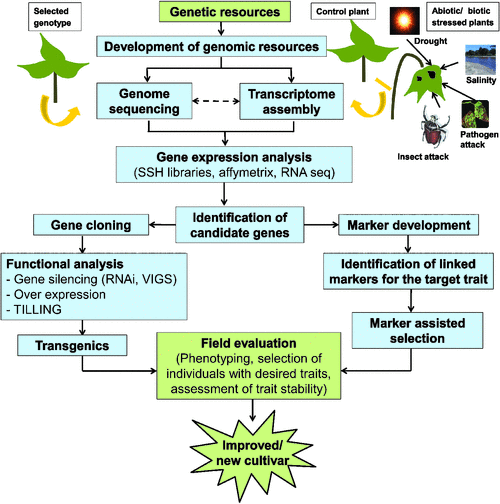
A major aim of genomic studies in plants is the identification of genes and pathways that affect crop production. Genome sequencing is fundamental to understand the genomic composition and gene repertoire of an organism; however, because of the high costs associated with sequencing a genome, initially only the genomes of model legumes were sequenced. An alternative approach to genome sequencing is targeted expressed gene sequencing. Therefore, in many crop legumes, efforts focussed on the development of cDNA libraries, the generation of expressed sequence tags (EST), gene expression analysis, and the in silico mining of functional information from EST datasets. An overview of functional genomics approaches for crop improvement is illustrated in Fig. 1.
|
In the absence of genome sequence data, EST collections produced by Sanger sequencing have proven extremely useful for many plant studies. EST databases provide basic sequence depositories for gene discovery and assist in comparative mapping. They also facilitate the identification of candidate genes for agronomic traits (Young and Bharti 2012). In legume species, extensive efforts have generated an abundance of ESTs from a range of tissues, including from plants challenged by stress. Today, more than 3 million legume ESTs are available, predominantly from soybean (1.5 million, Vodkin et al. 2004) followed by the model legumes M. truncatula (280 000, Cheung et al. 2006) and L. japonicus (242 000, Asamizu et al. 2004). Among crop legumes, cowpea contributed around 200 000 ESTs (Muchero et al. 2009), and common bean 114 139 (Blair et al. 2011). In the case of chickpea, cDNA libraries have been generated from plants under drought and salinity stress (Varshney et al. 2009a). In the case of pigeonpea, Fusarium wilt and sterility mosaic disease (SMD), responsive ESTs were generated (Raju et al. 2010). Sanger ESTs generated from stress-responsive tissues from selected key studies have been summarised in Table 1.

|
EST libraries have also been constructed using the suppression subtractive hybridisation (SSH) technique, and utilising this approach, ribosomal protein genes related to cold and salt stresses were cloned from soybean (Kim et al. 2004). In a different study, 372 high quality salt stress-responsive ESTs were generated from soybean SSH libraries (Li et al. 2012). In the case of chickpea, 477 drought-responsive ESTs were generated from root tissues (Buhariwalla et al. 2005). Deokar et al. (2011) also generated 3062 unigenes from SSH libraries of root and shoot tissues of contrasting drought-responsive genotypes in chickpea. In pigeonpea, 182 unique ESTs were generated from drought-stressed and unstressed pigeonpea seedlings using SSH (Qiao et al. 2012). Although the method can be technically demanding and labour intensive, the establishment of SSH libraries is a rewarding approach for the identification of candidate genes for a given stress.
Due to the availability of high-throughput and cost-effective NGS platforms such as the Illumina HiSeq (Illumina Inc, San Diego, CA, USA), GAIIx, MiSeq; Roche 454/FLX (454 Life Sciences, Branford, CT, USA); ABI SOLiD (Applied Biosystems, Carlsbad, CA, USA); and the Invitrogen Ion Proton (Invitrogen, Carlsbad, CA, USA), the sequencing of transcriptomes and genomes has become more efficient and economical (Varshney et al. 2009b; Edwards et al. 2013). The expansion of third generation sequencing technologies such as those of Pacific Biosciences (PacBio, Menlo Park, CA, USA), Oxford Nanopore Technologies (Oxford, UK) is expected to accelerate the large scale generation of genomic resources (Munroe and Harris 2010; Thudi et al. 2012). Several NGS platforms have already generated a vast set of transcript reads from a range of developing and stress-responsive tissues from a range of different crop legumes (Table 2).

|
The application of NGS technology has led to the production of transcriptome assemblies for chickpea (Hiremath et al. 2011; Garg et al. 2011a), pigeonpea (Dubey et al. 2011; Kudapa et al. 2012), peanut (Zhang et al. 2012), pea (Franssen et al. 2011) and lupin (Parra-González et al. 2012). In addition, the combination of reads generated by NGS platforms and Sanger ESTs has improved transcriptome assemblies, particularly in the context of contig length (Hiremath et al. 2011). For example, a transcriptome assembly based on FLX/454 sequencing together with Sanger ESTs comprised 103 215 tentative unique sequences (TUSs) with an average contig length of 459 bp (Hiremath et al. 2011). By analysing sequencing data from FLX/454 (~7 million reads), Illumina (~100 million tags) and Sanger (~150 000 ESTs) platforms, an improved assembly with 46 369 unigenes with an average contig length of 965 bp could be achieved (H Kudapa, S Azam, AG Sharpe, B Taran, R Li, B Deonovic, C Cameron, AD Farmer, RK Varshney, unpubl. data). In an effort to improve transcriptome assembly, researchers have compared the performance of different assemblers including CAP3, MIRA, TGICL, and Velvet, either alone or in combination (Garg et al. 2011b; Kudapa et al. 2012). The National Center for Genome Resources (NCGR) in cooperation with the USA Department of Agriculture (USDA)-supported Legume Information System (LIS, http://www.comparative-legumes.org, accessed 9 June 2013) offers a comprehensive collection of transcriptome assemblies for several legumes.
Whole-genome sequencing is fundamental to understand the genetic composition of an organism. The two model legume species M. truncatula and L. japonicus were first selected for sequencing in depth. Draft genome assemblies were published for Medicago (Young et al. 2011) and Lotus (Sato et al. 2008). The Medicago assembly captured ~94% of expressed genes, whereas the Lotus assembly represented 91% of the gene space (Sato et al. 2008).
Among crop legumes, the assembly of a soybean reference genome was fundamental and improved our current understanding of legume genomes generally. About 969.6 Mb of the 1115 Mb genome was assembled after generating eight times the whole-genome shotgun (WGS) data using Sanger sequencing (Schmutz et al. 2010). Recently, draft genome sequences of pigeonpea and chickpea have been reported, representing 73% (Varshney et al. 2012) and 74% (Varshney et al. 2013) of the respective genomes. A common bean genome sequence has also become available on the Phytozome data portal (http://www.phytozome.net/commonbean.php, accessed 13 June 2013). Collaborative projects involving ICRISAT are underway to generate the tetraploid (US-led initiative) and the diploid genomes (China-led initiative) of peanut. Efforts are also underway to assemble the genomes of mungbean (S Ha-Lee, pers. comm.), lentil, lupin and pea (D Edwards, pers. comm.). Details of the released genome sequences and the total number of genes identified from crop legume species are summarised in Table 3. We note that the genome sequencing projects revealed several genes involved in stress response. For example, 111 drought responsive genes in pigeonpea (Varshney et al. 2012) and 187 disease resistance genes in chickpea (Varshney et al. 2013) were identified from the genome sequence analysis. The stress responsive genes identified from genome sequencing projects could be of great value for dissecting candidate genes for important stresses in respective crop legumes.

|
In addition to the generation of reference genome sequences (Imelfort et al. 2009a; Edwards and Batley 2010); NGS can facilitate the re-sequencing of genomes to identify genomic variation such as SNPs (Imelfort et al. 2009b; Lorenc et al. 2012). In the case of soybean, 17 wild and 14 cultivated soybean genomes have been re-sequenced to date (Lam et al. 2010). This study revealed patterns of genetic variation between wild and cultivated soybeans and identified greater allelic diversity in wild soybeans, and a set of 205 614 SNPs have been identified for use in QTL mapping and association studies. In the model legume Medicago, the genomes of 26 diverse accessions were sequenced to identify and characterise sequence polymorphisms and linkage disequilibrium (LD). More than 3 million SNPs were detected and this study suggested that M. truncatula demonstrated greater diversity and less LD when compared with soybean (Branca et al. 2011). Ninety chickpea genomes have been re-sequenced, revealing 4.4 million variants (SNPs and INDELs). In addition, genetic diversity and phylogenetic analyses from this study highlighted the mixed use of desi and kabuli genotypes in the history of chickpea breeding (Varshney et al. 2013).
Genome re-sequencing is a powerful approach for detecting new alleles and haplotypes that can be used in genome-wide association studies (GWAS). As re-sequencing costs are continuously decreasing, re-sequencing-based allele discovery is expected to become more popular (Tuskan et al. 2011).
Gene discovery through functional genomics approaches
For the discovery of differentially expressed genes or candidate genes for a particular trait, the following two approaches have been used as follows.
Hybridisation-based gene expression profiling
Various microarray technologies have been found useful to unravel key biological processes. Generally, microarray platforms fall into a bewildering variety of architectures that are now mostly superseded by sequencing based approaches. In the context of this review, cDNA arrays and oligonucleotide-based chips are considered in some detail (Sreenivasulu et al. 2002). Complementary DNAs or oligonucleotides representing non-redundant sets of ESTs were immobilised on either nylon membranes or glass slides and effectively applied for gene expression analysis, especially in species for which only limited genome information was available. Several cDNA-based arrays have been applied in crop legumes. For example, in soybean, high density arrays based on 27 513 cDNA inserts that represented a low redundancy ‘unigene set’ were applied for a wide range of samples from various developmental stages, including disease-challenged and stress-exposed tissues (Vodkin et al. 2004). These arrays were also used to infer global gene expression patterns in mutant iso-lines. This study identified a set of candidate genes that respond to different stresses including drought, heat, flooding, herbicide application and various pathogens. In chickpea, 8098 probes corresponding to 2013 unigenes were immobilised onto a microarray and global gene expression profiles examined in roots during vascular wilt (Ashraf et al. 2009). Potential innate immune-responsive candidate genes involved in a complex regulatory network could be identified. A 768-feature microarray comprising chickpea cDNAs (559), grasspea cDNAs (156), lentil resistance gene analogues (RGAs) (41) and controls (12) were designed by Mantri et al. (2010) to explore abiotic stress-responsive transcripts in chickpea. The authors identified differentially expressed genes in contrasting genotypes (tolerant/ sensitive for drought, salinity and cold stresses). Furthermore, genes coding for various regulatory and functional proteins are now known, and the complex mechanisms of multi-gene control in abiotic stress responses partly deciphered. As a further spin-off, common genes expressed under different stresses suggested an activation of distinct gene batteries as a general phenomenon in stress responses (Cheong et al. 2002). Another platform for hybridisation of RNA and comparison of gene expression across tissues/genotypes is the Affymetrix gene chip. For legumes, the Affymetrix Array GeneChip platform is available for Medicago (http://www.affymetrix.com/estore/browse/products.jsp?productId=131472#1_1, accessed 29 May 2013) and soybean (http://www.affymetrix.com/estore/browse/products.jsp?productId=131507#1_1, accessed 29 May 2013). Several studies in model legumes have employed these GeneChip Genome Arrays for research into developmental or metabolic pathways (Pang et al. 2009; Verdier et al. 2013). In the case of soybean, the GeneChip Soybean Genome Array permitted the characterisation of genome-wide expression patterns, and identified drought-responsive candidate genes. GeneChip Soybean Genome arrays were also applied to other legume species. For instance, the response to root knot nematode infection of resistant cowpea genotype CB46 and a susceptible near-isogenic line (null-Rk) were investigated at 3 and 9 days post inoculation (Das et al. 2010). Furthermore, GeneChip Soybean Genome Arrays identified single feature polymorphisms (SFPs) in cowpea (Das et al. 2008) and pigeonpea (Saxena et al. 2011).
Sequencing-based expression profiling
Serial analysis of gene expression (SAGE) and its multiple variants allow to quantify global gene expression. In the original SAGE method, mRNA is oligo (dT)-trapped and reverse transcribed into cDNA, then a small sequence (‘tag’) is extracted from a defined position of each cDNA molecule. These small tags are ligated to form a long concatemeric chain that is cloned into a vector. Subsequently, these concatemers are sequenced. Although SAGE proved its usefulness, the size of the SAGE tag is too short to unequivocally identify the gene of origin. To overcome this problem, a variant of SAGE, called SuperSAGE was developed by Matsumura et al. (2005). If combined with one of the next-generation sequencing platforms, it is more precisely called deepSuperSAGE. This approach uses the type III-endonuclease EcoP15I of phage P1 to cut 26 bp long sequence tags from each transcript’s cDNA, expanding the tag-size and thereby the security of annotation. Quantification of a particular tag by automated counting provides the expression level of the corresponding transcript and also unravels novel expressed regions of the genome. By using SuperSAGE, Kahl et al. (2007) investigated salt- and drought- stress transcriptomes of chickpea and lentil by analysing 360 000 transcripts representing 40 000 unique mRNAs, and identified 3000 transcripts responding to these stresses. In another deepSuperSAGE application, 80 238 tags representing 17 493 unique transcripts from drought-stressed and non-stressed control roots in chickpea have been identified (Molina et al. 2008, 2011).
Massively parallel signature sequencing (MPSS) is yet another powerful technique for transcription profiling on a genome-wide scale (Brenner et al. 2000), though it is no longer competing with the NGS platforms. In this method, tagged PCR products produced from cDNA are amplified so that for each mRNA molecule ~100 000 of PCR products with a unique tag are produced. These tags are used to attach the PCR products to microbeads that avoid separate cDNA isolation, template processing and robotic procedures. Subsequently, after several rounds of ligation-based sequence determination using the type IIs restriction endonuclease BbvI, a sequence signature of ~16–20 bp is identified from each bead; routinely, 17 bp of high quality sequence is obtained. This procedure is performed in parallel, and ~1 million sequence signatures are obtained per experiment. However, because of its cost, the full potential of MPSS in the global expression profiling of the abiotic and biotic stress response is yet to be realised.
Although Sanger ESTs have also been used for digital gene expression (DGE) studies (Sreenivasulu et al. 2002), NGS platforms have greatly expanded genome wide sequence based gene expression analysis. The sequencing of RNA populations at an unprecedented depth and the quantification of the transcripts, though problematic, can be achieved through RNA-seq. The efficiency of Illumina-based DGE for the identification of differentially expressed genes was demonstrated by Hoen et al. (2008) by comparing RNA-seq with five different microarray platforms. This study concluded that deep sequencing provides a major advance in robustness, comparability and richness of expression profiling data. In soybean, DGE tag profiling was used to compare the transcriptional profiles between wild-type (CS) and a mutant isoline (CG) (Hunt et al. 2011; Wang et al. 2012). About 85 000 unique tags representing over 4.7 million DGE tags were generated (each from CS and CG) and applied to extend predicted gene models for the soybean genome. The datasets showed highly expressed genes as well as differentially expressed genes between young shoot tips CS and CG lines that encode proteins related to: ribosomes (70 different tags), protein biosynthesis/metabolism (35 tags), photosynthesis (34 tags), others (29 tags) and histones (28 tags) (Hunt et al. 2011). In the case of chickpea, Hiremath et al. (2011) observed 2974 TUSs with significant expression changes, of which 2823 could be associated with gene ontology annotations. Furthermore, expression patterns of many genes suggested their role in various pathways of secondary metabolism. In a different study, a wide range of expression levels were observed by mapping all reads onto a non-redundant set of chickpea transcripts, where the number of reads corresponding to each transcript ranged from 14 (0.16 reads per million, rpm) to 270 894 (3137 rpm), with an average of 1617 (18.7 rpm) (Garg et al. 2011a). This report identified 250 transcripts with root-, and 217 transcripts with shoot-specific expression. In the case of pigeonpea, significant differential expression was observed for 6673 to 11 518 TUSs for specific parental combinations (tolerant/sensitive for FW and SMD) (Dubey et al. 2011), and candidate FW- and SMD- responsive genes identified, which represent starting points to analyse biotic stress.
Of all the various methods of DGE, RNA-Seq is the most advanced technique for quantifying gene expression in crop legumes, especially when transcriptome assemblies and genome sequences become available. Most recently, a broad repertoire of greatly advanced techniques from proteomics, metabolomics and phenomics complement the aforementioned gene discovery suites.
Forward genetics based gene cloning
Gene cloning is an approach for isolating candidate genes that are functionally related to the trait of interest. A forward genetics approach for the identification of genes controlling a trait is positional cloning. Positional cloning per se may not conclusively identify target genes associated with a particular phenotype. It provides, however, useful genetic information that often requires support at the transcriptome (mRNA), proteome (proteins) and metabolome (metabolites) levels. The ultimate proof for the causative linkage of a gene in the target region linked to the trait of interest is complementation analysis (Langridge and Fleury 2011). In map-based cloning (MBC, a variant of positional cloning) the chromosomal location of the gene is identified through genetic mapping using molecular markers. Thanks to advances in sequencing and the genome-wide identification of sequence polymorphisms (e.g. SNPs), MBC became more accessible, and has been conducted in a range of crop species such as rice (Vij and Tyagi 2007) as well as in some legumes including soybean (Watanabe et al. 2009).
With ever faster and more and more accurate DNA sequencing technologies and the availability of large scale genomic resources (molecular markers) map based cloning limitations could be overcome. The published genome sequence assemblies for legumes such as soybean, chickpea, pigeonpea and common bean as well as the advanced DNA polymorphism detection will eventually make MBC of genes from crop legumes routine. The majority of the MBC projects in crop legumes have been applied in soybean. For example, the soybean phytochrome A gene (GmPhyA3) which modulates flowering time has been cloned using an MBC approach (Watanabe et al. 2009). In a different study, a candidate gene Ln controlling leaflet and seed number per pod was cloned with a combination of MBC and association study (Fang et al. 2013). The cloning and characterisation of Fusarium wilt resistance genes using MBC was demonstrated for chickpea (Huettel et al. 2002; Sharma and Muehlbauer 2007). Efforts are under way to clone genes from within a drought tolerance QTL region in chickpea (Thudi 2013). Some examples of MBC in crop legumes are depicted in Table 4.

|
Meng et al. (2007), using a comparative genomics approach, isolated and cloned GmNAC1 to GmNAC6 genes in soybean that encode cold stress-responsive factors. In common bean, Torres-Franklin et al. (2008) cloned the gene glutathione reductase (dtGR), a drought stress-responsive gene. Other stress-related genes have been identified and cloned with the above approaches in crop legumes including soybean, chickpea and cowpea (Sharma and Muehlbauer 2007; França et al. 2008; Fang et al. 2013). Some examples are shown in Table 4.
Validation of functional genes
After discovering trait associated genes by any of above mentioned approaches, the next step is their functional validation. Several approaches such as overexpression, RNAi, virus induced gene silencing (VIGS) and TILLING have been applied for this purpose.
Overexpression of genes
One of the most reliable methods of validation of isolated/cloned genes is to generate transgenics and assess the expression of the respective trait and this approach has been applied to validate the function of stress-responsive genes. In this procedure, success depends on the incorporation of the stress-responsive gene into the genome, and its expression. The lack of routine transformation protocols with high efficiency has been a constraint in crop legumes, mainly due to poor regeneration ability (especially via callus) and lack of compatible gene delivery methods. Many techniques (e.g. electroporation of intact tissues, silicon carbide whiskers) have been tested for gene delivery to the plant cell, and Agrobacterium-mediated and particle bombardment have been extensively employed for genetic transformation in several other crop plants. However, Agrobacterium-mediated transformation has low efficiency in grain legumes (Chandra and Pental 2003).
Furthermore, different pathways of regeneration vary in their amenability to different gene delivery techniques. Regeneration of plant tissues in vitro is through two pathways: ‘organogenesis’ and ‘embryogenesis’. Shoot buds are organised by concerted meristematic activity of several cells in organogenesis, whereas a single cell or a small cluster of cells undergo differentiation to produce somatic embryos similar to zygotic embryos in embryogenesis. However, the most prevalent mode of regeneration is via direct organogenesis in crop legumes and has been found to be most responsive in several crop legumes such as soybean (Kaneda et al. 1997) and pea (Jackson and Hobbs 1990).
Particle-gun mediated transformation has been used in some legume crops to generate transgenics although at low frequencies. Nevertheless good protocols with higher efficiency are already available in several legume crops, including chickpea (e.g. Acharjee et al. 2010), cowpea (e.g. Citadin et al. 2013), pigeonpea (e.g. Sharma et al. 2006), peanut (e.g. Bhatnagar-Mathur et al. 2007) and common bean (e.g. Aragão et al. 2013). Overall, the development of transgenics in some crop legumes, such as mungbean and lentil still remains a challenge. In addition, precise evaluation of the transgenic plant under stress conditions and understanding the physiological effect of the inserted gene(s) at the whole-plant level is also necessary in understanding overexpression studies (Bhatnagar-Mathur et al. 2009).
The majority of studies involving the overexpression of biotic or abiotic stress-responsive genes from either model or crop species were conducted in model plants such as Arabidopsis or Medicago, and only recently in some crop legumes. For the engineering of biotic stress resistance, the gene encoding α-amylase inhibitor αAI-1, a bruchid resistance factor from common bean, was overexpressed in other grain legumes including chickpea, pea, azuki bean and cowpea. The αAI-1 gene present in transgenic chickpea and cowpea under the control of a cotyledon-specific promoter provided resistance to several important bruchid pest species (Lüthi et al. 2010). A comparison of the post-translational modifications of αAI expressed in transgenic peas and chickpeas damaged by bruchids, with the processed forms of the same protein from several beans revealed microheterogeneity, with variations in the frequency of addition and variable processing of glycans, and in the C-terminal exopeptidase activity.
In the case of abiotic stress, the overexpression of Arabidopsis thaliana vacuolar H+-PPase (AVP1) in M. sativa lead to enhanced salt and drought tolerance. The trangenic plants accumulated more Na+, K+ and Ca2+ in their leaves and roots, and retained more water in the leaves during drought stress as compared with the wild-type plants (Bao et al. 2009). Similarly, overexpression of the pyrroline-5-carboxylate synthetase gene (P5CS) from Arabidopsis enhanced salt tolerance in chickpea (Ghanti et al. 2011). Very recently, Hanafy et al. (2013) reported enhanced tolerance to drought and salt stress in transgenic faba bean due to the heterologous expression of the PR10a gene, which encodes a pathogenesis related (PR) protein from potato.
In brief, overexpression of the candidate genes not only provides reliable validation of gene function, but may also lead to the development of improved lines in targeted crop species. Those improved, transgenic lines can be taken to greenhouse or field and exploited for enhancing yield and improving food or feed quality subject to biosafety procedures.
RNA interference (RNAi) and virus induced gene silencing (VIGS)
RNAi and VIGS are important approaches to validate the functions of candidate genes. In both of these methods, genes belonging to gene families are blocked or expressed across several tissues and developmental stages where antisense technologies fail to perform. The RNAi approach is sequence specific and can be targeted and controlled in tissue specific and time dependent manner. It is a popular approach for validating the function of candidate genes which have been identified on the basis of sequence similarity or through genetic mapping. This technology can be applied as an initial screen and subsequently validated by other methods (Small 2007).
RNAi-induced gene silencing is well established in model legumes and soybean. For example, in Medicago, RNAi was used to interfere with the RNA encoding PIN (auxin export facilitator) proteins responsible for nodule development. Reduced expression levels of root-specific PIN proteins produced plants with a reduced number of nodules, demonstrating the important role of PIN proteins in nodule development (Huo et al. 2006). In soybean, RNAi was employed to silence the gene encoding myo-inositol-1-phosphate (GmMIPS1), which plays an important role in regulating cellular metabolism and controlling growth. Seed development was not possible in lines in which the GmMIPS1 gene was silenced, demonstrating the correlation between GmMIPS1 gene expression and seed development (Nunes et al. 2006). Through RNAi-induced gene silencing is well established, VIGS may be a better approach in the long-term, due to its persistence during vegetative and in vitro propagation which, in turn, allows the generation of genotypically identical silenced plants.
VIGS has already been employed in soybean and pea. In soybean, a bean pod mottle virus (BPMV)-based system was used to identify genes participating in basal, resistance gene-mediated, and systemic immunity (Kachroo and Ghabrial 2012). In another study, the apple latent spherical virus (ALSV) vector was used to study gene function in the reproductive and early growth stages (emergence and cotyledon) in addition to the vegetative stages (Yamagishi and Yoshikawa 2009). The pea early-browning virus (PEBV) has been developed as a VIGS vector and used for functional analysis of several genes involved in the symbiosis. This study identified genes involved in symbiosis at the early and late growth stages of the plant (Grønlund et al. 2010). In summary, VIGS can be used as a forward or reverse genetics tool to validate the function of candidate gene(s) in transgenic plants as well as to characterise germplasm lines with differential expression of a gene with a desirable trait (Senthil-Kumar and Mysore 2011). However, the lack of appropriate vectors (specific for crop legumes), and efficient method for virus vector delivery may be limiting factors for the extensive applications of VIGS in crop legumes.
Targeting induced local lesions in genomes (TILLING)
Validation of genes through genetic transformation, RNAi or VIGS is a time consuming process in legumes, mainly due to lack of efficient transformation systems in legumes. This situation has promoted the application of TILLING to study gene function. In TILLING, candidate genes are screened across a mutant population (with point mutation), and line(s) with the mutation for the target gene are identified (McCallum et al. 2000). If the identified line exhibits the expected phenotype for the candidate gene, the function of the candidate gene is supported. The TILLING approach could be preferred over RNAi for irreversibly reducing or eliminating the target genes in commercial crop plants since it avoids genetic transformation and increases stability of the phenotype (Barkley and Wang 2008).
TILLING populations have been developed for several legumes. For example, in the model legumes, Medicago (12 000 M2 plants) (Rogers et al. 2009) and Lotus (4904 M2 lines) (Perry et al. 2009) mutant populations were developed for use in reverse genetics. In the case of crop legumes, over 3000 M3 lines were developed in common bean and evaluated with root nodulation tests by Porch et al. (2009). In peanut a TILLING population of 10 000 lines has been established, and a subset of this population investigated for allergenicity (Tadege et al. 2009). In chickpea, a TILLING population of ~3500 lines has been developed and is being used to identify candidate genes for drought tolerance (M. Thudi, pers. comm.). The use of NGS technologies for TILLING may increase the application of TILLING in crop legumes.
EcoTILLING is a variant of TILLING, except that its objective is to discover naturally occurring polymorphisms as opposed to experimentally induced mutations. Single nucleotide polymorphisms (SNPs), small insertions and deletions, and variations in microsatellite repeat number can be efficiently detected using the EcoTILLING technique. For example, in legumes this method has been used to develop molecular markers for cyst nematode candidate resistance genes in soybean (Liu et al. 2012). In mungbean, it has been proven to be a valuable method for detecting polymorphisms in a collection that was previously shown to have limited diversity (Barkley and Wang 2008).
Implications of functional genomics research on crop genetics and breeding
In recent years, significant progress has been made in developing genomic resources including genome sequences and transcriptome assemblies for a handful of crop legumes. These genomic tools may help to identify key factors involved in legume stress response. The availability of the complete genome or draft genome sequences of legume species and advances in sequencing and bioinformatics will accelerate gene discovery, particularly tolerance/resistance genes to abiotic/biotic stress in legumes (Edwards 2007). Though bioinformatics can predict potential gene function, it is important to validate gene function at the plant level, which can be done with overexpression, VIGS, RNAi, or TILLING, to name few. The outcome of experiments with these techniques can be complemented with metabolomics and proteomics. Once identified and validated, candidate genes enable the researcher to convert them into markers, to mine for superior alleles in germplasm collections, and to use them for a production of transgenics. Furthermore, identified and isolated candidate genes from one species can be channelled into comparative genomics studies of related species.
Acknowledgements
The authors are thankful to the Indo-German Science Technology Centre (IGSTC) and Australia-India Strategic Research Fund (AISRF) for financial support of the authors’ research presented in this study. The authors also thank Dadakhalandar Doddamani for his help during preparation of this manuscript. This work has been undertaken as part of the CGIAR Research Program on Grain Legumes. ICRISAT is a member of CGIAR Consortium.
References
Acharjee S, Sarmah BK, Kumar PA, Olsen K, Mahon R, Moar WJ, Moore A, Higgins TJV (2010) Transgenic chickpeas (Cicer arietinum L.) expressing a sequence-modified cry2Aa gene. Plant Science 178, 333–339.| Transgenic chickpeas (Cicer arietinum L.) expressing a sequence-modified cry2Aa gene.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXjtVSntbs%3D&md5=8cbb936b20301299fa93945bf8042ff5CAS |
Aragão FJ, Nogueira EO, Tinoco MLP, Faria JC (2013) Molecular characterization of the first commercial transgenic common bean immune to the Bean golden mosaic virus. Journal of Biotechnology 166, 42–50.
| Molecular characterization of the first commercial transgenic common bean immune to the Bean golden mosaic virus.Crossref | GoogleScholarGoogle Scholar | 23639387PubMed |
Asamizu E, Nakamura Y, Sato S, Tabata S (2004) Characteristics of the Lotus japonicus gene repertoire deduced from large-scale expressed sequence tag (EST) analysis. Plant Molecular Biology 54, 405–414.
| Characteristics of the Lotus japonicus gene repertoire deduced from large-scale expressed sequence tag (EST) analysis.Crossref | GoogleScholarGoogle Scholar | 15284495PubMed |
Ashraf N, Ghai D, Barman P, Basu S, Gangisetty N, Mandal MK, Chakraborty N, Datta A, Chakraborty S (2009) Comparative analyses of genotype dependent expressed sequence tags and stress-responsive transcriptome of chickpea wilt illustrate predicted and unexpected genes and novel regulators of plant immunity. BMC Genomics 10, 415
| Comparative analyses of genotype dependent expressed sequence tags and stress-responsive transcriptome of chickpea wilt illustrate predicted and unexpected genes and novel regulators of plant immunity.Crossref | GoogleScholarGoogle Scholar | 19732460PubMed |
Bao AK, Wang SM, Wu GQ, Xi JJ, Zhang JL, Wang CM (2009) Over expression of the Arabidopsis H+-PPase enhanced resistance to salt and drought stress in transgenic alfalfa (Medicago sativa L.). Plant Science 176, 232–240.
| Over expression of the Arabidopsis H+-PPase enhanced resistance to salt and drought stress in transgenic alfalfa (Medicago sativa L.).Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhsFagtbvN&md5=0a4aee81c94daa31c9e15b98d86aa9b0CAS |
Barkley NA, Wang ML (2008) Application of TILLING and EcoTILLING as reverse genetic approaches to elucidate the function of genes in plants and animals. Current Genomics 9, 212–226.
| Application of TILLING and EcoTILLING as reverse genetic approaches to elucidate the function of genes in plants and animals.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXotVehurw%3D&md5=9ddb1c2f1b0b8a1c24f2a03cbf5f7e41CAS | 19452039PubMed |
Bhatnagar-Mathur P, Devi MJ, Reddy DS, Lavanya M, Vadez V, Serraj R, Yamaguchi-Shinozaki K, Sharma KK (2007) Stress-inducible expression of At DREB1A in transgenic peanut (Arachis hypogaea L.) increases transpiration efficiency under water-limiting conditions. Plant Cell Reports 26, 2071–2082.
| Stress-inducible expression of At DREB1A in transgenic peanut (Arachis hypogaea L.) increases transpiration efficiency under water-limiting conditions.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhtlWrtr%2FI&md5=8b9804eb76ed6b4a824ab12e592e9fd6CAS | 17653723PubMed |
Bhatnagar-Mathur P, Rao JS, Vadez V, Sharma KK (2009) Transgenic strategies for improved drought tolerance in legumes of semi-arid tropics. Journal of Crop Improvement 24, 92–111.
| Transgenic strategies for improved drought tolerance in legumes of semi-arid tropics.Crossref | GoogleScholarGoogle Scholar |
Blair MW, Fernandez AC, Ishitani M, Moreta D, Seki M, Ayling S, Shinozaki K (2011) Construction and EST sequencing of full-length, drought stress cDNA libraries for common beans (Phaseolus vulgaris L.). BMC Plant Biology 11, 171
| Construction and EST sequencing of full-length, drought stress cDNA libraries for common beans (Phaseolus vulgaris L.).Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhtVyitb0%3D&md5=ba384a2a1ca527dfc00ac24ca54bb7b6CAS | 22118559PubMed |
Branca A, Paape T, Zhou P, Briskine R, Farmer AD, Mudge J, Bharti AK, Woodward JE, May GD, Gentzbittel L, Ben C, Denny R, Sadowsky MJ, Ronfort J, Bataillon T, Young ND, Tiffin P (2011) Whole-genome nucleotide diversity, recombination, and linkage-disequilibrium in the model legume Medicago truncatula. Proceedings of the National Academy of Sciences of the United States of America 108, E864–E870.
| Whole-genome nucleotide diversity, recombination, and linkage-disequilibrium in the model legume Medicago truncatula.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtl2qurzN&md5=e87e4709826b414f7014682fc51108dbCAS | 21949378PubMed |
Brenner S, Johnson M, Bridgham J, Golda G, Lloyd DH (2000) Gene expression analysis by massively parallel signature sequencing (MPSS) on microbead arrays. Nature Biotechnology 18, 630–634.
| Gene expression analysis by massively parallel signature sequencing (MPSS) on microbead arrays.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXkt12gur8%3D&md5=c192c44b1058c73ee16ad13f442ad42aCAS | 10835600PubMed |
Buhariwalla HK, Jayashree B, Eshwar K, Crouch JH (2005) Development of ESTs from chickpea roots and their use in diversity analysis of the Cicer genus. BMC Plant Biology 5, 16
| Development of ESTs from chickpea roots and their use in diversity analysis of the Cicer genus.Crossref | GoogleScholarGoogle Scholar | 16107212PubMed |
Chandra A, Pental D (2003) Regeneration and genetic transformation of grain legumes: an overview. Current Science 84, 381–387.
Chen X, Zhu W, Azam S, Li H, Zhu F, Li H, Hong Y, Liu H, Zhang E, Wu H, Yu S, Zhou G, Li S, Zhong N, Wen S, Li X, Knapp SJ, Ozias-Akins P, Varshney RK, Liang X (2013) Deep sequencing analysis of the transcriptomes of peanut aerial and subterranean young pods identifies candidate genes related to early embryo abortion. Plant Biotechnology Journal 11, 115–127.
| Deep sequencing analysis of the transcriptomes of peanut aerial and subterranean young pods identifies candidate genes related to early embryo abortion.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXjt1SqtL0%3D&md5=42750ef30bbd52970857dc40a932770cCAS | 23130888PubMed |
Cheong YH, Chang HS, Gupta R, Wang X, Zhu T, Luan S (2002) Transcriptional profiling reveals novel interactions between wounding, pathogen, abiotic stress, and hormonal responses in Arabidopsis. Plant Physiology 129, 661–677.
| Transcriptional profiling reveals novel interactions between wounding, pathogen, abiotic stress, and hormonal responses in Arabidopsis.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XkvV2jtrk%3D&md5=6deafbe926626fe9d239c923543d920cCAS | 12068110PubMed |
Cheung F, Haas BJ, Goldberg SMD, May GD, Xiao Y, Town CD (2006) Sequencing Medicago truncatula expressed sequenced tags using 454 Life Sciences technology. BMC Genomics 7, 272
| Sequencing Medicago truncatula expressed sequenced tags using 454 Life Sciences technology.Crossref | GoogleScholarGoogle Scholar | 17062153PubMed |
Citadin CT, Cruz ARR, Aragão FJL (2013) Development of transgenic imazapyr-tolerant cowpea (Vigna unguiculata). Plant Cell Reports 32, 537–543.
| Development of transgenic imazapyr-tolerant cowpea (Vigna unguiculata).Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXktVOnsbY%3D&md5=78ac9045ac0267bfb92373f61252f172CAS | 23306633PubMed |
Cook DR (1999) Medicago truncatula – a model in the making! Current Opinion in Plant Biology 2, 301–304.
| Medicago truncatula – a model in the making!Crossref | GoogleScholarGoogle Scholar | 1:STN:280:DyaK1MzptVeisA%3D%3D&md5=180ab183fe72f27b44d4cb5b77ed1968CAS | 10459004PubMed |
Das S, Bhat PR, Sudhakar C, Ehlers JD, Wanamaker S, Roberts PA, Cui X, Close TJ (2008) Detection and validation of single feature polymorphisms in cowpea (Vigna unguiculata L. Walp) using a soybean genome array. BMC Genomics 9, 107
| Detection and validation of single feature polymorphisms in cowpea (Vigna unguiculata L. Walp) using a soybean genome array.Crossref | GoogleScholarGoogle Scholar | 18307807PubMed |
Das S, Ehlers J, Close T, Roberts P (2010) Transcriptional profiling of root-knot nematode induced feeding sites in cowpea (Vigna unguiculata L. Walp.) using a soybean genome array. BMC Genomics 11, 480
| Transcriptional profiling of root-knot nematode induced feeding sites in cowpea (Vigna unguiculata L. Walp.) using a soybean genome array.Crossref | GoogleScholarGoogle Scholar | 20723233PubMed |
Deokar AA, Kondawar V, Jain PK, Karuppayil SM, Raju NL, Vadez V, Varshney RK, Srinivasan R (2011) Comparative analysis of expressed sequence tags (ESTs) between drought-tolerant and-susceptible genotypes of chickpea under terminal drought stress. BMC Plant Biology 11, 70
| Comparative analysis of expressed sequence tags (ESTs) between drought-tolerant and-susceptible genotypes of chickpea under terminal drought stress.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXlsVamsrc%3D&md5=ce2a493e6d02d0870e41543deb825d1bCAS | 21513527PubMed |
Dubey A, Farmer A, Schlueter J, Cannon SB, Abernathy B, Tuteja R, Woodward J, Shah T, Mulasmanovic B, Kudapa H, Raju NL, Gothalwal R, Pande S, Xiao Y, Town CD, Singh NK, May GD, Jackson S, Varshney RK (2011) Defining the transcriptome assembly and its use for genome dynamics and transcriptome profiling studies in pigeonpea (Cajanus cajan L.). DNA Research 18, 153–164.
| Defining the transcriptome assembly and its use for genome dynamics and transcriptome profiling studies in pigeonpea (Cajanus cajan L.).Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXntlGrtLk%3D&md5=401a477c81f44e59467ee8643711586cCAS | 21565938PubMed |
Dutta S, Kumawat G, Singh BP, Gupta DK, Singh S, Dogra V, Gaikwad K, Sharma TR, Raje RS, Bandhopadhya TK, Datta S, Singh MN, Bashasab F, Kulwal P, Wanjari KB, Varshney RK, Cook DR, Singh NK (2011) Development of genic-SSR markers by deep transcriptome sequencing in pigeonpea (Cajanus cajan (L.) Millspaugh). BMC Plant Biology 11, 17
| Development of genic-SSR markers by deep transcriptome sequencing in pigeonpea (Cajanus cajan (L.) Millspaugh).Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhvFant7o%3D&md5=d81ca4c17537f2bd5d15fd453504529fCAS | 21251263PubMed |
Edwards D (2007) Bioinformatics and plant genomics for staple crops improvement. In ‘Breeding major food staples’. (Eds MS Kang MS, PM Priyadarshan) pp. 93–106. (Blackwell: Oxford, UK)
Edwards D, Batley J (2010) Plant genome sequencing: applications for crop improvement. Plant Biotechnology Journal 8, 2–9.
| Plant genome sequencing: applications for crop improvement.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXptlaktw%3D%3D&md5=4ceef3ec62e2442a5957bc6bf4dbe04eCAS | 19906089PubMed |
Edwards D, Batley J, Snowdon R (2013) Accessing complex crop genomes with next-generation sequencing. Theoretical and Applied Genetics 126, 1–11.
| Accessing complex crop genomes with next-generation sequencing.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXkt1yntQ%3D%3D&md5=e871ed089ac0786381568d7bada0db98CAS | 22948437PubMed |
Fang C, Li W, Li G, Wang Z, Zhou Z, Ma Y, Shen Y, Li C, Wu Y, Zhu B, Yang W, Tian Z (2013) Cloning of Ln gene through combined approach of map-based cloning and association study in soybean. Journal of Genetics and Genomics = Yi Chuan Xue Bao 40, 93–96.
| Cloning of Ln gene through combined approach of map-based cloning and association study in soybean.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXpsVCrt7c%3D&md5=3c29f45fc4835bd2134ec027b0116935CAS | 23439408PubMed |
França MG, Matos AR, D’Arcy-Lameta A, Passaquet C, Lichtlé C, Zuily-Fodil Y, Pham-Thi AT (2008) Cloning and characterization of drought-stimulated phosphatidic acid phosphatase genes from Vigna unguiculata. Plant Physiology and Biochemistry 46, 1093–1100.
| Cloning and characterization of drought-stimulated phosphatidic acid phosphatase genes from Vigna unguiculata.Crossref | GoogleScholarGoogle Scholar | 18755595PubMed |
Franssen SU, Shrestha RP, Bräutigam A, Bornberg-Bauer E, Weber APM (2011) Comprehensive transcriptome analysis of the highly complex Pisum sativum genome using next generation sequencing. BMC Genomics 12, 227
| Comprehensive transcriptome analysis of the highly complex Pisum sativum genome using next generation sequencing.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXmtlKlsLc%3D&md5=07c2b13f2c897a6d140055fdc292db25CAS | 21569327PubMed |
Garg R, Patel RK, Tyagi AK, Jain M (2011a) De novo assembly of chickpea transcriptome using short reads for gene discovery and marker identification. DNA Research 18, 53–63.
| De novo assembly of chickpea transcriptome using short reads for gene discovery and marker identification.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXit1Wgsb4%3D&md5=e306135b6d060f4f5bc059d99ff96019CAS | 21217129PubMed |
Garg R, Patel RK, Jhanwar S, Priya P, Bhattacharjee A, Yadav G, Bhatia S, Chattopadhyay D, Tyagi AK, Jain M (2011b) Gene discovery and tissue-specific transcriptome analysis in chickpea with massively parallel pyrosequencing and web resource development. Plant Physiology 156, 1661–1678.
| Gene discovery and tissue-specific transcriptome analysis in chickpea with massively parallel pyrosequencing and web resource development.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtVOrur7J&md5=70eb8fa3d0c64066a83193c6cfab69e8CAS | 21653784PubMed |
Graham PH, Vance CP (2003) Legumes. Importance and constraints to greater use. Plant Physiology 131, 872–877.
| Legumes. Importance and constraints to greater use.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXisFemtb4%3D&md5=26557d60498601c32cfe15ab4e0f4f3cCAS | 12644639PubMed |
Grønlund M, Olsen A, Johansen EI, Jakobsen I (2010) Protocol: using virus-induced gene silencing to study the arbuscular mycorrhizal symbiosis in Pisum sativum. Plant Methods 6, 28–35.
| Protocol: using virus-induced gene silencing to study the arbuscular mycorrhizal symbiosis in Pisum sativum.Crossref | GoogleScholarGoogle Scholar | 21156044PubMed |
Guimarães PM, Brasileiro ACM, Morgante CV, Martins ACQ, Pappas G, Silva OB, Togawa R, Leal-Bertioli SCM, Araujo ACG, Moretzsohn MC, Bertioli DJ (2012) Global transcriptome analysis of two wild relatives of peanut under drought and fungi infection. BMC Genomics 13, 387
| Global transcriptome analysis of two wild relatives of peanut under drought and fungi infection.Crossref | GoogleScholarGoogle Scholar | 22888963PubMed |
Hanafy M, El-Banna A, Schumacher H, Jacobsen HJ, Hassan F (2013) Enhanced tolerance to drought and salt stresses in transgenic faba bean (Vicia faba L.) plants by heterologous expression of the PR10a gene from potato. Plant Cell Reports 32, 663–674.
| Enhanced tolerance to drought and salt stresses in transgenic faba bean (Vicia faba L.) plants by heterologous expression of the PR10a gene from potato.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXlvVelurg%3D&md5=d075f2a45eab39fad1d811f8fd0dbab3CAS | 23455709PubMed |
Handberg K, Stougaard J (1992) Lotus japonicus, an autogamous, diploid legume species for classical and molecular genetics. The Plant Journal 2, 487–496.
| Lotus japonicus, an autogamous, diploid legume species for classical and molecular genetics.Crossref | GoogleScholarGoogle Scholar |
Hiremath PJ, Farmer A, Cannon SB, Woodward J, Kudapa H, Tuteja R, Kumar A, Bhanuprakash A, Mulaosmanovic B, Gujaria N, Laxmanan K, Pooran MG, Polavarapu KK, Shah T, Srinivasan R, Lohse M, Xiao Y, Christopher DT, Cook DR, May GD, Varshney RK (2011) Large-scale transcriptome analysis in chickpea (Cicer arietinum L.), an orphan legume crop of the semi-arid tropics of Asia and Africa. Plant Biotechnology Journal 9, 922–931.
| Large-scale transcriptome analysis in chickpea (Cicer arietinum L.), an orphan legume crop of the semi-arid tropics of Asia and Africa.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtlemtrnF&md5=42f895a3e03d56bea765a5d12c9e10baCAS | 21615673PubMed |
Hoen PAC, Ariyurek Y, Thygesen HH, Vreugdenhil E, Vossen RHAM, Menezes RX, Boer JM, Ommen GB, Dunnen JT (2008) Deep sequencing-based expression analysis shows major advances in robustness, resolution and inter-lab portability over five microarray platforms. Nucleic Acids Research 36, e141
| Deep sequencing-based expression analysis shows major advances in robustness, resolution and inter-lab portability over five microarray platforms.Crossref | GoogleScholarGoogle Scholar |
Huettel B, Santra D, Muehlbauer F, Kahl G (2002) Resistance gene analogues of chickpea (Cicer arietinum L.): isolation, genetic mapping and association with a Fusarium resistance gene cluster. Theoretical and Applied Genetics 105, 479–490.
Hunt M, Kaur N, Stromvik M, Vodkin L (2011) Transcript profiling reveals expression differences in wild-type and glabrous soybean lines. BMC Plant Biology 11, 145
| Transcript profiling reveals expression differences in wild-type and glabrous soybean lines.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XnsFaitQ%3D%3D&md5=23e2802882088615aa57168aeedc19a7CAS | 22029708PubMed |
Huo X, Schnabel E, Hughes K, Frugoli J (2006) RNAi phenotypes and the localization of a protein: GUS fusion imply a role for Medicago truncatula PIN genes in nodulation. Journal of Plant Growth Regulation 25, 156–165.
| RNAi phenotypes and the localization of a protein: GUS fusion imply a role for Medicago truncatula PIN genes in nodulation.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XmsVSjs7k%3D&md5=6b7e1a38b3352fa0d675629b662a259aCAS | 19444321PubMed |
Imelfort M, Batley J, Grimmond S, Edwards D (2009a) Genome sequencing approaches and successes. In ‘Plant genomics Methods in molecular biology’. (Eds D Somers, P Langridge, JP Gustafson) pp. 345–358. (Humana Press: NY, USA)
Imelfort M, Duran C, Batley J, Edwards D (2009b) Discovering genetic polymorphisms in next generation sequencing data. Plant Biotechnology Journal 7, 312–317.
| Discovering genetic polymorphisms in next generation sequencing data.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXlvFOitLw%3D&md5=d4e847b5b7ed32c55cec868b925ce585CAS | 19386039PubMed |
Jackson JA, Hobbs SL (1990) Rapid multiple shoot production from cotyledonary node explants of pea (Pisum sativum L.). In Vitro Cellular & Developmental Biology 26, 835–838.
| Rapid multiple shoot production from cotyledonary node explants of pea (Pisum sativum L.).Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3cXmtVynur4%3D&md5=764fdc20329bb31d66f783ac6d9ae024CAS |
Jhanwar S, Priya P, Garg R, Parida SK, Tyagi AK, Jain M (2012) Transcriptome sequencing of wild chickpea as a rich resource for marker development. Plant Biotechnology Journal 10, 690–702.
| Transcriptome sequencing of wild chickpea as a rich resource for marker development.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhsVWgurfE&md5=3b5d598eea655d18e519ff9603d6f322CAS | 22672127PubMed |
Jia Y, Gu H, Wang X, Chen Q, Shi S, Zhang J, Ma L, Zhang H, Ma H (2012) Molecular cloning and characterization of an F-box family gene CarF-box1 from chickpea (Cicer arietinum L.). Molecular Biology Reports 39, 2337–2345.
| Molecular cloning and characterization of an F-box family gene CarF-box1 from chickpea (Cicer arietinum L.).Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhvVWmt7s%3D&md5=a7e97565af67afabbfa2bbc82edfea63CAS | 21667242PubMed |
Kachroo A, Ghabrial S (2012) Virus-induced gene silencing in soybean. In ‘Antiviral resistance in plants’. pp. 287–297. (Humana Press: NY, USA)
Kahl G, Molina C, Udupa SM (2007) Super SAGE: exploring the stress transcriptome in chickpea. In ‘Plant and animal genome XV conference’. P. W91. (Town and Country Convention Center: San Diego, CA, USA)
Kalavacharla V, Liu Z, Meyers BC, Thimmapuram J, Melmaiee K (2011) Identification and analysis of common bean (Phaseolus vulgaris L.) transcriptomes by massively parallel pyrosequencing. BMC Plant Biology 11, 135
| Identification and analysis of common bean (Phaseolus vulgaris L.) transcriptomes by massively parallel pyrosequencing.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsVaksr7O&md5=82108346f65802255d2542fd8f8bbb71CAS | 21985325PubMed |
Kaneda Y, Tabei Y, Nishimura S, Harada K, Akihama T, Kitamura K (1997) Combination of thidiazuron and basal media with low salt concentrations increases the frequency of shoot organogenesis in soybeans (Glycine max (L.) Merr.). Plant Cell Reports 17, 8–12.
| Combination of thidiazuron and basal media with low salt concentrations increases the frequency of shoot organogenesis in soybeans (Glycine max (L.) Merr.).Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXkt1OgsA%3D%3D&md5=a3798db9dfd09f7d4181bb6c755d23efCAS |
Kaur H, Shukla RK, Yadav G, Chattopadhyay D, Majee M (2008) Two divergent genes encoding L-myo-inositol 1-phosphate synthase1 (CaMIPS1) and 2 (CaMIPS2) are differentially expressed in chickpea. Plant, Cell & Environment 31, 1701–1716.
| Two divergent genes encoding L-myo-inositol 1-phosphate synthase1 (CaMIPS1) and 2 (CaMIPS2) are differentially expressed in chickpea.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtl2hs7zM&md5=5f912a0c3812bc070c9ad534a7a5cb2bCAS |
Kaur S, Cogan N, Pembleton L, Shinozuka M, Savin K, Materne M, Forster J (2011) Transcriptome sequencing of lentil based on second-generation technology permits large-scale unigene assembly and SSR marker discovery. BMC Genomics 12, 265
| Transcriptome sequencing of lentil based on second-generation technology permits large-scale unigene assembly and SSR marker discovery.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXmvVKqsrw%3D&md5=0fe0628a3126c832d25d1f0853ccfdb9CAS | 21609489PubMed |
Kim KY, Park SW, Chung YS, Chung CH, Kim JI, Lee JH (2004) Molecular cloning of low-temperature-inducible ribosomal proteins from soybean. Journal of Experimental Botany 55, 1153–1155.
| Molecular cloning of low-temperature-inducible ribosomal proteins from soybean.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXjs1Wrurg%3D&md5=fea71d60e615bb406820cfd902e01ff7CAS | 15020631PubMed |
Ghanti S, Sujata KG, Vijay Kumar BV, Nataraha Karba N, Janardhan Reddy K, Srinath Rao MS, Kavi Kishor PB (2011) Heterologous expression of P5CS gene in chickpea enhances salt tolerance without affecting yield. Biologia Plantarum 55, 634–640.
| Heterologous expression of P5CS gene in chickpea enhances salt tolerance without affecting yield.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtlWrtb%2FK&md5=8491388e25f77b03a11d5817b87e7abaCAS |
Kudapa H, Bharti AK, Cannon SB, Farmer AD, Mulaosmanovic B, Kramer R, Bohra A, Weeks NT, Crow JA, Tuteja R, Shah T, Dutta S, Gupta DK, Singh A, Gaikwad K, Sharma TR, May GD, Singh NK, Varshney RK (2012) A comprehensive transcriptome assembly of pigeonpea (Cajanus cajan L.) using Sanger and second-generation sequencing platforms. Molecular Plant 5, 1020–1028.
| A comprehensive transcriptome assembly of pigeonpea (Cajanus cajan L.) using Sanger and second-generation sequencing platforms.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhtlKlu7fL&md5=04247911ef8f93208f4b5ae6b28b0f79CAS | 22241453PubMed |
Lam HM, Xu X, Lui X, Chen W, Yang G, Wong FL, Li MW, He W, Qin N, Wang B, Li J, Jian M, Wang J, Shao G, Wang J, Sun SS, Zhang G (2010) Re-sequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nature Genetics 42, 1053–1059.
| Re-sequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsVWmurrJ&md5=d76c137275ffd60d86c1fbc71fd74d11CAS | 21076406PubMed |
Langridge P, Fleury D (2011) Making the most of ‘omics’ for crop breeding. Trends in Biotechnology 29, 33–40.
| Making the most of ‘omics’ for crop breeding.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhs1ajtrrL&md5=3f8367bc742afc71c5fd8fcf215eb762CAS | 21030098PubMed |
Li XP, Tian AG, Luo GZ, Gong ZZ, Zhang JS, Chen SY (2005) Soybean DRE-binding transcription factors that are responsive to abiotic stresses. Theoretical and Applied Genetics 110, 1355–1362.
| Soybean DRE-binding transcription factors that are responsive to abiotic stresses.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXkvFSru78%3D&md5=0edad30d87f53adf66ffe8c4539e229fCAS | 15841365PubMed |
Li L, Wang WQ, Wu CX, Han TF, Hou WS (2012) Construction of two suppression subtractive hybridization libraries and identification of salt-induced genes in soybean. Journal of Integrative Agriculture 11, 1075–1085.
| Construction of two suppression subtractive hybridization libraries and identification of salt-induced genes in soybean.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhtV2itr7E&md5=fc9e32e0d0938cb805e8e5a033f20b11CAS |
Libault M, Farmer A, Joshi T, Takahashi K, Langley RJ, Franklin LD, Xu D, May G, Stacey G (2010) An integrated transcriptome atlas of the crop model Glycine max, and its use in comparative analyses in plants. The Plant Journal 63, 86–99.
Liu S, Kandoth PK, Warren SD, Yeckel G, Heinz R, Alden J, Yang C, Jamai A, El-Mellouki T, Juvale PS, Hill J, Baum TJ, Cianzio S, Whitham SA, Korkin D, Mitchum MG, Meksem K (2012) A soybean cyst nematode resistance gene points to a new mechanism of plant resistance to pathogens. Nature 492, 256–260.
| A soybean cyst nematode resistance gene points to a new mechanism of plant resistance to pathogens.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhvVamtb3F&md5=8d1b707422ccb4ee22859c24c792bed8CAS | 23235880PubMed |
Lorenc MT, Hayashi S, Stiller J, Lee H, Manoli S, Ruperao P, Visendi P, Berkman PJ, Lai K, Batley J, Edwards D (2012) Discovery of single nucleotide polymorphisms in complex genomes using SGSautoSNP. Biology 1, 370–382.
| Discovery of single nucleotide polymorphisms in complex genomes using SGSautoSNP.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhvVSrsL%2FO&md5=adb5605a1449230465161a848244b1e9CAS |
Lüthi C, Álvarez-Alfageme F, Romeis J (2010) The potential of transgenic legumes in integrated bruchid management: assessing the impact on bruchid parasitoids. In ‘10th International working conference on stored product protection’. pp. 977–982. (Julius Kühn-Institut: Estoril, Portugal)
Mahajan S, Sopory SK, Tuteja N (2006) Cloning and characterization of CBL‐CIPK signalling components from a legume (Pisum sativum). FEBS Journal 273, 907–925.
| Cloning and characterization of CBL‐CIPK signalling components from a legume (Pisum sativum).Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28Xjs1Oltr0%3D&md5=a61d96bbc2ffd2948732e116fdae07f6CAS | 16478466PubMed |
Mantri NL, Ford R, Coram TE, Pang EC (2010) Evidence of unique and shared responses to major biotic and abiotic stresses in chickpea. Environmental and Experimental Botany 69, 286–292.
| Evidence of unique and shared responses to major biotic and abiotic stresses in chickpea.Crossref | GoogleScholarGoogle Scholar |
Matsumura H, Ito A, Saitoh H, Winter P, Kahl G, Reuter M, Kruger DH, Terauchi R (2005) SuperSAGE. Cellular Microbiology 7, 11–18.
| SuperSAGE.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXotlWkug%3D%3D&md5=9f2323b4bdf3ce4d8ebddf22e87e73a5CAS | 15617519PubMed |
McCallum CM, Comai L, Greene EA, Henikoff S (2000) Targeting induced local lesions IN genomes (TILLING) for plant functional genomics. Plant Physiology 123, 439–442.
| Targeting induced local lesions IN genomes (TILLING) for plant functional genomics.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXktlWnt7s%3D&md5=8327d6bd1cfe46c2cc9bb95d6c62b880CAS | 10859174PubMed |
Meng Q, Zhang C, Gai J, Yu D (2007) Molecular cloning, sequence characterization and tissue-specific expression of six NAC-like genes in soybean (Glycine max (L.) Merr.) Journal of Plant Physiology 164, 1002–1012.
| Molecular cloning, sequence characterization and tissue-specific expression of six NAC-like genes in soybean (Glycine max (L.) Merr.)Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhtVSmsLrM&md5=536e574ab44cff7a5c0b09140d9fd3d7CAS | 16919368PubMed |
Moe KT, Chung JW, Cho YI, Moon JK, Ku JH, Jung JK, Lee J, Park YJ (2011) Sequence information on simple sequence repeats and single nucleotide polymorphisms through transcriptome analysis of mungbean. Journal of Integrative Plant Biology 53, 63–73.
| Sequence information on simple sequence repeats and single nucleotide polymorphisms through transcriptome analysis of mungbean.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhs1aqtbc%3D&md5=458b46a268b1cea4d2c6828c1dd74c1aCAS | 21205180PubMed |
Molina C, Rotter B, Horres R, Udupa SM, Besser B, Bellarmino L, Baum M, Matsumura H, Terauchi R, Kahl G, Winter P (2008) SuperSAGE: the drought stress-responsive transcriptome of chickpea roots. BMC Genomics 9, 553
| SuperSAGE: the drought stress-responsive transcriptome of chickpea roots.Crossref | GoogleScholarGoogle Scholar | 19025623PubMed |
Molina C, Zaman-Allah M, Khan F, Fatnassi N, Horres R, Rotter B, Steinhauer D, Amenc L, Drevon J-J, Winter P, Kahl G (2011) The salt-responsive transcriptome of chickpea roots and nodules via deepSuperSAGE. BMC Plant Biology 11, 31
| The salt-responsive transcriptome of chickpea roots and nodules via deepSuperSAGE.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXis1Sgsbk%3D&md5=dd2e8c6a24f934886bb95e35d620d9c7CAS | 21320317PubMed |
Muchero W, Diop NN, Bhat PR, Fenton RD, Wanamaker S, Pottor M, Hearne S, Cisse N, Fatokun C, Ehlers JD, Roberts PA, Close TJ (2009) A consensus genetic map of cowpea (Vigna unguiculata (L) Walp.) and synteny based on EST-derived SNPs. Proceedings of the National Academy of Sciences of the United States of America 106, 18159–18164.
| A consensus genetic map of cowpea (Vigna unguiculata (L) Walp.) and synteny based on EST-derived SNPs.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVags7rP&md5=b97f2e0f7276556cc3bed43e2e95d6afCAS | 19826088PubMed |
Munroe DJ, Harris TJ (2010) Third-generation sequencing fireworks at Marco Island. Nature Biotechnology 28, 426–428.
| Third-generation sequencing fireworks at Marco Island.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXlslyqu7o%3D&md5=760bd9f45af58b2a58794e28c8b6dd26CAS | 20458306PubMed |
Nunes AC, Vianna GR, Cuneo F, Amaya-Farfán J, de Capdeville G, Rech EL, Aragão FJ (2006) RNAi-mediated silencing of the myo-inositol-1-phosphate synthase gene (GmMIPS1) in transgenic soybean inhibited seed development and reduced phytate content. Planta 224, 125–132.
| RNAi-mediated silencing of the myo-inositol-1-phosphate synthase gene (GmMIPS1) in transgenic soybean inhibited seed development and reduced phytate content.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XkvFCrtrs%3D&md5=663d27f1c1f24da1c7df00b00adfa2dcCAS | 16395584PubMed |
O’Rourke JA, Yang SS, Miller SS, Bucciarelli B, Liu J, Rydeen A, Bozsoki Z, Uhde-Stone C, Tu ZJ, Allan D, Gronwald JW, Vance CP (2013) An RNA-Seq transcriptome analysis of orthophosphate-deficient white lupin reveals novel insights into phosphorus acclimation in plants1. Plant Physiology 161, 705–724.
| An RNA-Seq transcriptome analysis of orthophosphate-deficient white lupin reveals novel insights into phosphorus acclimation in plants1.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXmvFKqs78%3D&md5=8b911f8153bf44db3484164ca11e0490CAS | 23197803PubMed |
Pang Y, Wenger JP, Saathoff K, Peel GJ, Wen J, Huhman D, Allen SN, Tang Y, Cheng X, Tadege M, Ratet P, Mysore KS, Sumner LW, Marks DM, Dixon RA (2009) A WD40 repeat protein from Medicago truncatula is necessary for tissue-specific anthocyanin and proanthocyanidin biosynthesis but not for trichome development. Plant Physiology 151, 1114–1129.
| A WD40 repeat protein from Medicago truncatula is necessary for tissue-specific anthocyanin and proanthocyanidin biosynthesis but not for trichome development.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVCjsbrF&md5=e541600d87cf9b2b78a9b4b7581964d3CAS | 19710231PubMed |
Parra-González LB, Aravena-Abarzúa GA, Navarro-Navarro CS, Udall J, Maughan J, Peterson LM, Salvo-Garrido HE, Maureira-Butler IJ (2012) Yellow lupin (Lupinus luteus L.) transcriptome sequencing: molecular marker development and comparative studies. BMC Genomics 13, 425
| Yellow lupin (Lupinus luteus L.) transcriptome sequencing: molecular marker development and comparative studies.Crossref | GoogleScholarGoogle Scholar | 22920992PubMed |
Peng H, Cheng HY, Chen C, Yu XW, Yang JN, Gao WR, Shi QH, Zhang H, Li JG, Ma H (2009) A NAC transcription factor gene of chickpea (Cicer arietinum), CarNAC3, is involved in drought stress response and various developmental processes. Journal of Plant Physiology 166, 1934–1945.
| A NAC transcription factor gene of chickpea (Cicer arietinum), CarNAC3, is involved in drought stress response and various developmental processes.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsValsLzI&md5=d0c39d5a3cca73d8174169df02a45bbaCAS | 19595478PubMed |
Perry J, Brachmann A, Welham T, Binder A, Charpentier M, Groth M, Haage K, Markmann K, Wang TL, Parniske M (2009) TILLING in Lotus japonicus identified large allelic series for symbiosis genes and revealed a bias in functionally defective ethyl methanesulfonate alleles toward glycine replacements. Plant Physiology 151, 1281–1291.
| TILLING in Lotus japonicus identified large allelic series for symbiosis genes and revealed a bias in functionally defective ethyl methanesulfonate alleles toward glycine replacements.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVCjsbjO&md5=6477d2386ae8073b5dfbac2f64a00671CAS | 19641028PubMed |
Porch TG, Blair MW, Lariguet P, Galeano C, Pankhurst CE, Broughton WJ (2009) Generation of a mutant population for TILLING common bean genotype BAT 93. Journal of the American Society for Horticultural Science 134, 348–355.
Qiao G, Wen X, Yu L, Ji X (2012) Identification of differentially expressed genes preferably related to drought response in pigeon pea (Cajanus cajan) inoculated by arbuscular mycorrhizae fungi (AMF). Acta Physiologiae Plantarum 34, 1711–1721.
| Identification of differentially expressed genes preferably related to drought response in pigeon pea (Cajanus cajan) inoculated by arbuscular mycorrhizae fungi (AMF).Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXpvFeqsb4%3D&md5=1d511db674a4f2110369681eb50bc6caCAS |
Rajput MK, Upadhyaya KC (2010) Isolation and characterization of stress induced Ty1-copia like retrotransposable elements in chickpea (Cicer arietinum L.). Molecular Biology 44, 693–698.
| Isolation and characterization of stress induced Ty1-copia like retrotransposable elements in chickpea (Cicer arietinum L.).Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXht1Kqt7zN&md5=a1bbd35cf5cc345af3c91e794a90fd00CAS |
Raju NL, Gnanesh BN, Lekha P, Jayashree B, Pande S, Hiremath PJ, Byregowda M, Singh NK, Varshney RK (2010) The first set of EST resource for gene discovery and marker development in pigeonpea (Cajanus cajan L.). BMC Plant Biology 10, 45
| The first set of EST resource for gene discovery and marker development in pigeonpea (Cajanus cajan L.).Crossref | GoogleScholarGoogle Scholar | 20222972PubMed |
Rogers C, Wen J, Chen R, Oldroyd G (2009) Deletion-based reverse genetics in Medicago truncatula. Plant Physiology 151, 1077–1086.
| Deletion-based reverse genetics in Medicago truncatula.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVCjsbrK&md5=4f09eafe7f3078aec744aec69e968ef3CAS | 19759346PubMed |
Romo S, Labrador E, Dopico B (2001) Water stress-regulated gene expression in Cicer arietinum seedlings and plants. Plant Physiology and Biochemistry 39, 1017–1026.
| Water stress-regulated gene expression in Cicer arietinum seedlings and plants.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXosFOis70%3D&md5=e432ca462bc7b1b30b0b03f26e08bd52CAS |
Sato S, Nakamura Y, Kaneko T, Asamizu E, Kato T, Nakao M, Sato S, Nakamura Y, Kaneko T, Asamizul E, et al (2008) Genome structure of the legume, Lotus japonicus. DNA Research 15, 227–239.
| Genome structure of the legume, Lotus japonicus.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXht12ht7zO&md5=814d5a76da0362fdef0f42dd800465f1CAS | 18511435PubMed |
Saxena RK, Cui X, Thakur V, Walter B, Close TJ, Varshney RK (2011) Single feature polymorphisms (SFPs) for drought tolerance in pigeonpea (Cajanus spp.). Functional & Integrative Genomics 11, 651–657.
| Single feature polymorphisms (SFPs) for drought tolerance in pigeonpea (Cajanus spp.).Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsV2lsrjM&md5=2399367ad6ced985bd4d1bb31078d872CAS |
Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, Nelson W, Hyten DL, Song Q, Thelen JJ, Cheng J, Xu D, et al (2010) Genome sequence of the palaeopolyploid soybean. Nature 463, 178–183.
| Genome sequence of the palaeopolyploid soybean.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXntVClsQ%3D%3D&md5=25ab7ea670a325318576566fa9c47bc4CAS | 20075913PubMed |
Sekhar K, Priyanka B, Reddy VD, Rao KV (2011) Metallothionein 1 (CcMT1) of pigeonpea (Cajanus cajan, L.) confers enhanced tolerance to copper and cadmium in Escherichia coli and Arabidopsis thaliana. Environmental and Experimental Botany 72, 131–139.
| Metallothionein 1 (CcMT1) of pigeonpea (Cajanus cajan, L.) confers enhanced tolerance to copper and cadmium in Escherichia coli and Arabidopsis thaliana.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXnt1Kqsb4%3D&md5=38de00393a0094a634de1c40a407903dCAS |
Senthil-Kumar M, Mysore KS (2011) New dimensions for VIGS in plant functional genomics. Trends in Plant Science 16, 656–665.
| New dimensions for VIGS in plant functional genomics.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsFOhu7%2FL&md5=cf5e00e04ff82a168a37b078132722d5CAS | 21937256PubMed |
Sharma KD, Muehlbauer FJ (2007) Fusarium wilt of chickpea: physiological specialization, genetics of resistance and resistance gene tagging. Euphytica 157, 1–14.
| Fusarium wilt of chickpea: physiological specialization, genetics of resistance and resistance gene tagging.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXpslaqtbg%3D&md5=5914778b8549ab0029d75549ba7d96ddCAS |
Sharma KK, Lavanya M, Anjaiah V (2006) Agrobacterium-mediated production of transgenic pigeonpea (Cajanus cajan L. Millsp) expressing the synthetic BT CRY1AB gene. In Vitro Cellular & Developmental Biology. Plant 42, 165–173.
| Agrobacterium-mediated production of transgenic pigeonpea (Cajanus cajan L. Millsp) expressing the synthetic BT CRY1AB gene.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28Xms1ymurg%3D&md5=3a6ff9f137b246e4b65e68d2d1f34c7eCAS |
Sharpe AG, Ramsay L, Sanderson LA, Fedoruk MJ, Clarke WE, Li R, Kagale S, Vijayan P, Vandenberg A, Bett KE (2013) Ancient orphan crop joins modern era: gene-based SNP discovery and mapping in lentil. BMC Genomics 14, 192
| Ancient orphan crop joins modern era: gene-based SNP discovery and mapping in lentil.Crossref | GoogleScholarGoogle Scholar | 23506258PubMed |
Shoemaker R, Keim P, Vodkin L, Retzel E, Clifton SW, Waterston R, Smoller D, Coryell V, Khanna A, Erpelding J, et al (2002) A compilation of soybean ESTs: generation and analysis. Genome 45, 329–338.
| A compilation of soybean ESTs: generation and analysis.Crossref | GoogleScholarGoogle Scholar | 11962630PubMed |
Small I (2007) RNAi for revealing and engineering plant gene functions. Current Opinion in Biotechnology 18, 148–153.
| RNAi for revealing and engineering plant gene functions.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXksFehu7s%3D&md5=7e450868a66d4a8e7960582cd8d96420CAS | 17287115PubMed |
Sreenivasulu N, Kishor PBK, Varshney RK, Altschmied L (2002) Mining functional information from cereal genomics – the utility of expressed sequence tags. Current Science 83, 965
Tachi H, Fukuda-Yamada K, Kojima T, Shiraiwa M, Takahara H (2009) Molecular characterization of a novel soybean gene encoding a neutral PR-5 protein induced by high-salt stress. Plant Physiology and Biochemistry 47, 73–79.
| Molecular characterization of a novel soybean gene encoding a neutral PR-5 protein induced by high-salt stress.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhsFamtbbP&md5=536f14d1c24e636cd2042aa63b76042dCAS | 19010689PubMed |
Tadege M, Wang TL, Wen J, Ratet P, Mysore KS (2009) Mutagenesis and beyond! Tools for understanding legume biology. Plant Physiology 151, 978–984.
| Mutagenesis and beyond! Tools for understanding legume biology.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVCjsb3J&md5=ac265466042b366030c4ffe8fc0d58caCAS | 19741047PubMed |
Thibivilliers S, Joshi T, Campbell KB, Scheffler B, Xu D, Cooper B, Nguyen HT, Stacey G (2009) Generation of Phaseolus vulgaris ESTs and investigation of their regulation upon Uromyces appendiculatus infection. BMC Plant Biology 9, 46
| Generation of Phaseolus vulgaris ESTs and investigation of their regulation upon Uromyces appendiculatus infection.Crossref | GoogleScholarGoogle Scholar | 19397807PubMed |
Thudi M (2013) Towards fine mapping of drought tolerance related QTL region in chickpea using genotyping by sequencing (GBS) approach. In ‘Plant and animal genome XXI conference’. pp. 11–15. (Town and Country Convention Center: San Diego, CA, USA)
Thudi M, Li Y, Jackson SA, May GD, Varshney RK (2012) Current state-of-art of sequencing technologies for plant genomics research. Briefings in Functional Genomics 11, 3–11.
| Current state-of-art of sequencing technologies for plant genomics research.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xisl2lsbk%3D&md5=4534f2b1367ca7e7e3d1fa0f988472f6CAS | 22345601PubMed |
Torres-Franklin ML, Contour-Ansel D, Zuily-Fodil Y, Pham-Thi AT (2008) Molecular cloning of glutathione reductase cDNAs and analysis of GR gene expression in cowpea and common bean leaves during recovery from moderate drought stress. Journal of Plant Physiology 165, 514–521.
| Molecular cloning of glutathione reductase cDNAs and analysis of GR gene expression in cowpea and common bean leaves during recovery from moderate drought stress.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXltlWms7k%3D&md5=af18b9e97c18bf2fd888789e7bbdaa75CAS | 17707549PubMed |
Tuskan G, Slavov G, DiFazio S, Muchero W, Pryia R, Schackwitz W, Martin J, Rokhsar D, Sykes R, Davis M, Studer M, Wyman C (2011) Populus resequencing: towards genome-wide association studies. BMC Proceedings 5, I21
| Populus resequencing: towards genome-wide association studies.Crossref | GoogleScholarGoogle Scholar |
Valliyodan B, Nguyen HT (2006) Understanding regulatory networks and engineering for enhanced drought tolerance in plants. Current Opinion in Plant Biology 9, 189–195.
| Understanding regulatory networks and engineering for enhanced drought tolerance in plants.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XhvVWrurw%3D&md5=091d63bee7635a0957809506a42460f1CAS | 16483835PubMed |
Varshney RK, Hiremath PJ, Lekha P, Kashiwagi J, Balaji J, Deokar AA, Vadez V, Xiao Y, Srinivasan R, Gaur PM, Siddique KHM, Town CD, Hoisington DA (2009a) A comprehensive resource of drought- and salinity- responsive ESTs for gene discovery and marker development in chickpea (Cicer arietinum L.). BMC Genomics 10, 523
| A comprehensive resource of drought- and salinity- responsive ESTs for gene discovery and marker development in chickpea (Cicer arietinum L.).Crossref | GoogleScholarGoogle Scholar | 19912666PubMed |
Varshney RK, Nayak SN, May GD, Jackson SA (2009b) Next generation sequencing technologies and their implications for crop genetics and breeding. Trends in Biotechnology 27, 522–530.
| Next generation sequencing technologies and their implications for crop genetics and breeding.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtVeitbbE&md5=a84efed0bcc8f3f430b7f8996a41a29dCAS | 19679362PubMed |
Varshney RK, Chen W, Li Y, Bharthi AK, Saxena RK, Schlueter JA, Donoghue MA, Azam S, Fan G, Whaley AM, et al (2012) Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers. Nature Biotechnology 30, 83–89.
| Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsVagu7%2FO&md5=ef626a7aab6281f0fb337da41fa84938CAS |
Varshney RK, Song C, Saxena RK, Azam S, Yu S, Sharpe AG, Cannon S, Baek J, Rosen BD, Tar’an B, et al (2013) Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nature Biotechnology 31, 240–246.
| Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXhsVymtrY%3D&md5=0db53d940f00eb902500954ef0e1eb06CAS | 23354103PubMed |
Verdier J, Torres‐Jerez I, Wang M, Andriankaja A, Allen SN, He J, Tang Y, Murray JD, Udvardi MK (2013) Establishment of the Lotus japonicus gene expression atlas (LjGEA) and its use to explore legume seed maturation. The Plant Journal 74, 351–362.
| Establishment of the Lotus japonicus gene expression atlas (LjGEA) and its use to explore legume seed maturation.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXlt1ags7w%3D&md5=785245da2ee45622c870e222d15b20aaCAS | 23452239PubMed |
Vij S, Tyagi AK (2007) Emerging trends in the functional genomics of the abiotic stress response in crop plants. Plant Biotechnology Journal 5, 361–380.
| Emerging trends in the functional genomics of the abiotic stress response in crop plants.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXlsF2ktbc%3D&md5=3e5a04102cef9746ff1e3a9027dbd4bbCAS | 17430544PubMed |
Vodkin LO, Khanna A, Robin Shealy R, Steven J, Clough SJ, Gonzalez DO, Philip R, Gracia Zabala G, Thibaud-Nissen F, Sidarous M, et al (2004) Microarrays for global expression constructed with a low redundancy set of 27 500 sequenced cDNAs representing an array of developmental stages and physiological conditions of the soybean plant. BMC Genomics 5, 73
| Microarrays for global expression constructed with a low redundancy set of 27 500 sequenced cDNAs representing an array of developmental stages and physiological conditions of the soybean plant.Crossref | GoogleScholarGoogle Scholar | 15453914PubMed |
Wang N, Khan W, Smith DL (2012) Changes in soybean global gene expression after application of lipo-chitooligosaccharide from Bradyrhizobium japonicum under sub-optimal temperature. PLoS ONE 7, e31571
| Changes in soybean global gene expression after application of lipo-chitooligosaccharide from Bradyrhizobium japonicum under sub-optimal temperature.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XivVWksbg%3D&md5=58305b63716b2b4f41ca1eee798be9feCAS | 22348109PubMed |
Watanabe S, Hideshima R, Xia Z, Tsubokura Y, Sato S, Nakamoto Y, Yamanaka N, Takahashi R, Ishimoto M, Anai T, Tabata S, Harada K (2009) Map-based cloning of the gene associated with the soybean maturity locus E3. Genetics 182, 1251–1262.
| Map-based cloning of the gene associated with the soybean maturity locus E3.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtFGktbfJ&md5=9a63230c8ffd42f102685758ccab73d3CAS | 19474204PubMed |
Wu N, Matand K, Wu H, Li B, Li Y, Zhang X, He Z, Qian J, Liu X, Conley S, Bailey M, Acquaah G (2013) De novo next-generation sequencing, assembling and annotation of Arachis hypogaea L. Spanish botanical type whole plant transcriptome. Theoretical and Applied Genetics 126, 1145–1149.
| De novo next-generation sequencing, assembling and annotation of Arachis hypogaea L. Spanish botanical type whole plant transcriptome.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXmslSksLg%3D&md5=54d6d74ccb1557d3fdeaebb747a87eb2CAS | 23338522PubMed |
Yamagishi N, Yoshikawa N (2009) Virus-induced gene silencing in soybean seeds and the emergence stage of soybean plants with apple latent spherical virus vectors. Plant Molecular Biology 71, 15–24.
| Virus-induced gene silencing in soybean seeds and the emergence stage of soybean plants with apple latent spherical virus vectors.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXptVGjtbs%3D&md5=54ccc8b57b37e0d42b70f79076f22877CAS | 19495995PubMed |
Young ND, Bharti AK (2012) Genome-enabled insights into legume biology. Annual Review of Plant Biology 63, 283–305.
| Genome-enabled insights into legume biology.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xos1amsL8%3D&md5=a71e6384f5cc5831809625c696f17ba9CAS | 22404476PubMed |
Young ND, Debellé F, Oldroyd GE, Geurts R, Cannon SB, Udvardi MK, Benedito VA, Mayer KFX, Gouzy J, Schoof H, et al (2011) The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature 480, 520–524.
Zhang J, Liang S, Duan J, Wang J, Chen S, Cheng Z, Zhang Q, Liang X, Li Y (2012) De novo assembly and characterisation of the transcriptome during seed development, and generation of genic-SSR markers in Peanut (Arachis hypogaea L.). BMC Genomics 13, 90
| De novo assembly and characterisation of the transcriptome during seed development, and generation of genic-SSR markers in Peanut (Arachis hypogaea L.).Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xot12rtr8%3D&md5=6b180c7ac06d066faa9b278e3128621aCAS | 22409576PubMed |
Zhu H, Choi H, Cook DR, Shoemaker RC (2005) Bridging model and crop legumes through comparative genomics. Plant Physiology 137, 1189–1196.
| Bridging model and crop legumes through comparative genomics.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXjslaqtbg%3D&md5=f892e3ea7ed0a7ec983a3b44abe53d27CAS | 15824281PubMed |