
Molecular homology and multiple-sequence alignment: an analysis of concepts and practice
David A. Morrison A D , Matthew J. Morgan B and Scot A. Kelchner CA Systematic Biology, Uppsala University, Norbyvägen 18D, Uppsala 75236, Sweden.
B CSIRO Ecosystem Sciences, GPO Box 1700, Canberra, ACT 2601, Australia.
C Department of Biology, Utah State University, 5305 Old Main Hill, Logan, UT 84322-5305, USA.
D Corresponding author. Email: david.morrison@ebc.uu.se
Australian Systematic Botany 28(1) 46-62 https://doi.org/10.1071/SB15001
Submitted: 2 February 2015 Accepted: 8 April 2015 Published: 10 September 2015
Journal Compilation © CSIRO Publishing 2015 Open Access CC BY-NC-ND
Abstract
Sequence alignment is just as much a part of phylogenetics as is tree building, although it is often viewed solely as a necessary tool to construct trees. However, alignment for the purpose of phylogenetic inference is primarily about homology, as it is the procedure that expresses homology relationships among the characters, rather than the historical relationships of the taxa. Molecular homology is rather vaguely defined and understood, despite its importance in the molecular age. Indeed, homology has rarely been evaluated with respect to nucleotide sequence alignments, in spite of the fact that nucleotides are the only data that directly represent genotype. All other molecular data represent phenotype, just as do morphology and anatomy. Thus, efforts to improve sequence alignment for phylogenetic purposes should involve a more refined use of the homology concept at a molecular level. To this end, we present examples of molecular-data levels at which homology might be considered, and arrange them in a hierarchy. The concept that we propose has many levels, which link directly to the developmental and morphological components of homology. Of note, there is no simple relationship between gene homology and nucleotide homology. We also propose terminology with which to better describe and discuss molecular homology at these levels. Our over-arching conceptual framework is then used to shed light on the multitude of automated procedures that have been created for multiple-sequence alignment. Sequence alignment needs to be based on aligning homologous nucleotides, without necessary reference to homology at any other level of the hierarchy. In particular, inference of nucleotide homology involves deriving a plausible scenario for molecular change among the set of sequences. Our clarifications should allow the development of a procedure that specifically addresses homology, which is required when performing alignment for phylogenetic purposes, but which does not yet exist.
Additional keywords: multiple alignment, nucleotide alignment, sequence homology.
Introduction
Published review articles about multiple-sequence alignment have almost always focussed on the algorithmic aspects of producing the alignment, such as efficiency and accuracy, but rarely on the biology. After a brief initial mention that sequence alignment has something to do with the biological concept of homology, this is usually the last time in the review that any strictly biological concept is noted. As a part of bioinformatics, the emphasis is strongly on the ‘informatics’ not the ‘bio’ (Wilke 2012). That is, alignment is commonly treated as a computational problem rather than a biological one, and seen as little more than a ‘bottleneck’ in sequence-processing pipelines. That leaves the homology aspect of multiple-sequence alignment still open for discussion.
Homology is a fundamental concept for inferring biological relationships and character evolution. It is central to comparative biology, especially evolutionary biology (and, thus, biology in general). Therefore, a large literature exists on the subject (see Hall 1994; Hoßfeld and Olsson 2005; Kleisner 2007; Pavlinov 2012), which is perhaps second only to that involving concepts of species.
Discussions about morphological homology apparently reached a consensus many years ago (Patterson 1982; Rieppel 1988; de Pinna 1991; Brower and Schawaroch 1996; Laubichler 2000; Cracraft 2005). However, several issues regarding homology of molecular characters remain unresolved (Mindell and Meyer 2001). For example, Patterson (1988) viewed molecular homology as a statistical concept, which has become the basis of most multiple-alignment programs, whereas other authors have emphasised the vital importance of congruence (synapomorphy on a phylogenetic tree diagram) as the critical test of homology (Mindell 1991; Doyle and Davis 1998).
Confusion over what constitutes homology in molecular data leads to very different approaches to data analysis, particularly sequence alignment. The distinct nature of two well known approaches will suffice to illustrate this point. In the first case, both direct optimisation (DO, Phillips et al. 2000) and statistical alignment (SA, Metzler and Fleissner 2009) are methods based on the idea that homology is impossible to know beforehand, and that alignment and tree-building are inter-dependent. One is, therefore, logically compelled to infer both the alignment and tree simultaneously under the same statistical model. This idea stems from an early paper by Sankoff et al. (1973).
In the second case, as most researchers do, statements of homology are made before the phylogeny estimate and remain unchanged during the inference process. In other words, the dataset is established, from which an estimate is made. The resulting tree topology is accepted as conditional on those initial homology statements. Biologically relevant information is used when proposing homologies of the nucleotides and when recognising gene orthology. If there is sufficient doubt about certain homology assessments, those ambiguous regions of the DNA alignment can be excluded before tree building, and only well supported gene orthologies are used in subsequent analyses.
Given the strong difference between these two approaches to alignment for phylogenetic purposes, and the persistence of each approach in algorithm development, it is high time we considered what is meant when we talk about molecular homology, and in particular ‘alignment of homologous DNA sequences’.
In this paper, we explore the various components of biological homology at conceptual levels ranging from the nucleotide to the organism. We then propose a hierarchical scheme of those components for many types of molecular data, and demonstrate which levels of homology are being addressed by each type in a phylogenetic analysis. We explicitly link these homology levels with nucleotide sequence alignment, which is the only level at which homology applies to the genotype, and is thus unproblematic as a concept representing inheritance. We also consider the practical aspects of generating hypotheses of homology when creating a sequence alignment.
Background to molecular homology and alignment
Preface
If homology is a concept central to comparative biology, why does it still need to be discussed? The reason dates back to the rather vague formulation of the term by Richard Owen (1843): ‘the same organ in different animals under every variety of form and function’ (p. 379). Since then, biologists have asked: what on earth does this actually mean? What is sameness? Is it phenotypic identity, genotypic identity, or both? Is it identity of physiological processes, identity of historical descent, or identity of a developmental program? The answer to each of these questions is ‘yes’, depending on your viewpoint. Well, we are phylogeneticists, and so to us homology has to do with sameness derived from a common historical origin. In modern terms, Hall (2007) notes: ‘a working definition of homology is the presence of the same feature in two [or more] organisms whose most recent common ancestor also possessed the feature’ (p. 473). This formulation is usually credited to Ray Lankester (1870) under the name homogeny: ‘depending simply on the inheritance of a common part’ (p. 42).
But the conceptual discussion does not end there, because Owen and Lankester were thinking in terms of morphology and anatomy, whereas modern biology includes topics related to genetics, such as genes and genotypes. Homology must apply to them, too, and so a conceptual framework is needed that allows us in practice to recognise homology of nucleotides, amino acids, protein domains, genes, and regulatory gene networks, among other things.
Interestingly, the evolutionary homology of nucleotides themselves is a topic that has rarely been addressed in detail, although a few papers have certainly considered its implications for alignment (e.g. Kjer 1995; Kelchner 2000; Morgan and Kelchner 2010). There has been some discussion of gene homology (Patterson 1988; Hillis 1994; Brigandt 2003; Freudenstein 2005; Haggerty et al. 2014), particularly with the recognition of various types of gene homology (orthology, paralogy, xenology). Similarly, the roles of convergent evolution and chance in producing similarity of amino acid sequences has long been recognised (e.g. Doolittle 1981; Simmons 2000; Galperin and Koonin 2012), as has the disconnection between gene homology and the control of morphological development (de Beer 1971; Meyer 1999; Wagner 2014). But in terms of the body of literature, the identity of nucleotides in a set of coding or non-coding DNA sequences has been a poor cousin, largely relegated to an ostensible role in computerised sequence alignment.
Sequence alignment
Sequence alignment is an odd topic. It is usually claimed to be conceptually important, yet in practice it is often treated merely as a tool. It is the vital first step of a phylogenetic analysis, but many consider the ‘real’ work to be building the tree. It is the core of database searching in molecular biology, but the main interest is in the resulting gene homologies, not the nucleotide homologies. And it is essential for molecular structure prediction (sometimes disconcertingly called ‘homology modelling’), but the structures themselves are really the principal goal.
Perhaps a large part of the problem arises from the community’s persistent focus on phylogenetic trees (Felsenstein 2004). We are often told that ‘multiple-sequence alignment is typically the first step in estimating phylogenetic trees’, with a strong implication that inaccuracies in the alignment can be tolerated provided that they do not affect the subsequent production of an accurate (or adequate) tree. However, if an alignment truly does represent hypotheses of homology among the characters, then the alignment itself is also a worthwhile goal.
Thus, although homology has been an important concept in phylogenetics, phylogeneticists have been mostly interested in the reconstruction of phylogenetic trees or, in the modern world, phylogenetic networks (Bapteste et al. 2013). At heart, however, a phylogenetic tree or network is simply a graphical representation of possible evolutionary relationships inferred from a set of homologous characters, and therefore the primary focus of phylogenetics is actually on homology itself. It is surprising, then, that little has been said in the literature about homology as it applies to nucleotide sequence data (Morrison 2015), in spite of the fact that modern phylogenetics is dominated by the use of those data. We will therefore focus on those data here.
In practice, for the study of DNA sequences, homologies are usually represented as a multiple-sequence alignment, in which (by convention) the rows are DNA sequences and the columns are putatively homologous nucleotides. In theory, a phylogeny is simply a graphical version of the same information contained in the tabular version of the sequence alignment (Mishler 2005). However, the alignment can be used for many other purposes than building a tree (see below).
Obviously, to be of proper use for phylogenetic inference, the multiple-sequence alignment needs to accurately represent homology among the nucleotides. Unfortunately, this is not easy to achieve, because we can neither make direct observations of homologies nor can we perform experiments to investigate them. Homology arises from chance evolutionary events that are historically unique, and there is no known algorithm for reliably identifying such events. Algorithms are considered important in science because they can provide objective and repeatable procedures for turning observations and inferences into data. It is, thus, unsurprising that the development of alignment procedures has focussed on the algorithmic aspects. In theory, we would like to apply the algorithmic paradigm to multiple-sequence alignment, but this has proven to be rather difficult in practice. The concepts of algorithm and homology seem to be incompossible.
To begin addressing this problem, we must first establish a lexicon. Although we do not wish to become mired in semantics, progress has been hindered by not having a community-wide understanding and terminology of molecular homology and its components, and thus we need to present one here.
Terminology
For simplicity, we will adopt this definition of a character from Platnick (1979): ‘a character consists of two or more different attributes (character states) found in two or more specimens that, despite their differences, can be considered alternate forms of the same thing (the character)’ (p. 542).
When dealing with DNA sequences, the only tangible objects are the four nucleotides (A, C, G, T), which are observed to form linear patterns within chromosomes (although the chromosomes themselves can be circular). These form the fundamental units on which the theoretical aspects of alignment and homology are based. They are aggregated into blocks of functional and non-functional loci, or more loosely ‘genes’.
In the conventional tabular presentation of a DNA multiple-sequence alignment, the taxa are the rows of the table, with one sequence per row, and the characters are the columns, with one character per column. The character states are the cells formed by the intersection of the rows and columns. Both taxa and characters are theoretical constructions, being the products of some operational definitions (de Pinna 1991; Brower and Schawaroch 1996).
The distinction between units and states is critically important, and a failure to distinguish them causes confusion, such as that which separates the two alignment approaches presented in the Introduction. The nucleotides are not intrinsically character states. A nucleotide (say an A) becomes a state only when it is assigned to a particular character. If it is assigned to a different character, then it is a different state, even though it is still the same nucleotide. This means that two units in different columns can appear ‘the same’ (e.g. if they are both As), even though they are different states.
Indeed, alignment at its most simple level is trying to decide which unit is which state. If there is nucleotide variation, then having four units allows a clear distinction of character states, whereas if there is little variation, then having only four units can make decisions problematic (cf. Stace 2005). This confusion between units and states is rarer in morphological studies because there are potentially many units not just four (although, see the example presented below). Complexity is usually important for assessing hypotheses of homology, which can be thought of as ‘1 : 1 correspondences between parts of complex entities in which a set of relations is preserved’ (Jardine 1967, p. 128).
Some people see no ontological distinction between characters and character states (e.g. Platnick 1979; Patterson 1982, 1988). In this view, a character state is merely a character at a less universal level of inclusiveness. There has been considerable philosophical debate surrounding this subject (Pleijel 1995; Wilkinson 1995; Hawkins et al. 1997; see the chapters in Scotland and Pennington 2000). However, there is an operational distinction between character and state that is important for our discussion here of sequence alignment. Essentially, assessment of primary homology has the following two steps (Brower and Schawaroch 1996; Hawkins et al. 1997): (1) comparative study of variation in features is used to define characters (the columns in the data matrix), and (2) characters are partitioned and coded as character states (cells within each column of the data matrix). Characters are then treated as logically independent (but not necessarily biologically independent), whereas character states are hierarchically related to each other.
The distinction between sequences as rows and characters as columns is vital in both theory and practice. It is important for theory because homology is about relationships among characters, whereas a phylogeny is about relationships among taxa (represented by nucleotide sequences). That is, homology does not apply to whole organisms but to parts of organisms, and yet, we use homologies to derive phylogenies of whole organisms. The distinction is important in practice because, to study homologies, we need to move the focus from the sequences as a contiguous string of nucleotides along a chromosome (the rows) to the evolutionary characters (the columns). The fundamental practical limitation of all current computer algorithms for multiple-sequence alignment is that they focus on the rows not the columns.
In a DNA multiple-sequence alignment, the observations are both the nucleotides and the ‘gaps’. That is, we observe that sequences of homologous loci or genes are not necessarily all of the same length (i.e. they often have different numbers of nucleotides). Thus, even though gaps are not tangible objects, as nucleotides are, they do represent observable phenomena that require explanation, just as do nucleotide matches and mis-matches. The only practical difference is that we do not observe the location(s) of the gaps relative to the nucleotides.
The commonly used alignment model consists solely of ‘substitutions’ and ‘indels’. Indels conceptually model length differences, whereas substitutions model mis-matches between sequences. This combined model is a mathematical simplification that allows us to identify where the gaps might be shared between sequences. The identification of indels is what most alignment programs have focussed on, in the sense that producing a successful multiple alignment has been seen as an attempt to get the indels in the same places as the gaps (i.e. to correctly reconstruct the indel history), so that any remaining discrepancies between the sequences are assumed to be substitutions.
The processes are the molecular mechanisms responsible for creating the DNA-sequence variation. For the observed gaps, these include insertions (addition of a novel subsequence), deletions (removal of an existing subsequence), translocations (removal of a subsequence and its insertion at another location) and duplications (copying of a subsequence), notably tandem repeats and inverted repeats. The processes responsible for creating the inferred mis-matches include point mutations (change of a single nucleotide), inversions (replacement of a subsequence by its reverse-complement) and transpositions (exchange of subsequences between locations). All of these processes can occur within a locus or gene; processes involving duplications, deletions, insertions, inversions and rearrangements can also comprise large blocks of nucleotides consisting of whole genes (i.e. they are both within-gene and between-gene processes). Tandem repeats are probably the most common cause of sequence-length variation within loci (e.g. Huntley and Clark 2007; Messer and Arndt 2007), and yet they are detected only poorly by most alignment programs. Furthermore, small inversions often go undetected because the programs do not look for them explicitly, and therefore interpret them as multiple adjacent substitutions (Kelchner and Wendel 1996; Kelchner and Clark 1997; Graham et al. 2000; Quandt and Stech 2005).
Note that evolutionary processes occur at the DNA level, rather than at other levels of genetic complexity. So, the molecular mechanisms listed here are properly restricted to nucleotide sequences. This is part of the distinction between genotype and phenotype, the latter being defined as the expression of a genotype in interaction with its environment (this distinction is credited to Johannsen 1909). Nucleotides are part of the genotype, whereas amino acids, protein domains, etc., are all part of a phenotype, not a genotype, even though they are molecular data. This idea (that, by definition, an amino acid is just as much a part of phenotype as is a forelimb) seems to be rarely appreciated, but it has an important role to play in the conception of molecular homology. The critical distinction is not between molecular and morphological data but between genotypic and phenotypic data.
Sequence identity refers to patterns of residues that are indistinguishable. It is the primary concept used during assembly of sequence contigs from short reads, where the reads are assumed to overlap based on an identically repeated pattern of nucleotides (Li and Homer 2010).
Sequence similarity is a model-based assessment of resemblance between residue patterns. Different models of similarity are used for practices such as database searching, secondary-structure modelling and prediction, and elucidation of sequence function.
Sequence homology refers to residue patterns that reflect descent from their occurrence in a common ancestor. It is, or should be, the primary concept of relationship when using sequences for phylogenetic purposes. In molecular biology, it is unfortunate that the word homology has long been used as a synonym for similarity (Margoliash 1969; Reeck et al. 1987). As discussed below, similarity is one of several criteria that can be used to help infer homology, but making the words synonymous confuses empirical measurements (similarity) with inferred conclusions (homology).
Alignments versus phylogenies
Inference of homology involves deriving a plausible scenario for molecular change among the set of sequences. This scenario may involve a different set of details for each character (alignment column) or it may involve events common to groups of characters (contiguous blocks of columns in the alignment). Once this scenario has been derived, reconstructing a phylogeny is simply a matter of drawing a connected line graph that reflects the scenario. That is, the series of character transformations are turned into an organismal genealogy. This is exactly what Ernst Haeckel (1866) was trying to do when he coined the word ‘phylogeny’; to him, the phylogeny constituted what we now call the character transformation series, whereas diagrams of organismal relationships were called ‘stammbaum’ (Dayrat 2003).
It seems rarely to be appreciated that a sequence alignment contains more evolutionary information than does a phylogeny. The tree or network is simply a diagrammatic summary of some of the tabular information contained in the alignment, with networks often showing more of the information in a dataset than a tree would display. Implied alignments derived from the direct optimisation procedure (Wheeler 2003; Giribet 2005) are another good example, where several alternative alignments reflect different evolutionary histories of the characters but can all produce the same phylogenetic tree. There is, thus, an asymmetry between alignments and phylogenies, rather than the symmetrical relationship implied by the usual notion of interchangeability of trees and alignments. That is, several alignments may imply a single tree, and a single tree may reflect several alignments.
Alignments represent hypotheses about the results of evolutionary scenarios among characters, whereas phylogenies represent hypotheses of phylogenetic relationships among taxa. Each alignment is, thereby, associated with a sequence of molecular events that lead from an ancestral sequence (which we do not know and usually do not need to know) to the sampled descendants, and the alignment should explicitly reflect these events. Aligned columns represent descendants from the ancestral nucleotide, and only such descendants should be aligned in the columns. This is where similarity-based alignments can fail, as the criterion of similarity can frequently align nucleotides when no homology is implied at all (sometimes referred to as over-alignment; Golubchik et al. 2007; Löytynoja and Goldman 2008). Moreover, similarity-based alignment assumes that all sequence variation occurs at random, whereas this variation actually arises from specific molecular mechanisms that occur with non-random frequency at non-random locations in the sequence (see Kelchner 2000, and below).
This means that multiple DNA alignments have practical uses independent of a phylogeny, as well as functions that act through the phylogeny (Assis 2015). Some examples of independent uses include the following:
-
de novo prediction of protein-coding genes and their introns, including paralogs and xenologs, as well as searches for non-coding RNAs;
-
analysis of spatially constrained structures, such as nucleotide pairing in RNA, intron and ITS secondary and tertiary structure, and α helices and β sheets in protein secondary structure;
-
discovery of functional motifs, co-varying sites, and conserved regions that have some biological relevance, such as regulatory regions or binding sites;
-
estimates of selection (or adaptive evolution), such as interrupted reading frames, disrupted active sites, and variable omega values (the ratio of non-synonymous substitutions to synonymous substitutions per site);
-
identification of species-specific DNA, such as might be useful for bar-coding.
Phylogeny-based estimates that can depend critically on the alignment comprise:
-
topology (including degree of resolution, and amount of reticulation), which is used for several purposes, such as quantifying speciation and extinction, taxonomic classification, and protein classification;
-
branch lengths, including patterns of molecular change that cause sequence variation, which are used for studies of evolution;
-
time estimates, as used in epidemiology and phylogeography;
-
inference of ancestral character states, as used in comparative biology and palaeontology.
Therefore, alignment matters, because all downstream analyses depend on it (Löytynoja 2012). As a single example, the existence of horizontal gene transfer (HGT) in plants was first proposed by Went (1971) based on phenotype data. However, the first such claim to receive widespread attention on the basis of nucleotide sequence data was by Bergthorsson et al. (2003). Part of their evidence concerned anomalous placements of several taxa in a phylogeny based on sequences of the rps11 gene. Unfortunately, the majority of the evidence for these placements came from sequence regions with inferred indels where the alignment was extremely uncertain. Even minor adjustments to the alignment change the phylogeny significantly, and the evidence for HGT disappears.
The formal demonstration that the topology of phylogenies can be seriously affected by the underlying alignment dates from Ellis and Morrison (1995), so that changes in the alignment algorithm can change the topology (Morrison and Ellis 1997; Hickson et al. 2000). This has been further explored by Wong et al. (2008) using simulations and Blackburne and Whelan (2013, and references therein) using empirical data.
This raises the issue of so-called ‘alignment-free’ methods in phylogenetics (Vinga and Almeida 2003), by which is meant phylogenetic tree-building without a multiple-sequence alignment. In particular, it has been argued that ‘next-generation phylogenomics must aspire to become more fully independent of multiple sequence alignment, while capturing as much homology signal as possible in the face of genome dynamics’ (Chan and Ragan 2013, p. 3). There are several methods proposed (Sims et al. 2009; Domazet-Loöo and Haubold 2011; Nelesen et al. 2012; Ren et al. 2013; Bonham-Carter et al. 2014), based on calculating pairwise evolutionary distances directly from unaligned sequences, or from non-sequence data. However, Höhl and Ragan (2007, p. 206) noted that ‘no alignment-free method that we examined recovers the correct phylogeny as accurately as does an approach based on maximum-likelihood distance estimates of multiply aligned sequences’ (but cf. Chan et al. 2014).
So even at the genomic level, nucleotide homology is still an important source of evolutionary information, and evolutionary information is best inferred from homologous characters. From this perspective, we now need to place nucleotide alignment in the broader context of molecular homology, which we cover in the next section. This broader context was once well described by Brigandt (2003) in the following terms: ‘In molecular biology the scientific aim is the study of biological processes at the molecular level and their explanation by means of mechanisms. The role of molecular homology is the inference of information about the molecular behavior of genes and proteins (and their parts), particularly in order to guide further experimental investigation and technological manipulation’ (p. 15).
Different types of homology
Components of homology
Homology is a hierarchical concept (Roth 1991; Donoghue 1992; Dickinson 1995; Abouheif 1997; Freudenstein et al. 2003), and thus it is context-sensitive and depends on the research program (Assis 2015). There are actually many concepts of ‘homology’ sensu lato (Brigandt 2003; Pavlinov 2012), which are related to different levels of biological organisation. Homology between features can simultaneously be present at one level of the hierarchy but absent at others. That is, homology at one level in the hierarchy does not actually necessitate homology at other levels. Furthermore, homology at different levels is usually detected by different criteria.
This situation arises because phylogenetic history has a strong hierarchical component, and characters that arose early in history are now more widespread among taxa than are characters that arose later. The homology of some characters, therefore, occurs at a more general level than that of others (i.e. they are more inclusive). The classic example is the comparison of bird wings and bat wings. These are homologous as forelimbs (structures), which are general throughout the tetrapods, but they are not homologous as wings (functions), because they represent independent modifications of those forelimbs in the ancestors of birds and bats.
In practice, then, homology is a multidimensional concept, with potentially different interpretations being relevant to different biological studies, such as evolution, function and development; different fields use the homology concept to pursue different theoretical and practical goals. Nevertheless, homology implies descent of similar features from a common ancestor in all cases, and is distinct from analogy, which implies similarity owing to convergence. Recognising different types of homology that represent different hierarchical levels is unproblematic, provided that the appropriate adjective is used to indicate the level of biological organisation (Fitch 2000).
The concept of hierarchical levels applies across all conceivable characters, including those of molecules. Indeed, observed molecular similarities could reflect homology at any of several hierarchical levels. Moreover, homologous molecules may be involved in biochemical processes that are analogous, and homologous processes may involve molecules that are analogous (Wray and Abouheif 1998). Evolution produces diversity as well as maintaining uniformity, and this makes homology assessment as tricky a business for molecules as it is for morphology.
In Table 1, we list some of the conceptual levels at which molecular homology has been used in the literature, and we will discuss each of these homology components in turn. In all cases, the word ‘homology’ does imply shared descent (i.e. similarity of features predates the evolutionary divergence of the taxa). However, non-homologous features at lower levels can combine to produce features at higher levels that are typically considered to be homologues, as discussed in the next section. Our indication of equivalent terms in Table 1 is for convenience of discussion, and does not imply that there is no conceptual difference between them.

|
Evolutionary homology (sometimes confusingly called phylogenetic homology) is the classical concept, used throughout the rest of this paper. Only nucleotides are inherited by chains of descent, either vertically from parent to offspring or horizontally by gene flow (e.g. hybridisation, lateral gene transfer). Therefore, evolutionary homology strictly applies only at the level of nucleotides (and loci and sequences, which are aggregations of nucleotides). It is, for this reason, that multiple-sequence alignment of DNA is so important in phylogenetics; it constitutes the only direct interpretation of homology hypotheses.
A crucial difference between evolutionary homology and the other listed uses of the word ‘homology’ is that the latter are amenable to experimental testing, whereas the former is not. Some of these tests are based on pattern analysis of character variation (such as character-state homology and organismal homology), but the others can be determined by manipulative experimentation. Evolutionary homology, by contrast, refers to chance evolutionary events that are historically unique, and we can neither make direct observations of these events, nor can we perform experiments to investigate them. Important as it is, evolutionary homology remains largely a theoretical (or idealistic) concept.
Character, character-state and taxic homologies
Turning to more practical matters, we note that it is important to recognise the operational distinction between character-state homology, character homology and organismal (or taxic) homology (Table 1). The operational distinction between the first two concerns the construction of a data matrix (Brower and Schawaroch 1996; Hawkins et al. 1997). Characters are identified as comparable features between organisms and are operationally defined as the columns of the alignment matrix. Character states are then entered into the columns as being identical or not among sequences. Both characters and identical character states are, thereby, treated as hypothetically homologous (i.e. resulting from shared derived descent).
Identifying positional homology in sequence data thus equates to primary character identification. It has been noted that the step of identifying primary character hypotheses proceeds differently in morphological and molecular phylogenetics; rather than establishing characters to be sampled and then observing their states (Hawkins et al. 1997), for sequence data the states (nucleotides) are observed before character definition (Doyle and Davis 1998). This creates an extra step, namely that of developing a character set out of a sea of observations. In practice, first we construct an alignment and, then subsequently determine so-called regions of ambiguous alignment.
Character-state homology for sequence data is thus, in the first instance, a model-based inference based on similarity. We observe certain distributions of nucleotides, and we optimise some concept of similarity (see below) to assign them as states of certain characters. We, thus, define characters (columns) and assign pre-observed units (nucleotides) to them, thereby inferring character states.
It then becomes possible to examine this primary assignment of states to characters on a phylogenetic tree. The character states will appear as synapomorphies, homoplasies or symplesiomorphies on the diagram. If we subsequently optimise the inferred character-state transformations on the phylogenetic tree, then we are introducing the concept that homology equals synapomorphy. Note that this designation can be proposed without confirmation that the tree diagram is an accurate representation of true phylogeny. This concept is exploited by both the direct-optimisation and statistical-alignment procedures, which simultaneously produce both trees and alignments.
These methods, DO and SA, are linked to the concept of organismal (or taxic) homology. In a phylogeny, homologous character states become features that characterise monophyletic groups, as discovered through the phylogenetic analysis (this is also called cladistic homology). In this sense, homology is defined only with reference to the phylogeny (Patterson 1982; Assis 2013, and references therein). Evolutionary homology (homology as the correspondence between features owing to common ancestry) is used as the inferred explanation for the discovered taxic homology (synapomorphy as a result of the phylogenetic analysis; de Pinna 1991; Mindell 1991; Brower and de Pinna 2012).
Taxic homology treats congruence on the phylogeny as the ultimate test of homology. However, mere congruence of characters alone cannot determine homology (Nixon and Carpenter 2012; Assis 2013; Farris 2014). Although homology implies synapomorphy, apparent synapomorphy does not necessarily imply homology. A well known example of this is so-called long-branch attraction, in which spurious similarity of character states among sequences of rapidly evolving taxa overcomes the expectation that apparent synapomorphy is most likely equivalent to phylogenetic truth (Bergsten 2005). In such cases, one needs an independent causal basis for the hypotheses of homology, involving theories of inheritance and development (Kelchner 2000; Rieppel 2004; Morgan and Kelchner 2010).
Operationally, we also need to recognise the hierarchical level at which the test of congruence operates. Systematic analysis can distinguish synapomorphic character-state distributions from homoplastic ones whenever the characters are held fixed. It is only character-state identity that can be said to be tested by congruence, and even then, only in reference to a true phylogeny. Where character-state distributions are found to be homoplastic, the test of congruence provides no information as to the level at which the homology assumption is incorrect. More importantly, where character-state distributions are found to be synapomorphic on a phylogeny, the test provides no clue about the nature or reliability of the character (Morgan and Kelchner 2010).
This approach differs from the traditional one, in which homologies are not optimised on a phylogeny. In most cases, the phylogeny is treated as a test of the homologies, not a definition of them. Many researchers would like a valid test of homology (see Rieppel and Kearney 2002) so that confidence may be improved for their phylogeny estimations. Such a test could be made in the context of a known phylogeny, which we hardly ever have. However, we do not actually need a test if we treat the phylogeny as only an estimation, not a known history.
Furthermore, what we also want is a test of the characters not just the character states (Rieppel and Kearney 2002; Richter 2005), that is, character analysis, not phylogenetic analysis. This is particularly true for nucleotide sequence alignments, where the definition of the characters (the alignment columns) is not always straightforward. This would involve a non-phylogenetic diagnosis of homology, so as to make homology discovery operational (Jardine 1967, 1969; Hawkins et al. 1997; Agnarsson and Coddington 2008). Indeed, if homology recognition is treated as a model-based process, then we are optimising possible criteria for discovering homologies (quantitative), rather than testing hypotheses (pass or fail; Morrison 2015).
Other levels of homology
Turning now to the other levels of the homology hierarchy, regional (or locus) homology (Table 1) recognises that blocks of contiguous nucleotides on a chromosome are typically descended from a common ancestor (i.e. nucleotides are rarely singletons with respect to their phylogenetic history). Indeed, De Laet (2014) explicitly distinguished ‘subsequence homology’ from ‘base-to-base homology’, noting that they are ‘two components of sequence homology that cannot be reduced to one another’. The sequence blocks can be rearranged owing to recombination and translocation, but they usually function as a unit, for example, as part of a protein-coding gene, an intron, a structural RNA, a transcribed spacer or a regulatory mi-RNA. Evolutionary homology applies directly to these loci, even though it may be difficult to apply this concept in practice (i.e. the region boundaries may be indeterminable).
Structural homology and functional homology (Table 1) refer to inheritance of molecular structures and functions from a common ancestor, irrespective of whether these are still controlled by homologous nucleotides in contemporary organisms. Owen’s (1843) original definition of homology referred to identity of organs irrespective of form or function, which appears to separate structure and function (and Owen apparently meant homology = similarity of structure, whereas analogy = similarity of function). However, there are separate and legitimate concepts for homology of structure and function based on shared descent (Love 2007), and traditionally structure has been regarded as the most reliable level at which to detect morphological homologies (Jardine 1969; Rieppel and Kearney 2002; Richter 2005; Agnarsson and Coddington 2008). Indeed, ‘homology’ is sometimes seen as solely being either structural or developmental (see below), rather than the larger hierarchy shown in Table 1.
Structure and function may be closely related for molecular data, as function usually determines molecular structure, and structure can often be used to identify function (Thompson and Poch 2005; Pei 2008). For example, the genes encoding many biomolecular systems and pathways are genomically organised in operons or gene clusters, and this arrangement can be used as evidence for gene homology (Medema et al. 2013). For molecules, it is commonly assumed that conservation of structure and function is more common than is convergence (this is almost a phenetic argument; Pavlinov 2012), although this always needs to be tested (Doolittle 1981; Simmons 2000; Galperin and Koonin 2012). Indeed, molecular biologists frequently list structural and functional homology as the most important alternative criteria to evolutionary homology when discussing sequence alignment, particularly for amino acid sequences.
For example, consider what appears to be the definition of sequence alignment most widely quoted on the Internet: ‘a sequence alignment is a way of arranging the sequences of DNA, RNA, or Protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences’ (usually credited to Mount 2004). As noted by Fitch (2000), ‘Life would have been simple if phylogenetic homology necessarily implied structural homology or either of them had necessarily implied functional homology. However, they map onto each other imperfectly’ (p. 231).
Moving on, genes are conventionally thought of as having detectable homology relationships (Table 1), and genes and gene products have occupied most of the discussion of molecular homology (Patterson 1988; Hillis 1994; Brigandt 2003; Freudenstein 2005). Genes have pre-cursor genes in ancestral organisms, and so there are inferred homologues among contemporary organisms. To the extent that genes represent blocks of homologous nucleotides, this idea is unproblematic, as are the concepts of orthology (evolutionary homology) and paralogy (gene duplication) (Fitch 1970). However, the concepts of orthology and paralogy may not always be clearly distinct in practice, owing to incomplete lineage sorting (Mallo et al. 2014), and detecting orthology without a phylogeny is problematic (Gabaldón 2008).
Fitch (2000) suggested that ‘there are no proven cases of genic analogy’ (p. 230), noting that gene variation usually involves gene orthology but functional analogy. There is no simple relationship between gene homology and nucleotide homology, however, because of several well known biological phenomena, including the following: recombination and gene conversion; translocation (e.g. exon shuffling); fusion, fission and domain replacement; and xenology and synology (i.e. horizontal gene flow; Mindell and Meyer 2001; Haggerty et al. 2014). These phenomena create genes that do not have nucleotide homology along the majority of their length. In such cases, we do not have gene homology sensu stricto, because only subsets of the gene are orthologous (e.g. domains of protein-coding genes). Nucleotide sequence similarity, then, becomes insufficient evidence to recognise orthology, and will produce mis-leading results (Thornton and DeSalle 2000).
Gene homology is important because only orthologous genes can be used to infer phylogenetic relationships among organisms. To this end, gene products are often grouped into families based on their inferred homology, so as to identify appropriate data sources. An alternate and more useful approach is to form databases around genic subsets, which are more likely to be orthologous. For example, the Pfam database (Finn et al. 2014) is intended to reflect homology of protein domains rather than complete genes, and the Rfam database (Burge et al. 2012) does the same for RNAs.
Finally, the concept of developmental homology (Table 1) is based on the recognition that sometimes identity of morphological characters is not the result of identity of the sets of genes that control their development. That is, sometimes non-homologous genes and gene networks can produce morphological structures that are usually considered to be homologues (Meyer 1999; Mindell and Meyer 2001), and this needs to be distinguished from cases where the developmental mechanisms have been inherited intact. Regulatory genes are rarely dedicated to a single developmental task, and only some of these roles will be conserved during evolution. Such genes can end up substituting for the role of some other non-homologous gene in a developmental pathway (called recruitment or co-option, leading to deep homology).
Developmental biologists, therefore, often prefer a process-oriented concept of homology, which they call biological homology, where homologous features are those sharing a set of developmental constraints. Iterative or serial homology within organisms has been incorporated into the definition, which is usually excluded from the concept of evolutionary homology. The pros and cons of this interpretation have been extensively discussed in the literature (Wagner 1989, 2014; Abouheif 1997; Wray and Abouheif 1998; Meyer 1999; Laubichler 2000, 2014; Brigandt 2003; Cracraft 2005; Rutishauser and Moline 2005; McCune and Schimenti 2012). Indeed, the terms syngeny (Butler and Saidel 2000) and homocracy (Nielsen and Martinez 2003) have been coined to describe morphological features that are organised through the expression of homologous gene networks, irrespective of whether those features are evolutionarily homologous or convergent.
Homology of molecular data
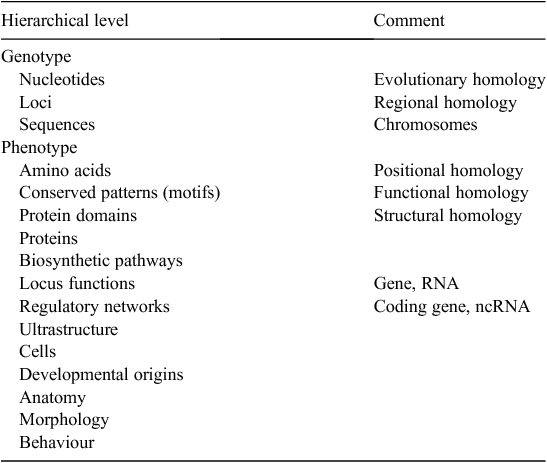
Data that require homology assessment will be expressed at multiple hierarchical levels in an organism, from nucleotides to amino acids, genes, gene functions, gene networks, developmental origins and morphological structures. Some examples of data levels at which homology might be considered are arranged in a simple hierarchy in Table 2. Features at lower levels in the hierarchy combine to generate features at higher levels.
|
There are two important points to recognise about this hierarchy. First, there is a distinction between genotype and phenotype. Second, there is no necessary 1 : 1 relationship between homology at different levels in the hierarchy. We will take each of these in turn.
Phenotype is the expression of a genotype in interaction with its environment. As noted above, only nucleotides and groups of nucleotides are part of the genotype. All other features are part of the phenotype, irrespective of whether they constitute molecular or morphological data. The importance of this distinction is that homology has a direct interpretation only at the level of nucleotides (and loci and sequences). Only here is homology necessarily simple, and homologues indivisible. We can theoretically provide a yes or no answer to the question ‘are these nucleotides descended by chains of inheritance from a common ancestral nucleotide?’ For phenotypes, homology is not necessarily as simple to interpret, and homologues are not always indivisible (see examples below). (However, we do not conclude from this that phenotype data are unimportant in phylogenetics; cf. Stace 2005.)
The key to homology is inheritance; a copy of the nucleotide at a particular position in the genome is inherited by daughter cells and organisms. So, genotypes are inherited whereas phenotypes are expressed. One does not inherit an amino acid sequence, one inherits a nucleotide sequence that can be translated into the amino acid sequence; one does not inherit a forelimb, one inherits a nucleotide sequence that codes for genes that control the development of the forelimb.
It is information, then, that is inherited (Roth 1991; States and Boguski 1991), and that information can be expressed in various ways. Indeed, many genotypes express the same phenotype (i.e. not all changes in a genotype are reflected in the phenotype). The redundancy of the genetic code, for example, has produced a case reported by Morrison (2006) of a sequence alignment in which there is a stretch of 143 amino acids that are conserved across all 10 taxa but only 284 of the 429 nucleotide alignment positions are conserved, leaving 145 (33.8%) variable positions. Similarly, high transition rates in Group II intron sequences allow extensive nucleotide substitution, while maintaining highly conserved secondary and tertiary RNA structures (Kelchner 2002).
So, sometimes only part of the inherited information is used for expression, or different pieces of information are combined for expression. This means that it is possible (indeed, quite common) that somewhere during evolution an arrangement of the information is changed, so that features that were once homologous are no longer ‘the same’ in some way. For example, different nucleotides might now code for a particular amino acid, different domains might make up a protein, different proteins might be involved in a biosynthetic pathway, or different gene networks might now control the development of a morphological structure.
This means that, for a phenotype (the expression of a genotype), homology may be complex, because there is no necessary 1 : 1 relationship to lower levels in the hierarchy (e.g. nucleotides : amino acids, domains : proteins, genes : development). At the level of phenotype, then, the idea that two things either are or are not homologous seems somewhat naïve, because there is no simple relationship between genotype and phenotype. For phenotypic features, homology means nothing more than that at least some part of the character-state information has descended from homologous DNA. In this sense, the concept that homology refers to descent from a single common ancestor is very limited, because most phenotypic levels of homology involve descent from multiple ancestors.
This idea leads to what has been called partial homology (Hillis 1994), or even degree of homology (note that this is distinct from the egregious ‘percentage homology’ that simply means similarity). Evolutionary homology at one level of the hierarchy does not necessarily imply evolutionary homology at other levels, although it will often do so. The levels of the hierarchy arise from combining units of information at lower levels, and there are many ways to combine those units. A well known example of such complex homology is chimaeric proteins from gene fusions that contain unrelated domains, thereby having only partial homology at the protein level (Kummerfeld and Teichmann 2005; Moore and Bornberg-Bauer 2012; Haggerty et al. 2014). Similarly, post-processing of a transcribed product can make the mRNA and tRNA different from the DNA, so that the RNA codes for something different from that indicated by the DNA alone (Maas 2012). In both cases, the transcribed or translated products have no simple 1 : 1 relationship to the DNA.
Perhaps the most straightforward example is that amino acids do not necessarily have a simple relationship to nucleotides because of, for instance, replication slippage during evolution. Consider the situation where a nucleotide is deleted at some time in the history of a protein-coding gene, while at the same time, a nucleotide is inserted somewhere nearby in the same sequence (Fig. 1). In the affected part of the sequence alignment, there is no longer a simple homology relationship between the amino acids and their coding nucleotides. The reading frame for the codons will be maintained, and, thus, there will be no evidence of insertions or deletions in the amino acid sequence. All of the amino acids will appear to align, even though some of the nucleotides do not align based on positional homology (i.e. the inserted and deleted nucleotides). In this sense, some of the amino acids would not be evolutionarily homologous at the nucleotide level (genotype), because they would be only partly coded for by homologous nucleotides.

|
However, under most phenotype levels of homology, the amino acids in this example would be considered homologous. The amino acid sequences would probably still be very similar, and so, at the level of amino acid, positional homology would still apply. Also, their functions would probably be maintained, and, thus, they would be homologous at the function level. The genes would also still be considered homologous, of course, although they are no longer coded for by nucleotides of which all are homologous. In this particular example, the amino acids and genes provide a framework within which the nucleotides can evolve, in the sense that they present constraints on the variation that can occur among the nucleotides; the functions of the amino acids and genes must be maintained, even though non-homologous nucleotides are involved.
These examples lead us to suggest that part of the problem in thinking about the relationship between multiple-sequence alignments and homology is the fact that so much of the work has involved amino acids and genes, where the connection between homology and alignment is not as direct as it is for nucleotides. Studies of the evolution of amino-acid sequences, for example, are still conducted in terms of a substitution and indel model (e.g. Ajawatanawong and Baldauf 2013; Chong et al. 2013), and it is the lack of adequate indel models that has been seen as a major limitation for the production of biologically realistic protein alignments (Anisimova et al. 2010).
Sequence-alignment procedures
Operations
It seems logical to consider that homologies are real, in the sense that we expect the characteristics of shared ancestors to be passed on to their descendants. However, we have no method for observing homologies directly, because they are the by-product of unique historical events. Homology exists independently of our ability to recognise it. Indeed, both homologies and phylogenies need to be ‘discovered’ within the phenotypic and genotypic data that we have accumulated about biological organisms. This is the distinction between the ontological definition of homology (characters sharing common ancestry) and the epistemological diagnosis of homology (some sort of observed shared similarity).
Comparative biology is based on studying the features of contemporary organisms, on the grounds that they will contain traces of their historical ancestry from which homology relations might be extracted, however imperfectly. As noted in the previous section, multiple-sequence alignment of nucleotides is an integral part of comparative biology because it is the only level of homology that directly represents the genotype.
In practice, we hypothesise that certain characteristics are homologous in a probabilistic sense – some homologies are more likely than are others. The main applied issue is how to devise the best set of hypotheses. Any operational procedure requires a quantitative notion of ‘best’, and it needs to be objective and repeatable. Because there is no single algorithm for coding characters and character states, morphological data are coded in many different ways in practice (Hawkins 2000). That is, there is no objective and repeatable methodology (although cf. Jardine 1967). However, for sequence alignment, this has traditionally been treated as a computational issue rather than a biological one.
When viewed algorithmically as a string-matching procedure (as it usually is in bioinformatics; Gusfield 1997; Pevzner 2000), the alignment process consists of shuffling the fundamental units back and forth to form states of different characters, the final arrangement being that which optimises some mathematical objective function. That is, operationally, multiple-sequence alignment consists of evaluating the probability of the nucleotides being a character state of each of the available characters: an A in one column is not the same as an A in any other column – they are states of different characters – and we need to decide among the possibilities.
In essence, this procedure is no different from trying to decide, for example, whether a particular plant structure is a leaf, a bract, a bracteole, a sepal or a petal. In the standard conception of floral morphology, these are all modified ‘leaves’ sensu lato, and they are therefore available as character choices (Rutishauser and Moline 2005). However, for any one organism, they can be present or absent in any combination, and deciding which ones are present is conceptually no different from trying to construct an alignment. In this sense, there is no fundamental difference, either theoretical or practical, between homology in phenotype studies and homology in genotype studies. Although it is rarely recognised as being so, ‘optimal sequence alignment’ is simply a restricted application of the algorithm that Jardine (1967) developed for morphological characters.
A major problem is that, in general, genotype homology is harder to assess than phenotype homology. In particular, molecular alignment is potentially harder than diagnosing morphological homology because the restricted nature of the units hampers comparison; mathematically, we cannot build much biological insight into a substitution matrix of four nucleotide states. However, in this paper we make a distinction among units, characters and character states because it will help us clarify some of the problems associated with homology assessment of nucleotide data, and how to work effectively with limits such as four states.
Some authors feel that the distinction among units, characters and character states is unnecessary (Patterson 1988; Freudenstein 2005) because the three concepts can be viewed as arbitrary levels of a nested hierarchy; units are nested within states, which are nested within characters. Such a view can be reinforced by the apparent arbitrariness of a program’s nucleotide shuffling during alignment. However, with our distinction of levels, that apparent arbitrariness becomes a focus for problem-solving an alignment. When homology is the objective of sequence alignment, the goal implies that we need a non-arbitrary process. How do we decide which A goes in which column (i.e. which unit forms which character state)? Each A looks the same. Ultimately, this explains the lack of programs for nucleotide-sequence alignment that use homology as their optimisation criterion.
This issue often does not apply for phenotypic characters, where there may be clear differences (Rieppel and Kearney 2002), and where we have already developed criteria for recognising homologues. We now need to consider how these criteria apply when studying sequences.
Sequence-homology criteria
Systematists have developed criteria for making decisions about potential homologies in an objective and (hopefully) repeatable manner (Patterson 1988), and Morrison (2015) has shown that these are directly applicable to nucleotide sequences. These criteria are as follows:
-
Similarity
-
-
Compositional = apparent likeness or resemblance between sequences (% similarity)
-
Topographical = apparent likeness or resemblance between sequences (second- and third-order structure of protein or RNA)
-
Functional = functional relationship to other characters in the same sequence (annotated function of the sequence in protein or RNA)
-
Ontogenetic = variation arising from the same molecular mechanism between sequences (inferred molecular mechanism creating the sequence variation)
-
-
Conjunction = possible within-genome copies of the same sequence (i.e. paralogy)
-
Congruence = agreement with other postulated homologies elsewhere in the same sequences (synapomorphy)
Traditionally, characters have been first proposed as homologous using the criteria of similarity and conjunction (together called primary homology), and then tested with the criterion of congruence (secondary homology; de Pinna 1991; Brower and Schawaroch 1996).
It is clear that these criteria have been incorporated singly into computerised procedures for producing multiple-sequence alignments, but rarely in combination. For example, compositional similarity is the criterion used by the most popular computer programs, such as CLUSTAL (Larkin et al. 2007), MAFFT (Katoh and Standley 2013) and Muscle (Edgar 2004). Topographical similarity is being invoked whenever structure-based alignments are produced, such as for RNA-coding sequences (e.g. PicXAA-R: Sahraeian and Yoo 2011; PMFastR: DeBlasio et al. 2012), or when nucleotide sequences are translated to amino acids before alignment (e.g. PROMALS: Pei and Grishin 2007). The use of complex nucleotide patterns, such as those of retrotransposons (transposable element insertions), notably short interspersed elements (SINEs; Kramerov and Vassetzky 2011), also fits into this category (Ray et al. 2006). Functional similarity is used for specialist studies of conserved motifs and binding sites (e.g. MEME: Bailey and Gribskov 1998; AlignACE: Roth et al. 1998). Ontogenetic similarity of nucleotide sequences is based on inferring the possible molecular processes that cause the observed sequence variation; the program Prank (Löytynoja and Goldman 2008) uses this criterion by distinguishing between insertions and deletions.
Conjunction as a criterion notes that homologous features cannot have multiple copies within the same organism, when assessing taxic homology. This makes the alignment of repeated subsequences problematic, because they constitute serial homology rather than taxic homology.
Congruence as a criterion involves the observation of repeated patterns of synapomorphy in a phylogeny. Among alignment algorithms, both DO (e.g. POY: Wheeler et al. 2015; MSAM: Yue et al. 2009; BeeTLe: Liu and Warnow 2012) and SA (e.g. BAli-Phy: Redelings and Suchard 2009; StatAlign: Arunapuram et al. 2013) try simultaneously to produce a multiple alignment and a phylogenetic tree, and thus attempt to optimise the criterion of congruence. In this sense, they are sometimes seen as maximising homology in sequence data (e.g. De Laet 2014). Programs that use iterative refinement of the alignment and guide tree also use congruence (Mindell 1991).
It is important to note that these criteria do not always agree with each other in their inferences of homology. Changes that occur during evolutionary history can weaken the connection between these criteria so that, for example, nucleotide homology inferred from structural similarity is no longer the same as nucleotide homology inferred from compositional similarity. It is, for this reason, that compositional similarity of the sequences is insufficient to establish gene orthology (Thornton and DeSalle 2000).
To make these criteria operational, we need to compare their inferences by evaluating the comparative evidence.
Making the criteria operational
Decisions regarding these criteria require making judgements about homology of the character states within each character. In practical terms, for a multiple sequence alignment, this means studying the relationships of the sequences across the rows within any one column of the alignment. However, current computerised sequence-alignment algorithms do not do this; instead, they evaluate the relationships of the sequences across the columns within the rows of the multiple alignment. They do this both during pairwise alignment algorithms and during the algorithms that braid the pairwise alignments together; the ‘multiple’ part of the alignment procedure consists of combining (horizontally) aligned pairs of sequences.
The essential problem, then, with current multiple-alignment algorithms is that they proceed horizontally rather than vertically. That is, the basis of the operation is the fact that the nucleotides are physically arranged as a string along a chromosome. However, for evolutionary purposes, the important idea is that each nucleotide position is a character shared with other sequences (as shown in Fig. 1), and this should be the basis of homology assessments.
This seems to be a fundamental operational difference between sequence assessment and other homology assessments. For morphological studies, comparative biology has always involved comparing what appear to be potentially homologous character states across multiple taxa. These comparisons are as detailed as is necessary to make a decision about homology (i.e. is it probable rather than merely possible?), and may involve detailed developmental studies. Comparative biology involving sequences, by contrast, has mostly done little more than assess patterns along pairs of sequences in terms of string matching. A multiple alignment is not simply a set of pairwise alignments braided together.
This means that alignment patterns are frequently missed that can easily be detected by looking at the alignment vertically rather than horizontally (see Fig. 1). This fact is addressed to some extent by algorithms that allow post hoc re-alignment. For example, Kim and Ma (2014) described an algorithm based on probabilistic consistency that provided small improvements in their simulation study. However, consistency is not the same thing as homology. Furthermore, even the application of the homology criteria can be problematic; for example, structure-based multiple-alignment algorithms are rarely >80% successful at identifying topographical homology (Letsch et al. 2010).
Humans are good at pattern matching, whereas computers currently are not (MacLeod 2008). Therefore, people can more easily detect mis-matched patterns (i.e. complex features that are difficult to implement in alignment algorithms). This is the simplest explanation for why biologists frequently ‘adjust the sequence alignment by eye’. A manual adjustment is currently performed by more than one-half of evolutionary biologists and more than three-quarters of phylogeneticists (Morrison 2009b). The homology assessments as produced by the computer program, working along the rows, are re-evaluated by looking at the patterns across the rows within the columns. This can also be seen as an attempt to move from an alignment that is based on simplistic substitution and indel modelling to one based on the molecular mechanisms that underlie sequence variation.
Unfortunately, these adjustments are currently performed manually; this is time-consuming, highly detailed work, and personal judgment may not be perfect, but at least there is an opportunity for it to be consciously based on homology as a character concept. A major improvement in alignment algorithms would obviously be achieved if this re-evaluation could be automated. This is particularly so when dealing with genomic datasets, where manual attention to issues of data quality is almost impossible. What we would need is a computerised procedure that will include all of the known criteria for homology assessment, but there are currently no mathematical models for doing this.
Morrison (2015) discussed several possible approaches to the problem, including the following:
-
try to reproduce the human approach to homology assessment, which is by homology hypotheses proposed based on similarity and conjunction, which are then tested with congruence;
-
search the nucleotide sequences for evidence of known molecular processes, and then optimise the combination of these to produce a set of optimal scenarios for the origin of the sequence variation;
-
evaluate the types of similarity independently as the criteria for alignment hypotheses, represent the hypotheses as a (large) set of local alignments, and then combine these local alignments into a global alignment;
-
use as a starting point a pre-existing curated and trusted alignment and then add new sequences to it, because this allows the high quality of the initial alignment to be maintained as the alignment grows in size;
-
use as a suitable starting point an alignment based on compositional similarity, and then modify it to represent a scenario of postulated homologies; this is apparently what is currently being undertaken manually by many practitioners.
The nature of sequence variation
Most computerised alignment methods model all sources of length variation as indels, and then treat indels as a type of substitution with a variable weight. Moreover, all of these methods mathematically treat both substitutions and indels as independent and identically distributed (IID) random variables. Simulation studies of alignment algorithms make the same IID assumption, by generating substitutions and indels at random in the simulated sequences (called stochastic modelling). The same issue applies to probabilistic assessments of alignment accuracy, which assume that alignment errors occur at random, whereas accuracy is likely to be related to the degree of sequence conservation. This is an all-pervading problem for computerised alignment procedures, because the IID assumption actually means that much (if not most) of the information about evolution is ignored.
The problem is that sequence variation occurs distinctly non-randomly and non-independently, both in space and time. A DNA sequence is not an arbitrary string of characters, but instead frequently codes for a macromolecule (e.g. protein, r-RNA, non-coding mi-RNA, intron, spacer) with specific biological constraints, so that contemporary nucleotide sequences are mosaics of conserved and non-conserved fragments with different properties (Kelchner 2000; Wuyts et al. 2001; Smit et al. 2009; Terekhanova et al. 2013). Molecular mechanisms operate differently in different types of gene loci and in different parts of the same locus, and differently again in non-transcribed regions. Often those mechanisms are a function of the nucleotide order itself, which can trigger a mutation event repeatedly at a single site (Kelchner and Wendel 1996; Kelchner 2000). Thus, observed sequence variation cannot be assumed to be either a random or an independent sample from the universe of all possible sequence variation.
The inherent limitation of contemporary computer algorithms is that the models are currently inadequate (Kelchner 2009; Morrison 2009b; Anisimova et al. 2010). They fail to model many of the important molecular mechanisms that cause sequence variation (for example, inversions, repeats), and even the parts they do model make unrealistic assumptions, such as IID. That is, all sequence mis-matches are modelled as IID substitutions (e.g. a four-base inversion is modelled as four independent substitutions) and all length variations are modelled as IID indels (e.g. a six-base tandem repeat is modelled using an affine cost for a variable-length indel). These factors combine to create a situation where many of the resulting empirical multiple-sequence alignments do not stand up to even casual scrutiny.
Such issues lead to bias in the sampling of the characters, sometimes called ascertainment bias. This bias is particularly manifest when practitioners exclude so-called ‘difficult to align’ regions of sequences, in an attempt to have only high-quality data in their alignment. These ‘difficult’ regions are not randomly distributed with respect to phylogenetic information, and so the attempt to have high-quality data can lead to poor-quality character sampling, and the resulting alignment and phylogeny are biased.
The multiple-alignment programs that are usually reported as performing best (see Nuin et al. 2006; Pais et al. 2014), such as MAFFT and ProbCons, work well because they deal most effectively with this non-randomness. The program Prank correctly separates insertions from deletions, but otherwise makes the IID assumption. SA algorithms treat indels and substitutions as separate parts of the model, but each part is assumed to be IID. The parsimony or likelihood analyses used by DO also make the IID assumption about synapomorphies. Current likelihood-based alignment algorithms try to allow for uncertainty in alignment when evaluating trees; however, their conceptual basis for uncertainty is stochastic variation, whereas the variation is anything but random, and the non-randomness contains valuable information about homologies.
Assessments of alignment accuracy also suffer from other potential problems, notably that the reference alignments (gold standards) commonly used (Wilm et al. 2006; Pei and Grishin 2007) are derived from biased samples of proteins and RNAs with a known structure (Kemena and Notredame 2009; Edgar 2010). These gold standards have, thus, focussed the methodological development of alignment algorithms towards the production of alignments that are correct only with respect to secondary structure (Notredame 2007; Aniba et al. 2010; Iantorno et al. 2014). This strengthens the criterion of topographical homology, but does nothing for the other criteria. No gold-standard database for homology alignment yet exists, although small empirical datasets have been created (Morrison 2009a).
Simulation has been strongly advocated as an alternative approach to evaluating alignment algorithms (Rosenberg and Ogden 2009). However, the results based on using artificial datasets seem to conflict with those based on the gold standards (Lassmann and Sonnhammer 2002, 2005; Löytynoja and Goldman 2008; Kemena and Notredame 2009), which suggests that current simulation models may be inadequate (Iantorno et al. 2014). The simulated data lack realism because the simulation models make the random and IID assumptions, whereas real sequence variation is not random or IID.
Other approaches to assessing alignments include consistency-based benchmarks (Lassmann and Sonnhammer 2005), based on the idea that different good aligners should tend to agree on a common alignment (namely, the correct one) whereas poor aligners might make different kinds of mistakes, thus resulting in inconsistent alignments. However, two wrongs do not make a right; that is, consistent methods may be collectively biased. Moreover, consistency is not independent of the set of methods used (some may be consistent with each other and not with others). There is also phylogenetic assessment of alignments (Dessimov and Gil 2010), which suggests that, given a reference tree, the more accurate the tree resulting from a given alignment is, then the more accurate the underlying alignment is assumed to be. However, this idea involves a false inversion of a proposition: accurate alignments may yield accurate trees, but we cannot conclude that therefore accurate trees must be based on accurate alignments.
The hierarchical nature of taxa
The hierarchical nature of homology is related to the hierarchical nature of biodiversity. This means that it will be more or less harder to assess homology at some taxonomic levels than at others. For instance, sequences may be easily alignable among species and yet very difficult to align among genera, let alone across a family. As a single example, the Legume Phylogeny Working Group (2013) noted that ‘Most legume systematists probably despair when examining a progressively broader taxonomic sample of ITS sequences. For example, this locus was barely alignable across the genus Vigna Savi. s.l. or the much smaller Leucaena’ (p. 232).
This particular characteristic of homology is not fundamentally different, either in theory or practice, for phenotype studies compared with genotype studies. Nevertheless, many researchers seem to expect that alignment algorithms will function equally well across all hierarchical levels of taxa, from intra-species all the way to kingdom. This expectation is unwarranted, because evidence for homology becomes obscured at greater evolutionary distances among taxa.
Unfortunately, in practice this situation often leads researchers to simply abandon certain genetic regions from their studies, solely because a single alignment cannot be produced across all of the sequences being studied. One frequently reads that ‘regions of ambiguous alignment were excluded’ or that ‘gaps were excluded’ from downstream phylogenetic analyses. However, it has been repeatedly shown empirically that this approach potentially loses valuable phylogenetic information (Simmons et al. 2001; Bapteste and Philippe 2002; Wrabl and Grishin 2004; Egan and Crandall 2008; Dwivedi and Gadagkar 2009; Dessimov and Gil 2010; Denton and Wheeler 2012).
The obvious way to deal with the situation is to take the hierarchical nature of homology into account explicitly, so that homology is assessed across sequences only where it can be applied in practice. This means subdividing sequence datasets when necessary. That is, the number of sequences being aligned may vary from one nucleotide region to another. Within each subdivision, high-quality alignments should be preserved across all of the included sequences. There are practical issues with this approach, of course, most notably how to define the subdivisions in an objective and repeatable manner.
If any one region cannot be aligned across all of the sequences, then the region will simply be presented as several consecutive subalignments, with each group of aligned sequences being offset horizontally from the others in a staggered manner (Barta 1997; see fig. 11 of Morrison 2006). This method preserves all of the available homology information within each subalignment, without falsely aligning non-homologues, and it will be a better practice than abandoning the information, as is the case when gapped regions are excluded from the final alignment.
This does not, of course, deny the potential existence of regions that are seemingly impossible to align, even between pairs of closely related species. Anyone who doubts this might like to look at the sequences of Helix 43 of the small-subunit rRNA of the Apicomplexan genus Plasmodium, which is massively elongated compared with sequences of related genera, apparently independently in each species.
Conclusions
Alignment is often viewed as simply a tool to get a phylogenetic tree; however, alignment for the purpose of phylogenetic inference is primarily about detecting and displaying homology. Unfortunately, molecular homology is rather vaguely defined and understood, despite its importance in the molecular age. Indeed, our focus on the computational issues associated with sequence alignment has overshadowed the much more fundamental issue of maximising character homology before a tree or network analysis.
Efforts to improve sequence alignment for phylogenetic purposes should involve a more refined use of the homology concept at a molecular level. Homology is a hierarchical concept, and there are actually many concepts of homology, which are related to different levels of biological organisation. Here, we have tried to present examples of molecular data levels at which homology might be considered, and arrange them in a logical hierarchy. In practice, we cannot expect that homology evaluation at any one level in the hierarchy automatically implies homology at other levels, although it frequently will do so. Importantly, there is no simple relationship between gene homology and nucleotide homology.
Multiple-sequence alignment, thus, needs to be based on aligning homologous nucleotides, without necessary reference to homology at any other level of the hierarchy. We need to recognise that each alignment is associated with a series or molecular events that have led from the ancestral condition of the sequence to the sampled condition among its descendents, and that these events often involve more than a single nucleotide (notably repeats and inversions). Inference of homology involves deriving a plausible scenario for molecular change among the set of sequences, based on whatever evidence is available, including compositional, topographical, functional and ontogenetic similarity, as well as congruence among characters.
Acknowledgements
Thanks to the Lorentz Center (Leiden) for travel assistance on two occasions. We also thank Mark Simmons and Karl Kjer for many comments that helped us clarify our explanations, and Thomas Borsch, Kai Müller and Dietmar Quandt for discussions over many years. D. A. Morrison thanks Akademikernas A-kassa and Trygghetsstiftelsen for funding. S. A. Kelchner was supported by an award from the National Science Foundation USA (DEB-1120856).
References
Abouheif E (1997) Developmental genetics and homology: a hierarchical approach. Trends in Ecology & Evolution 12, 405–408.Agnarsson I, Coddington JA (2008) Quantitative tests of primary homology. Cladistics 24, 51–61.
Ajawatanawong P, Baldauf SL (2013) Evolution of protein indels in plants, animals and fungi. BMC Evolutionary Biology 13, 140
Aniba MR, Poch O, Thompson JD (2010) Issues in bioinformatics benchmarking: the case study of multiple sequence alignment. Nucleic Acids Research 38, 7353–7363.
Anisimova M, Cannarozzi GM, Liberles DA (2010) Finding the balance between the mathematical and biological optima in multiple sequence alignment. Trends in Evolutionary Biology 2, e7
Arunapuram P, Edvardsson I, Golden M, Anderson JWJ, Novak Á, Sükösd Z, Hein J (2013) StatAlign 2.0: combining statistical alignment with RNA secondary structure prediction. Bioinformatics 29, 654–655.
Assis LCS (2013) Are homology and synapomorphy the same or different? Cladistics 29, 7–9.
Assis LCS (2015) Homology assessment in parsimony and model-based analyses: two sides of the same coin. Cladistics 31, 315–320.
| Homology assessment in parsimony and model-based analyses: two sides of the same coin.Crossref | GoogleScholarGoogle Scholar |
Bailey TL, Gribskov M (1998) Combining evidence using P-values: application to sequence homology searches. Bioinformatics 14, 48–54.
Bapteste E, Philippe H (2002) The potential value of indels as phylogenetic markers: position of trichomonads as a case study. Molecular Biology and Evolution 19, 972–977.
Bapteste E, van Iersel L, Janke A, Kelchner S, Kelk S, McInerney JO, Morrison DA, Nakhleh L, Steel M, Stougie L, Whitfield J (2013) Networks: expanding evolutionary thinking. Trends in Genetics 29, 439–441.
Barta JR (1997) Investigating phylogenetic relationships within the Apicomplexa using sequence data: the search for homology. Methods 13, 81–88.
Bergsten J (2005) A review of long-branch attraction. Cladistics 21, 163–193.
Bergthorsson U, Adams KL, Thomason B, Palmer JD (2003) Widespread horizontal transfer of mitochondrial genes in flowering plants. Nature 424, 197–201.
Blackburne BP, Whelan S (2013) Class of multiple sequence alignment algorithm affects genomic analysis. Molecular Biology and Evolution 30, 642–653.
Bonham-Carter O, Steele J, Bastola D (2014) Alignment-free genetic sequence comparisons: a review of recent approaches by word analysis. Briefings in Bioinformatics 15, 890–905.
Brigandt I (2003) Homology in comparative, molecular, and evolutionary developmental biology: the radiation of a concept. Journal of Experimental Zoology. Part B, Molecular and Developmental Evolution 299, 9–17.
Brower AVZ, de Pinna MCC (2012) Homology and errors. Cladistics 28, 529–538.
Brower AVZ, Schawaroch V (1996) Three steps of homology assessment. Cladistics 12, 265–272.
Burge SW, Daub J, Eberhardt R, Tate J, Barquist L, Nawrocki EP, Eddy SR, Gardner PP, Bateman A (2012) Rfam 11.0: 10 years of RNA families. Nucleic Acids Research 40, D226–D232.
Butler AB, Saidel WM (2000) Defining sameness: historical, biological, and generative homology. BioEssays 22, 846–853.
Chan CX, Ragan MA (2013) Next-generation phylogenomics. Biology Direct 8, 3
Chan CX, Bernard G, Poirion O, Hogan JM, Ragan MA (2014) Inferring phylogenies of evolving sequences without multiple sequence alignment. Scientific Reports 4, 6504
Chong Z, Zhai W, Li C, Gao M, Gong Q, Ruan J, Li J, Jiang L, Lv X, Hungate E, Wu C-I (2013) The evolution of small insertions and deletions in the coding genes of Drosophila melanogaster. Molecular Biology and Evolution 30, 2699–2708.
Cracraft J (2005) Phylogeny and evo-devo: characters, homology, and the historical analysis of the evolution of development. Zoology 108, 345–356.
Dayrat B (2003) The roots of phylogeny: how did Haeckel build his trees? Systematic Biology 52, 515–527.
de Beer GR (1971) ‘Homology: an Unsolved Problem.’ (Oxford University Press: Oxford, UK)
De Laet J (2014) Parsimony analysis of unaligned sequence data: maximization of homology and minimization of homoplasy, not minimization of operationally defined total cost or minimization of equally weighted transformations. Cladistics,
| Parsimony analysis of unaligned sequence data: maximization of homology and minimization of homoplasy, not minimization of operationally defined total cost or minimization of equally weighted transformations.Crossref | GoogleScholarGoogle Scholar | in press. [Published online 28 October 2014]
de Pinna MCC (1991) Concepts and tests of homology in the cladistic paradigm. Cladistics 7, 367–394.
DeBlasio D, Bruand J, Zhang S (2012) A memory efficient method for structure-based RNA multiple alignment. IEEE/ACM Transactions on Computational Biology and Bioinformatics 9, 1–11.
Denton JSS, Wheeler WC (2012) Indel information eliminates trivial sequence alignment in maximum likelihood phylogenetic analysis. Cladistics 28, 514–528.
Dessimov C, Gil M (2010) Phylogenetic assessment of alignments reveals neglected tree signal in gaps. Genome Biology 11, R37
Dickinson WJ (1995) Molecules and morphology: where’s the homology? Trends in Genetics 11, 119–121.
Domazet-Loöo M, Haubold B (2011) Alignment-free detection of local similarity among viral and bacterial genomes. Bioinformatics 27, 1466–1472.
Donoghue MJ (1992) Homology. In ‘Keywords in Evolutionary Biology’. (Eds E Fox Keller, E Lloyd) pp. 170–179. (Harvard University Press: Cambridge, MA)
Doolittle RF (1981) Similar amino acid sequences: chance or common ancestry? Science 214, 149–159.
Doyle JJ, Davis JI (1998) Homology in molecular phylogenetics: a parsimony perspective. In ‘Molecular Systematics of Plants II’. (Eds DE Soltis, PS Soltis, JJ Doyle) pp. 101–131. (Kluwer Academic Publishers: Dordrecht, Netherlands)
Dwivedi B, Gadagkar SR (2009) Phylogenetic inference under varying proportions of indel-induced alignment gaps. BMC Evolutionary Biology 9, 211
Edgar RC (2004) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5, 113
Edgar RC (2010) Quality measures for protein alignment benchmarks. Nucleic Acids Research 38, 2145–2153.
Egan AN, Crandall KA (2008) Incorporating gaps as phylogenetic characters across either DNA regions: ramifications for North American Psoraleeae (Leguminosae). Molecular Phylogenetics and Evolution 46, 532–546.
Ellis J, Morrison DA (1995) Effects of sequence alignment on the phylogeny of Sarcocystis deduced from 18S rDNA sequences. Parasitology Research 81, 696–699.
Farris JS (2014) Homology and misdirection. Cladistics 30, 555–561.
Felsenstein J (2004) ‘Inferring Phylogenies.’ (Sinauer Associates: Sunderland, MA)
Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, Sonnhammer ELL, Tate J, Punta M (2014) The Pfam protein families database. Nucleic Acids Research 42, D222–D230.
Fitch WM (1970) Distinguishing homologous from analogous proteins. Systematic Zoology 19, 99–113.
Fitch WM (2000) Homology: a personal view on some of the problems. Trends in Genetics 16, 227–231.
Freudenstein JV (2005) Characters, states and homology. Systematic Biology 54, 965–973.
Freudenstein JV, Pickett KM, Simmons MP, Wenzel JW (2003) From basepairs to birdsongs: phylogenetic data in the age of genomics. Cladistics 19, 333–347.
Gabaldón T (2008) Large-scale assignment of orthology: back to phylogenetics? Genome Biology 9, 235
Galperin MY, Koonin EV (2012) Divergence and convergence in enzyme evolution. The Journal of Biological Chemistry 287, 21–28.
Giribet G (2005) Generating implied alignments under direct optimization using POY. Cladistics 21, 396–402.
Golubchik T, Wise MJ, Easteal S, Jermiin LS (2007) Mind the gaps: evidence of bias in estimates of multiple sequence alignments. Molecular Biology and Evolution 24, 2433–2442.
Graham SW, Reeves PA, Burns ACF, Olmstead RG (2000) Microstructural changes in noncoding chloroplast DNA: interpretation, evolution, and utility of indels and inversions in basal angiosperm phylogenetic inference. International Journal of Plant Sciences 161, 83–96.
Gusfield D (1997) ‘Algorithms on Strings Trees, and Sequences: Computer Science and Computational Biology.’ (Cambridge University Press: Cambridge, MA, USA)
Haeckel E (1866) ‘Generelle Morphologie der Organismen.’ (Verlag von Georg Reimer: Berlin)
Haggerty LS, Jachiet P-A, Hanage WP, Fitzpatrick D, Lopez P, O’Connell MJ, Pisani D, Wilkinson M, Bapteste E, McInerney JO (2014) A pluralistic account of homology: adapting the models to the data. Molecular Biology and Evolution 31, 501–516.
Hall BK (Ed.) (1994) ‘Homology: the Hierarchical Basis of Comparative Biology.’ (Academic Press: San Diego, CA)
Hall BK (2007) Homology and homoplasy: dichotomy or continuum? Journal of Human Evolution 52, 473–479.
Hawkins JA (2000) A survey of primary homology assessment: different botanists perceive and define characters in different ways. In ‘Homology and Systematics: Coding Characters for Phylogenetic Analysis’. (Eds R Scotland, RT Pennington) pp. 22–53. (Taylor and Francis: London)
Hawkins JA, Hughes CE, Scotland RW (1997) Primary homology assessment, characters and character states. Cladistics 13, 275–283.
Hickson RE, Simon C, Perrey SW (2000) The performance of several multiple-sequence alignment programs in relation to secondary-structure features for an rRNA sequence. Molecular Biology and Evolution 17, 530–539.
Hillis DM (1994) Homology in molecular biology. In ‘Homology: the Hierarchical Basis of Comparative Biology’. (Ed. BK Hall) pp. 339–368. (Academic Press: New York)
Höhl M, Ragan MA (2007) Is multiple-sequence alignment required for accurate inference of phylogeny? Systematic Biology 56, 206–221.
Hoßfeld U, Olsson L (2005) The history of the homology concept and the ‘Phylogenetisches Symposium’. Theory in Biosciences 124, 243–253.
Huntley MA, Clark AG (2007) Evolutionary analysis of amino acid repeats across the genomes of 12 Drosophila species. Molecular Biology and Evolution 24, 2598–2609.
Iantorno S, Gori K, Goldman N, Gil M, Dessimoz C (2014) Who watches the watchmen? An appraisal of benchmarks for multiple sequence alignment. Methods in Molecular Biology 1079, 59–73.
Jardine N (1967) The concept of homology in biology. The British Journal for the Philosophy of Science 18, 125–139.
Jardine N (1969) The observational and theoretical components of homology: a study based on the morphology of the dermal skull-roofs of rhipidistian fishes. Biological Journal of the Linnean Society. Linnean Society of London 1, 327–361.
Johannsen W (1909) ‘Elemente der Exakten Erblichkeitslehre.’ (Gustav Fischer: Jena, Germany)
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution 30, 772–780.
Kelchner SA (2000) The evolution of non-coding chloroplast DNA and its application in plant systematics. Annals of the Missouri Botanical Garden 87, 482–498.
Kelchner SA (2002) Group II introns as phylogenetic tools: structure, function, and evolutionary constraints. American Journal of Botany 89, 1651–1669.
Kelchner SA (2009) Phylogenetic models and model selection for noncoding DNA. Plant Systematics and Evolution 282, 109–126.
Kelchner SA, Clark LG (1997) Molecular evolution and phylogenetic utility of the rpl16 intron in Chusquea and the Bambusoideae (Poaceae). Molecular Phylogenetics and Evolution 8, 385–397.
Kelchner SA, Wendel JF (1996) Hairpins create minute inversions in non-coding regions of chloroplast DNA. Current Genetics 30, 259–262.
Kemena C, Notredame C (2009) Upcoming challenges for multiple sequence alignment methods in the high-throughput era. Bioinformatics 25, 2455–2465.
Kim J, Ma J (2014) PSAR-Align: improving multiple sequence alignment using probabilistic sampling. Bioinformatics 30, 1010–1012.
Kjer KM (1995) Use of rRNA secondary structure in phylogenetic studies to identify homologous positions: an example of alignment and data presentation from the frogs. Molecular Phylogenetics and Evolution 4, 314–330.
Kleisner K (2007) The formation of the theory of homology in biological sciences. Acta Biotheoretica 55, 317–340.
Kramerov DA, Vassetzky NS (2011) Origin and evolution of SINEs in eukaryotic genomes. Heredity 107, 487–495.
Kummerfeld SK, Teichmann SA (2005) Relative rates of gene fusion and fission in multi-domain proteins. Trends in Genetics 21, 25–30.
Lankester ER (1870) On the use of the term homology in modern zoology, and the distinction between homogenetic and homoplastic agreements. Annals and Magazine of Natural History, series 4 6, 34–43.
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948.
Lassmann T, Sonnhammer ELL (2002) Quality assessment of multiple alignment programs. FEBS Letters 529, 126–130.
Lassmann T, Sonnhammer ELL (2005) Automatic assessment of alignment quality. Nucleic Acids Research 33, 7120–7128.
Laubichler MD (2000) Homology in development and the development of the homology concept. American Zoologist 40, 777–788.
Laubichler MD (2014) Homology as a bridge between evolutionary morphology, developmental evolution, and phylogenetic systematics. In ‘The Evolution of Phylogenetic Systematics’. (Ed. A Hamilton) pp. 63–85. (University of California Press: Berkeley, CA)
Legume Phylogeny Working Group (2013) Legume phylogeny and classification in the 21st century: progress, prospects and lessons for other species-rich clades. Taxon 62, 217–248.
Letsch HO, Kück P, Stocsits RR, Misof B (2010) The impact of rRNA secondary structure consideration in alignment and tree reconstruction: simulated data and a case study on the phylogeny of hexapods. Molecular Biology and Evolution 27, 2507–2521.
Li H, Homer N (2010) A survey of sequence alignment algorithms for next-generation sequencing. Briefings in Bioinformatics 11, 473–483.
Liu K, Warnow T (2012) Treelength optimization for phylogeny estimation. PLoS One 7, e33104
Love AC (2007) Functional homology and homology of function: biological concepts and philosophical consequences. Biology & Philosophy 22, 691–708.
Löytynoja A (2012) Alignment methods: strategies, challenges, benchmarking, and comparative overview. In ‘Evolutionary Genomics: Statistical and Computational Methods, Volume 1’. (Ed. M Anisimova) pp. 203–235. (Humana Press: New York)
Löytynoja A, Goldman N (2008) Phylogeny-aware gap placement prevents errors in sequence alignment and evolutionary analysis. Science 320, 1632–1635.
Maas S (2012) Posttranscriptional recoding by RNA editing. Advances in Protein Chemistry and Structural Biology 86, 193–224.
MacLeod N (2008) Understanding morphology in systematic contexts: 3D specimen ordination and 3D specimen recognition. In ‘The New Taxonomy’. (Ed. QD Wheeler) pp. 143–210. (CRC Press: Boca Raton, FL)
Mallo D, de Oliveira Martins L, Posada D (2014) Unsorted homology within locus and species trees. Systematic Biology 63, 988–992.
Margoliash E (1969) Homology: a definition. Science 163, 127
McCune AR, Schimenti JC (2012) Using genetic networks and homology to understand the evolution of phenotypic traits. Current Genomics 13, 74–84.
Medema MH, Takano E, Breitling R (2013) Detecting sequence homology at the gene cluster level with MultiGeneBlast. Molecular Biology and Evolution 30, 1218–1223.
Messer PW, Arndt PF (2007) The majority of recent short DNA insertions in the human genome are tandem duplications. Molecular Biology and Evolution 24, 1190–1197.
Metzler D, Fleissner R (2009) Sequence evolution models for simultaneous alignment and phylogeny construction. In ‘Sequence Alignment: Methods, Models, Concepts, and Strategies’, (Ed. MS Rosenberg) pp. 71–93. (University of California Press: Berkeley, CA)
Meyer A (1999) Homology and homoplasy: the retention of genetic programmes. In ‘Homology’. (Eds GR Bock, G Cardew) pp. 141–157. (Wiley: Chichester, UK)
Mindell DP (1991) Similarity and congruence as criteria for molecular homology. Molecular Biology and Evolution 8, 897–900.
Mindell DP, Meyer A (2001) Homology evolving. Trends in Ecology & Evolution 16, 434–440.
Mishler BD (2005) The logic of the data matrix in phylogenetic analysis. In ‘Parsimony, Phylogeny, and Genomics’. (Ed. VA Albert) pp. 57–70. (Oxford University Press: Oxford, UK)
Moore AD, Bornberg-Bauer E (2012) The dynamics and evolutionary potential of domain loss and emergence. Molecular Biology and Evolution 29, 787–796.
Morgan MJ, Kelchner SA (2010) Inference of molecular homology and sequence alignment by direct optimization. Molecular Phylogenetics and Evolution 56, 305–311.
Morrison DA (2006) Multiple sequence alignment for phylogenetic purposes. Australian Systematic Botany 19, 479–539.
Morrison DA (2009a) A framework for phylogenetic sequence alignment. Plant Systematics and Evolution 282, 127–149.
Morrison DA (2009b) Why would phylogeneticists ignore computerized sequence alignment? Systematic Biology 58, 150–158.
Morrison DA (2015) Is sequence alignment an art or a science? Systematic Botany 40, 14–26.
Morrison DA, Ellis JT (1997) Effects of nucleotide sequence alignment on phylogeny estimation: a case study of 18S rDNAs of Apicomplexa. Molecular Biology and Evolution 14, 428–441.
Mount DM (2004) ‘Bioinformatics: Sequence and Genome Analysis’, 2nd edn. (Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY)
Nelesen S, Liu K, Wang LS, Linder CR, Warnow T (2012) DACTAL: divide-and-conquer trees (almost) without alignments. Bioinformatics 28, i274–i282.
Nielsen C, Martinez P (2003) Patterns of gene expression: homology or homocracy? Development Genes and Evolution 213, 149–154.
Nixon KC, Carpenter JM (2012) On homology. Cladistics 28, 160–169.
Notredame C (2007) Recent evolutions of multiple sequence alignment algorithms. PLoS Computational Biology 3, e123
Nuin PAS, Wang Z, Tillier ERM (2006) The accuracy of several multiple sequence alignment programs for proteins. BMC Bioinformatics 7, 471
Owen R (1843) ‘Lectures on the Comparative Anatomy and Physiology of the Invertebrate Animals’. (Longman, Brown, Green, and Longmans: London)
Pais FS-M, Ruy PC, Oliveira G, Coimbra RS (2014) Assessing the efficiency of multiple sequence alignment programs. Algorithms for Molecular Biology; AMB 9, 4
Patterson C (1982) Morphological characters and homology. In ‘Problems of Phylogenetic Reconstruction’. (Ed. KA Joysey, AE Friday) pp. 21–74. (Academic Press: London)
Patterson C (1988) Homology in classical and molecular biology. Molecular Biology and Evolution 5, 603–625.
Pavlinov IY (2012) The contemporary concepts of homology in biology: a theoretical review. Biology Bulletin Reviews 2, 36–54.
Pei J (2008) Multiple protein sequence alignment. Current Opinion in Structural Biology 18, 382–386.
Pei J, Grishin NV (2007) PROMALS: towards accurate multiple sequence alignments of distantly related proteins. Bioinformatics 23, 802–808.
Pevzner P (2000) ‘Computational Molecular Biology: an Algorithmic Approach.’ (The MIT Press: Cambridge, MA)
Phillips A, Janies D, Wheeler W (2000) Multiple sequence alignment in phylogenetic analysis. Molecular Phylogenetics and Evolution 16, 317–330.
Platnick NJ (1979) Philosophy and the transformation of cladistics. Systematic Zoology 28, 537–546.
Pleijel F (1995) On character coding for phylogeny reconstruction. Cladistics 11, 309–315.
Quandt D, Stech M (2005) Molecular evolution of the trnLUAA intron in bryophytes. Molecular Phylogenetics and Evolution 36, 429–443.
Ray DA, Xing J, Salem A-H, Batzer MA (2006) SINEs of a nearly perfect character. Systematic Biology 55, 928–935.
Redelings BD, Suchard MA (2009) Robust inferences from ambiguous alignments. In ‘Sequence Alignment: Methods, Models, Concepts, and Strategies’. (Ed. MS Rosenberg) pp. 209–270. (University of California Press: Berkeley, CA)
Reeck GR, de Haën C, Teller DC, Doolittle RF, Fitch WM, Dickerson RE, Chambon P, McLachlan AD, Margoliash E, Jukes TH, Zuckerkandl E (1987) ‘Homology’ in proteins and nucleic acids: a terminology muddle and a way out of it. Cell 50, 667
Ren J, Song K, Sun F, Deng M, Reinert G (2013) Multiple alignment-free sequence comparison. Bioinformatics 29, 2690–2698.
Richter S (2005) Homologies in phylogenetic analyses: concept and test. Theory in Biosciences 124, 105–150.
Rieppel OC (1988) ‘Fundamentals of Comparative Biology.’ (Birkhäuser Verlag: Basel, Switzerland)
Rieppel O (2004) The language of systematics, and the philosophy of ‘total evidence’. Systematics and Biodiversity 2, 9–19.
Rieppel O, Kearney M (2002) Similarity. Biological Journal of the Linnean Society. Linnean Society of London 75, 59–82.
Rosenberg MS, Ogden TH (2009) Simulation approaches to evaluating alignment error and methods for comparing alternate alignments. In ‘Sequence Alignment: Methods, Models, Concepts, and Strategies’. (Ed. MS Rosenberg) pp. 179–207. (University of California Press: Berkeley, CA)
Roth VL (1991) Homology and hierarchies: problems solved and unresolved. Journal of Evolutionary Biology 4, 167–194.
Roth FP, Hughes JD, Estep PW, Church GM (1998) Finding DNA regulatory motifs within unaligned noncoding sequences clustered by whole-genome mRNA quantitation. Nature Biotechnology 16, 939–945.
Rutishauser R, Moline P (2005) Evo-devo and the search for homology (‘sameness’) in biological systems. Theory in Biosciences 124, 213–241.
Sahraeian SME, Yoo B-J (2011) PicXAA-R: efficient structural alignment of multiple RNA sequences using a greedy approach. BMC Bioinformatics 12, S38
Sankoff D, Morel C, Cedergren RJ (1973) Evolution of 5S RNA and the non-randomness of base replacement. Nature 245, 232–234.
Scotland R, Pennington RT (Eds) (2000) ‘Homology and Systematics: Coding Characters for Phylogenetic Analysis.’ (Taylor and Francis: London)
Simmons MP (2000) A fundamental problem with amino-acid-sequence characters for phylogenetic analyses. Cladistics 16, 274–282.
Simmons MP, Ochoterena H, Carr TG (2001) Incorporation, relative homoplasy, and effect of gap characters in sequence-based phylogenetic analyses. Systematic Biology 50, 454–462.
Sims GE, Jun S-R, Wu GA, Kim S-H (2009) Alignment-free genome comparison with feature frequency profiles (FFP) and optimal resolutions. Proceedings of the National Academy of Sciences of the United States of America 106, 2677–2682.
Smit S, Knight R, Heringa J (2009) RNA structure prediction from evolutionary patterns of nucleotide composition. Nucleic Acids Research 37, 1378–1386.
Stace CA (2005) Plant taxonomy and biosystematics: does DNA provide all the answers? Taxon 54, 999–1007.
States DJ, Boguski MS (1991) Homology and similarity. In ‘Sequence Analysis Primer’. (Eds M Gribskov, Devereux) pp. 89–157. (Oxford University Press: New York)
Terekhanova NV, Bazykin GA, Neverov A, Kondrashov AS, Seplyarskiy VB (2013) Prevalence of multinucleotide replacements in evolution of primates and Drosophila. Molecular Biology and Evolution 30, 1315–1325.
Thompson JD, Poch O (2005) Sequence alignment. In ‘Encyclopedia of Life Sciences’. (Wiley: New York)
Thornton JW, DeSalle R (2000) Gene family evolution and homology: genomics meets phylogenetics. Annual Review of Genomics and Human Genetics 1, 41–73.
Vinga S, Almeida J (2003) Alignment-free sequence comparison: a review. Bioinformatics 19, 513–523.
Wagner GP (1989) The biological homology concept. Annual Review of Ecology and Systematics 20, 51–69.
Wagner GP (2014) ‘Homology, Genes, and Evolutionary Innovation.’ (Princeton University Press: Princeton, NJ)
Went FW (1971) Parallel evolution. Taxon 20, 1–26.
Wheeler WC (2003) Implied alignment: a synapomorphy-based multiple-sequence alignment method and its use in cladogram search. Cladistics 19, 261–268.
Wheeler WC, Lucaroni N, Hong L, Crowley LM, Varón A (2015) POY version 5: phylogenetic analysis using dynamic homologies under multiple optimality criteria. Cladistics 31, 189–196.
Wilke C (2012) Bringing molecules back into molecular evolution. PLoS Computational Biology 8, e1002572
Wilkinson M (1995) A comparison of two methods of character construction. Cladistics 11, 297–308.
Wilm A, Mainz I, Steger G (2006) An enhanced RNA alignment benchmark for sequence alignment programs. Algorithms for Molecular Biology; AMB 1, 19
Wong KM, Suchard MA, Huelsenbeck JP (2008) Alignment uncertainty and genomic analysis. Science 25, 473–476.
Wrabl JO, Grishin NV (2004) Gaps in structurally similar proteins: towards improvement of multiple sequence alignment. Proteins 54, 71–87.
Wray GA, Abouheif E (1998) When is homology not homology? Current Opinion in Genetics & Development 8, 675–680.
Wuyts J, Van de Peer Y, De Wachter R (2001) Distribution of substitution rates and location of insertion sites in the tertiary structure of ribosomal RNA. Nucleic Acids Research 29, 5017–5028.
Xiao L, Sulaiman IM, Ryan UM, Zhou L, Atwill ER, Tischler ML, Zhang X, Fayer R, Lal AA (2002) Host adaptation and host-parasite co-evolution in Cryptosporidium: implications for taxonomy and public health. International Journal for Parasitology 32, 1773–1785.
Yue F, Shi J, Tang J (2009) Simultaneous phylogeny reconstruction and multiple sequence alignment. BMC Bioinformatics 10, S11