

Integration of genomic information into beef cattle and sheep genetic evaluations in Australia
Andrew A. Swan A B D , David J. Johnston A C , Daniel J. Brown A B , Bruce Tier A C and Hans-U. Graser A CA Animal Genetics and Breeding Unit (AGBU1), University of New England, Armidale, NSW 2351, Australia.
B The CRC for Sheep Industry Innovation, University of New England, Armidale, NSW 2351, Australia.
C The CRC for Beef Genetics Technology, University of New England, Armidale, NSW 2351, Australia.
D Corresponding author. Email: andrew.swan@une.edu.au
Animal Production Science 52(3) 126-132 https://doi.org/10.1071/AN11117
Submitted: 20 June 2011 Accepted: 1 November 2011 Published: 19 December 2011
Journal Compilation © CSIRO Publishing 2012 Open Access CC BY-NC-ND
Abstract
Genomic information has the potential to change the way beef cattle and sheep are selected and to substantially increase genetic gains. Ideally, genomic data will be used in combination with pedigree and phenotypic data to increase the accuracy of estimated breeding values (EBVs) and selection indexes. The first example of this in Australia was the integration of four markers for tenderness into beef cattle breeding values. Subsequently, the availability of high-density single nucleotide polymorphism (SNP) panels has made selection using genomic information possible, while at the same time creating significant challenges for genetic evaluation with regard to both data management and statistical modelling. Reference populations have been established in both the beef cattle and sheep industries, in which an extensive range of phenotypes have been collected and animals genotyped mainly using 50K SNP panels. From this information, genomic predictions of breeding value have been developed, albeit with varying levels of accuracy. These predictions have been incorporated into routine genetic evaluations using three approaches and trial results are now available to breeders. In the first, genomic predictions have been included in genetic evaluation models as additional traits. The challenges with this method have been the construction of consistent genetic covariance matrices, and a significant increase in computing time. The second approach has been to use a selection index procedure to blend genomic predictions with existing EBVs. This method has been shown to produce very similar results, and has the advantage of being simple to implement and fast to operate, although consistent genetic covariance matrices are still required. Third, in sheep a single-step analysis combining a genomic relationship matrix with a standard pedigree-based relationship matrix has been used to estimate breeding values for carcass and eating-quality traits. It is likely that this procedure or one similar will be incorporated into routine evaluations in the near future. While significant progress has been made in implementing methods of integrating genomic information in both beef and sheep evaluations in Australia, the major challenges for the future will be to continue to collect the phenotypes needed to derive accurate genomic predictions, and in managing much larger volumes of genomic data as the number of animals genotyped and the density of markers increase.
Introduction
Selection in the Australian beef cattle and sheep breeding sectors is often based on selection indexes for overall economic merit, and although significant genetic progress in mean index values has been reported by Barwick and Henzell (2005) in cattle and Swan et al. (2009) in sheep, increasing the rate of progress is essential for both industries to increase productivity and improve their competitive advantage. Genomic information offers the possibility of increasing the rate of progress by increasing selection accuracy at an earlier age (e.g. Sise and Amer 2009; Van Der Werf 2009; Van Eenennaam et al. 2011) and in the context of selection for economic merit, the potential impact of genomic information is greatest for breeding objective traits that are difficult or costly to measure. In the present paper, we describe how genomic information is currently being used in Australian beef cattle and sheep genetic evaluation systems to increase the accuracy of EBVs and selection indexes.
Genetic evaluation systems for beef cattle and sheep in Australia
Large industry wide genetic evaluations have been in place since the 1980s for beef cattle through BREEDPLAN (Graser et al. 2005) and since the 1990s for sheep through LAMBPLAN and MERINOSELECT (Brown et al. 2007). In beef cattle, separate evaluations are organised within a large number of breeds by breed societies using their databases that are maintained by the Agricultural Business Research Institute (ABRI) and use the BREEDPLAN evaluation software licenced to ABRI. Included are several temperate breeds that often combine data from Australian and New Zealand herds, and the tropically adapted breeds typically used in the north of Australia. These databases grow at a rate exceeding 130 000 calves per year from Australian herds.
The traits most commonly measured are early growth and live-animal ultrasound scanning of carcass traits. The traits influencing economic merit that will benefit most from genomic information are carcass traits, feed efficiency and female reproduction. The latter is particularly important in the Bos indicus-dominant Northern beef herd.
Sheep evaluations are run by Sheep Genetics, a business unit of Meat and Livestock Australia that maintains databases supporting two delivery systems, namely MERINOSELECT for Merino sheep and LAMBPLAN for terminal and maternal sire breeds. These are large systems, growing at a rate of 75 000–95 000 new animals per year for Merinos, 90 000–110 000 for terminal sire breeds and 40 000–45 000 for maternal sire breeds. The analyses are conducted by Animal Genetics and Breeding Unit using the OVIS evaluation software (Brown et al. 2007).
The terminal sire evaluation is across breed, with the dominant breeds being Poll Dorset, White Suffolk, Suffolk and Texel. Maternal sire evaluations are currently conducted separately but are moving to an across-breed system in which the dominant breeds will be Border Leicester and Coopworth. All of these populations can be considered multi-breed to varying degrees, with significant levels of crossbreeding identifiable. This has implications for genomic evaluation.
The traits most commonly measured in sheep include bodyweight and ultrasound scanning of muscle and fat on live animals (all breeds), and wool weight and quality (Merinos). These traits are recorded on young animals before selection age and therefore have breeding values of moderate to high accuracy. Genomic information will have little impact on these traits. There are several hard-to-measure traits where current accuracies of breeding values are low (and in some cases zero), including adult wool production, parasite resistance, reproduction, and carcass and eating-quality traits. Increasing the accuracies of these traits by using genomic information is where the largest gains in genetic progress can be made.
The development of genomic resources
Tests for individual gene markers for both disease and production traits have been commercially available in both industries for over a decade. Johnston and Graser (2010) showed the limitations of such tests in beef cattle, with the size and direction (phase) of effects needing to be established in each target population.
The use of individual marker tests has largely been superceded by the availability of high density SNP marker panels for both cattle and sheep. Use of these SNP panels makes genomic selection possible (Meuwissen et al. 2001), in which estimation of individual marker and quantitative trait loci (QTL) effects is not so important. SNP panels with 10K and then 50K markers have been used to genotype large numbers of animals in Beef CRC resource herds (Bindon 2001; Johnston et al. 2003), while a 50K panel has been used extensively in two sheep-resource flocks, namely the Sheep Genomics Project flock (Oddy et al. 2007) and the Sheep CRC Information Nucleus (Fogarty et al. 2007). Of the order of 8000 cattle and 10 000 sheep have been genotyped in these reference populations, and phenotypes have been recorded for a very wide range of traits, including those that are difficult or costly to measure. While the number of genotypes is increasing quite rapidly, collecting phenotypes can take much longer, particularly for sex-limited and later-age traits such as female reproduction and adult wool production.
The reference populations have been used to develop genomic predictions of genetic merit (genomic estimated breeding values, or GEBV), and to determine the accuracy of these predictions using data from validation populations comprising genotyped animals from outside the reference (although they may have close pedigree relationships with the reference population). The Beef CRC is generating validation populations, one of which includes 1450 BREEDPLAN sires across eight breeds, mostly genotyped with the 50K panel, but with a subset genotyped with the high-density 800K panel. In sheep, a validation population of 460 rams with highly accurate EBVs has been genotyped, and the correlation between GEBVs and EBVs for these sires has been used as an estimate of accuracy of genomic prediction. While this procedure can be used for traits that are in the genetic evaluation system, most of the difficult-to-measure traits of interest do not have EBVs and for these the accuracy of genomic prediction is determined by cross-validation, dividing the reference population into subsets to first develop the genomic prediction and then to determine its accuracy. This limits the accuracies achievable for these traits because fewer animals can be used to develop the prediction.
Work on the Beef CRC data is ongoing, but in a validation of commercially available GEBVs produced by Pfizer Animal Genetics for Australian Angus bulls, accuracies for growth and carcass traits were between 0.20 and 0.45, for calving ease between 0.21 and 0.24, and for feed intake between 0.01 and 0.22 (Johnston et al. 2010). Sheep CRC research has shown accuracies from 0.15 to 0.79 for wool traits in Merinos, with fleece weight and fibre diameter having accuracies >0.70, and from –0.07 to 0.57 for meat traits, depending on the breed (Daetwyler et al. 2010). These results show that accuracies are approaching levels that are promising for genomic selection in both species, but mainly for traits that are easy to measure. Hard-to-measure traits tend to have lower accuracies, through lack of phenotypes and the need to split reference populations for cross-validation, as discussed above. This situation will improve over time as the number of phenotypes increases.
As described above, a diverse range of breeds is used in both the beef cattle and sheep industries in Australia, so the ability to predict GEBVs across breeds is important to maximise return on investment. However, low accuracies have been observed when predicting across breeds. The current hope is that higher-density SNP panels will improve this situation, although it is not clear that this will be the case. With the recent availability of an 800K panel for cattle, key animals can be genotyped at the higher density and existing 50K genotypes can be imputed with high accuracy (Goddard and Hayes 2009). Higher-density information, including full sequence data, will become more common in both industries as it becomes less expensive.
One important difference between the two industries in Australia is that in beef, SNP panel genotyping of seedstock animals is being offered as a commercial product by private companies, but this has not happened yet in sheep. So far, in beef, the information has been returned to breeders as GEBVs, trademarked for example as Pfizer Molecular Value Predictions (MVPs), with the actual genotypes not available (at the time of writing MVPs were available only for the Angus breed). However, genotypes are available from research datasets such as the Beef CRC. Genetic evaluation models need the capacity to handle both types of data.
By contrast, in sheep, genotyping of seedstock animals has been partially funded by public research and development organisations that have also been responsible for the reference and validation resources described above. In 2010–2011, a pilot project was established in which breeders were invited to submit DNA samples on young rams. The aim of the project was to investigate the issues associated with running an efficient processing pipeline, including on-farm sampling, DNA extraction, genotyping, quality control, database storage and genetic evaluation. A second and larger-scale project sampling of more animals is planned for 2011–2012. In the early stages of the use of genomic information, the real benefit of the approach adopted by the sheep industry is that the complete SNP genotypes from all resources, reference, validation and pilot project have been combined in a single database. This has been a major advantage for the development of genetic evaluation methods.
While the volume of genomic information is rapidly expanding in both of these industries, it is clear that even as SNP genotyping density increases, there is an ongoing need for reference populations to increase the number of phenotypes on difficult to measure traits, and to cope with the likely decline in accuracy of genomic predictions over generations (Habier et al. 2010). With the Beef and Sheep CRC populations either no longer extant or coming to the end of their funding, both industries are exploring the possibility of establishing new information nucleus resources to fulfil this role.
Genetic evaluation methodology
Genomic selection has the potential to increase the rate of genetic gain by increasing the accuracy of EBVs at earlier ages by combining genomic information with pedigree and performance data in the genetic evaluation system. In the Australian beef cattle and sheep industries, these evaluations are large multi-trait animal-model BLUP systems (Graser et al. 2005; Brown et al. 2007). In this section we describe how genomic information has been included in these systems, either by merging GEBVs with other information during or after analysis, or by including genomic information directly in the analysis. These methods apply to the estimation of breeding values and accuracies, and once the necessary procedures have been implemented in evaluation models, they flow directly through to selection indexes such that calculation of indexes is unchanged.
Calculation of genomic predictions and their accuracies
For the first two methods, the preliminary step is to estimate GEBVs and the accuracy with which they predict the true breeding value (TBV). Several statistical models can be used to develop the necessary prediction equations (e.g. Moser et al. 2009) and most give similar results. GEBVs for sheep have been estimated using the so called ‘GBLUP’ method, in which a genomic relationship matrix (VanRaden 2008) is used in place of the usual pedigree-based relationship matrix in the BLUP mixed model equations. These analyses included data from the reference population, with GEBVs estimated for the animals of interest, such as validation sires or young rams, via their genomic relationships with the phenotyped reference animals (Daetwyler et al. 2010). While initial work on estimating GEBVs from the Beef CRC resources has commenced (e.g. Zhang et al. 2010) and is ongoing, GEBVs for beef cattle have been estimated by private companies as described above. Accuracies of GEBVs as predictors of TBV have been calculated in independent data sets, genotyping validation sires with high-accuracy EBVs. The correlation between their GEBVs and EBVs can be used as a measure of accuracy (Daetwyler et al. 2010).
Inclusion of GEBVs as additional traits
The first method tested was to add GEBVs to genetic evaluation models as additional traits (direct inclusion of GEBVs). With this method, modifications to the analysis software are minimal. The challenge has been to extend covariance matrices to accommodate the GEBV traits. Assuming heritabilities close to one, small values are used for residual variances, and residual covariances between traits are set to zero. In the genetic covariance matrix, GEBV variances can be set to r2σa2 where r is the accuracy of the GEBV as a predictor of TBV, and σa2 is the genetic variance of the target trait. Under this assumption, the covariance between the GEBV and the target trait is also r2σa2, given that the accuracy is the correlation between the GEBV and TBV. In practice, the covariances between GEBVs and all other traits can be estimated from data as described below.
The first application was the inclusion of a GEBV based on four markers in an evaluation to estimate breeding values for shear force of meat in Brahman cattle (Johnston et al. 2009). The model included three traits, including phenotypes for shear force and flight time, in addition to the GEBV. After establishing the covariances between the GEBV and other traits, implementation was straightforward.
A full-scale implementation of the approach was made in the Sheep Genetics Merino and terminal sire genetic evaluations with ~1.2 and 1.5 million animals, respectively, and 45 ‘phenotypic’ traits (i.e. the standard measured traits). In the Merino evaluation, GEBVs were available for eight of these phenotypic traits, for 195 validation sires and 79 pilot project rams. Accuracies had been estimated as simple correlations between GEBVs and EBVs of validation sires but were re-estimated in a series of bivariate REML analyses using GEBVs for validation sires as the first trait and the sires’ progeny records for the target traits extracted from the genetic evaluation databases as the second trait. Genetic correlations estimated from these models tended to be lower than the equivalent accuracies from the simple analysis. Genetic correlations between GEBV traits and all other phenotypic traits were also estimated using this method, allowing construction of a genetic covariance matrix for all traits. The same process was used to develop a model for the terminal sire evaluation, with GEBVs available for three traits on 331 validation sires and 153 pilot rams.
These models were run successfully, with the results in line with expectation in terms of variances of the genomically enhanced EBVs, correlations with original EBVs, and changes in accuracy. However, time taken to solve the equation system using the preconditioned conjugate gradient method (Tsuruta et al. 2001) increased by 7–10 times. This meant that it was impractical to use this approach in the routine evaluation system.
The same approach was used to include GEBVs (Pfizer MVPs) in the full BREEDPLAN analysis for the Angus breed, but it was possible to generate results only by including a single GEBV at a time, because convergence was again very slow using the standard preconditioned conjugate gradient equation solver. Research to overcome this problem by modifying the BREEDPLAN and OVIS solvers is required before this approach can be implemented.
Post-analysis combining of GEBVs and EBVs
Given the current impracticality of including GEBVs directly in the genetic evaluation in the current routinely used software for beef and sheep, post-analysis combining or ‘blending’ as described by Hayes et al. (2009) and Harris and Johnson (2010) was tested as an alternative. This is a selection-index approach which requires deregression of EBVs and GEBVs by their accuracies. The formula used was
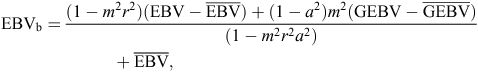
where EBVb is the blended EBV, r is the accuracy of the GEBV as a predictor of TBV as defined above, a is the accuracy of the EBV, m2 is the heritability of the GEBV, and 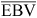 and
and 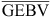 are the means of EBVs and GEBVs for animals with GEBVs. The accuracy of the EBVb is
are the means of EBVs and GEBVs for animals with GEBVs. The accuracy of the EBVb is
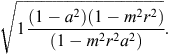
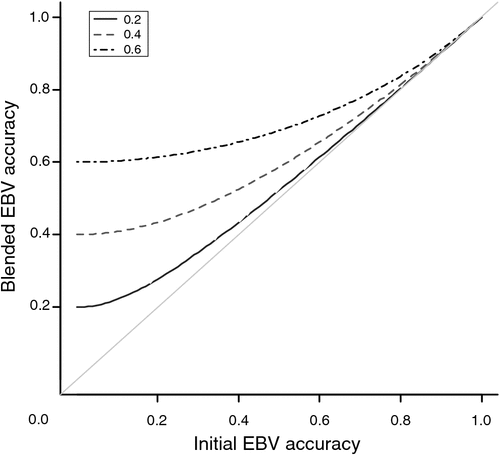
The relationship between the accuracy of blended breeding values and the accuracy of initial EBVs is shown in Fig. 1, demonstrating that the genomic information contributes more when the accuracy of EBVs is low.
|
Blending was implemented in sheep as a single-trait procedure combining EBVs with GEBVs for each target trait separately, for animals with GEBVs. Accuracies of EBVs for individual animals were taken from the routine run, while a single overall figure for accuracy of GEBVs was used for each trait (rather than using accuracies of GEBVs calculated for individual animals). Estimation of this accuracy was as described above. There was a close agreement between the EBVs and accuracies obtained from the blending and direct-inclusion approaches, with correlations between EBVs >0.94. Given this finding, it was decided to use blending to generate results in the pilot project, and these blended EBVs have been delivered to the breeders involved. A summary of the variation observed and accuracies for Merino rams from the pilot project is shown in Table 1. While an increase in accuracy is evident, this set of rams already had moderate to high accuracies, even without genomic information. Results for terminal sires are shown in Table 2. There was no increase in accuracy for bodyweight traits and a small increase in accuracy for eye muscle and fat depth, because of the low accuracies of GEBVs for terminal sire breeds.

|

|
This form of blending has also been implemented in beef for the Angus BREEDPLAN analysis, blending GEBVs (Pfizer MVPs) with BREEDPLAN EBVs only for animals with MVPs. As with sheep, blending is conducted for each trait separately, although traits without GEBVs can be blended with GEBVs for highly correlated traits. Examples of this are mature cow weight, where the EBV is blended with GEBVs for birthweight, weaning weight and carcass weight, and carcass rump fat depth where the EBV is blended with the GEBV for carcass rib fat depth. Results are shown in Table 3. Again, the impact of genomic information is limited because the animals tested had high initial accuracies and the accuracies of GEBVs were low.

|
A trend evident in Tables 1–3 is that the standard deviation of the blended EBVs is not always greater than the standard deviation of the initial EBVs. This is to some extent, unexpected, but could be because GEBVs have limited impact because of the high initial accuracy of EBVs, as discussed above. This requires further investigation once more data become available.
Single-step method
Ideally, all sources of information, pedigree, performance and genomic should be combined in a single analysis. This would accommodate animals with different levels of information; e.g. some animals may only have genotype, while others may have progeny-test records, and there are often pedigree links between different classes of animals. A single-step method which uses all information has been developed by Misztal et al. (2009) and Aguilar et al. (2010) and applied by Forni et al. (2011). The method involves replacing the pedigree-based inverse-relationship matrix used in the mixed-model equations with an inverse matrix which is the same for ungenotyped animals, but with the submatrix for genotyped animals replaced with G–1 – A22–1, where G is a genomic relationship matrix (VanRaden 2008) for genotyped animals and A22 is the pedigree-based relationship matrix for genotyped animals only.
The single-step method has been applied to six carcass and eating-quality traits in sheep. These traits have not been part of the routine genetic evaluations but have been recorded in the Sheep CRC information nucleus and some other historical research flocks which have used industry-recorded sires. The goal of the analysis was to estimate breeding values for sires used in the information nucleus and the young rams from the pilot project, such that they could be compared. The number of phenotypes ranged from 3500 to 6700, with 45–74% of these animals genotyped. EBVs and accuracies for the sires were similar to those from an analysis using the standard model based on pedigree and performance data only. The benefits of the single-step method were apparent in the young rams which only have genotypes (and varying levels of pedigree information). In these animals, accuracies increased by 0.14–0.24, compared with the standard model. However, these accuracies were higher than corresponding accuracies derived from validation analyses (H. D. Daetwyler, pers. comm.) and it is likely that they were overestimated because of the multi-breed nature of the data. This is an area requiring further research.
Implementation issues
The development of accurate genomic predictions requires large numbers of animals. Although our reference populations appear large (of the order of 10 000 for both cattle and sheep), they are made up of numbers of subpopulations based on breed, and in sheep, on cross-bred animals as well. Prediction equations that work across breed can be developed only with data from those breeds, and therefore, reference populations need to be representative of the major breeds of interest, while at the same time covering within breed diversity.
Furthermore, it is becoming apparent (Clark et al. 2011) that the accuracy of genomic predictions varies with the genetic distance of the predicted individual from the reference population. Considerably greater understanding of the relationship between genomic predictions and the TBVs is required if we are to use the genomic predictions most efficiently. Whatever the case, it is evident that reference populations need to include animals representing all breeds of interest for prediction, and that these animals are as highly related as possible to the selection candidates in the wider population.
The development of genomic predictions requires separate subpopulations for reference, validation and, to a certain extent, genetic evaluation. This has been a challenge for the beef and sheep industries in Australia. For example, commercially available GEBVs in beef cattle have been developed using genotypes from sires with high-accuracy BREEDPLAN EBVs. In establishing the accuracy of these predictions, Johnston et al. (2010) restricted validation analyses to data on grand progeny because these would have less influence on the sires’ EBVs than would progeny and own performance data. The drawbacks with this approach are that the reference and validation populations are often not strictly independent, and that there can be little data available for validation, especially for hard-to-measure traits. It is sometimes possible to exclude the phenotypes from the reference population when estimating EBVs for validation purposes, but obtaining sufficient data for accurate predictions remains a challenge.
In sheep the Information Nucleus, flock has been used to develop genomic predictions, but is also included in the routine genetic evaluations because one of the aims of the flock was to provide progeny test information on the sires used. Therefore, with the direct inclusion and blending of the methods, the reference data are included twice, first, in the form of phenotypic records, and second, through GEBVs.
The single-step method avoids this double counting issue, because genomic information is included directly in the model, without the need to establish accuracy. As shown by Aguilar et al. (2010), the method can be applied to large datasets with millions of animals phenotyped and tens of thousands of animals genotyped. It is likely, that a method of this type will be implemented in Australian beef cattle and sheep genetic evaluations in the near future. However, a full implementation in beef will require the genotypes of commercially tested animals to be made available.
A major practical challenge for including genomic information in genetic evaluations is to develop scalable data-management systems. Research and commercial interests are generating increasing numbers of genotypes with increasing density of SNPs, up to and including full sequence data. In addition, it will be necessary to cater for the situation where the genomic information consists of third-party GEBVs, potentially with multiple GEBVs for the same trait. Genetic covariance matrices need to be extended to include these GEBVs, and the challenge of performing these validation analyses should not be underestimated.
Conclusions
Genomic information is becoming available on an increasing number of young animals and has been succesfully incorporated into Australian beef cattle and sheep genetic evaluations, with breeders receiving the first genomically enhanced trial EBVs in late 2010. These enhanced EBVs are being produced using a blending approach, which has been shown to give reliable results. In the longer term, a single-step method will be implemented to include genotypes directly in the evaluation model where they are available, with third-party GEBVs included as additional traits where they are not.
There is an ongoing need in both industries to continue investment to maintain reference and validation populations that have a wide range of relevant phenotypic traits recorded to develop the accurate genomic predictions required to underpin genomic selection.
Acknowledgements
This work was supported by MLA Projects B.SGN.0127 and B.BFG.0050, the CRC for Sheep Industry Innovation and the CRC for Beef Genetics Technology.
References
Aguilar I, Misztal I, Johnson DL, Legarra A, Tsuruta S, Lawlor TJ (2010) A unified approach to utilize phenotypic, full pedigree, and genomic information for genetic evaluation of Holstein final score. Journal of Dairy Science 93, 743–752.| A unified approach to utilize phenotypic, full pedigree, and genomic information for genetic evaluation of Holstein final score.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXht1CjtbY%3D&md5=75eb50e2e10bf343c8ad2c5494204ac9CAS |
Barwick SA, Henzell AL (2005) Development successes and issues for the future in deriving and applying selection indexes for beef breeding. Australian Journal of Experimental Agriculture 45, 923–933.
| Development successes and issues for the future in deriving and applying selection indexes for beef breeding.Crossref | GoogleScholarGoogle Scholar |
Bindon BM (2001) Genesis of the Cooperative Research Centre for the Cattle and Beef Industry: integration of resources for beef quality research (1993–2000). Australian Journal of Experimental Agriculture 41, 843–853.
| Genesis of the Cooperative Research Centre for the Cattle and Beef Industry: integration of resources for beef quality research (1993–2000).Crossref | GoogleScholarGoogle Scholar |
Brown DJ, Huisman AE, Swan AA, Graser H-U, Woolaston RR, Ball AJ, Atkins KD, Banks RB (2007) Genetic evaluation for the Australian sheep industry. Proceedings of the Association for the Advancement of Animal Breeding and Genetics 17, 187–194.
Clark SA, Hickey JM, Van Der Werf JHJ (2011) The relative importance of information on unrelated individuals on the prediction of genomic breeding values. Proceedings of the Association for the Advancement of Animal Breeding and Genetics 19, 291–294.
Daetwyler HD, Hickey JM, Henshall JM, Dominik S, Gredler B, Van Der Werf JHJ, Hayes BJ (2010) Accuracy of estimated genomic breeding values for wool and meat traits in a multi-breed sheep population. Animal Production Science 50, 1004–1010.
| Accuracy of estimated genomic breeding values for wool and meat traits in a multi-breed sheep population.Crossref | GoogleScholarGoogle Scholar |
Fogarty NM, Banks RG, Van Der Werf JHJ, Ball AJ, Gibson JP (2007) The Information Nucleus – a new concept to enhance sheep industry genetic improvement. Proceedings of the Association for the Advancement of Animal Breeding and Genetics 17, 29–32.
Forni S, Aguilar I, Misztal I (2011) Different genomic relationship matrices for single-step analysis using phenotypic, pedigree and genomic information. Genetics, Selection, Evolution 43, 1
| Different genomic relationship matrices for single-step analysis using phenotypic, pedigree and genomic information.Crossref | GoogleScholarGoogle Scholar |
Goddard ME, Hayes BJ (2009) Genomic selection based on dense genotypes inferred from sparse genotypes. Proceedings of the Association for the Advancement of Animal Breeding and Genetics 18, 26–29.
Graser H-U, Tier B, Johnston DJ, Barwick SA (2005) Genetic evaluation for the beef industry in Australia. Australian Journal of Experimental Agriculture 45, 913–921.
| Genetic evaluation for the beef industry in Australia.Crossref | GoogleScholarGoogle Scholar |
Habier D, Tetens J, Seefried F-R, Lichtner P, And Thaller G (2010) The impact of genetic relationship information on genomic breeding values in German Holstein cattle. Genetics, Selection, Evolution 42, 5
| The impact of genetic relationship information on genomic breeding values in German Holstein cattle.Crossref | GoogleScholarGoogle Scholar |
Harris BL, Johnson DL (2010) Genomic predictions for New Zealand dairy bulls and integration with national genetic evaluation. Journal of Dairy Science 93, 1243–1252.
| Genomic predictions for New Zealand dairy bulls and integration with national genetic evaluation.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXitlOis78%3D&md5=675a2330d2e443ef6654d212df0cf0cfCAS |
Hayes BJ, Bowman PJ, Chamberlain AJ, Goddard ME (2009) Genomic selection in dairy cattle: progress and challenges. Journal of Dairy Science 92, 433–443.
| Genomic selection in dairy cattle: progress and challenges.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXit1Kju7s%3D&md5=ecfb717f26024e66360ffb500acb8d77CAS |
Johnston DJ, Graser H-U (2010) Estimated gene frequencies of GeneSTAR markers and their size of effects on meat tenderness, marbling, and feed efficiency in temperate and tropical beef cattle breeds across a range of production systems. Journal of Animal Science 88, 1917–1935.
| Estimated gene frequencies of GeneSTAR markers and their size of effects on meat tenderness, marbling, and feed efficiency in temperate and tropical beef cattle breeds across a range of production systems.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXmvVeqtrk%3D&md5=a9ebca6185b6c4ca4873e5623f955dd9CAS |
Johnston DJ, Reverter A, Burrow HM, Oddy VH, Robinson DL (2003) Genetic and phenotypic characterization of animal, carcass, and meat quality traits from temperate and tropically adapted beef breeds. 1. Animal measures. Australian Journal of Agricultural Research 54, 107–118.
| Genetic and phenotypic characterization of animal, carcass, and meat quality traits from temperate and tropically adapted beef breeds. 1. Animal measures.Crossref | GoogleScholarGoogle Scholar |
Johnston DJ, Tier B, Graser H-U (2009) Integration of DNA markers into BREEDPLAN EBVs. Proceedings of the Association for the Advancement of Animal Breeding and Genetics 18, 30–33.
Johnston DJ, Jeyaruban MG, Graser H-U (2010) Evaluation of Pfizer Animal Genetics HD 50K MVP calibration. AGBU Report November 2010. Available at http://agbu.une.edu.au/pdf/Pfizer_50K_September%202010.pdf [Verified 8 June 2011].
Meuwissen THE, Hayes BJ, Goddard ME (2001) Prediction of total genetic value using genome wide dense marker maps. Genetics 157, 1819–1829.
Misztal I, Legarra A, Aguilar I (2009) Computing procedures for genetic evaluation including phenotypic, full pedigree, and genomic information. Journal of Dairy Science 92, 4648–4655.
| Computing procedures for genetic evaluation including phenotypic, full pedigree, and genomic information.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtVKqtr3L&md5=120d7ea7af28a78389e5738c28aed4a4CAS |
Moser G, Tier B, Crump RE, Khatkar MS, Raadsma HW (2009) A comparison of five methods to predict genomic breeding values of dairy bulls from genome-wide SNP markers. Genetics, Selection, Evolution 41, 56
| A comparison of five methods to predict genomic breeding values of dairy bulls from genome-wide SNP markers.Crossref | GoogleScholarGoogle Scholar |
Oddy H, Dalrymple B, McEwan J, Kijas J, Haye B, van der Werf J, Emery D, Hynd P, Longhurst T, Fischer T, Ferguson D, Forage R, Cockett N, Nicholas F (2007) SheepGenomics and the International Sheep Genomics Consortium. Proceedings of the Association for the Advancement of Animal Breeding and Genetics 17, 411–417.
Sise JA, Amer PR (2009) SNP predictors to accelerate the rate of genetic progress in sheep. Proceedings of the Association for the Advancement of Animal Breeding and Genetics 18, 220–223.
Swan AA, Brown DJ, Banks RG (2009) Genetic progress in the Australian sheep industry. Proceedings of the Association for the Advancement of Animal Breeding and Genetics 18, 326–329.
Tsuruta S, Misztal I, Stranden I (2001) Use of the preconditioned conjugate gradeitn algorithm as a generic solver for mixed model equations in animal breeding applications. Journal of Animal Science 79, 1166–1172.
Van Der Werf JHJ (2009) Potential benefit of genomic selection in sheep. Proceedings of the Association for the Advancement of Animal Breeding and Genetics 18, 38–41.
Van Eenennaam AL, Van Der Werf JHJ, Goddard ME (2011) The value of using DNA markers for beef bull selection in the seedstock sector. Journal of Animal Science 89, 307–320.
| The value of using DNA markers for beef bull selection in the seedstock sector.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhvFSntb0%3D&md5=ce326c56a61e14687834889f4f8c3e53CAS |
VanRaden PM (2008) Efficient methods to compute genomic predictions. Journal of Dairy Science 91, 4414–4423.
| Efficient methods to compute genomic predictions.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtlajtLzO&md5=61b981a556c06d80137ef0165ddd4ae2CAS |
Zhang YD Tier B Hawken RJ (2010 ) Whole genome analysis of heifer puberty in Brahman cattle. Proceedings of the 9th World Congress on Genetics Applied to Livestock Production Communication 761 .
1AGBU is a joint venture of NSW Department of Primary Industries and the University of New England.