

ROWLEY REVIEW. Phylogeography: its development and impact in Australo-Papuan ornithology with special reference to paraphyly in Australian birds
Leo Joseph A C and Kevin E. Omland BA Australian National Wildlife Collection, CSIRO Sustainable Ecosystems, GPO Box 284, Canberra, ACT 2601, Australia.
B Department of Biological Sciences, University of Maryland, Baltimore County, 1000 Hilltop Circle, Baltimore, MD 21250, USA.
C Corresponding author. Email: leo.joseph@csiro.au
Emu 109(1) 1-23 https://doi.org/10.1071/MU08024
Submitted: 12 May 2008 Accepted: 18 September 2008 Published: 20 January 2009
Abstract
With examples from Australo-Papuan ornithology, we examine the technical and theoretical roots of molecular phylogeography and review its development. We describe the progression from ad hoc interpretation of gene trees in single species phylogeographic studies through to comparative phylogeography and currently advocated model-testing approaches. Mitochondrial DNA (mtDNA) sequences have provided most advances to date, although we demonstrate and advocate the future use of multilocus datasets analysed with coalescent methods. We examine interrelationships among speciation research, historical biogeography, phylogeography and landscape genetics. Mitochondrial paraphyly, in which individuals of one species or population have mtDNA that is more closely related to that of another than to their own, emerges in 44% of Australian studies to date as a common, important result in Australian avian phylogeography. Accordingly, we explore at length its most common causes and its impact on case studies in Australo-Papuan avian phylogeography. The impact of so much paraphyly on avian phylogeography and taxonomy is a major theme of the review. We suggest a full research agenda for avian phylogeography in the Australo-Papuan region that spans diverse topics: the need for more studies of pelagic birds, spatio-temporal links between New Guinea and Australia, island populations, testing of long-established biogeographical hypotheses, and integration of molecular and non-molecular datasets into integrated evolutionary understanding of species and populations. Studying the full continuum of divergences from landscape genetics, to phylogeography, to recently diverged species with evidence of paraphyly, to highly divergent species with many fixed differences will lead to a more complete understanding of the processes and patterns of avian evolution.
Introduction
Molecular phylogeography (hereafter ‘phylogeography’) is the study of how genetic diversity within a species has evolved (phylo-) and how it is organised across the geographical distribution of that species (-geography) (Avise et al. 1987). Here we trace the origins and development of phylogeography to address three interrelated objectives: (1) to understand phylogeography’s own recurring themes and conceptual debates; (2) to review and interpret how these issues relate to Australo-Papuan avian biology, biogeography and systematics; and (3) to suggest future phylogeographic research programs and agendas in the theatre of ornithology and the stage provided, mainly, by Australo-Papuan birds. A major theme to emerge is that morphological and molecular analyses of Australian birds are frequently at odds in how they suggest the evolutionary history of a species and the populations within it should be understood. Throughout we examine relationships among phylogeography, speciation research and other disciplines, such as historical biogeography (the study of how geological processes have shaped present-day species distributions) and landscape genetics (the study of present-day genetic structure within species at the scale of individual landscapes) (see also Avise and Walker 1998; Diniz-Filho et al. 2008). Our review focuses on Australo-Papuan avian phylogeography with an intended emphasis on Australia itself. Examples from outside Australia will provide context for some points.
Since the inception of phylogeography in 1987, a revolution has occurred in the study of DNA-level diversity within species and how it informs relationships among populations within and between species. Studies on birds have been prominent in this revolution (Avise and Walker 1998). Reviews of phylogeographic data for North American birds (Zink 1997) and South American and African montane birds (Roy et al. 1997) have appeared but no review has been tailored for ornithological audiences and certainly not for the southern hemisphere avian focus of Emu (see also Beheregaray 2008, Zink and Barrowclough 2008). Our intended audience is conservation managers and ornithologists interested in relationships at the species–population level interface. Much of this audience is increasingly frustrated with what is sometimes seen as counterintuitive, contradictory or inconclusive results from phylogeography and molecular data in general. We hope to clarify relevant issues, especially in an Australo-Papuan context. Also, ornithologists and Australo-Papuan ornithology have tackled questions on the spectrum from historical biogeography, taxonomy and hybrid zones through phylogeography to landscape genetics (Keast 1961; Ford 1987; Schodde and Mason 1999; Schodde 2006; Christidis and Boles 2008; review in Joseph 2008a). That literature is now a wealth of questions and hypotheses for the resources and tools of phylogeography to address. This review is timely because each study we consider is a piece in a larger jigsaw that informs us of the origins and history of the avifauna, as well as helping to determine where future work is needed (Box 1).

|
Phylogeography: an overview of its context and development
Phylogeography’s place in the spectrum from speciation to landscape genetics
Studies of speciation, historical biogeography, phylogeography and landscape genetics can be seen as points on a continuum that seek to understand evolutionary history of one or more species in relation to the landscape on which they have evolved. It is useful, therefore, to examine how they differ from each other. Figure 1 in Diniz-Filho et al. (2008) usefully depicts phylogeography’s relationships to these and other facets of evolutionary biology.
Speciation research often addresses how reproductive isolation evolves and how it is maintained. Price’s (2008) survey of speciation in birds does not index ‘phylogeography’. This is informative because it stresses speciation’s relationship to reproductive isolation, which is not a primary conceptual focus of phylogeography although it can be fruitfully inferred in some cases from gene trees. (Note here the debate over whether reproductive isolation is a cause or consequence of speciation – see Zink and McKay 2008.) Phylogeography can help define where in the landscape or between which populations of a species one might usefully look for incipient reproductive isolation. It offers patterns for further investigation, not a mechanism of speciation (Edwards 2008a, 2008b) and issues of tension between phylogeography and speciation research can be tested with phylogeographic methods (Knowles 2004). Examples are whether founder events are involved in speciation or whether reproductive isolation is a by-product of the gradual accumulation of species differences by genetic drift. Price (2008) argued the greater relevance of selection in generating reproductive isolation in avian speciation.
Historical biogeography studies the relationships between Earth history and species distributions. Its temporal framework is necessarily long-term but overlaps with phylogeography when single species are discussed. At the other end of the continuum, landscape genetics shares with phylogeography the question of how landscape features may explain major evolutionary disjunctions or events in the history of a species, but they differ in temporal and spatial scales. Landscape genetics analyses spatial genetic data without requiring that discrete populations be identified a priori. It is focused at the finest geographical scales of a species’ range. It addresses processes and patterns of gene flow and local adaptation through to how landscape characteristics structure populations (Manel et al. 2003). It usually studies individuals in geographical areas that are phylogenetically more closely related to each other than to individuals in other such units, as defined by phylogeography (i.e. within its phylogroups). The two have much in common when biological interest focusses on zones of contact between two phylogroups within a species. Then both are concerned with measuring gene flow across such zones. Phylogeography has a broader interest in that past gene flow and population size are of as much interest as current ones, whereas landscape genetics is concerned only with the present. Population genetics straddles phylogeography and landscape genetics. The microevolutionary process of how the genetic variation present in a common ancestor sorts into unique clusters (phylogroups, clades) in descendent populations or species is unique to these areas and generates the monophyly that higher level systematics and biogeography depend on. We explore this process at length in this review but it is, we suggest, a very real difference between microevolution and macroevolution. Speciation research, historical biogeography, phylogeography and landscape genetics, in our view, are continua, or different points, on a spectrum, not independent fields of study. A given question might be cast in terms of one or the other but full illumination of an evolutionary problem may require some or all of the different perspectives offered by these fields be considered. Australian examples of this interdependence that we will discuss are in studies of the Australian Magpie (Gymnorhina tibicen) (Hughes et al. 2001; Toon et al. 2007), the White-winged Fairy-wren (Malurus leucopterus) (Driskell et al. 2002), and blue-cheeked parrots of the Crimson Rosella (Platycercus elegans) complex (Joseph et al. 2008).
Origins: the late 1980s and early 1990s
Phylogeography has technical and theoretical roots. The late 1970s and early 1980s saw wide application of the technique of allozyme electrophoresis to answer species-level taxonomic questions with the genetic distance data that it generates. Studies of birds were prominent but found little within-species variation (Avise et al. 1980a, 1980b; Avise and Aquadro 1982; see Joseph and Hope 1984 for an Australian example). For questions of within-species variation, other kinds of data have occasionally proven useful, and still do, especially in organisms other than birds. Chromosomal variation in the Australian rock-wallabies (Petrogale spp.) is an outstanding example (Sharman et al. 1989). Allozymes are cost-effective and especially useful in the discovery of cryptic diversity (Hillis et al. 1996). But they are one step removed from the ultimate ideal for molecular data, DNA sequences, which have now largely supplanted them.
By 1987, theoretical and practical disconnects had grown between population genetics, which was concerned with heredity and microevolutionary processes, and systematics and palaeontology, which studied phylogeny and macroevolution. The stage was set for the profound theoretical impact of a seminal paper by Avise et al. (1987). They noted that several classic population genetics texts had not indexed ‘phylogeny’, ‘systematics’ or ‘speciation’ and that important texts in systematics could be read with only basic Mendelian and population genetics. Molecular evolution with its own grounding in genetics and phylogeny was an obvious way to link the study of microevolution and macroevolution. Critically, Avise et al. (1987) argued that phylogenetic reasoning could provide the basis for a dialogue between population genetics and systematics. They illustrated this by showing that a phylogenetic tree comprises family pedigrees or genealogies. Figure 1 illustrates this simple but profound concept. Their central thesis was that mitochondrial DNA (mtDNA) by virtue of properties discussed above provided a ‘liaison service for expanding communication between population geneticists and systematists’. They coined the term ‘intraspecific phylogeography’ because it unites phylogenetic relationships of mtDNA molecules within a species with the geographical distributions of those phylogenetic groupings.
|
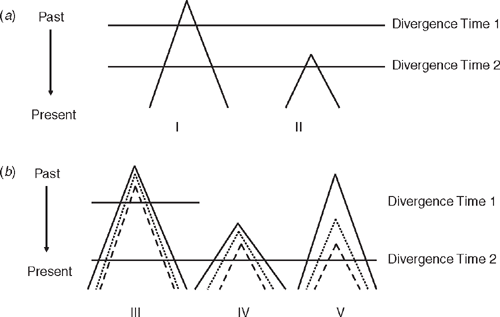
Thus began the era of building mtDNA gene trees. The primary currency of phylogeography has been a phylogenetic tree of mtDNA relationships among individuals and populations within a species. Where structure exists, the different clades within a species can be termed phylogroups. Such trees are overlain on maps of the landscape, and then the goal is to understand how the species and its component populations have evolved on that landscape. It has often been stressed that gene trees are embedded in the true species tree, which is the ultimate concern of phylogeny and phylogeography, and that they do not always correspond with each other, a concept illustrated in Fig. 2. We examine the reasons for discordance between gene trees and species trees in more detail later but it is important to emphasise the principle here. Maddison (1997) noted that ‘Phylogeny is more like a statistical distribution than a simple tree of discrete thin branches. It has a central tendency, but it also has a variance because of the diversity of gene trees. Gene trees that disagree with the central tendency are not wrong; rather, they are part of the diffuse pattern that is the genetic history.’ One can now estimate phylogenetic trees of populations and species from collections of gene trees, which are the real characters of trees at the level of species and populations (Maddison and Knowles 2006; Liu et al. 2008).
|
Explosion of phylogeography: the mid- to late 1990s
Avise et al.’s (1987) crystallisation of links between microevolution, macroevolution and geography came at a time ideally suited for profound impacts on systematists and population geneticists. This was because of two further developments that fuelled phylogeography’s growth. First was technical access to the genotype. By 1989, mtDNA variation could readily be quantified with Restriction Fragment Length Polymorphism (RFLP). In Australian ornithology, Ovenden et al. (1987), Edwards and Wilson (1990), Degnan and Moritz (1992), Austin et al. (1994) and Joseph and Moritz (1993a, 1993b, 1994) applied this technique to rosellas (Platycercus spp.), babblers (Pomatostomus spp.), white-eyes (Zosterops spp.), Short-tailed Shearwaters (Puffinus tenuirostris) and scrubwrens (Sericornis spp.), respectively. Degnan’s (1993a, 1993b) work on whiteyes was among the first demonstrations of the importance of using nuclear DNA (nDNA) and mtDNA. Direct sequencing of DNA has been more common since ~1992. Second was the development of software for phylogenetic and population genetics analyses of DNA sequences. There is now a bewildering array of packages, though only a few stalwarts do much of the published work (see reviews in Excoffier and Heckel 2006; Latch et al. 2006; Chen et al. 2007).
A revolution followed Avise et al. (1987). A 2007 search of the internet for citations of the paper showed 1173 citations. Similar searches on the word ‘phylogeography’ for the periods 1988–97 and 1998–2007, found 115 v. 3048 papers, respectively. Corresponding totals for ‘avian phylogeography’, though likely underestimates, show the same trend: 6 v. 129, respectively. Typically, many of these papers described mtDNA diversity within a species, estimated its phylogenetic structure and then overlaid that structure on the geography of the species’ range. Armed with this essentially correlative approach, inferences were made about how landscape features had shaped genetic diversity.
Two early key examples from Australian avian phylogeography showed how statistical approaches could take data interpretation beyond simple correlation. They also detected major phylogeographic breaks in this early era of phylogeography. First, Edwards’s (1993a) study of the Grey-crowned Babbler (Pomatostomus temporalis) was the first, large-scale, continent-wide phylogeographic study of an Australian vertebrate. Statistical testing demonstrated a phylogeographic break correlating with the previously recognised Carpentarian Barrier about the Gulf of Carpentaria. This set the scene for further phylogeographic work on grass-finches (Poephila spp.) (Jennings and Edwards 2005) and fairy-wrens (Malurus spp.) (Lee and Edwards 2008). Second, for birds of Australia’s Wet Tropics with differing degrees of specialisation to rainforest, Joseph et al. (1995) used permutation tests to show statistically significant associations between habitat specialisation and structure of gene trees. They concluded that the more habitat-specialised a species is to rainforests, the earlier it would have been isolated into separate subpopulations by the Pleistocene’s increasingly severe cycles of aridity. They also detected a phylogeographic break in rainforest species north of Cairns across what is known as the Black Mountain Barrier (see Hugall et al. 2002).
Comparative phylogeography
By 1998, the merit of comparative phylogeography, the study of phylogeographies of unrelated species that share the same distribution, was being stressed: if unrelated species with similar distributions shared the same phylogeographic structure, then a common extrinsic, environmental cause could be sought. The journal Molecular Ecology devoted an issue to comparative phylogeography (Bermingham and Moritz 1998).
Comparative phylogeographic studies of birds of Australo-Papuan rainforests have been done on scales ranging from New Guinea and Australia together (Joseph et al. 2001; Norman et al. 2002, 2007) to within eastern Australia generally (Joseph et al. 1993; Joseph and Moritz 1994; Nicholls and Austin 2005; Nicholls et al. 2006), to within Wet Tropics rainforests (Joseph et al. 1995). They have clarified histories of habitat specialisation and long-standing systematic issues.
Observation of two co-distributed but unrelated species having the same branching pattern but at different depths may seem to offer compelling evidence that they evolved that structure at very different times. This could equally well be explained, however, by differentiation within the two having occurred at the same time but with different ancestral population sizes in the different pairs of populations (Edwards and Beerli 2000). The smaller population will sort more quickly and might be misinterpreted as having diverged at a different time than the larger populations. This emphasises the relationship between phylogeography’s goal of describing patterns of phylogroups within species on one hand and understanding the processes that generate those patterns on the other hand. An example is how population size interacts with genetic diversity. Larger populations require more time for ancestral variation to sort into groups that correspond with population divergence. This reiterates differences between population and gene divergences (Fig. 2). Perhaps most crucially, the link is stressed between understanding and estimation of phylogeographic structure and the need to understand past and present demographic processes. This is why data from multiple loci, not just mtDNA, will be increasingly important if, as Zink and Barrowclough (2008) argued, modern evolutionary genetics is to integrate phylogeography and evolutionary process. The value of multilocus data is illustrated in Fig. 3 and we return to it throughout the review. Also, Cook and Crisp (2005) argued that environmentally induced bias in dispersal direction could also derail phylogeographic reconstruction. This raises how gene divergences are analysed, and a key part of this in recent years has been coalescent theory (Kingman 2000), which we now briefly examine.
|
Coalescence and multiple loci
At the same time as the explosion of phylogeography in the mid-1990s, two major shifts occurred in how DNA sequence data are gathered and analysed. In classical population genetics, one measured gene frequencies in populations, and microevolution was seen as a shift in gene frequencies in a population over time. In coalescent theory, the genetic diversity in a sample of individuals in one or more populations is traced back to its common ancestor along the branches of a phylogenetic tree estimated from present-day diversity. The mathematics of this approach allows estimates of population divergence times, ancestral and present-day population sizes, and ancestral and present migration rates. A full review of coalescent theory is beyond our scope. We will reiterate two important points made by Edwards and Beerli (2000). First is that when present-day genetic variation (allelic diversity) is sampled within and between two or more populations of a species, diversity traces to a single allele that was present in the population that was, in turn, ancestral to the present-day, descendent populations. That ancestral population necessarily had some allelic diversity, which will manifest itself as some coalescent depth to its gene trees. Therefore, gene divergences used to estimate population divergences that are often of prime interest will precede the population divergence. Care is needed in analyses to acknowledge this (Fig. 2; Jennings and Edwards 2005). Discrepancy between the times of divergence of genes and populations will be greater for a larger ancestral population. A second point was increased awareness of the value of data from multiple loci to counteract the stochasticity that can arise from working with a single gene (Fig. 3). Stochastic sorting of lineages has the potential to generate artefactual geographical structuring in single genes, leading to over-interpretation of geographical history when studies are limited to a single gene (Wilkins 2004; Kuo and Avise 2005). This is particularly a concern when inference is from a single species or where multiple species do not have congruent geographical breaks. Thus, testing for concordance across loci within and among species can be at least as important as screening across concordantly distributed species. This is because congruent patterns across species and different loci rather than across species for one locus are more likely to describe common historical events (Avise 2000; Jennings and Edwards 2005; Brito 2007).
Statistical phylogeography, model-testing and nested clade analysis
A legitimate criticism of phylogeography was that ad hoc inferences of phylogeographic datasets and their gene trees are little more than qualitative guesses. These guesses may or may not accurately capture the biogeographical and temporal features of a species’ history (Knowles and Maddison 2002). For example, a simple underlying population history is often assumed, such as simple divergence of two undivided populations of constant size. But what of more complex and realistic histories such as sequentially diverging populations with varying migration rates between them? In response, Templeton et al. (1995) introduced Nested Clade Analysis (NCA), a protocol for rigorously repeatable interpretations of phylogeographic datasets. It integrated geographical and genetic data from specimens in a study to determine, for example, where vicariance could be supported or rejected, where dispersal was needed to explain the data, and so on. Although widely seized upon by phylogeographers, NCA is controversial (Knowles and Maddison 2002; Templeton 2004). Petit (2008a) argued that NCA is fatally flawed in generating false-positive results at unacceptable rates and more debate predictably ensued (Garrick et al. 2008a; Knowles 2008; Petit 2008b; Templeton 2008). Although used widely in avian phylogeography outside Australo-Papua and within Australia for groups other than birds, it has simply not been used in many analyses of this region’s birds. Joseph and Wilke (2007) used it as a minor, complementary analysis in examining diversity in three species widespread across the Australian arid zone.
Statistical phylogeography was advocated in response to perceived problems with NCA (Knowles and Maddison 2002). Its basic tenet is that phylogeographic data should only be interpreted against a priori models. That is, one should infer specific biological processes with explicit reference to stochastically derived expectations (Knowles 2004; Knowles and Carstens 2007; Knowles et al. 2007), a basic procedure in science after all. Some key challenges in this approach are to define hypotheses simple enough that they can be discriminated with the data available, yet still capture the essence of the biologically interesting problem; to determine how complex a model can be fit without making overly simplified assumptions that might potentially affect the accuracy of the conclusions; and to develop more testable and biologically relevant hypotheses by incorporating external data, including information from other disciplines. Garrick et al. (2008b) demonstrate the power of an approach that combines NCA and statistical phylogeography.
Mitochondrial monophyly and paraphyly – their impact on phylogeography
We now examine the key concepts of monophyly and paraphyly in phylogeographic data. This sets the scene for closer examination of results and challenges in Australo-Papuan avian phylogeography. We will stress that at the species–population level interface, one is capturing the dynamics of what happens to an ancestral gene pool at different snapshots in time as daughter species split from each other and as the relevant processes, which we will discuss, unfold. At higher taxonomic levels when one is looking at neutral markers, these processes will often have run to completion and are not so confounding as in phylogeography. Thus, working within and between species can blur conventional taxonomic boundaries. This explains our emphasis on these processes in this review.
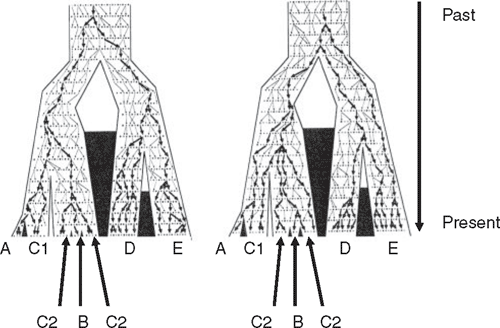
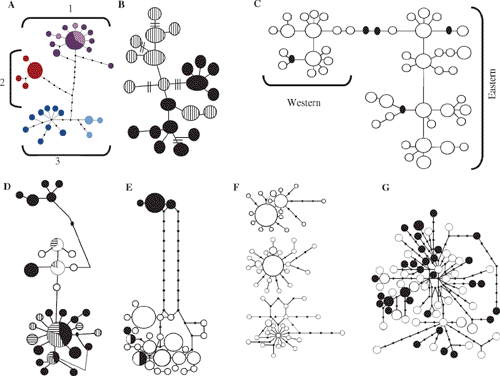
Intuitively, and from the viewpoint of many field workers, a reasonable and common a priori expectation is that in a phylogeographic study DNA sequences for a given species should have DNA unique to that species, at least for DNA neutral to selection. Any DNA sequence from Chestnut Teal (Anas castanea), for example, might reasonably be expected to be unique to Chestnut Teal, a Grey Teal (Anas gracilis) sequence should be similarly unique to Grey Teal, and so on. Stated more robustly, we expect that DNA from any individual Chestnut Teal will be more closely related to other individuals of its species than to DNA of any individual of any other species: Chestnut Teal DNAs will be reciprocally monophyletic with respect to other species, and vice versa. Yet phylogeographic studies often reject reciprocal monophyly and find discordance between gene trees and species trees (Fig. 2). Australian examples include white-eyes (Degnan and Moritz 1992), scrubwrens (Joseph and Moritz 1993a, 1993b), White-winged Fairy-wren complex (Driskell et al. 2002) and woodswallows (Artamus spp.) (Joseph et al. 2006) (Fig. 4). DNA ‘intermixing’ between closely related species is more strictly termed paraphyly (Fig. 2). It occurs when DNAs of individuals of a species or population are more closely related to DNAs of individuals that clearly belong to another species or population (Page and Holmes 1998).
|
Early molecular studies of phylogeography and species relationships often had at most a few representatives of each species. Avise et al. (1990) found that some Mallards (Anas platyrhynchos) have mtDNA more similar to American Black Ducks (Anas rubripes) than to other Mallards. In fact, Mallard mtDNA is paraphyletic with mtDNA of several Anas species including the Pacific Black Duck (A. superciliosa). Mallards on a London pond could have mtDNA more closely related to that of a Pacific Black Duck in Sydney than to that of others on that same London pond (see Omland 1997; Johnson and Sorenson 1999). However, as sample sizes increased, patterns emerged showing mtDNAs of closely related species often to be paraphyletic not reciprocally monophyletic. This is increasingly common in the literature. Joseph and Wilke (2004, 2006) urged that it be seen not as failure of molecular data to be a magic wand that would solve long-standing, intransigent taxonomic issues. Instead, they argue, interesting new biological questions should be the focus.
Funk and Omland (2003) surveyed paraphyly in published mtDNA studies of more than 2000 animal species. Of these, 23% were not monophyletic in their mtDNA trees. The survey included 331 species of birds, of which 17% were paraphyletic or polyphyletic in their mtDNA trees. Levels of mtDNA non-monophyly may be higher in Australia because eight of the 18 non-pelagic species (44%) surveyed for the present review (Table 1; see also Jonsson et al. 2008) are paraphyletic. This level of paraphyly cautions that exact congruence between molecules and morphology or between gene trees and species trees almost should not even be expected in phylogeography when one is at the interface between species and populations. Rather than dismiss paraphyly as impenetrable, we note the insight it brings to knowledge of a species’ history and biology. Accordingly, we now discuss two main causes of DNA paraphyly relevant to phylogeography and their impact on understanding of species limits. They are hybridisation and incomplete lineage sorting (ILS), and we stress the need to consider whether one is discussing paraphyly between species or among populations within a species. We exclude frequency-dependent selection and heterozygote advantage, which population genetics theory long ago showed can lead to shared genetic variation between species (see full review of causes in Funk and Omland 2003).

|
Causes of paraphyly I(a) – recent hybridisation between species
Hybridisation between bird species is common (Grant and Grant 1992; Price 2008). It results in flow or introgression of genes from one species into another and thus paraphyly of at least one of the species. A simple Australian example concerns the habitat-generalist White-browed Scrubwren (Sericornis frontalis) and the rainforest-restricted Atherton Scrubwren (S. keri). The 2.8% divergence between most mtDNA sequences of these species implies divergence from a common ancestor on the order of approximately 1 million years ago (Joseph and Moritz 1993a). One of four Wet Tropics individuals of frontalis caught together in sclerophyll woodland had mtDNA identical (0.0% divergence) to most keri sequences. The simplest explanation of this is that a hybridisation event at an unknown time in the past but likely too recent for mutation to have generated new diversity introduced mtDNA from keri into frontalis.
Arguably as interesting as the detection of hybridisation itself are fresh biological questions arising from phylogeography. The White-browed Scrubwren is a rainforest bird wherever the habitat occurs in its range except in the Wet Tropics. There, the White-browed Scrubwren is confined to sclerophyllous associations, secondary growth rainforest and rainforest edges, whereas the Atherton Scrubwren is confined to rainforest. With molecular data showing that hybridisation has occurred, the ecological and demographic dynamics between these two species at the rainforest–sclerophyll ecotone should be studied.
Causes of paraphyly I(b) – past hybridisation between species
More complicated is the inference and understanding of older hybridisation events, here defined as having occurred thousands or hundreds of thousands of years ago. Phylogeography of Australian white-eyes (Zosterops spp.) is a strong example. Degnan and Moritz (1992) used mtDNA data in the widespread Silvereye (Z. lateralis) and the more restricted tropical northern Yellow White-eye (Z. luteus). Their mtDNAs were paraphyletic, with one luteus haplotype, labelled i, being more closely related to haplotypes e, f, g and h that were in many lateralis individuals than to other luteus. Conversely, of course, those lateralis haplotypes e, f, g and h were more closely related to luteus haplotype i than to many lateralis haplotypes. However, no haplotypes were shared between the two species, and these intermixed haplotypes are 0.3–0.7% divergent from each other. Assuming a rate of divergence in mtDNA of 1.6–2.0% per million years (Fleischer et al.1998; also see Lovette 2004; Ho 2007; Weir and Schluter 2008), one infers that the hybridisation that caused this paraphyly likely occurred on the order of hundreds of thousands of years ago. More work on this subject is in progress.
Degnan and Moritz (1992) argued against ILS as the explanation for their data because large sequence divergences were involved. The remaining luteus sampled (haplotypes j and k) are nearly 4% divergent from the others sampled from the two species (again implying 2–3 million years of separate evolution for these mitochondrial lineages and likely for these species). It is unlikely that any ancestral mtDNA polymorphism would be retained for such a period. Data from the nuclear genome provide additional tests of the past hybridisation hypothesis (Degnan 1993a). Recent ongoing hybridisation and recent speciation with ILS could both result in many nuclear genes being shared between species (with identical or similar genotypes intermixed in the two species). Degnan (1993a) reported data from two nuclear genes and found that they showed generally fixed differences and over 0.6% divergence between the two species. However, one luteus did carry a haplotype found in lateralis, implying the possibility of some recent or ongoing gene flow. Although the evidence supports older introgression as the cause of mtDNA paraphyly between these two species, further work could reveal much additional complexity with which to test further the ancient hybridisation hypothesis. This work is underway using an expanded sample of luteus that has since been collected (n = 6 in earlier studies), more dense geographical sampling (especially near the western contact zone between the species), multiple loci, and currently available analytical methods especially coalescent approaches. Rigorous statistical analyses are needed to reject the possibility of ILS (Peters et al. 2007; see below). We now review how ILS occurs and provide likely Australian examples.
Causes of paraphyly II – incomplete lineage sorting within and between species
Phylogeography of the widespread Australian Magpie (Gymnorhina tibicen) was studied by Toon et al. (2007). At the Last Glacial Maximum 18 000 years ago, Magpies could easily have been continuously distributed across present-day Tasmania and the Australian mainland. Later isolation of Tasmania likely would have caused complete cessation of gene flow between Tasmania and mainland populations. Diversity of the ancestral population would have been shared in both daughter populations for a long time following isolation (see Omland et al. 2006). This could explain Toon et al.’s (2007, fig. 2) finding of haplotypes shared among Tasmanian and mainland Magpies. Had genetic study been done during the early post-isolation period it would show alleles and diversity that were in the ancestor. That is, it would detect paraphyly of Tasmanian populations with respect to the mainland populations and vice versa. A first interpretation might be of ongoing gene flow. However, recent divergence and the retention of ancestral diversity is a sufficient explanation in this example. The lineages have not ‘sorted out’ to where Tasmanian and mainland populations are reciprocally monophyletic, or unique. Thus there is ILS. After a period of time, likely to be on the order of thousands of years, genetic drift and relevant demographic variables such as variance in number of female offspring complete the lineage sorting and reciprocal monophyly could be attained. The larger the original ancestral population and the two daughter populations were, the longer that the ancestral polymorphism will be present and the longer that ancestral polymorphism will be retained. Thus, larger ‘effective population sizes’ will increase the duration of the process of lineage sorting. Although mtDNA has a lower effective population size (see above), complete sorting is hardly instantaneous.
ILS is an important, ubiquitous process that has been involved in every speciation event on Earth. Therefore, DNA paraphyly between species even after all gene flow between them has ceased is neither extraordinary nor special and phylogeographers should expect it. Examples of ILS of mtDNA in birds are numerous (e.g. Table 1). In each, the evolutionary process has been captured at a snapshot in time when variation in two daughter lineages that our taxonomy may recognise as species still reflects that of their most recent common ancestor. Discordance between phenotype and genotype results.
Biological insights from paraphyly
We have stressed that the taxonomic level at which one examines paraphyly can profoundly affect its analysis and interpretation. Within a species it usually means that gene flow between populations or that a relatively recent hybridisation event has been detected. Between species, it can mean a trace of ancient hybridisation remains or that two species have evolved recently. In this section, we now explore different insights from paraphyly at different taxonomic levels. We have also seen (Table 1) that paraphyly emerges as a common finding in phylogeographic studies of Australian birds. So, we now consider the interesting range of biological insights it offers (see also Avise and Walker 1998).
Levels of paraphyly: historical and contemporary processes
Edwards (1993b) applied coalescent-based methods to interpret paraphyly among island and mainland populations of the Grey-crowned Babbler. He favoured the explanation of ongoing gene flow rather than diversity retained in common from purely historical association of ancestral populations. Similarly in another babbler, Hall’s Babbler (P. halli), paraphyly was of interest because it suggested novel demographic insights (Miura and Edwards 2001).
A spectacular example of paraphyly at the interspecific level is in phylogeography of continental populations of woodswallows (Joseph et al. 2006). Coupled with the examples above and in Table 1, it hints not that there is gene flow between the two species involved but that widespread Australian birds have maintained large effective population sizes historically, not just currently.
The mtDNA haplotypes of the White-browed Woodswallow (A. superciliosus) and Masked Woodswallow (A. personatus) were almost completely randomly intermixed. Closest mtDNA relatives of many superciliosus haplotypes are personatus haplotypes, and vice versa. The haplotype network for these two woodswallows (Fig. 4) is among the most complicated yet found in birds. Two aspects of the data suggest that ILS, not hybridisation or gene flow between the two species, best explains the findings. First, the two species shared no haplotypes whereas recent gene flow should result in some interspecific sharing of haplotypes. Second, many weakly divergent haplotypes existed within each species. Inferences are that both species must have very large effective population sizes, that their common ancestor had a large enough population size to maintain a high number and diversity of haplotypes, and that both species evidently have retained or added to this diversity. With large effective population sizes, the process of lineage sorting will slow dramatically, and even more rapidly evolving parts of mtDNA, such as the control region, can retain ancestral polymorphisms and paraphyly, likely for many hundreds of thousands of years (see Neigel and Avise 1986 for modelling of this process). However, multiple nuclear loci and coalescent methods are needed to test further whether ILS is necessary and adequate to explain the data.
The woodswallow data exemplify a point in the series of stages predicted to occur by Omland et al. (2006) as mtDNA diversity of two diverging populations undergoes lineage sorting. It is a stage predicted to involve no sharing of haplotypes yet near complete paraphyly. This is stage II of intermediate divergence or ‘neotypy’ because none of the old ancestral haplotypes are shared between species.
This example suggests a further point. Phylogeographic studies of northern hemisphere birds commonly infer that Pleistocene ice-sheets that blanketed the landscape caused major population bottlenecks and erosion of genetic diversity and that glaciation generally influenced present-day structure. For North American examples see Zink (1997), Boulet and Gibbs (2006), and Ruegg et al. (2006), and for Eurasian examples see Yang et al. (2005), Pavlova et al. (2006), and Brito (2007). Widespread Australian birds studied to date, however, are showing patterns ranging from deep to shallow nucleotide diversity at a range of geographical scales across sometimes vast parts of the continent (Table 1). This suggests that they have maintained relatively large effective population sizes despite cycles of Pleistocene glaciation. Pleistocene climatic fluctuations were global but perhaps being under ice had a more dramatic effect firstly on habitats and population sizes of animals and secondly on eradicating genetic diversity than the drying, cooling and habitat contraction into smaller and more isolated patches as appears to have occurred in Australia. Perhaps many tropical, subtropical, arid and, more generally, southern hemisphere land areas may have maintained larger effective population sizes. If so, they may be more likely to have maintained ancestral polymorphisms paraphyletically with close relatives. We await systematic comparisons of different continents and habitat types to test whether high levels of mtDNA paraphyly are consistent across Australian birds, and for birds outside of the Palaearctic and Nearctic in general. Byrne et al. (2008) discuss this in a review of arid zone phylogeographic data worldwide.
Paraphyly and biological insights: complexity in the present and past
Rhymer et al.’s (2004) analysis of paraphyly in the Pacific Black Duck cautions against finding complexity in the present (paraphyly) while invoking simplicity in the past (reciprocal monophyly). Mitochondrial DNA diversity in Australian and New Zealand subspecies of Pacific Black Duck (n = 21 and 34, respectively) involved group I only from New Zealand and group II with New Zealand and Australian samples. Rhymer et al. (2004) argued this to be evidence of gene flow and grounds to question the merit of recognising two subspecies. The results are compatible, however, with little or no recent gene flow between Australia and New Zealand if only because the Australian sample would likely have missed group I if it is rare in Australia (see Wiens and Servedio 2000). Gene flow between Australia and New Zealand would result in both groups I and II in both places. Recalculating the analyses of molecular variance (AMOVAs) with the two groups as genotypes, we found that the two regions explain 20% of the variation. Three haplotypes shared between Australia and New Zealand in relatively internal positions of the haplotype network are more likely to be ancestral haplotypes (Castelloe and Templeton 1994; Omland et al. 2006), suggesting the possibility of earlier connections. Even with thorough sampling, inference of population history on single locus data is especially problematic when there are important conservation implications.
Interesting questions remain in this case. Might there have been paraphyly in the past? Need group I represent pure historical New Zealand and group II pure historical Australia? Plausible alternative scenarios to explain how two deep mitochondrial groups could arise in this species are easy to conceive, such as groups I and II having been in Australia and New Zealand but group I went extinct in Australia during Pleistocene climatic fluctuations. Statistical analyses of multiple loci are needed to reject any such scenario and to find models of best fit.
The exciting complexity of evolutionary processes operated in the past as well as in the present. Although most studies reviewed here (Table 1) were limited to gene-tree analysis of mtDNA, this last case demonstrates problems when emphasis is on single-gene reciprocal monophyly for informing conservation decisions, determining species and subspecies limits, and understanding the complex evolutionary histories of Australasian bird species.
Paraphyly in phylogeographic studies of Australian birds: frequency and causes
Table 1 reviews published studies of the phylogeography and speciation of Australian birds. Among the non-pelagic continental taxa, 18 species were tested for mitochondrial paraphyly (studies that did not include any outgroups do not test for paraphyly and were excluded). Eight of these 18 species (44%) revealed species-level paraphyly in mtDNA, much higher than the 16.7% reported by Funk and Omland (2003) in their survey of mtDNA paraphyly among 331 species of bird worldwide. Despite the small number of Australian species assayed, the difference in frequency is significant (Fisher’s exact test, P < 0.01). A caveat is that the current survey did not include strictly phylogenetic studies of genera and families in which two or more individuals of one species were sampled. Many such species were included in Funk and Omland (2003), some with as few as four congeners sampled and thus having a very low chance of revealing paraphyly. The criteria of the worldwide survey may have artificially lowered the level of paraphyly it could detect. Although future studies comparing different continents and climate zones using the same criteria will be interesting, the 44% paraphyly we report for Australian birds is so much higher that it seems worth exploring further.
Funk and Omland (2003) reviewed factors that could influence levels of mtDNA paraphyly. One important cause is ‘incomplete taxonomy’ – paraphyly arising from what should be a single species having been ‘oversplit’, or, conversely, two species mistakenly being treated as one. For example, if only one species were recognised for Masked and White-browed Woodswallows, mtDNA paraphyly would disappear. This would be warranted if all other data supported one species, and there was evidence of simply two ‘morphs’ that randomly interbreed, but it has been argued that that is not the case (Joseph et al. 2006). Does mitochondrial paraphyly indicate inadequate taxonomy? The answer is ‘perhaps’ if one uses only a criterion of mtDNA monophyly to determine species limits. This would be contrasted with using it to recognise an Evolutionarily Significant Unit (see Moritz 1994). Using a ‘mitochondrial species concept’ can result in taxonomies that are misleading for reconstructing evolutionary history, for understanding behaviour and ecology, and for determining conservation priorities (see Crandall et al. 2000). We do not think that any of the examples of species level paraphyly in Australian birds result from inadequate taxonomy. Rather, we believe the mtDNA paraphyly in most or all cases will indicate either hybridisation or ILS, thus reflecting just one aspect of the history of each of these species. Ornithology’s attempts throughout the last century to find one perfect species concept have failed. There is increasing recognition of the importance of multiple criteria including plumage, morphometrics, song, mtDNA, nDNA, interbreeding, and others, when determining species limits (e.g. de Queiroz 1998, 2007; Helbig et al. 2002; Sites and Marshall 2004).
Paraphyly between species and hypothesis testing
Closer examination of paraphyly in mtDNA between two species is warranted. ILS and ancient hybridisation are possible explanations but independent data from multiple nuclear loci are needed to evaluate statistically these two causes of species-level paraphyly.
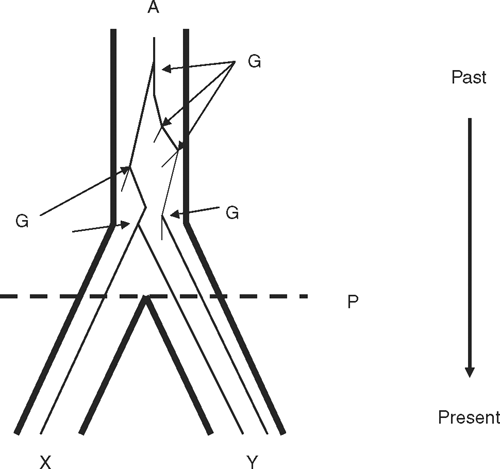
Increasingly, the program IM (‘isolation with migration’; Hey and Nielsen 2004; Hey 2005) (Fig. 5) is being used for this. Its underlying principle is that shared polymorphism can result from ILS as well as hybridisation and the resultant gene flow. Either model could be fitted to a given dataset, but it is more realistic to estimate simultaneously the likelihood of both. IM simultaneously estimates divergence between two populations and the rate of gene flow between them. Importantly, this coalescent approach incorporates the stochasticity of mutation and genetic drift when calculating parameters, which in turn are associated with populations rather than genes (Brito and Edwards 2008). Thus, the key output of IM, in Bayesian parlance, is credibility intervals for each parameter – the 90% highest posterior densities (HPDs). Joseph et al. (in press) apply this to paraphyly among Grey and Chestnut Teal and conclude that ILS is the more likely explanation of the data (see non-Australian example in Peters et al. 2007). More generally, multiple loci and rigorous coalescent methods can help understand paraphyly and a wide range of other topics in phylogeography and recent speciation.
|
To summarise, inference of the cause of mitochondrial paraphyly in extreme examples can be straightforward. So too, then, will be implications for speciation and species limits. Shared haplotypes in spite of otherwise deep mitochondrial divergences between species suggests recent hybridisation (scrubwren example). No shared haplotypes with generally very shallow divergences suggests ILS (woodswallow example). Discrimination between the two processes can be harder when they produce very similar patterns, such as when hybridisation occurred more distantly in the past or when speciation has occurred very recently. Two very different processes, ancient hybridisation and relatively recent speciation, can produce similar patterns. For such cases, rigorous analyses are needed, and seemingly straightforward examples should warrant examination with multiple loci and coalescent methods (e.g. Peters et al. 2007; Lee and Edwards 2008; Joseph et al., in press).
Paraphyly: multiple nuclear loci and coalescence
Analyses of multiple nDNA loci to understand mtDNA paraphyly is not without difficulties. Female birds, for example, are more strongly affected by hybrid infertility (Haldane’s rule; see Price 2008), so mtDNA is less likely than nDNA to cross species boundaries in birds (Brumfield et al. 2001; Allen and Omland 2003; Funk and Omland 2003). Furthermore, lower effective population size of mtDNA means it will generally sort to monophyly much more rapidly than does nDNA (Moore 1995; Palumbi et al. 2001). So mtDNA remains an ideal marker with which to begin studies of phylogeography and speciation in birds. The problem is that any one locus is subject to stochasticity, selection and unique events (Figs 2 and 3). Use of multiple loci is imperative for fully understanding evolutionary history (e.g. Knowles 2004; Peters et al. 2007), although it is legitimate to distinguish this more all-embracing task from that of simply estimating phylogeographic structure in mtDNA (Zink and Barrowclough 2008). Turning to the nuclear genome is the crucial next step not because one rapidly evolving nuclear gene can replace mtDNA, but because many independent nuclear loci can complete the evolutionary picture of populations and species. Each one may have variation that can be used to study evolutionary history, thus complementing, testing and adding to data from mtDNA. In coalescent analyses the power to obtain more accurate estimates of population divergence time and to identify loci under selection comes with more independent loci (see Edwards and Beerli 2000; Akey et al. 2004; Zink and Barrowclough 2008).
Trends and directions in Australo-Papuan avian phylogeography
Understanding the recent evolutionary history of Australo-Papuan birds should benefit from points discussed above, such as testing alternative hypotheses, increased and focussed geographical sampling, coalescent analyses and the use of many loci to improve sampling of stochastic variation between loci (e.g. Jennings and Edwards 2005). Their implementation should become easier with improved access to the nuclear genome (Backström et al. 2008) and the next generation of DNA sequencing (Ellegren 2008; Hudson 2008). Our last goal is to help set and define a research agenda for Australo-Papuan phylogeography.
Relationships between Australia and New Guinea
New Guinea and Australia share many species and have many others that are closely related. Study of relationships among these species and groups demonstrates the interrelatedness we have described of historical biogeography, speciation research and phylogeography. Driskell and Christidis (2004), Edwards (1993a), Norman et al. (2002), Joseph et al. (2001) and Murphy et al. (2007) illustrated this in studies of honeyeaters (Meliphagidae), Grey-crowned Babblers, logrunners (Orthonyx spp.) and the Sooty Owl (Tyto tenebricosa), and the Palm Cockatoo (Probosciger aterrimus), respectively. Yet the potential of work of this sort has barely been tapped. Phylogeographic analyses of groups found only in New Guinea are even fewer, but work on New Guinean pitohuis (Pitohui spp.) (Dumbacher and Fleischer 2001) provided insights into the biology of plumage mimicry.
Origins and maintenance of phenotypic differentiation: selection v. history
Geographical variation in plumage and morphology within Australian bird species has overwhelmingly been attributed to vicariant origins in historical allopatry followed by population expansion leading to secondary contact and hybrid zones (Keast 1961; Ford 1987; Schodde and Mason 1999). The alternative of geographically structured variation being in response to selection along environmental gradients has certainly been championed (Ford 1981) and occasionally argued (Wooller et al. 1985; Schodde and Mason 1999) but has rarely been rigorously tested.
Several Australian phylogeographic studies have addressed this. Geographical variation in back colour of the Australian Magpie has long been ascribed to historical allopatry of populations that evolved variously into black- or white-backed and variable forms and reflected as such in subspecific taxonomy (e.g. Schodde and Mason 1999). This suggests, in eastern Australia for example, that there was isolation between northern and southern ancestral populations. Work from the laboratory of Jane Hughes, however, has suggested that the major historical breaks in the species have been east to west, and that back colour is maintained through selection (see Baker et al. 2000; Toon et al. 2003, 2007). In the Singing Honeyeater (Lichenostomus virescens), geographical variation in size and intensity of plumage markings had been attributed either to origins of differentiation in historical allopatry followed by secondary contact (Schodde and Mason 1999) or as a selective response to environmental gradients (Wooller et al. 1985). Consistent with the latter hypothesis, mtDNA diversity showed no structure across the range of the species and recovered a signal of a recent population expansion (Joseph and Wilke 2007). Similar discordance between strongly structured geographical diversity in plumage and unstructured mtDNA diversity has been found in the western clade of the Australian Ringneck (Barnardius zonarius) (Joseph and Wilke 2006) and the eastern subspecies of the Splendid Fairy-wren (Malurus splendens) (Kearns et al. 2008). Recent population expansions estimated to have occurred within the Pleistocene in response to climatic fluctuations have been predicted for many years (Keast 1961; Schodde 1982) and have emerged commonly in phylogeographic studies of widespread Australian birds to date (Driskell et al. 2002; Joseph et al. 2002, 2006; Joseph and Wilke 2006, 2007; Kearns et al. 2008). Increased use of datasets with multiple loci should see an improvement in clarifying the relative roles of selection and purely historical isolation in generating and maintaining differentiation.
Hybrid zones, biogeographical barriers and modes of speciation
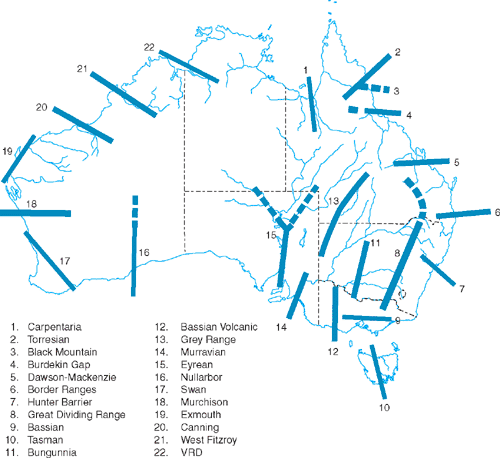
The literatures of these topics are intertwined, and Fig. 5 shows hybrid zones and putative historical biogeographical barriers recognised for Australian birds (see Keast 1961; Ford 1974a, 1987; Schodde and Mason 1999). Only a few phylogeographic studies have tested this rich literature, however.
The Carpentarian Barrier (Macdonald 1969; see Fig. 5) separates populations of many species on Cape York Peninsula and eastern Australia from those west of the Gulf of Carpentaria (Jennings and Edwards 2005). In the Grey-crowned Babbler, a hybrid zone has been recognised as occurring across it (Schodde and Mason 1999). Phylogeographic analysis of this species showed the Barrier’s location to be concordant with that of a major phylogeographic break within the species (Edwards 1993a). Gene flow across the region of the Barrier was assumed to be low because of the mtDNA monophyly for each subspecies on either side of the Barrier. Little sampling was done within the region of the Barrier and this warrants closer study.
In a further study of the Carpentarian Barrier, Jennings and Edwards (2005) developed assays for 30 anonymous nuclear markers and performed coalescent-based analyses in a study of the Black-throated Finch (Poephila cincta) of Cape York Peninsula and the Kimberley and Top End forms of Long-tailed Finch (P. a. acuticauda and P. a. hecki, respectively). They found that P. cincta diverged from the P. a. acuticauda–P. a. hecki lineage across the Gulf of Carpentaria (the Carpentarian Barrier of Macdonald 1969), ~700 000 years before the present (YBP), whereas P. acuticauda diverged from P. a. acuticauda and P. a hecki across Kimberley–Arnhem Land ~500 000 YBP (confidence intervals strongly support Pleistocene timing for both of these divergences).
Similarly, the Eyrean Barrier in southern Australia (Ford 1974a; Schodde 1982; Fig. 5) has been recognised as an agent of vicariance giving rise to eastern and western differentiates in the Australian Ringneck and the Splendid Fairy-wren (Ford 1987). Phylogeographic studies of these two species (Joseph and Wilke 2006; Kearns et al. 2008) each detected major phylogeographic breaks into two clades that are essentially concordant with the position of the Eyrean Barrier. Interestingly, some gene flow across the Eyrean and the Carpentarian Barriers is suggested by intermediate plumage phenotypes but mtDNA is entirely that of one of the putative parental forms. Studies of avian hybrid zones beyond Australia have detected similar asymmetry in mtDNA and discordance between mtDNA and nuclear markers (Parsons et al. 1993; Brumfield et al. 2001; Joseph et al. 2003; Gay et al. 2007).
Figure 5 shows much potential for molecular tests of hypotheses in earlier literature about diversity within and across putative refugia, barriers and hybrid zones (see Ford 1987; Schodde and Mason 1999; Schodde 2006). Nonetheless, as noted earlier, new and interesting biological questions do arise from phylogeographic data gathered to date (Driskell et al. 2002; Joseph et al. 2006, 2008; Kearns et al. 2008; Lee and Edwards 2008). Examples are the biological dynamics within hybrid zones (Australian Ringnecks and Splendid Fairy-wrens) and how reproductive biology and ecology impacts the movement of genetic markers across and through these zones (Crimson Rosella group). Brumfield et al. (2001) expressed this succinctly, noting that a focus on diagnostic phenotypic markers can create a self-fulfilling notion that hybrid zones are black holes into which genes enter but never emerge. Multiple neutral markers coupled with phenotypic data, on the other hand, can show that hybrid zones are evolutionary conduits for neutral and adaptive markers. We reiterate that paraphyly in mtDNA diversity, especially in these zones, should be welcomed for the new biological insights it can bring to the species being studied.
Island populations
Islands are the easiest sites at which to sample pelagic birds, which are conspicuous by their relative dearth in this review (Table 1; Austin et al. 1994; Abbott and Double 2003a, 2003b; Peck and Congdon 2004). Abbott and Double (2003a, 2003b) illustrated the value to albatross taxonomy of phylogeographic data obtained by sampling at island breeding colonies. Island populations should continue to provide excellent opportunities to see evolution in action and to estimate demographic parameters such as rates of gene flow between them and nearby mainlands. Degnan (1993b) and Degnan and Moritz (1992) argued that the Heron Island population of the Silvereye (Z. l. chlorocephala) had been founded and presumably differentiated within the last few thousand years but that current gene flow from the mainland is rare. Clegg et al. (2002) further discussed colonisation of Pacific Ocean islands by Silvereyes (Zosterops lateralis) and documented sequential founder events. Work on Grey-crowned Babblers on Melville Island and the mainland has been cited already (Edwards 1993b).
The potential is great for integrating molecular and non-molecular datasets in island and mainland populations. In the White-winged Fairy-wren (Driskell et al. 2002; Doucet et al. 2004) phylogeography complements genetic and microstructural analysis of blue–black plumage variation. Based on mtDNA, blue mainland populations in Western Australia are more closely related to one of the two nearby black island populations, that on Dirk Hartog Island, than to eastern Australian mainland blue populations. Further, it is not clear that the two island black populations are each other’s closest mitochondrial relatives. Conversely, there are no molecular data to complement study of evolution in vocalisations on Rottnest Island near Perth (Baker et al. 2006).
Work to date on Kangaroo Island and the nearby South Australian mainland shows the breadth of evolutionary insight from island–mainland comparisons. Building on biogeographical analysis of Kangaroo Island (Abbott 1974), work on the host–parasite biology of birds and their ticks (Kleindorfer et al. 2006) and adaptive divergence in morphology in the Superb Fairy-wren (Malurus cyaneus) between island and mainland (Schlotfeldt and Kleindorfer 2006) elucidates much interesting evolution. That work, however, awaits complementary phylogeographic analyses of divergence times and spatial analysis of diversity within and between mainland and island populations. Analyses within and between the phenotypically divergent Kangaroo Island and mainland populations of the Crimson Rosella group hint at other complexity (Joseph et al. 2008).
Despite the preceding, few island populations of Australian birds have been studied phylogeographically. In addition to examples already mentioned, rewarding examples are likely to come from phylogeographic analyses of island v. mainland birds and comparisons between birds and other animals, such as: on King and Flinders Islands compared with Tasmania; offshore islands of the Kimberley region compared with northern Western Australia; Groote Eylandt, the Sir Edward Pellew Group, Wellesley Islands, including Mornington Island in the Gulf of Carpentaria, compared with mainland Northern Territory and Queensland; and the small archipelagos off the south-western and southern coasts of South Australia and Western Australia, respectively, compared with the adjacent mainland.
Integrating phylogeography and palaeoclimatic modelling of distributions
To date, palaeoclimatic modelling of past distributions of Australo-Papuan birds has not been reconciled with what is ‘retrodicted’ of their past distributions from phylogeography. Reconciling palaeodistributions of species-pairs that still share ancestral polymorphism, such as Masked and White-browed Woodswallows or Grey and Chestnut Teal (Joseph et al., in press) should be illuminating. Examples of the approach are from South American (Peterson and Nyári 2007) and North American (Ruegg et al. 2006) birds, Australian frogs (McGuigan et al. 1998) and land snails (Hugall et al. 2002) as well as reviews by Kozak et al. (2008) and Swenson (2008). In light of climate change, predicting change in distributions of bird species and their phylogeographic units will be increasingly relevant.
Australo-Papuan avian phylogeography and DNA barcoding
The phylogeographic and recent speciation studies of birds reviewed here provide tantalising suggestions of differences between Australian birds and those elsewhere, especially temperate North America and Eurasia. It seems likely that widespread Australian birds, at least of arid and semi-arid environments, may often maintain larger effective population sizes, develop and retain more genetic diversity within species, be more likely to retain ancestral polymorphisms between species, and generally show high levels of interspecific mtDNA paraphyly. These general trends for widespread Australian birds will need to be tested with more species, more samples per species, and more loci. However, if consistent, these trends suggest caution in the use of mtDNA barcoding (Hebert et al. 2004) to describe biodiversity.
DNA barcoding in its strict sense uses one gene to identify unknown tissue samples and discover new species. In animals, effort has focussed on ~650 base pairs (bp) of the mtDNA cytochrome c oxidase subunit I (COI) gene. North American birds have been used to demonstrate apparent utility of barcoding (Hebert et al. 2004; Kerr et al. 2007; see also Tavares and Baker 2008). In general, the goals of barcoding will be achieved when mtDNA paraphyly is rare (see Moritz and Cicero 2004). Identification of unknown samples can be effective despite some paraphyly if the taxa involved have been very well sampled and the extent and causes of specific cases of paraphyly are well understood. However, discovery of new taxa in birds is problematic given the high incidence of paraphyly in birds (Funk and Omland 2003; this review). Serious problems arise in using some threshold amount (e.g. 2.3% sequence divergence; Kerr et al. 2007) to diagnose new species. Penhallurick and Wink (2004) unintentionally provided a spectacular example of these problems (see also Omland et al. 2000, 2006; Rheindt and Austin 2005). Even more problematic would be to suggest that some species-pairs perhaps should be synonymised because they diverged from each other recently and are below some threshold level of divergence (Kerr et al. 2007). Reducing taxonomic decisions to whether a genetic distance is above or below some arbitrary level is bad biology and ignores the complexity of the evolutionary process that molecular tools can describe and uncover.
In sum, DNA barcoding will succeed in identifying unknown individuals to species in many groups of organisms in many places. It remains to be seen whether it would be as successful with many Australian birds. The combination of the birds’ historical patterns of speciation with the possibility of relatively high long-term population sizes may not result in conditions that will facilitate simple barcoding.
Phylogeography’s impact on taxonomy: a hint of future debates
An area of concern to ornithologists and conservation managers is the interplay between phylogeography and taxonomy (see also Joseph 2008b). Numerous examples from Australian ornithology reviewed here show that one-to-one correspondence between units defined by molecular phylogeography (phylogroups) and subspecies defined by phenotype-based taxonomy, and sometimes even species, should be abandoned as a default expectation (Zink 2004). They can be expected to occur in environments that have been stable for long periods, as in the Wet Tropics (Joseph et al. 1995), in island-like populations isolated in inland ranges (Christidis et al. 2008), and eastern Australian mesic environments (Nicholls and Austin 2005; Nicholls et al. 2006). But in populations and environments moulded by the climatic fluctuations of the Pleistocene and where variance among gene trees can be expected to be high (Fig. 3), non-correspondence is probably to be expected more often. This is an interesting consequence of Avise et al.’s (1987) call to apply phylogenetic reasoning of macroevolution to microevolution. Whereas in macroevolution one can expect to be able to recover clearly diagnosable units and establish their relationships to each other, it may be far more challenging to do so in microevolution if sorting of ancestral polymorphism is still far from complete. Coalescent methods designed to treat the population, rather than the gene, as the unit of analysis and an emphasis on whether gene flow is occurring will greatly aid this effort.
If named taxa are not monophyletic for mtDNA, are they invalid? Should new taxa be named only when molecular and morphological data are at hand? We answer, ‘No’, provided one acknowledges what different datasets can and cannot achieve and reveal about the organism and its history. Phenotypic differentiation, presumably often driven by natural selection even if drift in allopatry is also involved (Price 2008) and possibly indicative of reproductive isolation, can evolve more rapidly than mtDNA can sort to reciprocal monophyly, for example. We have noted instances of this and argued that rejection of established taxonomy is not always necessary (e.g. natural history data in case of the White-browed and Masked Woodswallows). Conversely, Abbott and Double (2003a) marshal a case for recognising Shy (Thalassarche cauta) and White-capped (Thalassarche steadi) Albatrosses as species despite only a minor difference in mtDNA. Joseph (2002) named a distinctive species of South American parakeet, the Madeira Parakeet (Pyrrhura snethlageae), only on morphological grounds. Later, mtDNA paraphyly of this taxon with respect to another was shown but was ascribed to recent divergence, not necessarily incorrect taxonomy (Ribas et al. 2006). That hypothesis now needs testing.
An alternative approach would be that molecular phylogeographic data must be obtained and that taxonomy, at least subspecies, should reflect deep historical units (phylogroups) estimated through the clearly imperfect marker of mtDNA data (Zink 2004). A major theme we have addressed is that this strongly cladistic view is unrepresentative of biological reality at the species and population level where one deals with incompletely sorted DNA in phenotypically distinct entities. Also, interpretation of monophyly in mtDNA can be confounded by population history and sampling (Crandall et al. 2000; Edwards et al. 2005; Rosenberg 2007; Edwards 2008b). It is tempting to think that taxa should be underlain by corresponding genetic units. If one looked hard and long enough that could be achieved. But such taxonomy would lose much biological information inherent in the phenotype (e.g. the woodswallows). Knowles and Carstens (2007) note that it would take more than 1 million years after speciation before species would be delimited under a strict reciprocal monophyly criterion if 15 loci were sampled in species with an effective population size of 100 000 and assuming one generation a year. If avian speciation involves selectively driven divergence as much as Price (2008) argued, then decisions based on neutral DNA divergence will tend to be too conservative (i.e. will fail to recognise species if the taxa have recently originated) (see Edwards et al. 2005; Joseph et al. 2006). It is thus clear that species can be named without a requirement of mtDNA monophyly.
We share a view with many outside ornithology that quantum level change is needed in the way we think about taxonomy. For us, this means not just the need to explore new naming conventions such as the PhyloCode (http://www.ohiou.edu/phylocode/, accessed 8 October 2008) but also the need for managers and theoreticians alike to discuss change in what we expect taxonomic names to mean, what they can mean in a Linnaean system, and what we expect taxonomy to achieve below the generic level. Molecular and phenotypic data are two different ways of slicing one cake. Ideally, they would converge on intuitive expectations we discussed above (i.e. Chestnut Teal have Chestnut Teal DNA). That they often do not is because at the population level one is examining snapshots in time of evolutionary processes that will eventually lead to the kind of clear-cut reciprocal monophyly that we are more familiar with at higher taxonomic levels but which may not have done so at the time we are examining them. Endless debates about species concepts, which in ornithology at least are inextricably tied up in this issue, are exactly that: endless and unresolvable. They are different ways to slice a cake and are often paralleled by the molecular–phenotypic dichotomy. The debates suffer from use of one word, ‘species’, to describe different concepts that are then discussed at cross purposes. For an entrée to the large literature on species concepts in birds see Helbig et al. (2002), Remsen (2005), Watson (2005), Garnett and Christidis (2007) and de Queiroz (2007, especially fig. 1).
We further conclude that the question of how many subspecies are in a species, or even some questions of species or subspecies status, is often not where primary biological interest lies, interest in reproductive isolation notwithstanding. Again, for an entrée into relevant literature with reference to Australia see Ford (1974b), Schodde and Mason (1999), Zink (2004), and Phillimore and Owens (2006). Subspecies will always be useful descriptors of observable geographical variation, which is itself a result of evolution that we should manage and conserve. With respect to subspecies especially, we are sceptical, however, of what external morphology can convey about evolutionary history for reasons outlined long ago by Wilson and Brown (1953). Phylogeography of Australian birds clearly show that molecules and morphology are often discordant in determining where putative historical breaks should be considered to occur, at least in widespread species (Driskell et al. 2002; Joseph and Wilke 2006; Toon et al. 2007; Joseph et al. 2008; Lee and Edwards 2008). Sometimes molecular data show that the question itself is inappropriate: Johnson et al. (2005) argue from molecular data that the Cape Verde Kite, sometimes considered the rarest raptor in the world, does not even exist!
Clearly, we need names for reasons ranging from discussion of evolution through to conservation and management (Garnett and Christidis 2007). But that need does not equate with default worth of the species v. subspecies question. In comparison, the history of a population and historical v. contemporary relationships among populations are arguably more relevant to our understanding of evolution and conservation, which for that matter, is nothing if not our attempt to maintain evolutionary processes. We hope that readers will come to think about some of the perspectives on populations that we have described. They relate to what has driven species and subspecies classifications since Darwin’s theory of evolution by natural selection brought new dimensions to Linnaean taxonomy. That a nomenclatural system designed 200 years ago cannot always easily account for what we have learned of evolution from molecular phylogeography ought not be a surprise. On the contrary, the rejuvenation it brings is exciting! It challenges conservation and management of bird populations, not just theoretical aspects of study.
Finally, phylogeography examines neutral markers to determine present structure and population history. To fully understand evolution, we also need to understand selection. Neutral markers must be distinguished from those under influence of selection. Developments such as pyrosequencing, the nuclear marker set of Backström et al. (2008), and gene-chip technology (Cheviron et al. 2008) hold promise here. Phylogeographic studies of Australian birds have already hinted at a role for selection in some groups (e.g. Australian Ringneck, Singing Honeyeater). With genomic data from the chicken (International Chicken Genome Sequencing Consortium 2004) and Zebra Finches (Taeniopygia guttata) (Stapley et al. 2008) already being studied, avian equivalents of studies such as Akey et al. (2004) in humans are emerging (Axelsson et al. 2008).
Whither phylogeography? A glimpse into the future of Australian avian phylogeography
Phylogeography’s major theoretical and practical challenges are to derive data from many loci, not just mtDNA, analyse them with the power of coalescence theory in a model-testing framework, and so estimate the history of populations not just the history of genes sampled from them. Mitochondrial DNA will continue being a powerful marker for estimating population structure and phylogeographic patterns, whereas multilocus nuclear data will be necessary to integrate structure with demographic processes that have generated it, such as gene flow, coalescence times and population growth (Zink and Barrowclough 2008). Ways of meeting these challenges will involve robust model-testing (Knowles and Carstens 2007). The hypothesis-testing approach is neatly illustrated in an Australian bird by Lee and Edwards (2008) and in non-Australian birds by Congdon et al. (2000) and Peters et al. (2007, 2008).
Insight into how these challenges may play out comes from papers such as Jennings and Edwards (2005) already described. It is increasingly straightforward to isolate multiple nuclear loci in birds; see, for example, Backström et al.’s (2008) set of primers for 242 nDNA regions in most birds. Developed with genomic knowledge of several birds, especially the chicken (International Chicken Genome Sequencing Consortium 2004) and an Australian bird, the Zebra Finch (Stapley et al. 2008), the genome of which is now online, these markers offer high levels of variation at single sites in DNA sequences (single nucleotide polymorphisms or SNPs) that could be used for a wide range of studies from hybrid zones to phylogenies. Finally, the next generation of DNA-sequencing technology, for example, pyrosequencing, which can sequence hundreds of millions of base pairs in days, will likely have an impact on the study of natural populations and the rate with which empirical datasets can catch-up with theory in the study of natural populations of birds (Ellegren 2008; Hudson 2008). These and other methods such as gene-chip technology will allow study of loci under the effect of selection together with those neutral to selection. More complete evolutionary histories of natural populations should result.
The apparent messiness that paraphyly brings from molecular phylogeography to speciation studies in Australian birds also presents opportunities. One is to use the history of species in Australia to test generalisations developed in the temperate northern hemisphere. Second, when species retain much of the genetic diversity present in a common ancestor, one can look back in time and understand past evolutionary events (Edwards and Beerli 2000). In contrast, if recent bottlenecks had reduced genetic diversity to one or a few haplotypes, then most of the information about the histories of species would have been erased.
Phylogeographic study of Australia’s birds, coupled with the continent’s well-documented history (see Hugall et al. 2002; Byrne et al. 2008), is a theatre for applying theoretical and technical developments in phylogeography reviewed in the first part of this paper. Pelagic birds are a little-studied group in which molecular data should enhance and interestingly confound understanding of population structure and demographics. Whether inland mountain ranges have been arid-zone refugia should be tested with a priori models of population structure. Do generalisations emerge within and between different groups of birds for correlations of molecular and morphological diversity or will all cases need to be considered individually? Understanding of phylogeography, speciation and hybridisation will be increased by intensive study of some examples in Table 1 and the framework provided by Fig. 5. Most bird species in Australo-Papua have not been studied phylogeographically but we acknowledge that studies are underway in many. Few, if any, species have been sampled across their entire geographical range, but good tissue collections are growing for Australian and New Guinean birds. With large sample sizes, multiple loci and coalescent approaches, there will be many exciting avenues for using Australian birds in advancing important questions in systematics and evolution, as we have attempted to show. The foundation laid by the last 15 years of molecular phylogeography, and the morphologically based foundation of biogeography and systematics that it grew on, will enable research to continue building on the fine work of earlier Australian ornithologists (Keast 1961; Schodde and Mason 1999; Schodde 2006; Christidis and Boles 2008).
Note added in proof
Christidis et al. (2008) report deep, structured divergence between populations of the Short-tailed Grasswren (Amytornis merrotsyi) isolated in inland ranges of the arid zone.
Acknowledgements
We especially thank Scott Edwards for his detailed comments on two drafts. We also thank Gaynor Dolman, Michael Double, and anonymous reviewers for critically commenting on a draft, and David Winkler for discussion. We thank our colleagues and collaborators for their help in many projects,especially J. Peters and T. Wilke. Kevin E. Omland (KEO) is funded by an US National Science Foundation, Systematics Program CAREER Grant (DEB – 0347083). KEO’s contribution was made while on sabbatical leave at the Australian National University, Canberra, supported by a Visiting Fellowship from the School of Botany and Zoology (BoZo). KEO thanks William Foley, Andrew Cockburn, Mike Crisp, and other members of the Crisp Laboratory and BoZo for interesting discussions. Responsibility for views expressed, of course, lies with L. Joseph and K. E. Omland.
Abbott, I. (1974). The avifauna of Kangaroo Island and causes of its impoverishment. Emu 74, 124–134.
Avise, J. C. , and Aquadro, C. F. (1982). A comparative summary of genetic distances in vertebrates. Evolutionary Biology 15, 151–185.
Christidis, L. , Horton, P. , and Norman, J. (2008). Subspeciation in the Short-tailed Grasswren (Amytornis merrotsyi, Maluridae). Emu 108, 275–282.
| Crossref | GoogleScholarGoogle Scholar |
de Queiroz, K. (2007). Species concepts and species delimitation. Systematic Biology 56, 879–886.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Ho, S. Y. W. (2007). Calibrating molecular estimates of substitution rates and divergence times in birds. Journal of Avian Biology 38, 409–414.
Joseph, L. (2008b). Book Review: Systematics and Taxonomy of Australian Birds by Les Christidis and Walter E. Boles. Emu 108, 365–368.
| Crossref | GoogleScholarGoogle Scholar |
Nicholls, J. A. , and Austin, J. J. (2005). Phylogeography of an east Australian wet-forest bird, the satin bowerbird (Ptilonorhynchus violaceus), derived from mtDNA, and its relationship to morphology. Molecular Ecology 14, 1485–1496.
| Crossref | GoogleScholarGoogle Scholar | CAS | PubMed |
Palumbi, S. R. , and Cipriano, F. (1998). Species identification using genetic tools: the value of nuclear and mitochondrial gene sequences in whale conservation. Journal of Heredity 89, 459–464.
| Crossref | GoogleScholarGoogle Scholar | CAS | PubMed |
Rand, D. M. (2001). The units of selection on mitochondrial DNA. Annual Review of Ecology and Systematics 32, 415–448.
| Crossref | GoogleScholarGoogle Scholar |
Ruegg, K. , Hijmans, R. J. , and Moritz, C. (2006). Climate change and the origin of migratory pathways in the Swainson’s Thrush, Catharus ustulatus. Journal of Biogeography 33, 1172–1182.
| Crossref | GoogleScholarGoogle Scholar |
Sharman, G. B. , Close, R. L. , and Maynes, G. M. (1989). Chromosome evolution, phylogeny and speciation of rock wallabies (Petrogale : Macropodidae). Australian Journal of Zoology 37, 351–363.
| Crossref | GoogleScholarGoogle Scholar |
Zink, R. M. (2004). The role of subspecies in obscuring avian biological diversity and misleading conservation policy. Proceedings of the Royal Society of London. Series B: Biological Sciences 271, 561–564.
| Crossref | GoogleScholarGoogle Scholar |

Zink, R. M. , and Barrowclough, G. (2008). Mitochondrial DNA under siege in avian phylogeography. Molecular Ecology 17, 2107–2121.
| Crossref | GoogleScholarGoogle Scholar | CAS | PubMed |
Zink, R. M. , and McKay, B. (2008). Book Review: Speciation in Birds by Trevor Price. Auk 125, 504–505.
| Crossref | GoogleScholarGoogle Scholar |