

Living Radical Polymerization by the RAFT Process – A Second Update
Graeme Moad A B , Ezio Rizzardo A B and San H. Thang A BA CSIRO Molecular and Health Technologies, Bag 10, Clayton South, Victoria 3169, Australia.
B Corresponding authors. Email: graeme.moad@csiro.au; ezio.rizzardo@csiro.au; san.thang@csiro.au

Graeme Moad obtained his B.Sc. (Hons) and Ph.D. from the Adelaide University in the field of organic free radical chemistry. After undertaking post-doctoral research at Pennsylvania State University in the field of biological organic chemistry he joined CSIRO in 1979 and is currently a chief research scientist and a research team leader. Dr Moad is author or co-author of 147 papers in refereed journals, co-inventor of 32 patent families (eight relate to the RAFT process) and co-author of the book ‘The Chemistry of Radical Polymerization’. His papers have been cited more than 9000 times and his h-index is 46. His research interests lie in the fields of polymer design and synthesis (radical polymerization, reactive extrusion, polymer nanocomposites) and polymerization kinetics and mechanism. |

Ezio Rizzardo is a graduate of the University of New South Wales and he received his Ph.D. from the University of Sydney for his studies on the photochemistry of organic nitro compounds. He joined CSIRO in 1976 after postdoctoral research on the synthesis of biologically active organic compounds at Rice University, RIMAC, and the Australian National University. His CSIRO research has focussed on developing methods for controlling free radical polymerization. For this he has received a number of awards including the RACI Australian Polymer Medal and the CSIRO Chairman’s Gold Medal. Ezio is a CSIRO Fellow and a Fellow of the Australian Academy of Science. |

San H. Thang completed his B.Sc. (Hons) in 1983 and Ph.D. in 1987 both from Griffith University. He joined CSIRO in 1986 as a Research Fellow, and then moved to ICIAustralia Research Group in December 1987 to undertake the challenge of industrial research. He returned to CSIRO in late 1990 and currently is a Senior Principal Research Scientist at Molecular and Health Technologies where his research focuses primarily on the interface between organic and polymer chemistry. San is a leading expert in the field of free radical chemistry and polymer chemistry. He is a co-inventor of the living radical polymerization by reversible addition–fragmentation chain transfer (RAFT process). |
Australian Journal of Chemistry 62(11) 1402-1472 https://doi.org/10.1071/CH09311
Submitted: 29 May 2009 Accepted: 4 August 2009 Published: 20 November 2009
Abstract
This paper provides a second update to the review of reversible deactivation radical polymerization achieved with thiocarbonylthio compounds (ZC(=S)SR) by a mechanism of reversible addition–fragmentation chain transfer (RAFT) that was published in June 2005 (Aust. J. Chem. 2005, 58, 379–410). The first update was published in November 2006 (Aust. J. Chem. 2006, 59, 669–692). This review cites over 500 papers that appeared during the period mid-2006 to mid-2009 covering various aspects of RAFT polymerization ranging from reagent synthesis and properties, kinetics and mechanism of polymerization, novel polymer syntheses and a diverse range of applications. Significant developments have occurred, particularly in the areas of novel RAFT agents, techniques for end-group removal and transformation, the production of micro/nanoparticles and modified surfaces, and biopolymer conjugates both for therapeutic and diagnostic applications.
Introduction
Radical polymerization is one of the most widely used processes for the commercial production of high-molecular-weight polymers.[1] The emergence of techniques for implementing reversible deactivation radical polymerization (RDRP), which serve to impart living characteristics to the process, has provided a new set of tools for polymer chemists that allow control over the polymerization process while retaining much of the versatility of conventional radical polymerization. It is no longer a formidable task to apply radical polymerization to the synthesis of blocks, stars, or other polymers of complex architecture. New materials with the potential of revolutionizing a large part of the polymer industry continue to appear. The polymerization techniques that are receiving greatest attention are nitroxide-mediated polymerization (NMP), atom transfer radical polymerization (ATRP), and reversible addition–fragmentation chain transfer (RAFT).
There remains some controversy over the use of the terms ‘living’ and ‘controlled’ in describing processes for radical polymerizations such as ATRP, NMP, or RAFT.[2] The recommendation of the International Union of Pure and Applied Chemistry (IUPAC), that a living polymerization is ‘a chain polymerization from which irreversible chain transfer and irreversible chain termination (deactivation) are absent’, precludes use of the adjective ‘living’ in referring to these processes.[3] ‘Controlled’, when used by itself, is also contrary to IUPAC recommendations. It is incorrect to use ‘controlled’ in an exclusive sense to indicate a particular form of polymerization because the word has an established, much broader, usage. Use of the terms ‘controlled living’, ‘controlled/living’, ‘pseudo-living’, and ‘quasi-living’ is also discouraged. An IUPAC task group has recently recommended the term reversible deactivation radical polymerization (RDRP) to describe polymerizations, such as ATRP, NMP, or RAFT, that entail equilibria between active and dormant chains.[4] This term is not intended to have any connotations as to the fraction of living chains that might be present in a particular polymerization process.
It remains acceptable to use the term ‘living radical polymerization’ to describe a hypothetical process in which termination is indeed absent. It is in this context that we use ‘living radical polymerization’ in the title of this review and the previous articles of this series. We do not intend to imply that termination is absent from any polymerizations described herein. Many systems do display the observable characteristics normally associated with living polymerizations and in a few cases, termination, while undeniably present, is undetectable using current techniques.
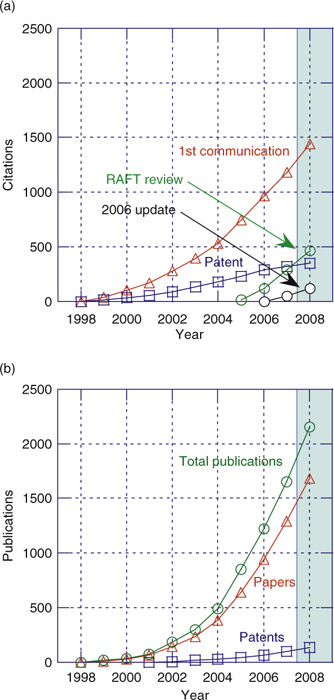
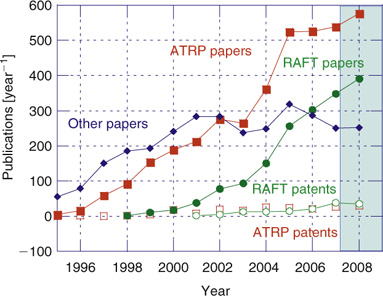
The increasing importance of RAFT is illustrated by Fig. 1, which shows the cumulative citations for our first communication on RAFT with thiocarbonylthio compounds,[5] the first RAFT patent,[6] and the previous reviews in the Australian Journal of Chemistry.[7,8] It should be noted that not all papers on RAFT polymerization cite these sources, nor are all of the papers citing these documents directly relevant to RAFT polymerization. Fig. 2 shows that the remarkable growth in publication of papers covering all forms of RDRP has continued unabated. The total number of papers that mention ‘RAFT polymerization’ has more than doubled over the period 2006–2009 with more than 1000 papers being published, and approximately one-third of papers on RDRP now pertain to the concept ‘RAFT polymerization’.
|
|
This review is primarily intended to cover the literature on RAFT polymerization that has appeared since publication of the update review published in the Australian Journal of Chemistry in late 2006.[8] We also refer to some earlier papers that were not included in that or the earlier reviews. Work cited in the previous reviews[7,8] is only mentioned again where necessary to put the more recent work in context. The past 2 years has seen the publication of further general reviews detailing the RAFT process which include works by Moad, Rizzardo, and Thang,[9–12] a Handbook of RAFT Polymerization,[13] and a highlight article by Barner-Kowollik and Perrier[14] on the future of RAFT. Reviews on specific areas include the kinetics and mechanism of RAFT polymerization,[15–18] the use of RAFT to probe the kinetics of radical polymerization,[19,20] the use of RAFT in organic synthesis,[21] amphiphilic block copolymer synthesis,[22,23] the synthesis of end functional polymers,[24] the synthesis of star polymers and other complex architectures,[25,26] the use of trithiocarbonate RAFT agents,[27] the use of xanthate RAFT agents (MADIX),[28] polymerization in heterogeneous media,[29–32] RAFT polymerization initiated with ionizing radiation,[33] polymer synthesis in aqueous solution,[34–37] surface and particle modification,[38,39] synthesis of self assembling and/or stimuli responsive polymers,[36,40] RAFT-synthesized polymers in drug delivery,[22,41] and other applications of RAFT-synthesized polymers.[30,42,43] The process is also given substantial coverage in most recent reviews that, in part, relate to polymer synthesis, living or controlled polymerization, or novel architectures. Some of these documents are referred to in subsequent sections of this review.
Mechanism of RAFT
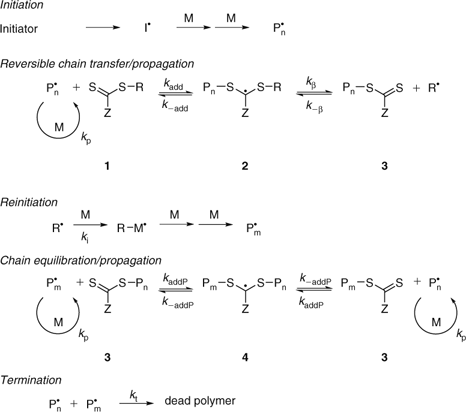
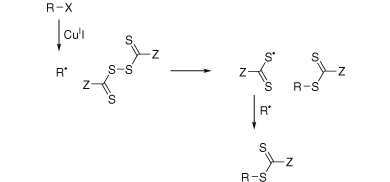
The key feature of the mechanism of RAFT polymerization with thiocarbonylthio compounds, as proposed in our first communication on the subject,[5] is the sequence of addition–fragmentation equilibria shown in Scheme 1. Initiation and radical–radical termination occur as in conventional radical polymerization. In the early stages of the polymerization, addition of a propagating radical (Pn•) to the thiocarbonylthio compound [RSC(Z)=S (1)] followed by fragmentation of the intermediate radical provides a polymeric thiocarbonylthio compound [PnS(Z)C=S (3)] and a new radical (R•). Reaction of this radical (R•) with monomer forms a new propagating radical (Pm•). Rapid equilibrium between the active propagating radicals (Pn• and Pm•) and the dormant polymeric thiocarbonylthio compounds (3) provides equal probability for all chains to grow and allows for the production of narrow dispersity polymers. When the polymerization is complete (or stopped), most of the chains retain the thiocarbonylthio end-group and can be isolated as stable materials.
|
The reactions associated with RAFT equilibria shown in Scheme 1 are in addition to those (i.e., initiation, propagation, transfer, and termination) that occur during conventional radical polymerization. In an ideal RAFT process, the RAFT agent should behave as an ideal transfer agent. Thus, as with radical polymerization with conventional chain transfer, the kinetics of polymerization should not be directly affected by the presence of the RAFT agent beyond those affects attributable to the differing molecular weights of the reacting species. Radical–radical termination is not directly suppressed by the RAFT process. Living characteristics are imparted when the molecular weight of the polymer formed is substantially lower than that which would be formed under the same conditions but in the absence of a RAFT agent, and is such that the number of polymer molecules with RAFT agent-derived ends far exceeds the number formed as a consequence of termination. Many RAFT polymerizations stray from this ideal. Although the basic mechanism shown in Scheme 1 is generally not disputed, much debate continues on the detailed kinetics of the RAFT process, the rapidity with which the various equilibria are established, and what side reactions might occur to complicate the process in specific circumstances.
Mechanisms for retardation in RAFT polymerization mediated by, in particular, dithiobenzoate RAFT agents continue to attract significant interest. Mass spectrometry has been used to provide evidence to support the occurrence of intermediate radical termination during RAFT polymerization of acrylates,[44] styrene (St),[45,46] and N-acryloylmorpholine (NAM)[47] with dithiobenzoate RAFT agents. A recent paper[48] purports to suggest a possible compromise between the ‘slow fragmentation’ and ‘intermediate radical termination’ models for retardation in RAFT polymerization with dithiobenzoate RAFT agents. According to the proposed model intermediate radical termination occurs, but only for initiator-derived or oligomeric species (chain length <2).[48,49]
Further details of the so-called ‘missing reaction step’ mechanism for retardation in RAFT polymerization with dithiobenzoate RAFT agents have also been published.[50] Some support for this mechanism comes from studies on 2,2′-azoisobutyronitrile-α-13C-initiated polymerization of St.[51]
Kinetic analysis together with Monte Carlo simulation of the reaction of alkoxyamines with dithiobenzoates (22, 49) was investigated as a means of estimating rate constants for addition and fragmentation.[52] However, side reactions proved to be an issue when applying the method.
The initialization process (consumption of the initial RAFT agent) in RAFT (co)polymerization has been studied by 1H NMR spectroscopy for St/maleic anhydride (St/MAH) with RAFT agents 22 and 24,[53] methyl acrylate (MA) with RAFT agents 22, 24, and 92,[54] and of vinyl acetate (VAc) and N-vinylpyrrolidone (NVP) with xanthates 207, 209, and 211.[55] For many of these systems so-called selective initialization was observed in which there is substantial conversion of the initial RAFT agent into a single unit species before conversion into higher molecular weight chains. The effect is attributed to the rate constant of addition of the ‘R’ radical to MA being substantially slower than that for propagation.[56] The course of the RAFT polymerization of butyl acrylate (BA) and St with cumyl (22) and cyanoisopropyl dithiobenzoate (24) was followed by electron spin resonance (ESR) spectroscopy.[57] 13C NMR spectroscopy was used to follow the initiation of St polymerization with AIBN-α-13C and with cumyl (22) and cyanoisopropyl (24) and benzyl dithiobenzoates (61) and with cyanoisopropyl dodecyl trithiocarbonate (121).[51] An unexpected finding was the observation of 13C chemically induced nuclear polarization (CIDNP) for the ketenimine formed by cage recombination of the AIBN-derived cyanoisopropyl radicals. Selective initialization was observed for the more active RAFT agents 22, 24, and 121 (with good ‘R’ leaving group). However, with benzyl dithiobenzoate (61) (poor ‘R’ leaving group relative to propagating species), consumption of the initial RAFT agent is slow with respect to the rate of polymerization and higher molecular weight species are formed from the inception of RAFT polymerization.
The rate constant for chain transfer (ktr) for a RAFT agent is given by the expression in Eqn 1 and depends on the rate of addition of the propagating radical to the RAFT agent and a coefficient (φ) which describes how the intermediate radical (2) partitions between starting materials and products – refer to Scheme 1:
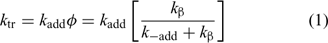
The transfer agent-derived radical (R•) is also partitioned between adding to monomer and reacting with the macro-RAFT agent (3). We, therefore, define a rate constant associated with this reaction (k–tr) as shown in Eqn 2.
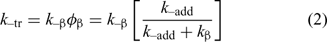
Knowledge of the partition coefficients φ and φβ (note φβ = 1 – φ) and C–tr is required for an understanding of RAFT agent activity. The high reactivity of RAFT agents is such that C–tr is seldom zero. Transfer constants measured by methods which include an assumption that C–tr is zero or that kβ is zero will typically underestimate the actual Ctr. These values should be called apparent transfer constants Ctrapp. In some cases, values of Ctr may be higher than Ctrapp by several orders of magnitude.[56] An indication that the reverse reaction is important is the dependence of Ctrapp on the RAFT agent concentration and on monomer conversion.[56,58,59] The spread in literature values for Ctrapp may be attributed to this effect. Note that the situation is simplified for the case of macro-RAFT agents in homopolymerization where, notwithstanding effects of chain length, the forward and reverse reactions are the same (Ctr = C–tr) and the partition coefficient φ is 0.5.
For less active RAFT agents (Ctr ≤ 1), transfer constants for RAFT agents may be determined using the usual methods (e.g., the Mayo method) with little loss of accuracy. Experimental values of kinetic parameters associated with the RAFT process (addition rate constants (kadd), fragmentation (kβ, k–add) rate constants, and forward (Ctr = ktr/kp), reverse (C–tr = k–tr/ki) and apparent transfer constants (Ctrapp)) in the literature through 2006 were summarized by Moad et al.[9] Data for Ctrapp, Ctr, and C–tr that has appeared since that review are summarized in Table 1. A new method of estimating k* = 1/Ctrapp based on fitting oligomer molecular weight distributions was recently reported.[60]

|
Instrumentation and reactor design to allow direct on-line monitoring of the RAFT polymerization by viscometry, ultraviolet-visible spectrophotometry (with a photodiode array detector), refractive index, and multi-angle light scattering has been described and applied to study the time/conversion dependence of molecular weight for the polymerization of BA with RAFT agent 154 as a function of the RAFT agent concentration.[61]
The effect of the monomer feed composition on Ctrapp for RAFT agents in butyl acrylate (BA)/St and methyl methacrylate (MMA)/BA copolymerization has also been studied.[62] It was proposed that effective transfer constants in copolymerization ![]() Ctr
Ctr![]() might be estimated on the basis of the reactivity ratios and the homo (Ctr,nn) and cross transfer constants (Ctr,nm).
might be estimated on the basis of the reactivity ratios and the homo (Ctr,nn) and cross transfer constants (Ctr,nm).
RAFT-Related Processes
Polymerization of VAc and similar monomers mediated by organocobalt porphyrin complexes is proposed to involve a RAFT-like mechanism called associative degenerative chain transfer.[74–76] A reversible coupling mechanism is generally proposed for polymerizations in the presence of bis(acetylacetonate)cobalt(ii) complexes. The process has been applied to VAc miniemulsion polymerization[77] and in the synthesis of poly(vinyl acetate)-block-poly(vinyl pyrrolidone) (PVAc-b-PNVP)[78,79] and PVAc-b-PSt.[80] Cobalt-mediated St polymerization is not believed to involve a reversible deactivation mechanism but rather is believed to be a conventional polymerization initiated by the PVAc organocobalt species.
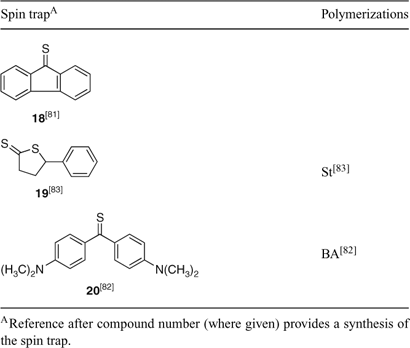
Ab initio calculations have been used as a tool in the design of thioketones as spin traps for controlling radical polymerization by an addition–fragmentation process.[81] The thiofluorenone 18 was proposed as a compound suitable for investigation with high radical affinity, appropriate addition–fragmentation equilibrium constant, and a low propensity to undergo copolymerization (Table 2). The diarylthioketone 20 was found to offer some control over BA polymerization.[82]
|
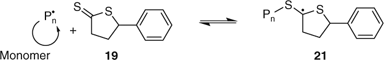
The thiolactone 19 was proposed to control the polymerization of St by a similar addition–fragmentation process (Scheme 2).[83] The mechanism as shown seems unlikely since, for control, most of the propagating species would need to be present as the dormant species 21. ESR experiments showed that a high concentration of radicals were not present during polymerization. The mechanism would suggest that the final product should also have structure 21.
|
Choice of RAFT Agents
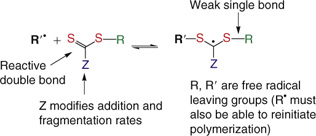
A wide variety of thiocarbonylthio RAFT agents (ZC(=S)SR, 1) have been reported. A broad summary of these and the factors that influence the choice of RAFT agent for a particular polymerization was presented in previous reviews.[1,7,12] The effectiveness of the RAFT agent depends on the monomer being polymerized and is determined by the properties of the free radical leaving group R and the group Z, which can be chosen to activate or deactivate the thiocarbonyl double bond of the RAFT agent 1 and modify the stability of the intermediate radicals 2 and 4. For an efficient RAFT polymerization (Scheme 1, Fig. 3):
|
-
The initial RAFT agents 1 and the polymer RAFT agent 3 should have a reactive C=S double bond (high kadd).
-
The intermediate radicals 2 and 4 should fragment rapidly (high kβ, weak S–R bond in the intermediate) and give no side reactions.
-
The intermediate 2 should partition in favour of products (kβ ≥ k–add).
-
The expelled radicals (R•) must efficiently re-initiate the polymerization (ki > kp).
The properties of RAFT agents are often discussed in terms of the value of the equilibrium constants (K) associated with radical addition to the thiocarbonylthio compound. Rates of addition are typically high (kadd~106–108 M–1 s–1). Thus a high equilibrium constant generally implies a low fragmentation rate for the radical adduct and an increased likelihood for retardation and/or side reaction involving this species. Values of K do not, by themselves, provide sufficient information to predict the ability of a RAFT agent to control polymerization.
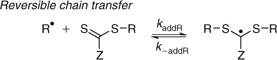
In a given RAFT polymerization, there are at least four equilibrium constants that need to be considered.
-
KP (= kaddP/k–addP) associated with the main equilibrium.
-
K (= kadd/k–add) and Kβ = (k–β/kβ) associated with the pre-equilibrium.
-
KR (= kaddR/k–addR) associated with the reaction of the expelled radical with the initial RAFT agent (Scheme 3).
|
There are other equilibrium constants to consider if penultimate group effects are significant. Ab initio methods have been used for the prediction and rationalization of substituent effects on RAFT agent activity.[16,84]
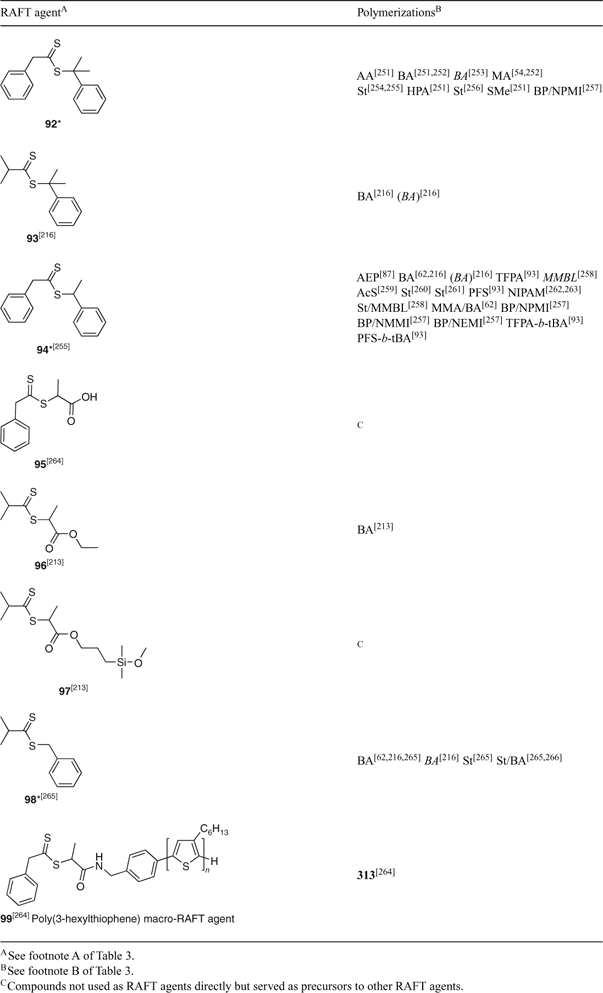
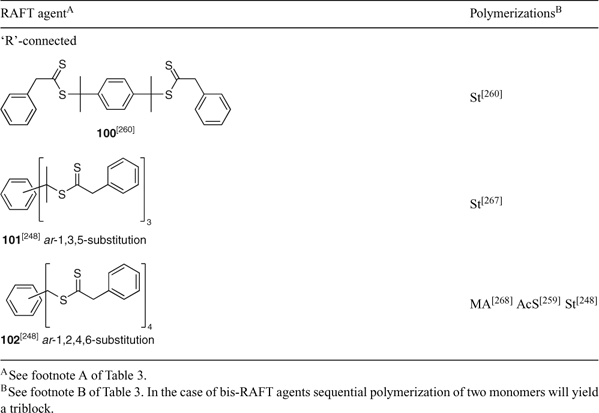
A summary of ‘new’ RAFT agents and new polymerizations in which they and pre-existing RAFT agents have been applied are shown in Tables 3–15. The tables include some RAFT agent/monomer combinations that provide poorer molecular weight control and/or dispersity ÐM > 1.4. These are generally indicated by the monomer being in parentheses. Often these have been investigated in order to provide an understanding of the mechanism and to allow construction of guidelines for the choice of RAFT agent.

|

|
|
|

|

|

|

|

|

|

|

|

|
The data included in Tables 3–15 show that tertiary-dithiobenzoates (in particular, 22–32) continue to be the most popular RAFT agents for synthesizing polymers based on 1,1-disubsituted methacrylate or methacrylamide monomers. The use of the corresponding trithiocarbonates 118–125 has also been promoted in this context.[27] Secondary aromatic dithioesters with R = –CHPh(CN) (42) and –CHPh(CO2R) (43–48) and analogous trithiocarbonates 144, 145, and 146–148, respectively, have been shown to have utility in controlling polymerization of methacrylates.[27] Dithiobenzoates 24 and 25 and the trithiocarbonates 121 and 125 are now commercially available from Strem Chemicals. RAFT agents are also available from Sigma-Aldrich and from Monomer-Polymer & Dajac Labs.[85]
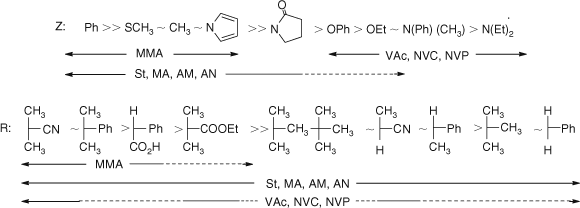
Fig. 4, based on that from our previous reviews,[7–10] provides a general summary of how to select the appropriate RAFT agent for particular monomers. Note should be made of the dashed lines in the chart. Although some control might be achieved with these monomer RAFT agent combinations, the molecular weight distribution may be broad or there may be substantial retardation or prolonged inhibition. This proviso has been omitted in some representations of this data in the literature.
|
In synthesizing block copolymers by sequential monomer addition the order in which the blocks are prepared is important. The propagating radical derived from the macro-RAFT agent must be a reasonable free radical leaving group with respect to that formed by the monomer being polymerized.
RAFT agents such as dithioesters (Z = aryl or alkyl) or trithiocarbonates (Z = alkylthio) suitable for controlling polymerization of ‘more-activated’ monomers (MAMs), e.g., MMA, St, MA, acrylamide (AM), acrylonitrile (AN) inhibit or retard polymerizations of ‘less activated’ monomers (LAMs), e.g., VAc, NVP, and N-vinylcarbazole (NVC). Similarly RAFT agents suitable for controlling polymerizations of LAMs such as N,N-dialkyl- or N-alkyl-N-aryl dithiocarbamates and xanthates tend to be ineffective with MAMs.
The reduced effectiveness of the dithiocarbamate RAFT agents with MAMs relates to their lower reactivity towards radical addition and consequent lower transfer constants.[58] The double-bond character of the thiocarbonyl group is reduced by the contribution of zwitterionic canonical forms, which localize a positive charge on the nitrogen and a negative charge on the sulfur.[58,421] However, the tendency of dithioesters or trithiocarbonates to inhibit polymerization of LAMs is a consequence of the poor radical leaving group ability, with respect to the ‘R’ radical, of propagating species with a terminal LAM unit. Dithiocarbamates that possess electron-withdrawing groups adjacent to nitrogen or where the nitrogen lone pair is part of an aromatic ring system are effective with MAMs[58,421] but inhibit polymerizations of LAMs. Fluorodithioformates were proposed as universal RAFT agents but their application remains largely unproven.[417, 422] One consequence of this has been that the direct synthesis of narrow dispersity polyMAM-block-polyLAM is difficult or not possible using the conventional range of conventional RAFT agents.
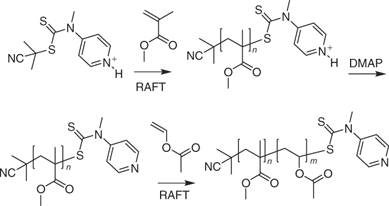
A new class of stimuli-responsive RAFT agents that can be switched to offer good control over polymerization of both MAMs and LAMs and a route to polyMAM-block-polyLAM has been reported.[416] N-(4-pyridinyl)-N-methyldithiocarbamates (Table 14) are effective with LAMs and in the presence of a strong acid, the protonated form of the N-(4-pyridinyl)-N-methyldithiocarbamates provide excellent control over the polymerization of MAMs.[416] The process is illustrated in Scheme 4 for the preparation of poly(methyl methacrylate)-block-PVAc. Thus in the first step the protonated RAFT agent (formed by adding 4-toluenesulfonic acid) is used to control the polymerization of MMA to form PMMA. This macro-RAFT agent is then neutralized in situ by adding a stoichiometric amount of N,N-dimethylaminopyridine (DMAP). RAFT polymerization of VAc then provided the desired block copolymer. Other approaches to the synthesis of polyMAM-block-polyLAM are mentioned in the section on Block Copolymers below.
|
It has also been reported that macro-RAFT agents derived from N,N-dialkylacrylamides are difficult to chain extend with N-alkylacrylamides.[302] Bivigou-Koumba et al.[286] reported that strong retardation was observed in extending PSt macro-RAFT agents with N-isopropylacrylamide (NIPAM), whereas chain extension of PNIPAM macro-RAFT with St was successful. In both cases slow initiation by the macro-RAFT agent-derived propagating species was suggested as a possible explanation. However, Wong et al.[218] reported that it was difficult to chain-extend PDMAM macro-RAFT agent with St and that it was important to make the PSt macro-RAFT agent first.
Synthesis of RAFT Agents
Primary and secondary trithiocarbonates, xanthates, and dithiocarbamate RAFT agents are commonly synthesized in high yield by reaction of the appropriate carbodithioate salt with an alkylating agent.[8,27,283] Skey and O’Reilly[283] advocated the use of acetone as solvent and phosphate as base and have applied the method to a wide range of RAFT agents (see Tables above). Surprisingly, in view of previous reports, they indicate that the method is also effective for the synthesis of some RAFT agents with tertiary ‘R’ (115 and 143).
Symmetrical trithiocarbonates are frequently synthesized from the corresponding halide, and carbon disulfide in the presence of a base. Aoyagi and Endo[290] have shown that this process can be performed on benzyl halides, and stoichiometric carbon disulfide at low temperature (40°C) in air with potassium bicarbonate as base to provide near quantitative yields of the corresponding dibenzyl trithiocarbonates (111–114).
RAFT agents can be synthesized from ATRP initiators as shown in Scheme 5. Dithioesters, dithiocarbamates, and xanthates were prepared in high yields by an atom transfer radical addition–fragmentation (ATRAF) process, using stoichiometric amounts of alkyl halides and bis(thiocarbonyl) disulfides in the presence of ATRP catalysts and, optionally, Cu metal as a reducing agent.[423] A similar procedure was described previously by Wager et al.[424]
|
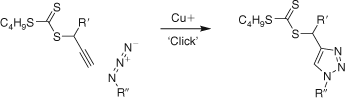
The synthesis of RAFT agents having a triazolinylmethyl leaving group are synthesized by a copper-catalyzed 3 + 2 cycloaddition reaction between an azide and a propargyl thiocarbonylthio compound (Scheme 6).[339] The triazolinyl trithiocarbonates 153 and 163 were effective in controlling polymerizations of St and BA, while the triazolinyl xanthate 225 was able to control polymerizations of NVP and VAc.
|
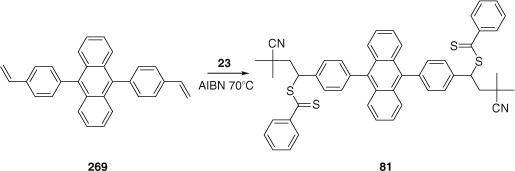
New RAFT agents can be synthesized by insertion of a single monomer unit into a RAFT agent.[425] The success of this process relies on the addition of R′ to the monomer being substantially faster than subsequent monomer additions and requires a very active RAFT agent such that the number of monomer additions per activation cycle is greater than 1. It requires selective initialization (see section on Mechanism of RAFT above). The methodology was applied in the synthesis of the bis-RAFT agent 81 (Scheme 7) which was used in the synthesis of light harvesting polymers.[244] Another recent example of this strategy is its application to 4-(tert-butoxy)styrene (tBS) to provide a route to mono-hydroxy end-functional PSt.[133]
|
The process has been adapted to provide a method for end-functionalizing macro-RAFT agents by addition of a single monomer unit to form a new macro-RAFT agent. Recent examples of this involve attachment of maleic anhydride (MAH) unit to a PSt macro-RAFT agent[186] and a single maleimide unit to a poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA) macro-RAFT agent.[426]
Characterization of RAFT-Synthesized Polymers
Most papers on RAFT polymerization contain information on the characterization of RAFT-synthesized polymers and/or the RAFT process. We consider here papers where the characterization of RAFT-synthesized polymers by spectroscopic or chromatographic methods is a primary focus.
A comparison of electrospray ionization (ESI) and matrix-assisted laser desorption–ionization time-of-flight (MALDI-TOF) mass spectrometries for characterizing PSt synthesized with dithiobenzoate 60 and AIBN initiation has been reported.[46] The relative merits of these techniques for characterizing PSt made by ATRP, NMP, and reverse iodine transfer polymerization (RITP) was also examined.[46] A MALDI-TOF mass spectrometry study of PSt with dithiobenzoate RAFT agents 69 and 70 has claimed to provide evidence of intermediate radical termination.[45] The application of ESI mass spectrometry to PSt with labile chain ends is facilitated by use of silver(i) tetrafluoroborate salt. The method was applied in identifying products and mapping reaction pathways in forming PSt with 92,[254] PMA with 92,[252] and PAA and PNIPAM with 103,[269] and in forming star polymers from MA[268] and AcS[259] with 102.
Dithiobenzoate RAFT agents and macro-RAFT agents were found to very effectively quench the fluorescence of coumarone derivatives and acenaphthalene units.[215,244,427] No quenching was observed for the RAFT-synthesized polymers from which the thiocarbonylthio end-group had been removed by aminolysis[215] or radical-induced reduction.[244]
On-line monitoring of RAFT polymerization by NMR and other methods is mentioned above in the section Mechanism of RAFT.
RAFT synthesized polymers, in particular PSt or PMMA with dithiobenzoate chain ends, have been found to be unstable to storage in cyclic ether solvents (tetrahydrofuran).[428] Thus PMMA-dithiobenzoate was converted into PMMA-hydroperoxide within 7 days. PSt-dithiobenzoate was more slowly converted into a mixture of products in which the dithiobenzoate end-group had been converted into the corresponding sulfine or thionoester end-groups or was lost and the polymer left with epoxy and unsaturated end-groups. The end-group degradation was attributed to the presence of tetrahydrofuran hydroperoxide in the solution. Much slower degradation was also observed in dichloromethane solution. The result has implications with respect to storage of samples for gel permeation chromatography where tetrahydrofuran is commonly used as solvent/eluent. We have also observed that RAFT-synthesized polymers (not just dithiobenzoates) can be unstable in tetrahydrofuran solution and recommend that only peroxide-free solvent is used and that samples, if they must be kept, are stored under an inert atmosphere and protected from light.
Polymerization Kinetics
Kinetic simulation is frequently used as a tool to correlate experimental data with theoretical models.[429] The use of RAFT polymerization and kinetic simulation with Predici have been applied to determine chain length dependent termination rate constants in radical polymerizations (the RAFT-CLD-T method) has been previously reviewed.[19]
Criteria for selecting conditions for the determination of termination rate constants by the RAFT-CLD-T method have been proposed and justified by kinetic simulation.[430] The RAFT-CLD-T method has been used to determine chain length dependent termination rate constants in MMA polymerization in various conversion regimes up to high conversion[119,120] and termination rate constants during formation of star polymers based on MMA, MA, and St.[20,256]
In the SP-PLP-NIR-RAFT method, polymerization is induced by a single laser pulse (SP) and the resulting decay in monomer concentration, cM, is monitored by NIR spectroscopy with a time resolution of microseconds. The presence of a RAFT agent ensures the correlation of radical chain length and monomer-to-polymer conversion.[281,429] The method has been applied to determine chain length dependent kt in MA,[281] BA,[429] and DA[281] polymerizations.
A kinetic simulation was used to model RAFT polymerization controlled by bis-RAFT agents.[431] It was demonstrated theoretically that the use of such RAFT agents allows production of linear polymers with an extremely low level of dead chains. This is a consequence of it being statistically unlikely to produce chains with zero RAFT end-groups.
Reaction Conditions (Initiator, Temperature, Pressure, Solvent, Lewis Acids)
The general guideline for choosing initiator concentrations for RAFT polymerization is that the mole ratio of RAFT agent to amount of initiator decomposed should be >10:1 and be such that the molecular weight obtained in a control experiment (i.e., same conditions without RAFT agent) is at least 10-fold higher than the desired molecular weight.[7] It must be remembered that for every pair of radicals generated, a pair of radicals will terminate to provide dead polymer impurity.
RAFT polymerizations with either 24 or 254 as RAFT agent were successfully carried out at room temperature using benzoyl peroxide/N,N-dimethylaniline as a redox initiator.[410]
A detailed study of the RAFT polymerization of MA with a trithiocarbonate RAFT agent (157) and AIBN initiator at 50°C has appeared.[344] The variables explored were: ratio of RAFT agent to initiator (1:5 to 1:0.1), ratio of RAFT agent to monomer (500:1 and 100:1), monomer concentration (bulk and 33.3 wt-% monomer in solvent) and solvent (toluene, DMF, MEK). For this system, good control was achieved under all conditions studied.
Several papers have appeared on the protocol for conducting high-throughput RAFT polymerization.[114,115,251] High throughput methods have been applied to synthesize libraries of methacrylate polymers (MAA, various oligo(ethylene glycol) methacrylates and copolymers of MAA with PEGMA,[108] MMA, DMAEMA and block and statistical copolymers,[114] and copolymers of DMAEMA with PEGMA[113]) for evaluating the composition dependence of the lower critical solution temperature (LCST).
RAFT polymerization of ‘polar’ monomers (MMA,[432] MA,[432,433] VAc,[433] DADMAC,[434] and NIPAM[304]) were reported to be substantially accelerated by microwave heating. Less but still significant acceleration was observed for St polymerization.[432,433,435] It is expected that monomers with a higher dielectric constant will be more effectively heated by microwave irradiation. The microwave effect, particularly with MMA and MA, was suggested to be greater than expected for an effect of temperature alone,[432] although an alternate explanation has not yet been provided. A more recent study on MMA polymerization[109] indicates that the polymerization kinetics are independent of whether microwave or conventional heating processes are used. The advantage of a microwave reactor is that it allows RAFT polymerization to be readily conducted at high temperatures in superheated monomer. Thus, RAFT polymerizations of MMA were carried out at 120, 150, and 180°C in the absence of an added radical initiator.
Several studies on photo-initiated RAFT polymerization have appeared.[270,275,285,309,413,436] RAFT polymerization of St derivatives can be carried out at near ambient temperature (30°C) with 115 as RAFT agent using 365 nm irradiation and (2,4,6-trimethylbenzoyl)-diphenylphosphine oxide as photoinitiator.[309] The same photoinitiator with 420 nm irradiation was used in ambient temperature RAFT polymerization of the UV sensitive cinnamate derivative 298[436] and for polymerization of BA and MA under solar irradiation (>400 nm) at 20°C.[270]
The use of ionizing radiation to initiate RAFT polymerization has been reviewed.[33,437] Other studies on gamma-initiated RAFT polymerization include the polymerization of MA and BA,[252] polymerization of NIPAM[269] and AA[269,354] in aqueous media, BA miniemulsion polymerization,[253] St/MAH/NVP terpolymerization,[288,289] MA polymerization with dithiocarbamate RAFT agents[405] and the grafting of styrenic monomers to cellulose[167,438] and t-butyl acrylate onto ethylene propylene rubber.[439]
Further experiments have been reported on RAFT polymerization in the presence of a Lewis acid to control tacticity (110/AM/Y(OTf)3;[285] 94/NIPAM/Sc(OTf)3;[262] 94/NIPAM/Y(OTf)3;[262] 103/NIPAM/Y(OTf)3;[271] and 119/MMA/Sc(OTf)3[88]). The disappearance of the RAFT agents (cumyl dithiobenzoate 22 or cyanoisopropyl dithiobenzoate 24) in solution at 60°C in the presence of a Lewis acid (Sc(OTf)3) was followed by NMR spectroscopy.[88] These RAFT agents underwent significant decomposition within 1 h and were completely converted into a complex mixture of products within 16 h. RAFT agent instability provides an explanation for the poor control over molecular weight distribution observed in earlier attempts to achieve control over tacticity of PMMA.[88] Other RAFT agents, including dithiobenzoate 39, cyanoisopropyl methyl trithiocarbonate 119, and cyanoisopropyl 1-pyrrolecarbodithioate 239, and the PMMA macro-RAFT agents formed with use of these RAFT agents were found to be stable in the presence of Lewis acid over a 16 h period at 60°C.[88] RAFT polymerization of MMA in the presence of 119 and Sc(OTf)3 provided simultaneous RAFT-controlled molecular weight and molecular weight distribution (Ð < 1.3 at > 95% conversion), Lewis acid-controlled tacticity with mm:mr:rr = 12:44:44 for a mole ratio Sc(OTf)3/MMA of 1:8.5 at 60°C [compared with mm : mr : rr = 4 : 33 : 63 in the absence of Sc(OTf)3] and a significantly enhanced rate of polymerization.[88] The effect of the Lewis acid on tacticity was the same as that observed in the absence of RAFT agent.
The rate of RAFT polymerization of MA with 241 is substantially enhanced in the presence of alumina, which is attributed to the alumina acting as a Lewis acid (Fig. 5).[399]
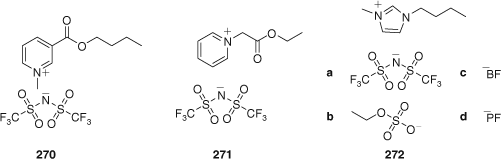
|
RAFT polymerization of St was successfully carried out in biodegradable ionic liquids 270 or 271 (RAFT agent 92, 90°C, ACHN initiator) (Fig. 5).[255] The polymerization kinetics were similar to those for toluene solution. Note that previous attempts to polymerize St in conventional imidazolium ionic liquids were unsuccessful. The outcome of RAFT polymerization of MMA with 24 or 115 in imidazolium ionic liquids (272a–d) is dependent on the counteranion with best results being obtained with 272a and 272d and relatively poor control being seen with 272b or 272c as solvent.[122]
RAFT polymerization of cyclodextrin host–guest complexes has been performed. Precipitation was observed during RAFT-polymerization of the cyclodextrin host–guest complex of styrene.[356] However, homopolymerization of a cyclodextrin host–guest complex of the adamantyl derivative 346 remained homogeneous allowing synthesis of the amphiphilic block copolymers P346-block-PDMAM in aqueous solution.[358]
RAFT Polymerization in Heterogeneous Media
Several reviews relating to the use of RAFT in heterogeneous media have appeared.[29,440–442] Examples of RAFT polymerizations in heterogeneous media are included in Tables 3–15 and are distinguished in the tables by the monomer appearing in italics.
The use of macro-RAFT agents as stabilizers in ‘surfactantless’ emulsion polymerization,[145,181,225,287,300,310,341,393,443–446] miniemulsion polymerization,[124,181] suspension polymerization[381] and non-aqueous dispersion polymerization in both organic media,[188] and in supercritical CO2[353,389,447,448] has gained popularity. Particle nucleation and growth during RAFT emulsion polymerization of St and BA mediated by macro-RAFT agents of various compositions has been studied by calorimetry.[341] More hydrophilic amphipathic macro-RAFT agents (e.g., AA 10 units–St 10 units) were thought to be promising for producing latex products with best control over particle number and particle size distribution. In most cases this work has involved use of a hydrophilic or an amphiphilic macro-RAFT agent. However, macro-RAFT agents based on dextran[393] or poly(ethylene oxide) (PEO)[124,181,444] have also been exploited.
There have been several papers on the use of the RAFT process in the dispersion polymerization of St,[353] MMA,[447,448] and NVP[389] in supercritical CO2 making use of macro-RAFT agents as stabilizer.
Kinetic simulation of the RAFT miniemulsion polymerization was performed to investigate the polymerization rate[449] and molecular weight distribution.[450] It was found that the polymerization rate increases with a reduction in the particle size. However, for particles with particle diameter (Dp) < 100 nm, the statistical variation in monomer concentration among the particles should not be neglected and may slow the polymerization rate.[449] The variation in monomer concentration between particles may also cause a broadening of the molecular weight distribution by causing different molecular weights of polymer to be formed.[450]
RAFT Polymer Syntheses
Polymer syntheses by RAFT polymerization are summarized in Tables 3–15. Only systems that require separate comment are mentioned here or in subsequent sections.
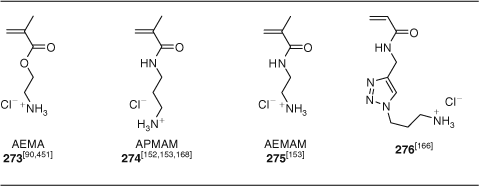
RAFT technology is generally incompatible with unprotected primary or secondary amine functionality. Recently, several groups have reported that the polymerization of monomers with primary amine functionality (273–276) (Table 16) can be effectively controlled with the monomer as the ammonium salt and with no or minimal loss of the RAFT end-group.[90,152,153,166] Some of the more exotic monomer subjected to RAFT polymerization are included in the tables that follow. They include methacrylates (Table 17), acrylates (Table 18), methacrylamides (Table 19), acrylamides derived from amino acids (Table 20), other acrylamides (Table 21), St derivatives (Table 22), and vinyl monomers (Table 23). Monomers of the above classes with reactive functionality are in Tables 14–26. Macromonomers are considered in the section Polymer Brushes/Graft Copolymers/Comb Polymers/Surface Modification.
|

|

|
|
|

|

|
|
|
|

|
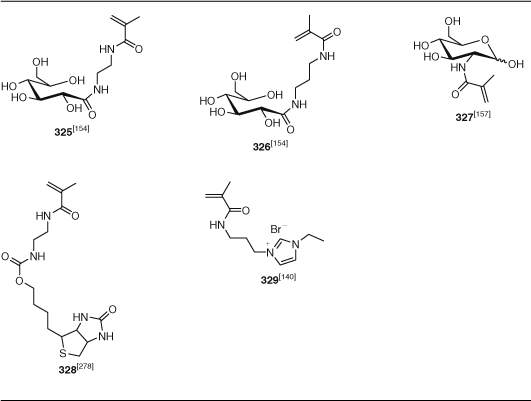
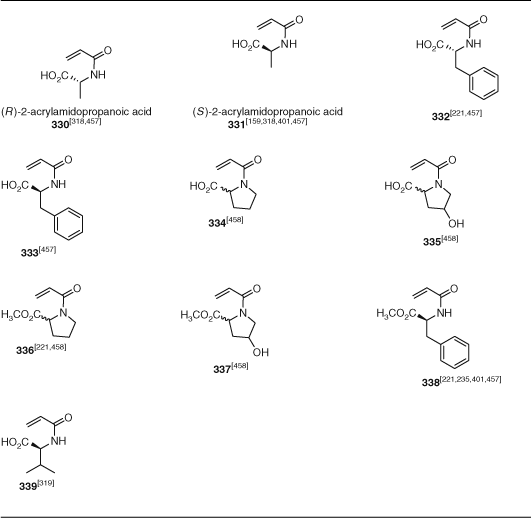
Other functional monomers to receive attention include fluorinated methacrylates (280–281, PFMA, TFPMA), acrylates (318, TFPA), and styrenes (PFS), monomers with betaine (292–293), thiirane (301), boronic acid (343), and pyridyl disulfide (391) and a wide range of other functionalities. The number of examples of synthesis of glycopolymers using RAFT polymerization has substantially expanded. These have been prepared by the polymerization of methacrylate 285–291, acrylate 319, St 356, acrylamide 342–340, or methacrylamide 325–326 glycomonomers. There has also been significant interest in RAFT polymerization of acrylamides derived from amino acids (330–339; Table 20).
Use of ‘click chemistry’ in polymer chemistry has recently attracted much attention.[461–464] The clickable functionality may be present in the monomers or on the Z or R groups of the RAFT agent (see Block Copolymers). The clickable monomers are incorporated as homopolymer blocks as a precursor to a polymer brush, or copolymerized to provide sites for attachment of functionality or for crosslinking.
A significant number of these papers concern the combination of RAFT and azide–alkyne 1,3-dipolar cycloaddition. Azide- and alkyne-functional monomers are given in Table 24. The importance of protecting alkyne-functional monomers (371, 373) (and RAFT agents) as the trimethylsilyl derivative has been stressed by some authors. However, some have been used unprotected with apparently minimal side reactions, which is attributed to the alkyne being much less reactive towards radical addition than the (meth)acrylate double bond. Azide-functional polymers have also been prepared from 3-chloropropyl acrylate polymers, which are converted to 3-azidopropyl acrylate copolymer post polymerization by reaction with sodium azide.[338,346]
‘Active ester’ monomers that have been subject to RAFT polymerization are 379–386 shown in Table 25.[465] These active ester groups undergo facile reaction with, in particular, substrates with primary amine groups. Other monomers with reactive functionality that have been successfully (co)polymerized include 391 (with thiol reactive functionality)[161,162] and 392 (with protected aldehyde functionality) (Table 26).[97,98]
Copolymers of monomers with reactive functionality, such as 371–392 (Tables 24–26), have potential application as scaffolds for bioconjugation. This and other applications are considered below in the section Polymer Brushes/Graft Copolymers/Comb Polymers/Surface Modification.
End-Functional Polymers and End-Group Transformations
Processes for thiocarbonylthio end-group removal or transformation post RAFT polymerization continue to attract significant interest.
Radical-Induced Reduction
Several new papers have appeared on radical-induced reduction that allows the thiocarbonylthio group of a RAFT-synthesized polymer to be replaced with a hydrogen atom.[94,244,395,469] It was found that the end-groups of the (meth)acrylic polymers, e.g., PBA and PMMA were more readily reduced than those of PSt and the efficiency of hydrogen atom donors increases in the series toluene < propan-2-ol < triethylsilane < triphenylsilane ~ tris(trimethylsilyl)silane ~ N-ethylpiperidine hypophosphite < tri-n-butylstannane.[94] The use of hypophosphite salts, in particular, N-ethylpiperidine hypophosphite, overcomes reagent toxicity and workup issues associated with the use of stannane and silane H-donors. For organic-soluble polymers workup is simplified to a water wash. Radical-induced reduction with tri-n-butylstannane was used to remove xanthate chain ends from PVAc (Table 27).[395]

|
Addition–Fragmentation Coupling
Another popular method for end-group removal/transformation involves heating the RAFT-synthesized polymer with a large excess of a radical initiator, most often an azo compound.[470,471] Recent examples include removal of dithioester or trithiocarbonate end-groups from polymers with a terminal methacrylate, acrylate, methacrylamide, acrylamide unit, or styrene (Table 28).
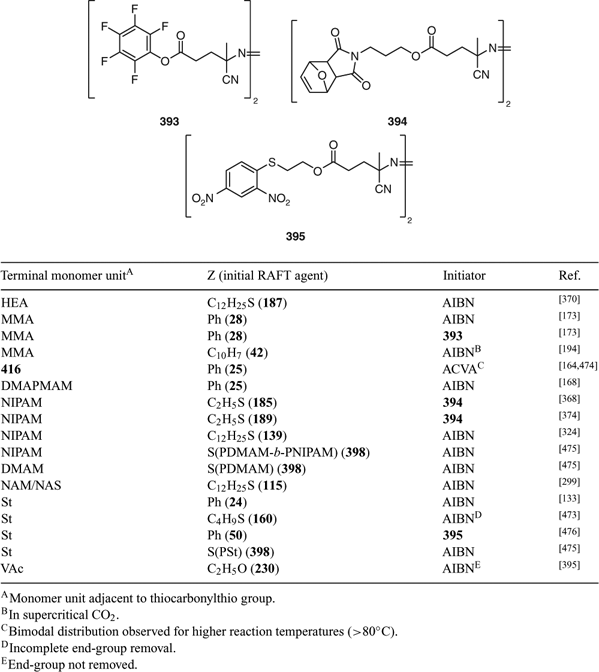
|
It was reported that bimodal distributions are obtained with a MA polymer when temperatures higher than 80°C were used.[164] The problem was attributed to the higher radical flux that results at higher temperatures. We found that with this method, while successful for removing the chain ends of polymers with a terminal MA unit, it was not possible to obtain complete removal of dithiobenzoate or butyl trithiocarbonate ends from polymers with a terminal BA[472] or St unit,[472,473] even when using a very large molar excess (up to 100-fold) of initiator (AIBN) and extended reaction times. The method was not effective for removing xanthate chain ends from PVAc.[395] The issues with incomplete end-group removal are most likely associated with the cyanoisopropyl radical (a very good radical leaving group) not being effective in displacing the propagating species.
A further cautionary note when using this method to introduce functionality by end-group transformation, is that the reaction of (for example) cyanoisopropyl radicals with propagating radicals may involve disproportionation as well as the intended combination. For example, the combination/disproportionation ratio (ktd/ktc) for cyanoisopropyl reacting with PSt propagating radical is 0.61.[1]
The use of peroxide initiators (didodecyl peroxide, dibenzoyl peroxide) in end-group removal has also been explored in the context of using such initiators in radical-induced reduction.[94] These initiators appear generally more effective than azo initiators, for example, AIBN, in achieving end-group removal. However, the complication of termination by self reaction of propagating species is much more pronounced.[94]
Thermolysis
The thermal stability of RAFT-synthesized PMMA has been studied by several groups.[116,121] PMMA with a methyl trithiocarbonate end-group undergoes loss of that end-group at ~180°C, at least in part, by homolysis of the C–CS2SCH3 bond and subsequent depropagation.[121] In contrast, PMMA with a dithiobenzoate end appears more stable. Only the end-group is lost at ~180°C and the dominant mechanism is proposed to be a concerted elimination process analogous to that involved in the Chugaev reaction (Scheme 8).[121]
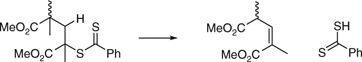
|
Thermal elimination of the end-groups from PSt or PBA with secondary xanthate end-groups occurs at relatively low temperatures. It is proposed that the elimination proceeds selectively to produce a polymer with a thiol chain end also by a Chugaev mechanism (Scheme 9).[469] In contrast butyl trithiocarbonate end-groups are cleaved from RAFT-synthesized PSt[473,477] and PBA[477,478] at relatively high temperatures with elimination of a thiol and formation of an unsaturated end-group.
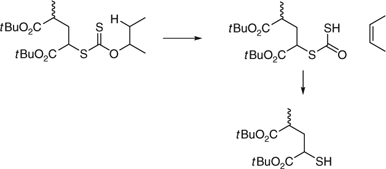
|
Aminolysis/Hydrolysis/Ionic Reduction
A summary of the use of aminolysis or ionic reduction (typically use of borohydride) is provided in Table 29. It was reported that aminolysis of PNIPAM trithiocarbonate proceeds quantitatively to the thiol when conducted in the presence of a small amount of a phosphine reducing agent (TCEP).[371,372] Two groups have compared aminolysis with borohydride reduction for PSt dithiobenzoate.[117,205] One study[205] compared (a) sodium borohydride in aqueous THF, (b) sodium borohydride in toluene with phase transfer catalysis, (c) lithium triethylborohydride in THF, and (d) propylamine in THF. The reaction (a) with sodium borohydride in aqueous THF was slow yet yielded the cleanest product. Aminolysis with propylamine (d) gave a substantial yield of the disulfide. The other reductions (b) and (c) gave, in addition, other unidentified byproducts. In the second study it was reported that disulfide formation during borohydride reduction or during was a problem even in the presence of a phosphine, P(Bu)3.[117]

|
Several groups have found that disulfide formation can be prevented if the thiol formed can be allowed to react in situ.[117,130,373,479,480] For example, end-group reduction can be performed in the presence of a Michael acceptor (to do an in-situ thiol-ene reaction).[117,130,479] This methodology has been applied to the synthesis of three-armed stars by an in-situ thiol-ene reaction with a triacrylate[130] and has been applied to directly make biopolymer conjugates.[479]
A recent study shows that dithioester and trithiocarbonate RAFT agents [R–SC(S)Z] undergo a radical-catalyzed reaction with thiols that results in oxidation of the thiol to a disulfide and concomitant reduction of the RAFT agent to R–H.[481] The relative rates of reaction suggested that dithioester RAFT agents should be preferred and symmetrical trithiocarbonates avoided when preparing polymers with terminal thiol functionality by RAFT polymerization followed by nucleophilic cleavage of the thiocarbonylthio group. This would minimize the production of non-functional and coupled polymers.
Aminolysis of PMMA-dithiobenzoate, as well as being complicated by coupling to provide the disulfide, is further complicated by the thiol undertaking a backbiting reaction to give a thiolactone chain end (Scheme 10).[183,480] Aminolysis in the presence of S-methyl methanethiosulfonate is reported to quantitatively produce PMMA with methanedithio chain ends even in non-degassed media (Scheme 10).[480] Functional methanethiosulfonates can be used in similar fashion to make the corresponding end-functional polymers. Thus, S-but-3-ynyl methanethiosulfonate was used to prepare an alkyne end functional polymer for use in ‘click’ chemistry.[89]
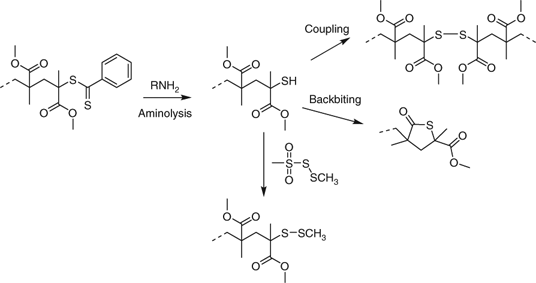
|
In other examples of this strategy, aminolysis of PNIPAM-trithiocarbonate was performed in the presence of 2,2′-dithiodipyridine to form a polymer with a pyridyl disulfide end-group in very high yield,[479] similarly, PNVP-xanthate was protected with Ellman’s reagent (see section on Block Copolymers).[396] These polymers can be used to regenerate a polymer with a thiol end-group under mild conditions and/or can be used to make biopolymer conjugates by a disulfide exchange process.
The issue of removal of byproducts following end-group removal can be resolved through use of a functional reagent (e.g., a functional amine)[482] and or a Z-functional RAFT agent[483] to simplify polymer purification.
Click Reactions
Use of ‘click chemistry’ in polymer chemistry has recently attracted much attention.[461–464] The RAFT process can be used to synthesize polymers with clickable moieties at the chain ends through the use of RAFT agents with appropriate functionality on ‘Z’ or ‘R’. A significant number of these papers concern the combination of RAFT and azide–alkyne 1,3-dipolar cycloaddition. Many RAFT agents with azido-functionality (27, 137, 148, 181, 218, 220) or acetylene-functionality (26, 46, 134) have appeared. Ladmiral et al.[335] have posted a warning that azides also undergo 1,3-dipolar cycloaddition with common monomers (MMA, MA, NIPAM, and St were studied) and that this can occur under polymerization conditions. The use of lower reaction temperatures in polymerization can minimize this issue.
Other click reactions are:
-
The thiol-ene reaction. Thiol-functional polymers are made as described above under Aminolysis/Hydrolysis/Ionic Reduction. Ene-functional RAFT agents are also possible. However, the potential for reaction during RAFT polymerization needs to be considered. Maleimide functionality may be introduced as described under Addition–Fragmentation Coupling above.
-
The active ester-amine reaction. RAFT agents with active ester functionality on ‘R’ include: 28, 31, 133.[173]
-
The pyridyl disulfide-thiol reaction. RAFT agents with pyridyl disulfide functionality on ‘Z’ include: 181–184.
RAFT agents and macro-RAFT agents with electron withdrawing ‘Z’ (62, 265) have been shown to undergo hetero-Diels–Alder reactions with suitable dienes (Scheme 11).[209,223,224,418,419,484,485] The process has also been developed as a route to block copolymers[223,418] and star polymers.[224,418,484]
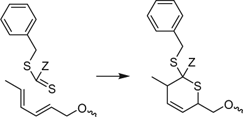
|
Other End-Group Modification Processes
PNVP chain ends formed with xanthate RAFT agents are susceptible to hydrolysis and can be converted into aldehyde chain ends (Scheme 12).[379] The aldehyde chain ends can be used for bioconjugation. End group stability may also be an issue during RAFT polymerization of NVP particularly at higher reaction temperatures (>60°C).
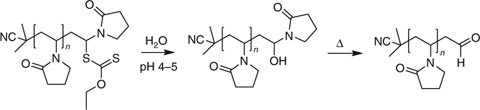
|
Macro-RAFT agents can be selectively end-functionalized by addition of a single monomer unit to form a new macro-RAFT agent. The strategy involves selection of monomer or reaction conditions such that there is no polymerization. Recent examples involve attachment of MAH to a PSt macro-RAFT agent[186] and a single maleimide unit to a PDMEAMA macro-RAFT agent.[426]
Statistical/Gradient Copolymers
It should be noted that, due to the effects of compositional drift, most copolymers prepared by RAFT polymerization generally fall under the description of gradient copolymers. Many examples of copolymer synthesis are included in the Tables above.
Kinetic simulations of monomer sequence distributions formed in RAFT polymerization have been performed.[266,486] Shot polymerizations were simulated to show the potential of using the RAFT process to produce copolymers with customized sequence distributions.[486] Stereogradient polymers (polymers in which the tacticity varies along the chain length) have been prepared by RAFT polymerization of TPMMA.[92]
RAFT copolymerizations of alk-1-enes with MAH have been shown to provide alternating copolymers with controlled molecular weights and relatively narrow molecular weight distributions.[314]
Block Copolymers
Examples of block copolymer synthesis by sequential; monomer addition are included in Tables 3–15. See also the section on Choice of RAFT agents for discussion on the importance of choosing which monomer to polymerize first when using this approach.
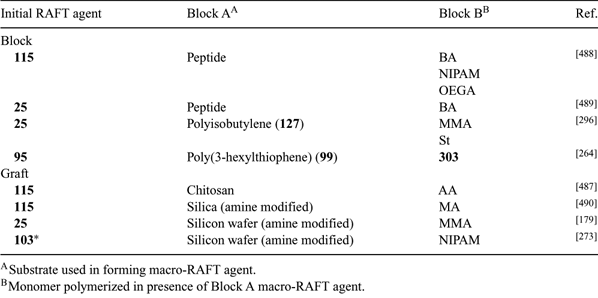
Block copolymers based on polymers formed by other polymerization mechanisms can be made by first preparing an end functional pre-polymer that is converted into a macro-RAFT agent by end-group transformation. This macro-RAFT agent is then used in the preparation of the desired block copolymer. Many examples of this strategy have now been reported. Macro-RAFT agents identified in Tables 3–15 include PEO macro-RAFT agents (33, 34, 78, 83, 126, 178, 183, 226, 227, 229, 230), PLA macro-RAFT agents (179, 180), a PE macro-RAFT agent (38), a PIB macro-RAFT agent (127), poly(3-hexylthiophene) macro-RAFT agents (99, 186), a poly(dimethylsiloxane) macro-RAFT agent (187), and a polyhedral oligomeric silsesquioxane (POSS) macro-RAFT agent (176).
Recently, there has also been substantial interest in the synthesis of block and graft copolymers (conjugates) with biological polymers. One approach has been to attach a RAFT agent functionality to the biopolymer so as to form a biopolymer macro-RAFT agent. Thus, BSA was converted into a macro-RAFT agent through reaction of the BSA thiol group with a pyridyl disulfide functional RAFT agent 183[366] or the maleimide functional RAFT agent 136.[329] These macro-RAFT agents were then used to synthesize conjugates through polymerization of NIPAM in aqueous media without denaturing the BSA.
A frequently used approach to the synthesis of macro-RAFT agents is the carbodiimide-assisted coupling of carboxy-functional RAFT agents with amino- or hydroxy-functionality in the (macro)molecule of interest (e.g., Schemes 13 and 14). The same approach may be used in forming graft copolymers to polyfunctional substrates such as chitosan.[487] For additional examples see Table 30 and our previous reviews.[7,8]
|
|
|
Block copolymers are also synthesized by coupling the RAFT-synthesized polymer with another polymer through functionality on either the ‘R’ or ‘Z’ groups. This is the most commonly exploited approach to make protein and other biopolymer conjugates.[491–494] Note that use of functionality on ‘Z’ leaves the thiocarbonylthio group as a potentially degradable link at the block juncture. This may or may not be advantageous depending on the application.
In making biopolymer conjugates, the biopolymer can be attached to a synthetic polymer having appropriate reactive functionality introduced through the use of a functional RAFT agent or attached post-RAFT polymerization by end-group transformation. Methods used include:
-
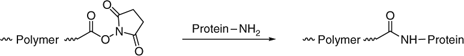
the use of active ester end-groups to react with amino-functional proteins. RAFT agents with active ester functionality on ‘R’ include 28, 30, 31, and 133 (Scheme 15).
-
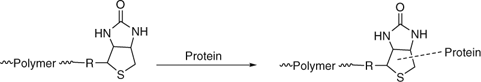
pyridyl disulfide end-groups with thiol functional proteins. RAFT agents with pyridyl disulfide functionality on ‘Z’ include 105[282] and 181–184 (Scheme 16).
-
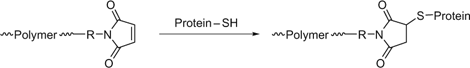
thiol-ene chemistry with thiol functional proteins. Maleimide functionality may be introduced post polymerization with use of addition–fragmentation coupling to replace a trithiocarbonate end-group (Scheme 17).[368,374]
-
azide–alkyne click chemistry. The stragegy requires attachment of alkyne (or azide) functionality to the protein. Azide-functional RAFT agents used in this context include 137[331,332] and 181 (Scheme 18).[364]
-
the attachment of ligands that show specific protein binding, for example, that of biotin with streptavidin (Scheme 19). RAFT agents incorporating biotin include 56[211,212] and 185.[368] The biotin moiety has also been attached post polymerization by azide–alkyne click chemistry[364] or by thiol-ene chemistry.[412]
-
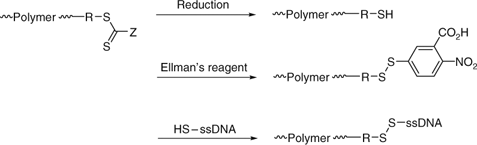
conversion of thiocarbonylthio groups of RAFT-synthesized polymers into thiol functionalities which are then used for conjugation. The thiol-end-groups were protected by reaction with Ellman’s reagent [5,5′-dithiobis-(2-nitrobenzoic acid)] which also activates the thiol for reaction with a thio functional biopolymer (ssDNA, peptide) by disulfide exchange (Scheme 20).[396]
|
|
|
|
|
|
|
|
Most of these same approaches have also been used in making other (non-biopolymer) block, star, and graft copolymers.
The synthesis of block copolymers by a combination of ATRP and RAFT has been reported. Several groups[495,496] have reported processes where a first block comprising a ‘more activated monomer’ is prepared by ATRP and then a second block comprising a ‘less activated monomer’ is prepared by RAFT (e.g., PtBA-b-PVAc, Scheme 21[495] or PSt-b-PNVP[496]). Sequential ATRP and RAFT has also been used to prepare block copolymer brushes.[497]
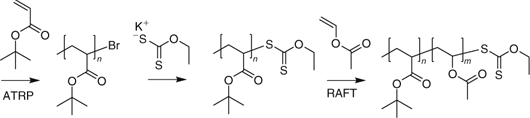
|
Matyjaszewski and coworkers[498] used a compound that combined a RAFT agent and an ATRP initiator functionality in the one molecule to synthesize PVAc-b-PMA (Scheme 22). Tong et al.[499] used a similar approach in the synthesis of PSt-b-PVAc. Other permutations of this approach were also explored.[498] The methodology was unsuccessful for PMMA-b-PVAc which was attributed to inefficient initiation of VAc polymerization by the PMMA propagating radical.
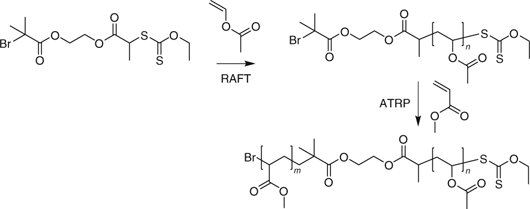
|
Multiblock Copolymers
There have been several new reports on the synthesis of multiblock copolymers by RAFT polymerization using poly-RAFT agents (polymers with in chain thiocarbonylthio functionality). One example is the poly(dimethylsiloxane) multiblock made using 396 as shown in Scheme 23.[500] Other examples are mentioned in Table 32.
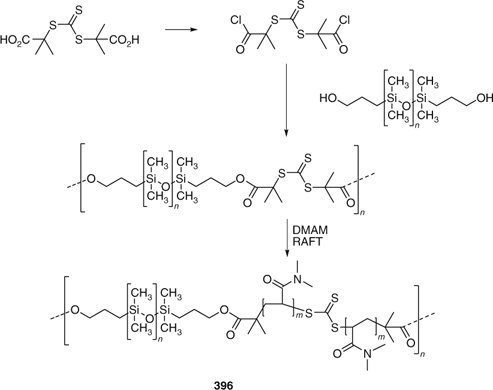
|

|
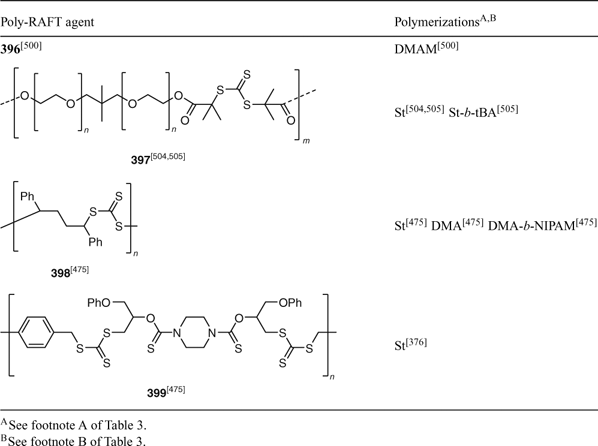
|
Further examples of cyclic trithiocarbonates have been reported as precursors to poly-RAFT agents (Table 33). Trithiocarbonate 400 was used to prepare multiblock copolymers of P4VP and PSt by sequential monomer addition.[501,502] RAFT polymerization with the cyclic trithiocarbonates (400–402) provides polymers of high dispersity (e.g., ÐM ~ 6 for PSt prepared with 402) with multiple trithiocarbonate units per chain, although individual segments have relatively low dispersity (ÐM < 1.3). Low temperature polymerization of MA or St in the presence of the cyclic xanthate (403) produced mixtures of cyclic and linear polymer that contain only one residue from 403 per chain.[503]

|
Another RAFT-based approach to multiblock copolymers is based on oxidative coupling of telechelic polymers with thiol end-groups.[196,240–242,260] The telechelics are synthesized by RAFT polymerization making use of a bis-RAFT agent with subsequent end-group transformation to form the bis-thiol. The procedure was also applied to synthesize cyclic polymers by using high dilution conditions.[260]
Star Polymers
The literature on the synthesis of star polymers using the RAFT process continues to grow rapidly. In this section we consider syntheses that begin with a substrate containing multiple thiocarbonylthio groups of appropriate design, a multi-RAFT agent, and ‘grafting-to’ syntheses that involve coupling of RAFT-made polymers to a core or arms of defined structure. Formation of star nano- or microgels by copolymerization with a divinyl monomer, by self-assembly and crosslinking of block copolymers or by growth from a crosslinked polymer or a nanoparticle core is covered in the next section, Microgels and Nanoparticles.
Use of Multi-RAFT Agents
The multi-RAFT agent may in principle be a small organic compound, an organometallic complex, a dendrimer, a hyperbranched species, a macromolecular species, a particle or indeed any moiety that possesses multiple thiocarbonylthio groups. Of these, macromolecular species and particles are considered in the section on Polymer Brushes below. Recent examples of star cores are shown in Table 4 and Table 6 (dithioesters), Table 9 (trithiocarbonates), and Table 11 (xanthates).
The first publications in this field[6] recognized two limiting forms of star (or graft/brush copolymer) growth depending on the orientation of the thiocarbonylthio group with respect to the core.
In the first strategy, the propagating radicals are linear chains that dissociate from the core. ‘Z’-connected RAFT agents (404, Scheme 24) are employed. The advantage of this strategy is that by-products from star–star coupling are unlikely. The thiocarbonylthio functionality is retained at the core of the star. A potential disadvantage of the ‘propagation away from core’ strategy is that reactions that cleave the thiocarbonylthio groups (e.g., aminolysis and thermolysis) cause destruction of the star structure (i.e., loss of the arms, Scheme 24). This feature can also be used to advantage in developing supported polymer syntheses or degradable network polymers (see Polymer Networks). A further potential issue is that the thiocarbonylthio functionality may become sterically inaccessible as the polymerization proceeds.
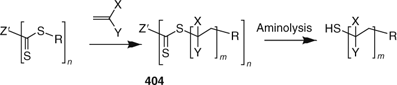
|
In the second strategy most propagating radicals remain attached to the core and ‘R’-connected RAFT agents (405, Scheme 25) are used. Most thiocarbonylthio functionality remains on the periphery of the star. However, a linear macro-RAFT agent is released to the polymerization medium by the RAFT process. Since propagating radicals are attached to the core, termination by star–star coupling is a complication. Because the thiocarbonylthio groups are end-groups, they can be cleaved (e.g., by aminolysis) without destroying the star structure (Scheme 25).
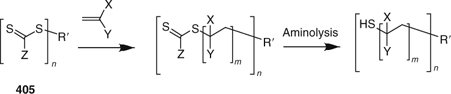
|
The advantages and disadvantages of the two approaches are considered in detail in several recent papers.[20,248,256,259,268,377,509–511] It is clear that the relative importance of the various factors mentioned above depends strongly on the particular monomer and RAFT agent used.
The leaving group ability of ‘R’ has been shown to have a significant effect on the molecular weight distribution of the arms. For ‘Z’-connected PSt stars formed with multi-RAFT agents (type 405) with R′ = benzyl (a relatively poor radical leaving group) the dispersities for the cleaved arms distribution is relatively broad even though the dispersity of the star polymers is narrow.[377] Substantially lower dispersities for the cleaved arms are seen for the similar RAFT agent with R′ = phenylethyl.[377] It is known that RAFT agents with R = phenylethyl have transfer constants an order of magnitude higher than those with R = benzyl.[56]
Stars in which the arms are connected to the core through ionic bonding have been described.[512,513] The approach has been exploited in synthesizing mikto-arm star polymers.[513] Thus, the PSt macro-RAFT agent 406 (Fig. 6) was used to make a mikto-arm star with PSt and PNIPAM arms. The ionic bonded arms were stable during RAFT polymerization and in solution surviving characterization by gel permeation chromatography. However, the PNIPAM arms were readily released from the core by treating the polymer with acid.[502]
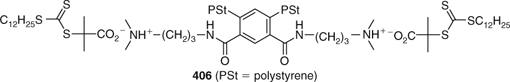
|
Star Polymers by ‘Grafting-To’
Various combinations of RAFT, ATRP, click, ring opening polymerization, and other methods have been used to synthesize star polymers, including mikto-arm stars.[174,223,224,330,418,514] Hetero-Diels–Alder coupling was used to synthesize a 12-armed star as shown in Scheme 26 from a star polymer precursor prepared by ATRP and a PSt macro-RAFT agent.[418]
|
Microgels and Nanoparticles
In this section we consider direct formation of polymer microgels or nanoparticles in a solution polymerization process or by self-assembly and crosslinking of block copolymers. Surface functionalization of nanoparticles by RAFT polymerization is considered in the next section. Formation of particles and nanoparticles by heterogeneous polymerization (emulsion and miniemulsion polymerization) is discussed above.
The synthesis of nanoparticles by block copolymer self assembly and crosslinking of micelles so formed has been reported by several groups. Examples are included in Table 34.

|
Polymeric nanoparticles were prepared through the chain collapse of linear polymers driven by noncovalent cross-linking of copolymers that contain dendritic self-complementary hydrogen-bonding monomers (283 and 284).[197] The behaviour observed was strongly dependent on the level of incorporation of the dendritic monomers.
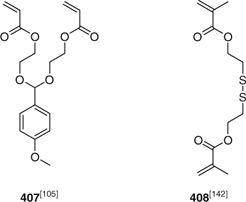
One of the more popular methods for forming core-crosslinked star polymers (microgels, nanogels) involves (co)polymerization of a divinyl monomer using some form of reversible deactivation polymerization.[516,517] Examples of this strategy in combination with RAFT polymerization include the synthesis of drug delivery vehicles involving use of an acid cleavable crosslinker 407[105] or the biodegradable crosslinker 408[142] (Fig. 7) and the synthesis of PSt star polymers with active ester groups on the periphery by making use of RAFT agent 133.[325] Other recent examples of applying this strategy are included in Table 35.
|

|
The formation of so-called capsosomes (liposome-compartmentalized polymer capsules) has been described. The process involved liposomes being sandwiched between a cholesterol-modified poly(l-lysine) precursor layer and a RAFT-synthesized poly(methacrylic acid)-co-(cholesteryl methacrylate) capping layer.[295]
RAFT polymerization may be used to grow arms on crosslinked cores formed by RAFT or other processes. For example, hyperbranched poly(glycerol) core functionalized with an azo-initiator and an ATRP initiator functionality was used to sequentially form arms by RAFT polymerization and ATRP to provide a mikto-arm structure.[519]
Polymer Brushes/Graft Copolymers/Comb Polymers/Surface Modification
Grafting-From Processes
The grafting-from approach involves attachment of a RAFT agent functionality to a surface and using this material as a macro-RAFT agent in polymerization. A variant on this approach is to attach initiator functionality which is then used to generate a macro-RAFT agent in situ during a RAFT polymerization.
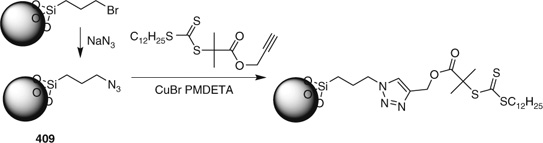
The synthesis of polymer–silica nanocomposites has been reviewed.[520] A variety of methods have been used to synthesize silica-bound RAFT agent functionality for surface-initiated grafting (Table 36). Several papers have appeared on the synthesis of brushes on silica nanoparticles using combinations of click chemistry and RAFT.[326,327,521] Ranjan et al.[326,521] reported on three variants. In the first ‘grafting-to’ approach, polymers made with RAFT agent were coupled to azide-functional silica particles. In the second ‘grafting-from’ approach polymers were grown from the silica particle bound RAFT agent (Scheme 27). In the third ‘tandem’ approach a click reaction and a RAFT polymerization step were performed simultaneously.

|
|
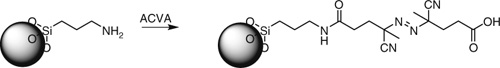
Mesoporous silica beads modified with an azo initiator (Scheme 28) were used to initiate RAFT polymerization of NIPAM mediated by benzyl dithiobenzoate.[219] The beads were evaluated as a thermoresponsive chromatographic support.
|
Other substrates to which the ‘grafting-from’ approach has been applied include silicon wafers,[179,213,524] poly(silsesquioxane),[210,228] polyhedral oligomeric silsesquioxane (POSS),[350,360,361] indium-tin oxide (ITO) surfaces,[176] carbon nanotubes,[525] titania particles,[526,527] clay,[528–530] cadmium sulfide nanoparticles,[531] cellulose,[532–535] and PVDF membranes.[536]
A RAFT agent with a pendent norbornene was polymerized by ROMP and the product used in RAFT polymerization to form a comb with P(St/MAH)-b-PSt arms.[323] Monomers with ATRP initiator functionality (323) have been subjected to RAFT polymerization and the polymers formed used in ATRP to form comb polymers.
RAFT agent functionalized silicon wafers were prepared by treatment of the wafer with RAFT agents having methoxysilane (59, 84, or 97),[213] or active ester (31)[179] functional groups on ‘R’ or with TMSPMA, AIBN, and phenylethyl dithiobenzoate (49) in a one-step process.[537] Silicon wafers have also been modified with potassium O-ethylxanthate and this was used to form surface-initiated MMA grafts.[524] The result is surprising because xanthates are generally not useful for controlling MMA polymerization.
Radiation-induced graft polymerization is believed to involve formation of initiating species on the substrate, which then initiate polymerization. Thus, further examples of a grafting-from approach are gamma-initiated grafting of styrene[438] or SSO3Na[167] from cellulose, and tBA from commercial ethylene propylene copolymer[439] in the presence of RAFT agent 92.
Grafting-To Processes
One use of RAFT polymerization in the ‘grafting-to’ approach involves the synthesis of polymers with reactive end-groups or block structures by RAFT polymerization, which are then self-assembled and/or bonded to a particle, surface, or other structure. This approach has been used to make inorganic (TiO2, SnO2 or ZnO) nanorods[102] or CdSe/ZnS quantum dots[538] with PAA-b-PSt,[539] P(AEME-co-328-co-342)[278] or semiconductor (P364 or P365) arms, gold nanoparticles with PSt,[476] PSt-b-P2VP,[198] PDMAEMA, PAA, or PSt,[91] P(340-co-AM) or P(341-co-AM),[239] PPEGA-b-PNIPAM,[217] PMMA-b-PNIPAM,[189] or P(DEGMA-co-tBA), P(DEGMA-co-tBA)-b-PGMA, PGMA, PSt[398] grafts, magnetic iron with PNIPAM grafts,[305] silver nanoparticles with PSt grafts,[409] chitosan with PAA grafts,[487] and silicon wafers with NIPAM grafts.[273] Silicon wafers and silica particles have also been modified with multiple layers of an amine-functional polymer, polyethyleneimine or poly(allylamine hydrochloride), respectively, and PAA-b-PSSO3Na in a layer-by-layer assembly process.[354]
A second way that RAFT is applied in a ‘grafting-to’ approach involves the use of RAFT copolymerization to synthesize a functional scaffold to which other polymers and or functionality are subsequently attached.
RAFT polymerization has been used to synthesize the scaffolds for attachment of biopolymers by copolymerization of monomers shown in Table 24 (azide or alkyne functionality), Table 25 (active ester functionality, e.g., Scheme 29) for attachment of therapeutic agents,[317] peptides,[160,317] or DNA,[347] or Table 26 (e.g., pyridyl disulfide functionality (391), Scheme 30) for attachment of thiol residues of proteins (e.g., BSA),[162] or drug molecules.[161] The production of such scaffolds by RAFT and other RDRP processes for post polymerization modification has recently been reviewed.[540]
|
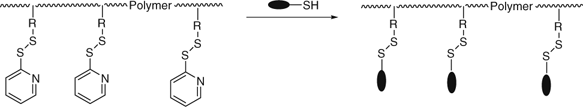
|
The general approach has wide application and has, for example, been used in the synthesis of photochromic polymers by coupling of spiropyran moieties to RAFT-synthesized P(BMA-co-HEMA).[106]
Grafting-Through Processes
This approach requires attaching monomer functionality to the substrate which may be:
-
A polymer chain (Table 37). Macromonomers used in RAFT polymerization include the valine-proline-glycine-valine-glycine (VPGVG) pentapeptide macromonomer 416,[163,164] the oligo(2-ethyl-2-oxazoline) macromonomer 417,[112] and the polycaprolactone (PCL) macromonomer 419.[363] The pH-sensitive dexamethasone-containing peptide macromonomer 420 has been synthesized and copolymerized with HPMA.[274] The conjugate was said to be an effective anti-arthritis therapy.
-
Particles and surfaces. Nanocomposite materials have been prepared by the grafting-through polymerization of acrylate-[527] or methacrylate-functional[541] titania particles or methacrylate-functional silica particles.[542]
|
The process of formation of polymers that contain active ester functionality or pyridyl disufide functionality as substrates for grafting-to processes (see above) can be considered as additional examples of grafting-through processes.
Polymer Networks
Polymer networks can be formed by crosslinking RAFT-synthesized homopolymers, block copolymers,[95,243,370] or star polymers[267] or can be formed directly by a RAFT (co)polymerization in te presence of crosslinking monomer (e.g., a divinyl monomer, ethylene glycol dimethacrylate, methylene-bis-acrylamide).[544,545] A wide variety of crosslinking processes have been explored. Polymer networks have applications in the controlled release of, for example, pharmaceuticals. Physically crosslinked hydrogel networks can be prepared by the assembly of RAFT-synthesized triblock copolymers.[86,272,546–548] The structures can be tuned to absorb either hydrophilic or hydrophobic substrates and to swell in specific media or in response to various stimuli such as pH, temperature, ionic strength, etc. by appropriate monomer selection.
Networks based on stars[267] or triblock copolymers[546] with trithiocarbonate linking groups can be degraded by aminolysis to form a polymer with thiol end-groups which is a precursor to a redox-responsive network that can be formed (through disulfide crosslinking) and degraded by addition of an oxidizing- or a reducing-agent, respectively.[267,546]
Other applications are in the area of
-
photo-embossing.[549] Photo-embossed surface relief structures with significantly improved aspect ratios were prepared by addition of a RAFT agent (23) to a polymerization mixture of benzyl methacrylate and dipentaerythritol penta/hexa-acrylate. The RAFT photo-embossing process also proved less sensitive to air.
-
production of molecularly imprinted monolithic chromatographic columns.[550] The monoliths were formed by copolymerization of MMA and ethylene glycol dimethacrylate in the presence of RAFT agent 110 and a template molecule. The use of RAFT polymerization allows better control over pore size distribution and provided columns with better resolution.
-
polymer network liquid crystals.[551] Devices were formed by polymerization of hexanediol diacrylate in the presence of PSt macro-RAFT agents of various molecular weights and the nematic liquid crystal on an ITO glass substrate.
Conclusions
In the past two and a half years we have seen a substantial expansion in the number of papers on applications of RAFT polymerization. These applications cover areas ranging from biomedical to electronics and photonics to coatings and rheology control agents. At the same time, we have not seen any reduction in the number of papers that explore RAFT polymerization as a technique but rather we have seen an increase as people seek both to improve the process and further define the intimate details of the RAFT mechanism.
Abbreviations
Abbreviations: AcS: 4-acetoxystyrene, AA: acrylic acid, AAEMA: 2-(acetoacetoxy)ethyl methacrylate, AEMA: 2-aminoethyl methacrylate hydrochloride (273), AEMAM: N-(2-aminoethyl)methacrylamide hydrochloride (275), AEP: 2-(acryloyloxy)ethyl phosphate, AIBN: azoisobutyronitrile, ACHN: azobis(1-cyclohexanenitrile), ACVA: azobis(cyanovaleric acid), AM: acrylamide, AMA: allyl methacrylate, AMPS: 2-acrylamido-2-methylpropanesulfonic acid sodium salt (AMPS), AN: acrylonitrile, APMAM: N-(3-aminopropyl)methacrylamide hydrochloride (274), ATRP: atom transfer radical polymerization, BA: butyl acrylate, BAM: N-butylacrylamide, BP: β-pinene, CA: cholesteryl methacrylate (322), CIDNP: chemically induced nuclear polarization, CMS: 4-(chloromethyl)styrene, CMA: cholesteryl methacrylate (309), CPA: 3-chloropropyl acrylate, DA: dodecyl acrylate, DADMAC: diallyldimethylammonium chloride, Dc: dec-1-ene, DEAM: N,N-diethylacrylamide, DEGMA: (diethylene glycol monomethyl ether) methacrylate, DEHEA: di(ethylene glycol) 2-ethylhexyl ether acrylate, ÐM: molar-mass dispersity = ratio of weight average to number average molecular weight,[552] DMAM: N,N-dimethylacrylamide, DMAEA: 2-(dimethylamino)ethyl acrylate, DMAEMA: 2-(dimethylamino)ethyl methacrylate, DMAPMAM: N-(3-(dimethylamino)propyl)methacrylamide hydrochloride, DPAEMA: 2-(diisopropylamino)ethyl methacrylate, EA: ethyl acrylate, EAA: ethyl α-acetoxyacrylate, E: ethene, EEA: ethoxyethyl acrylate, EGMA: (ethylene glycol monomethyl ether) methacrylate, EHA: 2-ethylhexyl acrylate, EMAM: N-ethyl-N-methylacrylamide, EVC: N-ethyl-3-vinylcarbazole, EVE: ethyl vinyl ether, GMA: glycidyl methacrylate, HEA: 2-hydroxyethyl acrylate, HEMA: 2-hydroxyethyl methacrylate, HMA: hexylmethacrylate, HMS: 4-(hydroxymethyl)styrene, HPMAM: N-hydroxypropyl methacrylamide, iBA: isobornyl acrylate, iBMA: isobutyl methacrylate, Ip: isoprene, LAM: less activated monomer (includes vinyl monomers such as VAc, NVP, and NVC), LMA: dodecyl methacrylate, MA: methyl acrylate, MADIX: Macromolecular Design by Interchange of Xanthate, MAEP: 2-(methacryloyloxy)ethyl phosphate, MAH: maleic anhydride, MAM: more activated monomer (includes styrenes, acrylates, acrylamides), MMA: methyl methacrylate, MMBL: γ-methyl-α-methylene-γ-butyrolactone (290), MPC: methacryloyloxyethyl phosphorylcholine (292), MVK: methyl vinyl ketone, NAM: N-acryloylmorpholine, NAPM: N-acryloyl-l-proline methyl ester, NAPAM: N-acryloyl-l-phenylalanine methyl ester, NAP: N-acryloylpyrrolidine, NAS: N-acryloyloxysuccinimide, NEMI: N-ethylmaleimide, NIPAM: N-isopropylacrylamide, NIPMAM: N-isopropylmethacrylamide, NMMI: N-methylmaleimide, NMP: nitroxide mediated polymerization, NPMI: N-phenylmaleimide, NMS: N-methacryloyloxysuccinimide, NVC: N-vinylcarbazole, NVCL: N-vinylcaprolactam, NVP: N-vinylpyrrolidone, NVPI: N-vinylphthalimide, PAM: N-propylacrylamide, PA: propargyl acrylate (372), PAA: α-propylacrylic acid (2-methylenepentanoic acid), PE: polyethylene, PEG: poly(ethylene glycol) monomethyl ether, PEGA: (poly(ethylene glycol) monomethyl ether) acrylate, PEGMA: (poly(ethylene glycol) monomethyl ether) methacrylate, PEO: poly(ethylene oxide), PFMA: pentafluorophenyl methacrylate, PFS: pentafluorophenylstyrene, PIB: polyisobutylene, PLA: poly(lactic acid), PMA: propargyl methacrylate, POSS: isobutyl polyhedral oligomeric silsesquioxane, PVK: phenyl vinyl ketone, RAFT: reversible addition fragmentation chain transfer, RDRP: reversible deactivation radical polymerization, SB: 4-(3-butenyl)styrene, siRNA: short interfering ribonucleic acid, SMe: 4-methylstyrene, ssDNA: single stranded deoxyribonucleic acid, SSO3Na: sodium styrene-4-sulfonate, St: styrene; TBAM: N-tert-butylacrylamide, TFPMA: 2,2,3,3-tetrafluoropropyl methacrylate, TFPA: 2,2,3,3-tetrafluoropropyl acrylate, THPA: tetrahydropyran, TMSEMA: 2-(trimethylsilyloxy)-ethyl methacrylate, TMSPMA: 3-(trimethoxysilyl)propyl methacrylate, tBA: tert-butyl acrylate, tBS: 4-(tert-butoxy)styrene, TMAEMA: 2-(trimethylamonium)ethyl methacrylate, TMAPMA: 3-(trimethylamonium)propyl methacrylate, TPMMA: triphenylmethyl methacrylate (302), VAc: vinyl acetate, VB: vinyl butyrate, VBA: 4-vinylbenzoic acid, VBSC: 4-vinylbenzenesulfonyl chloride, VBDA: (4-vinylbenzyl)dimethylamine, VBTAC: (ar-vinylbenzyl)trimethylammonium chloride (351), VBTPC: (4-vinylbenzyl)trimethylphosphonium chloride (359), 2VP: 2-vinylypyridine, 4VP: 4-vinylpyridine. Polymer abbreviations are formed by adding the suffix ‘P’ to the corresponding monomer abbreviation. Thus PMMA: poly(methyl methacrylate).
[1]
[2]
T. R. Darling,
T. P. Davis,
M. Fryd,
A. A. Gridnev,
D. M. Haddleton,
S. D. Ittel,
R. R. Matheson,
G. Moad,
E. Rizzardo,
J. Polym. Sci., Part A: Polym. Chem. 2000, 38, 1706.
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
(John Wiley & Sons: Verlag).
[12]
[13]
[14]
C. Barner-Kowollik,
S. Perrier,
J. Polym. Sci., Part A: Polym. Chem. 2008, 46, 5715.
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
