

Single-molecule Photoswitching and Localization1
Sebastian van de Linde A , Steve Wolter A and Markus Sauer A BA Biotechnology and Biophysics, Julius–Maximilians University Wuerzburg, Am Hubland, 97074 Wuerzburg, Germany.
B Corresponding author. Email: m.sauer@uni-wuerzburg.de
Australian Journal of Chemistry 64(5) 503-511 https://doi.org/10.1071/CH10284
Submitted: 29 July 2010 Accepted: 4 November 2010 Published: 30 May 2011
Abstract
Within only a few years super-resolution fluorescence imaging based on single-molecule localization and image reconstruction has attracted considerable interest because it offers a comparatively simple way to achieve a substantially improved optical resolution down to ∼20 nm in the image plane. Since super-resolution imaging methods such as photoactivated localization microscopy, fluorescence photoactivation localization microscopy, stochastic optical reconstruction microscopy, and direct stochastic optical reconstruction microscopy rely critically on exact fitting of the centre of mass and the shape of the point-spread-function of isolated emitters unaffected by neighbouring fluorophores, controlled photoswitching or photoactivation of fluorophores is the key parameter for resolution improvement. This review will explain the principles and requirements of single-molecule based localization microscopy, and compare different super-resolution imaging concepts and highlight their strengths and limitations with respect to applications in fixed and living cells with high spatio-temporal resolution.
Introduction
Fluorescence microscopy allows the direct observation of cellular processes in a relatively non-invasive fashion with molecular specificity and high temporal resolution in three dimensions.[1] Driven by the availability of efficient fluorescent dyes, semiconductor nanocrystals, and fluorescent proteins (FP), as well as the development of refined labelling strategies, almost any protein of interest can be specifically labelled.[2–6] Because of the wave nature of light, however, the spatial resolution is normally limited to about half of the wavelength of the light in the imaging plane.[7] That is, conventional fluorescence microscopes do not provide insight into the structural organization of vital protein assemblies and machineries with a size of a few tens of nanometers. Only recently have methods emerged that enable super-resolution imaging with substantially improved optical resolution near molecular scales.[8–15] This has been achieved by temporal control of fluorescence emission using deterministic approaches such as in stimulated emission depletion (STED),[16–18] structured illumination,[19–21] and conceptually analogue methods,[22–24] or stochastic approaches relying on wide-field single-molecule detection and localization as in photoactivated localization microscopy (PALM),[25,26] fluorescence photoactivation localization microscopy (FPALM),[27,28] stochastic optical reconstruction microscopy (STORM),[29,30] direct stochastic optical reconstruction microscopy (dSTORM),[31,32] and related methods.[33–41]
This review covers the stochastic methods that isolate the fluorescence emission of individual fluorophores from one another using distinguishable optical characteristics with a focus on the dSTORM concept (Fig. 1). Because several excellent reviews have already been published[8–15] we focus on the description of the basic requirements necessary for photoswitching and localization of individual fluorophores. We introduce the general concepts behind single-molecule based two- (2D) and three-dimensional (3D) localization microscopy methods, outline different point-spread-function (PSF) fitting approaches, and compare and discuss advantages and limitations of reversibly photoswitchable and photoactivatable fluorophores related to specific labelling, labelling density, and dynamic experiments in living cells.
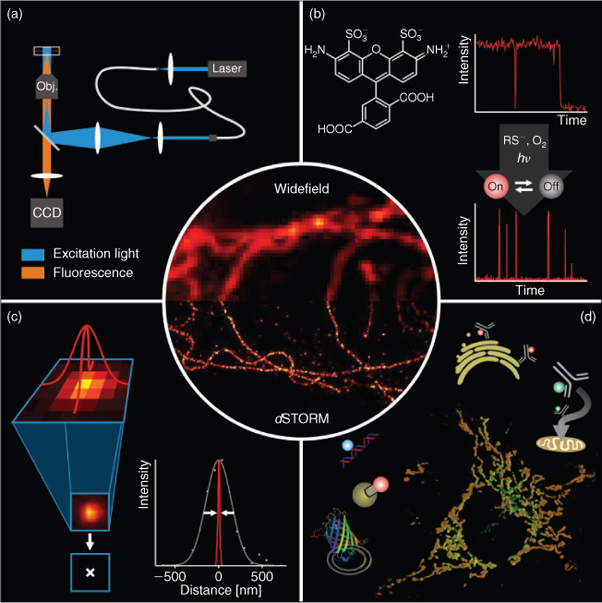
|
Fluorescence detection with a high signal-to-background ratio is the key for precise localization of the emission pattern of individual molecules. For negligible background noise the localization precision depends only on the number of collected photons N and on the standard deviation of the true PSF (σ) and can be approximated by σ/√N.[42,43] Given the fact that it is possible to detect thousands of fluorescence photons from a single organic fluorophore before it photobleaches, localization of individual fluorophores with one nanometer accuracy is feasible and was used successfully to monitor molecular motor dynamics.[44,45] From this point of view reversibly photoswitchable synthetic organic fluorophores in combination with STORM and dSTORM methods emerge as very attractive because they survive under moderate excitation conditions for prolonged time periods, can emit thousands of photons, and exhibit tunable photoswitching rates. However, to date specific labelling of proteins inside of cells with reversibly photoswitchable organic fluorophores is mainly limited to antibody-targeting in fixed cells. On the other hand, FP, whose reactivation with violet light after apparent photobleaching was shown for the first time more than 10 years ago,[46] have the advantage that they allow direct genetic labelling of proteins and dynamic measurements in living cells.
Underlying Concept of Single-molecule Based Localization Microscopy
To achieve super-resolution fluorescence imaging, fluorophore emission has to be separated in time. Whereas in the STED case, the phase mask defines the coordinates of fluorescence emission (predefined in space by the zero-node), single-molecule based localization microscopy methods randomly separate the emission of individual fluorescent molecules in time. This is achieved by stochastic activation of individual fluorophores, single-molecule detection using a wide-field fluorescence microscope equipped with a sensitive charge coupled device (CCD) camera, and precise position determination (localization), i.e., fitting of ideal PSF to the measured photon distributions. As long as the distance between individual fluorophores enables the unaffected analysis of the different emission spots, i.e., individual fluorophores are spaced further apart than the distance resolved by the microscope (>λ/2 on the CCD camera), the standard error of the fitted position can be made arbitrarily small by collecting more photons and minimizing noise factors.[43–45]
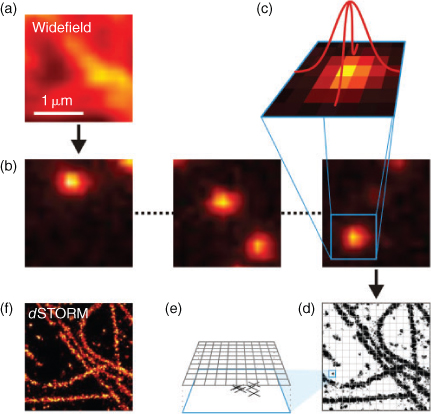
The principle procedure for single-molecule based localization microscopy is shown in Fig. 2. A target structure is densely labelled with photoswitchable or photoactivatable fluorophores, the majority of which have to be transferred to the non-fluorescent state. In the case of photoactivatable fluorophores, the fluorophores essentially reside in their non-fluorescent state before activation. If synthetic organic fluorophores are used, the majority of the fluorophores has to be converted into a metastable dark state upon irradiation with light. During the actual experiment, only a subset of single emitters is switched on stochastically per image. At any time the density of activated fluorophores should be low enough to allow the isolated localization of individual fluorophores. Activation and localization is repeated many times, and a final image with superior resolution is reconstructed from all single-molecule positions determined. Here, a lateral resolution of <50 nm is commonly achieved experimentally.[25,27,32,36,47]
|
For optimal data analysis (i.e., signal recognition and fitting) the signal-to-background ratio should be as high as possible. In general, super-resolution imaging methods based on single-molecule localization minimize fluorescence contributions from the background using an oil-immersion objective with high numerical aperture and total internal reflection fluorescence microscopy. However, it has to be pointed out that any wide-field fluorescence microscopy configuration (e.g., epi-illumination) can be used as long as the signal-to-background ratio is high enough to allow precise localization of the fluorescence signal of individual molecules. For example, illumination by a highly inclined and thin beam likewise enables single-molecule detection with a high signal-to-background ratio in the cytoplasm or nucleus of cells.[48]
As excitation sources, any continuous light source such as gas lasers or semiconductor lasers can be used provided that the excitation power is high enough, i.e., ≥20 mW for efficient excitation and read-out of single-molecule fluorescence signals and photoswitching. Dependent on the method used, excitation intensities in the range of less than 1 up to several tens of kW cm–2 are experimentally applied for read-out, photoswitching, or photobleaching of the fluorophores. Fluorescence detection is commonly performed using electron-multiplying charge coupled device (EMCCD) cameras with quantum yields of 80–90% in the visible range at frame rates of up to 1 kHz. The CCD chip collects the incoming photons in pixels with a typical physical size of 16 × 16 μm2 or 24 × 24 μm2. Thus, when a photon is collected by a pixel, information about the exact location of that photon is lost and all that is measured is the location of the pixel. To preserve most of the position information in the fluorescence signal data a pixel size of ∼2.3 times smaller than the Abbe resolution limit of the optical system should be used. Thus, for visible light the fluorescence signal should be imaged ensuring an image pixel size of 80–150 nm using appropriate magnification.
The CCD chip is used to record a time series of images with different activated fluorophore subsets, i.e., a video sequence containing several thousands of images is recorded. The number of images necessary to reconstruct a subdiffraction-resolution image with satisfying resolution is determined by the structure and varies typically between 4000 and 100 000 images. This series is compressed to a single super-resolved image by a computer in three stages: first, the number and location of fluorescent emissions is determined in each image, second, the most likely fluorophore position is computed for each emission, and third and finally the density of emission positions in all images combined is colour-coded to give a super-resolved image (Fig. 2). In the first stage, the computational task is to roughly find all positions where fluorophores have emitted, so that the position of each emission is approximated to a precision of ∼200 nm and the number of emissions estimated. With that information, more precise fitting is performed on each of the candidate positions to find the emitter position (localization) with nanometer precision. Here, three methods are commonly used: least-squares fitting, faster-than-least-squares methods, and maximum-likelihood estimation. Least-squares fitters operate by determining the sum of squared deviations from an ideal PSF and aim to reduce this sum by computing its gradients with respect to parameter change and following the gradient to a local minimum. The reader should note how this approach is quite tolerant to noise, but reliant on the initial position found in the first stage. Least-squares fitters have for a long time been considered the most precise and can be computed in real-time (considerably faster than the acquisition) on common PC.[49]
Before such real-time capability was available, several less precise methods had been developed to speed up computation.[43,50,51] With growing computational power, recent articles questioned whether the encountered noise may be adequately described with Gaussian statistics, which is the assumption underlying least-squares fitting, and used maximum likelihood estimators for Poisson statistics instead.[52,53]
Finally, a super-resolved image is reconstructed from this set of fitted emission positions by spanning a fine pixel grid (often 10 nm or less, much finer than the camera pixel array) and counting how many emissions were localized within or close to each pixel (Fig. 2). Colour-coding these counts yields an image that shows, with high accuracy, the number and positions of emissions (localizations) and thereby with a small statistical error the distribution of fluorescent probes in the sample.
Photoswitching and Photoactivation of Fluorophores
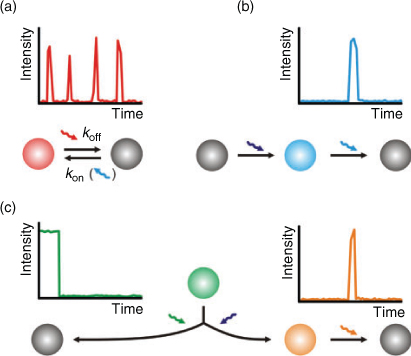
All photoswitchable fluorophores, including irreversible photoactivation and reversible photoswitching, exhibit a fluorescent on-state, a non-fluorescent off-state, and a transition between those states. In the past years, a huge amount of photoswitchable fluorophores has been developed including photochromic compounds,[54–56] conventional organic fluorophores,[31,35,36,40,47,57] caged fluorescent dyes,[58] push–pull fluorophores,[59] and a variety of photoswitchable FP.[4,60–62] In the following, we restrict our considerations to irreversible photoswitching of photoactivatable or photoconvertible FP and reversible photoswitching of FP and synthetic organic fluorophores since they are currently the most promising candidates for single-molecule based localization microscopy. Because both types of irreversible photoswitchable proteins, photoactivatable and photoconvertible FP, are ‘activated’ from an inactive to an active form, they are termed PA-FP.
Single molecule-based super-resolution imaging requires individual fluorophores to be turned ‘on’ and ‘off’ sequentially, either by photoswitching, photoactivation, irreversible photobleaching, or by any other stochastic photophysical process (Fig. 3). Compared with reversibly photoswitchable organic fluorophores, PA-FP[4,25–28] are superior concerning fluorescence labelling because they can be genetically fused to target proteins and endogenously expressed in cells and organelles. Moreover, the genetic expression of PA-FP ensures that the specificity and efficiency of protein labelling approaches nearly 100%, a level impossible to achieve by chemical staining with synthetic fluorophores. Finally PA-FP are much smaller than fluorophore-labelled antibodies, permitting higher labelling density in biological samples and allowing for higher imaging resolution according to the Nyquist criterion.[63] The number of detectable photons of PA-FP is <1000, i.e., typically a few hundred photons before they bleach, whereas organic synthetic fluorophores can emit >1000 photons per cycle enabling a higher localization precision.[14,43,64] Furthermore, PA-FP have to be photobleached upon readout before the next subset of fluorophores can be activated. Therefore, data acquisition, i.e., the frame rate, is commonly limited to 10–25 Hz.[26] Since reversibly photoswitchable fluorophores can be imaged repeatedly without irreversible photobleaching at higher switching rates, overall orders of magnitude more photons are detected and dynamic processes even of small individual cellular structures can be observed for longer time scales with high frame rates.[65]
|
Photoswitching and Photoactivation Mechanisms
Common to all FP is the tertiary structure of amino acids, the so called β-barrel that protects the inner fluorophore from the chemical environment. PA-FP can be converted either from a non-fluorescent state into a fluorescent state (off → on; PA-GFP or PamCherry1) or from a fluorescent state into a red-shifted fluorescent state (green–red photoconversion; EOS-FP, mEOS, Dendra2) upon activation with appropriate light. The required wavelength for photoactivation or photoconversion is commonly around 400 nm. In the case of PA-GFP the photoactivated form results from a UV-induced decarboxylation of the Glu222 side chain of the fluorophore,[66] whereas in green–red photoconversion for EOS-FP a light-induced cleavage in the peptide backbone generates the red fluorescent form.[67] Once activated the red form of EOS-FP can be read-out upon excitation at 570 nm until it photobleaches. The reader should note that the term ‘red’ stems from the fact that the fluorescence is red-shifted, whereas fluorescence occurs in most cases in the yellow to orange spectroscopic range. One advantage of using photoconvertible FP is that an area of interest for super-resolution imaging can be preselected by imaging the inactive form at another wavelength. Other FP can be photoswitched reversibly between a fluorescent on- and a non-fluorescent off-state. For example Dronpa can be switched reversibly between a bright on- and non-fluorescent off-state by applying 400 nm light for activation (on-switching) and 488 nm for read-out (off-switching).[68,69]
Synthetic fluorophores have been thoroughly optimized for photostability. Furthermore, the chemical environment has been engineered to enable long lasting single-molecule imaging with reduced fluorescence intermittencies.[70,71] The detailed understanding of the photophysics behind photobleaching pathways paved the way for the controlled generation of non-fluorescent states in standard synthetic fluorophores and their use in single-molecule based super-resolution imaging. In contrast to synthetic fluorophores, PA-FP reside in a non-fluorescent state at the beginning and have to be activated upon irradiation with light. Thus, the density of fluorophores can be controlled arbitrarily by adjusting the activation light intensity. Synthetic fluorophores, however, reside in their fluorescent state and the majority of the fluorophores has to be converted into the non-fluorescent state at the beginning of the experiment. Assuming a cellular pattern with 1000 fluorophores within a diffractive limited region (DLR) necessitates that only one fluorophore is fluorescent at the same time, which requires, at a given lifetime τon for the fluorescent on-state, an off-state lifetime τoff >1000 × τon. With respect to a minimum integration time of common sensitive EMCCD cameras of 1 ms (according to a maximum frame rate of 1 kHz) off-states with a lifetime of ∼1 s or longer are required and it becomes obvious that triplet states with much shorter lifetimes do not represent a suitable platform for subdiffraction-resolution imaging. However, triplet states of synthetic fluorophores with typical lifetimes of several microseconds at room temperature can be used as the initial state for follow-up reactions to generate stable off-states. Thus, only very low concentrations of electron donors and acceptors (micromolar concentrations) or high concentrations of otherwise inefficient electron transfer partners are suited to selectively quench the triplet state of fluorophores to produce charged intermediates such as radical ions with a long lifetime.[31,41,72]
In dSTORM millimolar concentrations of thiols such as β-mercaptoethylamine, glutathione (GSH), or dithithreitol in aqueous solvents at pH ∼7–8 are used to reduce the triplet state of commercially available ATTO and Alexa Fluor fluorophores and generate stable non-fluorescent dark states, e.g., radical ions, with lifetimes of several hundred milliseconds to seconds.[31,72,73] The fluorescent state of the rhodamine and oxazine fluorophores is quantitatively recovered upon oxidation by molecular oxygen naturally present in aqueous solvents at concentrations of 200–250 μM at room temperature,[31] or photoinduced in an oxygen depleted solution for cyanine dyes.[32]
The Role of Stable Non-fluorescent Off-states of Fluorophores
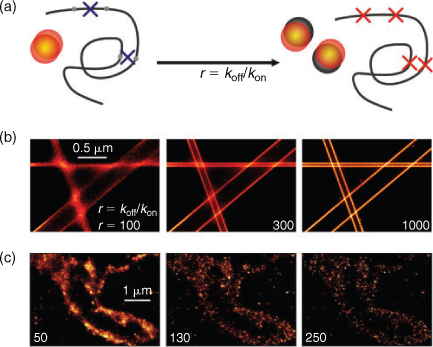
The detection and analysis of individual emitters from a densely labelled structure comprises that the majority of fluorophores must be non-fluorescent, while only a sparse subset of fluorophores is allowed to be fluorescent at any time. As a consequence and because of the stochastic nature of photoswitching, fluorophores used for super-resolution imaging must exhibit a long-lasting non-fluorescent off-state, e.g., a stable radical ion or other charged intermediate state as used in the dSTORM concept. Photoactivatable or photoconvertible FP as used in PALM and FPLAM generally exhibit thermally very stable off-states and can be switched on (‘activated’) upon irradiation with light of specific wavelength (in most cases ∼400 nm) and intensity. Thus, the density for single-molecule based localization microscopy can be precisely adjusted. In contrast, most photoswitchable synthetic fluorophores are fluorescent at the beginning of the experiment and have to be converted with rate koff to a non-fluorescent off-state to ensure single-molecule localization before the ground state is repopulated with rate kon. Here the switching rates and thus the length of the on-state and off-state of a fluorophore, i.e., the lifetimes τon and τoff, directly control the effective spot density and thus affect the ability to resolve a structure with a given fluorophore density NF.[29,32,40,74,75] The ability to resolve a structure can be best described introducing the ratio of the two rates or lifetimes, respectively, r = koff/kon = τoff/τon.
An increase of this ratio enhances the ability to localize more fluorophores correctly, i.e., as individual single-molecule events. Basically, the rate ratio can be increased by shortening τon or increasing τoff but it has to be considered that τon controls the maximum reasonable frame rate. Increasing the excitation intensity will enforce koff and thus shorten τon, whereas τoff can be prolonged when no or less reactivation intensity is used or the oxidation efficiency is reduced, e.g., by depleting oxygen from the buffer by an oxygen scavenging system.
Off-state lifetimes of photoswitchable synthetic fluorophores have been reported to vary from 10 to 100 ms[36,40] to several seconds.[41,47,57,72] For metastable off-states with a short τoff, high irradiation intensities must be applied to reduce τon and to generate a sufficient ratio r, e.g., r = 100 for τon = 1 ms and τoff = 100 ms. In the case of off-state lifetimes in the range of seconds a sufficiently high ratio r can be achieved with comparably long on-times and corresponding low irradiation intensity, which is very important for live cell applications.[65] For fixed cells, however, τon can be further decreased by applying higher irradiation intensities to increase r and the imaging speed (i.e., the frame rate). Currently only small areas of a sample can be imaged on a 128 × 128 pixel chip with the highest possible imaging speed of an EMCCD camera of ∼1 kHz, and hence the chip read-out time limits the acquisition speed for investigation of larger areas or whole cells.
Because of the stochastic nature of photoswitching, multi-fluorophore events can occur in single-molecule based localization microscopy experiments (Fig. 4). In other words, there is a chance that two or more fluorophores are fluorescent at the same time within a DLR. Consequently, the localization of the sum of the individual PSF introduces errors or artificial localizations, respectively.[74] To avoid multi-fluorophore localizations a photon threshold and a geometrical inspection of the PSF can be performed because it is expected that overlapping PSF of differently localized individual emitters produce an unsymmetrical resulting signal distribution. However, here it has to be mentioned that geometrical inspection of the PSF is inappropriate for 3D measurements where astigmatism is used to derive axial information (as described below) and fluorescence intensity is a poor parameter for quantification as each emitter might contribute differently to the resulting overlapping photon distribution. Hence, a certain fraction of false localized events is always present in the super-resolved image. However, the fraction of artificial or false localization can be kept low with appropriate control of the number of fluorophores residing in the fluorescent state and becomes higher when the photoswitching rates are set inappropriately, i.e., for low r. Using activatable FP each protein is tagged with a single PA-FP, whereas in the case of reversibly photoswitchable synthetic fluorophores often multiple labelled antibodies are used as fluorescent labels. Thus, the labelling density increases and an accordingly higher ratio has to be applied to be able to resolve the structure (Fig. 4c).
|
Furthermore, it is important to mention that for those concepts using reversibly photoswitchable fluorophores such as dSTORM, each fluorophore is generally localized multiple times. Therefore, structural information is not lost when a two- or multi-fluorophore event is recognized and discarded from further analysis because it is likely that the same fluorophore is localized again individually. In addition, even for inappropriately set photoswitching rates, a structure can be resolved as long as every fluorophore is localized individually at least once and the error-rate for discarding multi-fluorophore events (which increases with decreasing r) is kept low.
For the use of photoactivatable fluorophores in concepts like PALM or FPALM, two-spot events can occur stochastically or if the irradiation intensity for activation is set too high. Then the localization information is irretrievably lost or a false localization is made. Since the structural resolution is controlled by the labelling density, lost localizations decrease the achievable resolution. These considerations indicate that the density of fluorophores residing in their fluorescent state is easier to control in PALM or FPALM, whereas STORM or dSTORM exhibit the advantage that every fluorophore can be localized multiple times.
Live Cell Super-resolution Imaging
Related to specific fluorescence labelling of proteins in living cells, the use of PA-FP is advantageous because they can be co-expressed genetically to almost any target protein. Even though all biological processes are subject to dynamics on different time scales, PALM and FPALM have so far also been mainly used for the imaging of cellular structures in fixed cells. This can be understood considering the interrelation of temporal and spatial resolution underlying all super-resolution imaging concepts. Dependent on the structure investigated several hundred to thousands of images have to be measured and processed to reconstruct an image with subdiffraction resolution. Taking into account the facts that the kinetics of the photoswitching process determines the temporal resolution and PA-FP have to be localized and photobleached in each cycle, only low frame rates can be applied. Thus, live cell PALM or FPALM remains restricted to relatively slow processes such as the dynamics of adhesion complexes occurring on time scales of several tens of seconds to minutes.[26]
However, reversibly photoswitchable fluorophores are brighter than FP and can be imaged repeatedly without irreversible photobleaching at higher switching rates (because the photoswitching rates are tunable). Therefore, orders of magnitude more photons can be detected and dynamic processes can be observed for longer time scales with higher frame rates. Their photoswitching, however, until recently required specific chemical cocktails often involving oxygen depletion.[29,32,40] This restricted super-resolution imaging with reversibly photoswitchable fluorophores to fixed cells. Very recently, however, it was shown that synthetic fluorophores of the Alexa Fluor and ATTO family can be used for super-resolution imaging according to the dSTORM concept in the presence of millimolar concentrations of GSH under physiological conditions.[31,72] The tripeptide GSH is the most abundant low-molecular-weight thiol protectant and antioxidant in mammalian biology. The thiol groups are kept in a reduced state at millimolar concentration in animal cells.[76] Thus standard synthetic fluorophores can be used for dSTORM in living cells: GSH reduces the triplet state and forms stable negatively charged intermediate species with a lifetime of several hundreds of milliseconds to several seconds under physiological conditions whereas the fluorescent state is recovered upon oxidation of the negatively charged intermediates by molecular oxygen. These findings raise hopes that standard synthetic fluorophores can be used in combination with chemical tags for live cell super-resolution imaging.
Chemical tags provide a surrogate to FP in which a genetically encoded polypeptide tag is labelled with a modular synthetic fluorophore.[2,6,77] In addition to SNAP-, CLIP-tags and others, trimethoprim (TMP) chemical tags can be used advantageously for high-specific labelling of proteins in living cells.[78,79] The TMP-tag, which is based on the high-affinity non-covalent (and recently engineered covalent[80]) interaction between Escherichia coli dihydrofolate reductase (eDHFR) and TMP, is one of the few chemical tags able to label intracellular proteins in live cells with high a signal-to-noise ratio. Because eDHFR has a molecular weight (18 kDa) two-thirds that of GFP and is a stable, monomeric protein, eDHFR is an attractive protein chemical tag. The TMP-fluorophore conjugates have no apparent toxicity, can be synthesized in a few steps, and are readily cell permeable, reflecting the use of TMP clinically as an antibiotic.
In fact, it has been shown that the TMP-tag is suitable for stochastic single-molecule based localization microscopy according to the dSTORM method that combines the advantage of genetic encoding with a high photon-output photoswitchable synthetic organic fluorophore such as ATTO 655. The method ensures imaging of the dynamics of human histone protein H2B in living HeLa cells under physiological conditions at resolutions of ∼20 nm.[65] Applying a frame rate of 50 Hz in combination with a sliding window algorithm[81] the dynamics of nucleoprotein complexes were studied with nanometer spatial resolution and a temporal resolution of several seconds. A sliding window algorithm is used to generate dSTORM movie sequences consisting of super-resolved images each reconstructed from a subset of n images, e.g., a dSTORM movie sequence from subsets of 1000 subsequent frames moving the window always by 100 frames. Whereas the sliding window procedure does not increase the time resolution of the method, it enables the construction of video sequences from single-molecule localization data. The sliding window dSTORM method applied in live cell H2B imaging revealed movement of ∼3 nm s–1 for interphase nuclei and emphasizes the importance of the method for dynamic live cell super-resolution imaging with small tailored synthetic organic fluorophores.[46] Higher temporal resolutions can theoretically be easily achieved accelerating photoswitching kinetics by application of higher excitation intensities. However, as in all live cell experiments the excitation has to be carefully balanced between optimal photon flux and cell survival to enable the measurement of biologically valuable and usable information.
Three-dimensional Super-resolution Fluorescence Imaging
Contemporary to the introduction of super-resolution imaging based on single-molecule localization, progress has been reported on extending the principle to three dimensions. Three-dimensional super-resolution imaging has important applications, but seems at first hard to implement because wide-field excitation does not directly give information about fluorophore position and the cameras used for detection are inherently 2D. All of the ideas put forward for 3D imaging, therefore, try to encode information about the z-position of an emitting fluorophore in a modified PSF. Besides the use of double-helical PSF to resolve the 3D localization of single fluorophores,[82] interferrometric approaches,[83] and virtual volume super-resolution microscopy[64] have achieved impressive axial resolutions. Because of their relatively easy implementation the following chapter focuses on 3D super-resolution imaging based on introducing defocusing, i.e., dual or multi-focal-plane imaging[84–90] or astigmatism into the image (Fig. 5).[91–93]
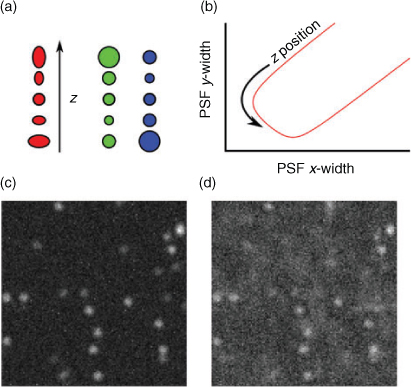
|
Three-dimensional astigmatism is implemented by inserting a cylindrical lens in the detection path and thereby separating the object planes for the x and y components, resulting in different defocussing behaviour depending on the z position of an emitter. To give an example, assume that the x object plane is at zx = –1 (i.e., objects with z position –1 are most sharp in x) and the y object plane is at zy = 1. Only objects at z = 0 will appear round on the camera, while objects at negative z coordinates are defocussed more in y than in x and therefore appear elongated in y, with the elongation ratio indicating the z position. Taking the shape of the signal into account in the PSF model function and the fitting procedure, a calibration curve for known z offsets can be obtained and the z information can be extracted. Dual-focal-plane methods achieve the same effect with two cameras or two areas of the same camera with different object planes. In this scheme, objects appear differently sized on the two cameras, with each detector being equivalent to one width in the astigmatism scheme (Fig. 5).
While this idea is easy enough to realize, all of the proposed schemes for 3D imaging do, however, introduce considerable problems with emission density, i.e., the density of fluorophores emitting concurrently. First, the excitation volume for 3D samples is considerably larger than for 2D samples, but the available detector area is the same. Second, the small detector area is burdened with larger and unsymmetrical photon distributions complicating PSF fitting. Third, the larger PSF means spreading the detected photons over a larger spatial area, resulting in a higher uncertainty in the precise localization of the lateral position. Finally, when labelling a target structure with FP or fluorophores, a minimum number of fluorescent probes is required to resolve the structure according to signal theory. The required density of fluorescent probes for super-resolution imaging can be estimated by the Nyquist–Shannon sampling theorem.[63] As an example, to obtain a structural resolution of 20 nm in one dimension, fluorophores have to be positioned at least every 10 nm corresponding to a labelling density of ∼104 fluorophores μm–2. For 3D super-resolution imaging the situation is even worse: to achieve a structural resolution of 20 nm in all three axes the labelling density has to be in the order of 106 fluorophores μm–3. The labelling density thus corresponds to a fluorophore concentration in the millimolar range which certainly alters cellular function and can induce side effects, e.g., the formation of non-fluorescent fluorophore dimers.
Conclusions
Within just a few years super-resolution imaging based on single-molecule localization has seen tremendous growth and has been successfully applied to study various cellular structures in fixed and living cells with quite recently unforeseeable optical resolution. The success story was permitted by its simple realization: a standard inverse fluorescence microscope equipped with a high numerical oil-immersion objective, continuous wave lasers providing a power of several mW, and a sensitive CCD camera are sufficient to initiate controlled photoswitching, photoconversion, and photoactivation of fluorophores and detect their single-molecule signals with high signal-to-background ratio. Complex 2D and 3D structures can be reconstructed using high labelling densities of appropriate fluorophores and photoswitching conditions in combination with efficient fitting algorithms.
In addition to the analysis of continuously labelled structures, single-molecule based localization microscopy also has great potential to analyze protein distributions and membrane clustering.[94–97] The understanding and control of fluorophore photophysics and efforts in protein engineering enables multi-colour super-resolution imaging with PA-FP and almost any organic synthetic fluorophore. However, even though there is ample scope for further technical improvements concerning camera frame and photoswitching rates that might enable dynamic super-resolution imaging of fast cellular processes, one should be aware of the fact that the photoinduced formation of reversible and irreversible off-states are likely to produce reactive side products. Therefore, the significance of live cell super-resolution imaging will also in the future critically depend on the experimental conditions adjustable by the excitation intensity and the labelling density.
References
[1] J. B. Pawley, Handbook of Biological Confocal Microscopy, 3rd edn 2006 (Springer: Heidelberg).[2] I. Chen, A. Y. Ting, Curr. Opin. Biotechnol. 2005, 16, 35.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXhsFShs7g%3D&md5=3a67950c56d79307f6deeec381561577CAS | 15722013PubMed |
[3] B. N. Giepmans, S. R. Adams, M. H. Ellisman, R. Y. Tsien, Science 2006, 312, 217.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XjtlOjs70%3D&md5=6a74988b32e46995ea249df363ef8f8dCAS | 16614209PubMed |
[4] J. Lippincott-Schwartz, G. H. Patterson, Trends Cell Biol. 2009, 19, 555.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtlChurnM&md5=3d0463d88c283a4f60f7edb20e874a33CAS | 19836954PubMed |
[5] X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir, S. Weiss, Science 2005, 307, 538.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXmslOhtw%3D%3D&md5=be5fc27e060de79d88469c92ddadd0e3CAS | 15681376PubMed |
[6] L. W. Miller, V. W. Cornish, Curr. Opin. Chem. Biol. 2005, 9, 56.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXhtV2ksrk%3D&md5=ae8686ea141afbee7afe46d14287d484CAS | 15701454PubMed |
[7] E. Abbe, Arch. Mikrosk. Anat. 1873, 9, 413.
| Crossref | GoogleScholarGoogle Scholar |
[8] M. Fernández-Suárez, A. Y. Ting, Nat. Rev. Mol. Cell Biol. 2008, 9, 929.
| Crossref | GoogleScholarGoogle Scholar | 19002208PubMed |
[9] M. Heilemann, P. Dedecker, J. Hofkens, M. Sauer, Laser Photonics Rev. 2009, 3, 180.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXksVWntLY%3D&md5=ca90b91a31642c7825be7b5daa46fa3bCAS |
[10] S. W. Hell, Science 2007, 316, 1153.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXls1KitL4%3D&md5=34a9954e7dc71d5622e72c8c0348447bCAS | 17525330PubMed |
[11] S. W. Hell, Nat. Methods 2009, 6, 24.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhsFCku73L&md5=0e5474b498a3f5636847b773ca86d14cCAS | 19116611PubMed |
[12] B. Huang, M. Bates, X. Zhuang, Annu. Rev. Biochem. 2009, 78, 993.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXos1Ghu7c%3D&md5=f35ec4a7eaa10bba698e3bf4f2299a5cCAS | 19489737PubMed |
[13] N. Ji, H. Shroff, H. Zhong, E. Betzig, Curr. Opin. Neurobiol. 2008, 18, 605.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXmtVKksL4%3D&md5=d4e95125dfa16604a79e8122dd505ae9CAS | 19375302PubMed |
[14] G. Patterson, M. Davidson, S. Manley, J. Lippincott-Schwartz, Annu. Rev. Phys. Chem. 2010, 61, 345.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXmt1ahtLg%3D&md5=27d4776d7c1a0c861c2490333d4a427bCAS | 20055680PubMed |
[15] B. A. Wilt, L. D. Burns, E. T. Wei Ho, K. K. Ghosh, E. A. Mukamel, M. J. Schnitzer, Annu. Rev. Neurosci. 2009, 32, 435.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXpsV2nu7Y%3D&md5=a485e554e957aceb8a36379437d58158CAS | 19555292PubMed |
[16] M. Dyba, S. W. Hell, Phys. Rev. Lett. 2002, 88, 163901.
| Crossref | GoogleScholarGoogle Scholar | 11955234PubMed |
[17] S. W. Hell, J. Wichmann, Opt. Lett. 1994, 19, 780.
| Crossref | GoogleScholarGoogle Scholar | 1:STN:280:DC%2BD1MjgsFGhsA%3D%3D&md5=acea88239f8930ad64d20e4af2908b23CAS | 19844443PubMed |
[18] K. I. Willig, S. O. Rizzoli, V. Westphal, R. Jahn, S. W. Hell, Nature 2006, 440, 935.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XjsVWktLY%3D&md5=06af46a7bd41d1b947afa5884facf7f8CAS | 16612384PubMed |
[19] M. G. L. Gustafsson, J. Microscopy 2000, 198, 82.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXktVOgt7o%3D&md5=0f7e41bf29735366529417716fecf9a5CAS |
[20] P. Kner, B. B. Chhun, E. R. Griffis, L. Winoto, M. G. Gustafsson, Nat. Methods 2009, 6, 339.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXltFymt7g%3D&md5=1da04b5ffdcc16d9eee6ffe6b88fd602CAS | 19404253PubMed |
[21] L. Schermelleh, P. M. Carlton, S. Haase, L. Shao, L. Winoto, P. Kner, B. Burke, M. C. Cardoso, D. A. Agard, M. G. Gustafsson, H. Leonhardt, J. W. Sedat, Science 2008, 320, 1332.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXmslWgtbw%3D&md5=15666575083cb9892d67f82f7613ac41CAS | 18535242PubMed |
[22] P. Dedecker, J. Hotta, C. Flors, M. Sliwa, H. Uji-i, M. B. Roeffaers, R. Ando, H. Mizuno, A. Miyawaki, J. Hofkens, J. Am. Chem. Soc. 2007, 129, 16132.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhtlOgsr3P&md5=fae5e1db04c243e79f13ae078eb3eb7dCAS | 18047340PubMed |
[23] J. Enderlein, Appl. Phys. Lett. 2005, 87, 095105.
[24] S. Hennig, S. van de Linde, M. Heilemann, M. Sauer, Nano Lett. 2009, 9, 2466.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXmtVGrsLg%3D&md5=85242e3fe12e4559ebd07c0059cb1437CAS | 19453186PubMed |
[25] E. Betzig, G. H. Patterson, R. Sougrat, O. W. Lindwasser, S. Olenych, J. S. Bonifacino, M. W. Davidson, J. Lippincott-Schwartz, H. F. Hess, Science 2006, 313, 1642.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XpsVOktL0%3D&md5=4090b503b73d57012c93b841414d9827CAS | 16902090PubMed |
[26] H. Shroff, C. G. Galbraith, J. A. Galbraith, E. Betzig, Nat. Methods 2008, 5, 417.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXlt1ahtbc%3D&md5=3dcb26bbfa588b8c29030bc3d4c66f68CAS | 18408726PubMed |
[27] S. T. Hess, T. P. Girirajan, M. D. Mason, Biophys. J. 2006, 91, 4258.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28Xht1Khur%2FN&md5=db2d5224e68cf7ba60cb0d5dad3c06ebCAS | 16980368PubMed |
[28] S. T. Hess, T. J. Gould, M. V. Gudheti, S. A. Maas, K. D. Mills, J. Zimmerberg, Proc. Natl. Acad. Sci. USA 2007, 104, 17370.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXht1ymtrjE&md5=5e95b50d0c6bc9442ed2bb7a71929ac5CAS |
[29] M. Bates, B. Huang, G. T. Dempsey, X. Zhuang, Science 2007, 317, 1749.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhtVGmsLnE&md5=1cc234141dfb68efc28178e49ecefa81CAS | 17702910PubMed |
[30] M. J. Rust, M. Bates, X. Zhuang, Nat. Methods 2006, 3, 793.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XpvVCmtLY%3D&md5=9aec11cf7b482ee17c23cc020e8bb0f9CAS | 16896339PubMed |
[31] M. Heilemann, S. van de Linde, A. Mukherjee, M. Sauer, Angew. Chem. Int. Ed. 2009, 48, 6903.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtVOrtrrO&md5=80c41eb22437cd757c0724e341aee101CAS |
[32] M. Heilemann, S. van de Linde, M. Schuttpelz, R. Kasper, B. Seefeldt, A. Mukherjee, P. Tinnefeld, M. Sauer, Angew. Chem. Int. Ed. 2008, 47, 6172.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXpvFSgtro%3D&md5=33d16b96bf19b3c15f24ff280e96b771CAS |
[33] J. S. Biteen, M. A. Thompson, N. K. Tselentis, G. R. Bowman, L. Shapiro, W. E. Moerner, Nat. Methods 2008, 5, 947.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtlSktrfI&md5=35d42770e28ae10e1822b60b004d34e8CAS | 18794860PubMed |
[34] C. Flors, J. Hotta, H. Uji-i, P. Dedecker, R. Ando, H. Mizuno, A. Miyawaki, J. Hofkens, J. Am. Chem. Soc. 2007, 129, 13970.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXht1Sgtb%2FK&md5=235ef24a9756d5f8b3a2bc195b5d71d0CAS | 17956094PubMed |
[35] C. Flors, C. N. Ravarani, D. T. Dryden, ChemPhysChem 2009, 10, 2201.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtV2itrzE&md5=d47eace13ddd394094a3c86516c0b019CAS | 19554598PubMed |
[36] J. Fölling, M. Bossi, H. Bock, R. Medda, C. A. Wurm, B. Hein, S. Jakobs, C. Eggeling, S. W. Hell, Nat. Methods 2008, 5, 943.
| Crossref | GoogleScholarGoogle Scholar | 18794861PubMed |
[37] M. Gunkel, F. Erdel, K. Rippe, P. Lemmer, R. Kaufmann, C. Hormann, R. Amberger, C. Cremer, Biotechnol. J. 2009, 4, 927.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXos1aqsbo%3D&md5=0e45d43ca07ba62b1383562db2d967e1CAS | 19548231PubMed |
[38] M. B. Roeffaers, G. De Cremer, J. Libeert, R. Ameloot, P. Dedecker, A. J. Bons, M. Buckins, J. A. Martens, B. F. Sels, D. E. De Vos, J. Hofkens, Angew. Chem. Int. Ed. 2009, 48, 9285.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVKhtr3E&md5=687a3218f7436311a49b790fbc10129eCAS |
[39] A. Sharonov, R. M. Hochstrasser, Proc. Natl. Acad. Sci. USA 2006, 103, 18911.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XhtlChu77N&md5=d332a96b56c096ca5fabc94943a667f5CAS |
[40] C. Steinhauer, C. Forthmann, J. Vogelsang, P. Tinnefeld, J. Am. Chem. Soc. 2008, 130, 16840.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhsVWrtbnP&md5=3971dc8369d11783363c7b2651b2c374CAS | 19053449PubMed |
[41] J. Vogelsang, T. Cordes, C. Forthmann, C. Steinhauer, P. Tinnefeld, Proc. Natl. Acad. Sci. USA 2009, 106, 8107.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXmvVWmsLw%3D&md5=37069903a39f4a98c8adeb5453dda1e7CAS |
[42] M. K. Cheezum, W. F. Walker, W. H. Guilford, Biophys. J. 2001, 81, 2378.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXntlSnt70%3D&md5=ceeb7c08b2705a6e666f981d83c4ffc5CAS | 11566807PubMed |
[43] R. E. Thompson, D. R. Larson, W. W. Webb, Biophys. J. 2002, 82, 2775.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XjtlKjtrY%3D&md5=2eed52844261778fcc75ef022781c067CAS | 11964263PubMed |
[44] A. Yildiz, J. N. Forkey, S. A. McKinney, T. Ha, Y. E. Goldman, P. R. Selvin, Science 2003, 300, 2061.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXkvVOrsLg%3D&md5=00e312b77e1a5c1ec7be02923491e6b6CAS | 12791999PubMed |
[45] A. Yildiz, P. R. Selvin, Acc. Chem. Res. 2005, 38, 574.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXisVyms74%3D&md5=eb501a39c06e0cb0fddf4eedc0935addCAS | 16028892PubMed |
[46] R. M. Dickson, A. B. Cubitt, R. Y. Tsien, W. E. Moerner, Nature 1997, 388, 355.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXkvVCjtbo%3D&md5=3e1370b61da47722aeeb9de69fa18b3eCAS | 9237752PubMed |
[47] M. Bates, T. R. Blosser, X. Zhuang, Phys. Rev. Lett. 2005, 94, 108101.
| Crossref | GoogleScholarGoogle Scholar | 15783528PubMed |
[48] M. Tokunaga, N. Imamoto, K. Sakata-Sogawa, Nat. Methods 2008, 5, 159.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhsV2ks7c%3D&md5=1c46d8b9d211403330708c5d88131532CAS | 18176568PubMed |
[49] S. Wolter, M. Schuttpelz, M. Tscherepanow, S. Van de Linde, M. Heilemann, M. Sauer, J. Microscopy 2010, 237, 12.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtFKkt7o%3D&md5=f6c158c9961e0f58553606f4c1fd846fCAS |
[50] P. N. Hedde, J. Fuchs, F. Oswald, J. Wiedenmann, G. U. Nienhaus, Nat. Methods 2009, 6, 689.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtF2kurbN&md5=27895ae2ae627d1f82099472607ec417CAS | 19789527PubMed |
[51] R. Henriques, M. Lelek, E. F. Fornasiero, F. Valtorta, C. Zimmer, M. M. Mhlanga, Nat. Methods 2010, 7, 339.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXlsVWjsLk%3D&md5=b72c9788c5d8d111679dfd9b0650efe4CAS | 20431545PubMed |
[52] K. I. Mortensen, L. S. Churchman, J. A. Spudich, H. Flyvbjerg, Nat. Methods 2010, 7, 377.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXkt1aqu7k%3D&md5=0dcf504e60d317a8f941365adbc10a0aCAS | 20364147PubMed |
[53] C. S. Smith, N. Joseph, B. Rieger, K. A. Lidke, Nat. Methods 2010, 7, 373.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXkt1aqsrY%3D&md5=3da012ca7c3bfc6a5d82936c1f3b8e29CAS | 20364146PubMed |
[54] T. Fukaminato, T. Sasaki, T. Kawai, N. Tamai, M. Irie, J. Am. Chem. Soc. 2004, 126, 14843.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXoslSgsbs%3D&md5=38fa2fe5c5a7c68e5ff4c2610deb8189CAS | 15535710PubMed |
[55] M. Irie, T. Fukaminato, T. Sasaki, N. Tamai, T. Kawai, Nature 2002, 420, 759.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XpsFygtrg%3D&md5=c212acdde5c276d84b2367ed87b18dabCAS | 12490936PubMed |
[56] B. Seefeldt, R. Kasper, M. Beining, J. Mattay, J. Arden-Jacob, N. Kemnitzer, K. H. Drexhage, M. Heilemann, M. Sauer, Photochem. Photobiol. Sci. 2010, 9, 213.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsVGqtLs%3D&md5=ea12b392c8dcecbe56004815e4aea9ddCAS | 20126797PubMed |
[57] M. Heilemann, E. Margeat, R. Kasper, M. Sauer, P. Tinnefeld, J. Am. Chem. Soc. 2005, 127, 3801.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXhslajsLw%3D&md5=938bdbd4b6e2e98450d76d69ebe0e764CAS | 15771514PubMed |
[58] V. N. Belov, C. A. Wurm, V. P. Boyarskiy, S. Jakobs, S. W. Hell, Angew. Chem. Int. Ed. 2010, 49, 3520.
| 1:CAS:528:DC%2BC3cXlsFCntLs%3D&md5=de406c8028999bc50e6c3df862f98866CAS |
[59] S. J. Lord, N. R. Conley, H. L. Lee, R. Samuel, N. Liu, R. J. Twieg, W. E. Moerner, J. Am. Chem. Soc. 2008, 130, 9204.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXns1elur8%3D&md5=fdf87f0c7c5124048e423a619eb9c198CAS | 18572940PubMed |
[60] M. Andresen, A. C. Stiel, J. Folling, D. Wenzel, A. Schonle, A. Egner, C. Eggeling, S. W. Hell, S. Jakobs, Nat. Biotechnol. 2008, 26, 1035.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtVygtLnP&md5=b9e0aff8ebaa8d5be601ba17f5226210CAS | 18724362PubMed |
[61] N. C. Shaner, G. H. Patterson, M. W. Davidson, J. Cell Sci. 2007, 120, 4247.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXns1GktQ%3D%3D&md5=667308ae26e142fd31da0c9769a133bcCAS | 18057027PubMed |
[62] J. Wiedenmann, F. Oswald, G. U. Nienhaus, IUBMB Life 2009, 61, 1029.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVyrtrzJ&md5=91428d2fee691f6f53ed237e5b186994CAS | 19859977PubMed |
[63] C. E. Shannon, Proc. Inst. Radio Eng. 1949, 37, 10.
[64] J. Tang, J. Akerboom, A. Vaziri, L. L. Looger, C. V. Shank, Proc. Natl. Acad. Sci. USA 2010, 107, 10068.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXnslGlt70%3D&md5=f65debdaeaf7f1cf4a0e831318879418CAS |
[65] R. Wombacher, M. Heidbreder, S. van de Linde, M. Sheetz, M. Heilemann, V. W. Cornish, M. Sauer, Nat. Methods 2010, 7, 717.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXpvVWgsLo%3D&md5=3b3513c648f5cc8950d34b24582e696cCAS | 20693998PubMed |
[66] J. N. Henderson, R. Gepshtein, J. R. Heenan, K. Kallio, D. Huppert, S. J. Remington, J. Am. Chem. Soc. 2009, 131, 4176.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXjtVSjs7c%3D&md5=b2a4675152894034e3224f21de169322CAS | 19278226PubMed |
[67] K. Nienhaus, G. U. Nienhaus, J. Wiedenmann, H. Nar, Proc. Natl. Acad. Sci. USA 2005, 102, 9156.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXlvF2kurk%3D&md5=5e3e5013df299a040c5553d7e72d0407CAS |
[68] S. Habuchi, P. Dedecker, J. Hotta, C. Flors, R. Ando, H. Mizuno, A. Miyawaki, J. Hofkens, Photochem. Photobiol. Sci. 2006, 5, 567.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XltlKqsLo%3D&md5=6f8dda35add12c24fbdad4554bd68753CAS | 16761085PubMed |
[69] S. Habuchi, R. Ando, P. Dedecker, W. Verheijen, H. Mizuno, A. Miyawaki, J. Hofkens, Proc. Natl. Acad. Sci. USA 2005, 102, 9511.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXmsVaktro%3D&md5=db08fd4c07b62ae7e0c712ef39b97b8dCAS |
[70] R. Kasper, B. Harke, C. Forthmann, P. Tinnefeld, S. W. Hell, M. Sauer, Small 2010, 6, 1379.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXosVyqt74%3D&md5=f7f8248fd478360a36822974cfabf65fCAS | 20521266PubMed |
[71] J. Vogelsang, R. Kasper, C. Steinhauer, B. Person, M. Heilemann, M. Sauer, P. Tinnefeld, Angew. Chem. Int. Ed. 2008, 47, 5465.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXovFymuro%3D&md5=04b516867f8b64b6c356145985d86088CAS |
[72] S. van de Linde, R. Kasper, M. Heilemann, M. Sauer, Appl. Phys. B 2008, 93, 725.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtl2qtLzL&md5=d9b09efa05097421e3f0bad72625c809CAS |
[73] S. van de Linde, U. Endesfelder, A. Mukherjee, M. Schuttpelz, G. Wiebusch, S. Wolter, M. Heilemann, M. Sauer, Photochem. Photobiol. Sci. 2009, 8, 465.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXjvV2mtrY%3D&md5=d0917ecbf85985c2d104f6b4e118b62cCAS | 19337659PubMed |
[74] S. van de Linde, S. Wolter, M. Heilemann, M. Sauer, J. Biotechnol. 2010, 149, 260.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXht1Wju7rM&md5=4fa9cfea83cfb4fd7b7918aca6c328deCAS | 20176060PubMed |
[75] T. Cordes, M. Strackharn, S. W. Stahl, W. Summerer, C. Steinhauer, C. Forthmann, E. M. Puchner, J. Vogelsang, H. E. Gaub, P. Tinnefeld, Nano Lett. 2010, 10, 645.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsFKksL7K&md5=41c7848a94b8c9187f8abf46a0807b73CAS | 20017533PubMed |
[76] H. Sies, Free Radical Biol. Med. 1999, 27, 916.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXns1ymsrw%3D&md5=af23bb02173ea1b254b6e9262bfff5d7CAS |
[77] H. M. O’Hare, K. Johnsson, A. Gautier, Curr. Opin. Struct. Biol. 2007, 17, 488.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhtFSltbvK&md5=6e238fa57c68ed6959e9283d5f4e88b3CAS | 17851069PubMed |
[78] N. T. Calloway, M. Choob, A. Sanz, M. P. Sheetz, L. W. Miller, V. W. Cornish, ChemBioChem 2007, 8, 767.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXls1artrc%3D&md5=d66b1e8cc2bf99f5a43a63a0f5a8a291CAS | 17378009PubMed |
[79] L. W. Miller, Y. Cai, M. P. Sheetz, V. W. Cornish, Nat. Methods 2005, 2, 255.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXisFejtbk%3D&md5=35664b2f749f6b8d3ca4b43a45ea7646CAS | 15782216PubMed |
[80] S. S. Gallagher, J. E. Sable, M. P. Sheetz, V. W. Cornish, ACS Chem. Biol. 2009, 4, 547.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXnvFWktLc%3D&md5=7b708f95e42a4f23b7bd4246d544619fCAS | 19492849PubMed |
[81] U. Endesfelder, S. van de Linde, S. Wolter, M. Sauer, M. Heilemann, ChemPhysChem 2010, 11, 836.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXjtFygsr4%3D&md5=2e6e678aacd35b708cc5f8906eb456fcCAS | 20186905PubMed |
[82] S. R. Pavani, M. A. Thompson, J. S. Biteen, S. J. Lord, N. Liu, R. J. Twieg, R. Piestun, W. E. Moerner, Proc. Natl. Acad. Sci. USA 2009, 106, 2995.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXivFyqtb4%3D&md5=b082b689872eeef69146d7b3540e4aecCAS |
[83] G. Shtengel, J. A. Galbraith, C. G. Galbraith, J. Lippincott-Schwartz, J. M. Gillette, S. Manley, R. Sougrat, C. M. Waterman, P. Kanchanawong, M. W. Davidson, R. D. Fetter, H. F. Hess, Proc. Natl. Acad. Sci. USA 2009, 106, 3125.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXivFyqu7w%3D&md5=e3f830bdef997b7821a09949094b9f0bCAS |
[84] M. F. Juette, T. J. Gould, M. D. Lessard, M. J. Mlodzianoski, B. S. Nagpure, B. T. Bennett, S. T. Hess, J. Bewersdorf, Nat. Methods 2008, 5, 527.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXmsVOjtL8%3D&md5=f1f082adf216557a5019d92067bb1b0bCAS | 18469823PubMed |
[85] S. Ram, P. Prabhat, J. Chao, E. S. Ward, R. J. Ober, Biophys. J. 2008, 95, 6025.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXotFGm&md5=be4bdc20fb6418d8dbd9ea3bc52c6a47CAS | 18835896PubMed |
[86] G. J. Schütz, M. Axmann, H. Schindler, Single Mol. 2001, 2, 69.
| Crossref | GoogleScholarGoogle Scholar |
[87] G. J. Schütz, G. Kada, V. P. Pastushenko, H. Schindler, EMBO J. 2000, 19, 892.
| Crossref | GoogleScholarGoogle Scholar | 10698931PubMed |
[88] M. Speidel, A. Jonas, E. L. Florin, Opt. Lett. 2003, 28, 69.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXotFGgtw%3D%3D&md5=306b73f4979b55f512c69976e6f2c325CAS | 12656488PubMed |
[89] E. Toprak, H. Balci, B. H. Blehm, P. R. Selvin, Nano Lett. 2007, 7, 2043.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXms1equ7c%3D&md5=93483de248c41d5c099ede4df4d2c326CAS | 17583964PubMed |
[90] A. M. van Oijen, J. Köhler, J. Schmidt, M. Müller, G. J. Brakenhoff, Chem. Phys. Lett. 1998, 292, 183.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXks1Wms7Y%3D&md5=4a2d51e848855e5fa412c4766a3f0e04CAS |
[91] L. Holtzer, T. Meckel, T. Schmidt, Appl. Phys. Lett. 2007, 90, 053902.
| Crossref | GoogleScholarGoogle Scholar |
[92] B. Huang, W. Wang, M. Bates, X. Zhuang, Science 2008, 319, 810.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhsFGhs7s%3D&md5=7ab4eda1d2d72a502430d665b19662a0CAS | 18174397PubMed |
[93] H. P. Kao, A. S. Verkman, Biophys. J. 1994, 67, 1291.
| Crossref | GoogleScholarGoogle Scholar | 1:STN:280:DyaK2M7htFSlug%3D%3D&md5=a7eaa4cddd82e53c30b6597027561249CAS | 7811944PubMed |
[94] D. Baddeley, I. D. Jayasinghe, L. Lam, S. Rossberger, M. B. Cannell, C. Soeller, Proc. Natl. Acad. Sci. USA 2009, 106, 22275.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXmtlahtw%3D%3D&md5=64c7d6ed23c5f38ba83c4ba9cad0e54fCAS |
[95] D. Greenfield, A. L. McEvoy, H. Shroff, G. E. Crooks, N. S. Wingreen, E. Betzig, J. Liphardt, PLoS Biol. 2009, 7, e1000137.
| Crossref | GoogleScholarGoogle Scholar | 19547746PubMed |
[96] D. M. Owen, C. Rentero, J. Rossy, A. Magenau, D. Williamson, M. Rodriguez, K. Gaus, J Biophotonics 2010, 446.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtValt7bO&md5=6834fe03a6a6b03e9039a6b732796ae2CAS | 20148419PubMed |
[97] S. van de Linde, M. Sauer, M. Heilemann, J. Struct. Biol. 2008, 164, 250.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtlyltLfE&md5=b69204924274df9a2e62514060cd036eCAS | 18790061PubMed |
1 This Review is published alongside a paper by Herten and coworkers as part of a proposed Research Front on Single Molecule Spectroscopy which did not reach fruition.