

On the Origins of Nitroxide Mediated Polymerization (NMP) and Reversible Addition–Fragmentation Chain Transfer (RAFT)*
Ezio Rizzardo A C and David H. Solomon B CA CSIRO Materials Science and Engineering, Clayton South, Melbourne, Vic. 3169, Australia.
B Department of Chemical & Biomolecular Engineering, The University of Melbourne, Parkville, Melbourne, Vic. 3010, Australia.
C Corresponding authors. Email: ezio.rizzardo@csiro.au; davids@unimelb.edu.au

Ezio Rizzardo (right) graduated with first class honours in applied organic chemistry from the University of New South Wales and was awarded a Ph.D. in organic chemistry by the University of Sydney in 1969 for his studies on the photochemistry of organic nitro compounds. He joined David Solomon’s group at CSIRO in 1976 after postdoctoral work on the synthesis of biologically active compounds with Richard Turner at Rice University (Houston), Sir Derek Barton at RIMAC (Boston), and Arthur Birch at the ANU (Canberra). His research at CSIRO has focused on the development of methods for understanding and controlling polymerization processes. He is the co-author of some 200 journal papers, which to date have received over 13000 citations, and co-inventor on 44 worldwide patents. He is a Fellow of the Royal Society of London, the Australian Academy of Science and the Australian Academy of Technological Sciences and Engineering, and the recipient of the Australian Polymer Medal, the CSIRO Chairman’s Gold Medal and the Australian Government’s Centenary Medal for contributions to society and polymer science. David Solomon (left) graduated from the University of New South Wales with a Ph.D. in 1959 and a D.Sc. in 1968; he also holds a Diploma in Chemistry from Sydney Technical College (1950). During his studies, he worked for Dulux and this is where his interest in the chemistry of radical polymerization began. He was seconded to work at ICI Paints Division in 1959 and returned to Dulux to set up their polymer research group. In 1963, he joined CSIRO and set up the polymer research section. He recruited Ezio into this group and together they developed techniques to better understand radical chemistry and eventually living radical systems. David was also the principal inventor of the world’s first polymer banknote. He was President of the Royal Australian Chemical Institute (RACI) from 1979 to 1980. He is a Fellow of the Royal Society of London, the Australian Academy of Science, the Australian Academy of Technological Sciences and Engineering (Foundation Fellow), the RACI and the Institute of Chemical Engineers. He has received numerous awards, including Member of the Order of Australia, D.App.Sc. (Honoris Causa) (University of Melbourne), and is the sole recipient of Australia’s Bicentennial Science Achievement Award. The Polymer Division of the RACI has recognized Dave’s contributions to polymer science with the annual Solomon Lecture given by a prestigious polymer scientist. |
Australian Journal of Chemistry 65(8) 945-969 https://doi.org/10.1071/CH12194
Submitted: 13 April 2012 Accepted: 17 May 2012 Published: 5 July 2012
Abstract
The early experiments on radical polymerization, which were to lead to a study of nitroxide trapping of the initiation step and the interest in defect groups, particularly the macromonomers formed by termination by disproportionation, are discussed. Results from the nitroxide trapping clearly show that the initiation step ranges from simple clean addition to the head of the monomer, to complex addition/abstraction reactions. Careful selection of the monomer/initiation system is emphasized with particular reference to two common monomers, styrene and methyl methacrylate, and two initiating radicals, t-butoxy and benzoyloxy. The discovery of nitroxide mediated polymerization (NMP) from observations made during the nitroxide trapping work is reported and the ability to have a living radical system demonstrated with numerous examples. Similarly, the study of the copolymerization of macromonomers, formed by disproportionation of the propagating chains, is discussed with the discovery of β-scission and an early form of addition–fragmentation reported. The evolution of reversible addition–fragmentation chain transfer (RAFT) to a highly versatile and commercially attractive radical system is reported and the detailed chemistry behind the discovery of this living radical system discussed. Both NMP and RAFT enable the synthesis of structures not previously possible by radical polymerization and in some cases not possible by any other process.
Introduction
Today free radical polymerization is one of the most studied fields in polymer chemistry and the majority of laboratories will be employing some form of living radical polymerization (LRP) either:
-
NMP – nitroxide mediated polymerization,
-
ATRP – atom transfer radical polymerization, or
-
RAFT – reversible addition–fragmentation chain transfer polymerization
Some may be using variants or combinations of these techniques to produce exciting molecular structures not previously possible.
NMP and RAFT were discovered by CSIRO in the Division of Applied Organic Chemistry and its successors in laboratories at Fishermen’s Bend and Clayton, Victoria. This article gives a history of the research group in CSIRO and the reasons why the research started. It also seeks to highlight the rewards that can stem from seeking a basic understanding of the science underpinning industrial processes.
Whilst this highlight focusses almost entirely on the work of the CSIRO group, it should be noted that numerous researchers around the world have contributed greatly to both processes (NMP and RAFT) since the CSIRO discoveries. Indeed, the number of publications on NMP and RAFT is now of the order of 2000 and 3000, respectively.
Background
Free radical polymerization is the most diverse means of synthesizing polymers; it can be carried out under relatively undemanding conditions, is suitable for a wide range of monomers, and can even be conducted in the presence of water. Hence it is a highly desirable choice for industrial applications. However in the 1960s radical polymerization lacked the ability to readily prepare controlled structures, such as block, graft, or star copolymers, and to precisely control the molecular architecture such as molecular weight and molecular weight distribution. A possible alternative was the much more demanding ionic polymerization which did offer the possibility of narrow polydispersity and different structures such as block copolymers. The big difference in mechanism between radical and ionic polymerization was in the termination step; Szwarc had coined the term ‘living’ to describe anionic systems, where there was no self-termination step.[1] In the ionic system a second monomer could be added, after the first was polymerized, and a block copolymer formed. In radical systems the highly reactive radicals rapidly self-terminate to dead polymer chains. The accepted mechanisms in the 1960s for radical and ionic polymerizations are shown as (a) and (b) respectively in Scheme 1.[2] The inset in Scheme 1 also shows the two different types of linkages that can be formed, either head–tail (H–T) or head–head (H–H).
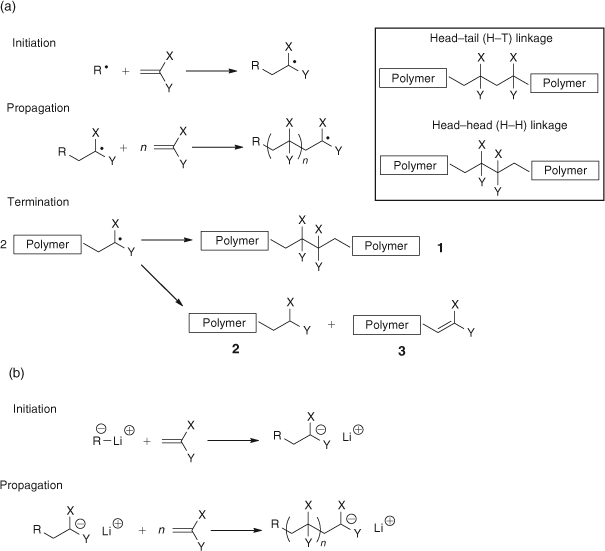
|
The reasons advanced for H–T addition in both radical and ionic polymerizations were based on thermodynamic considerations; the radical or ion stability will be, in order, tertiary (most stable), secondary, and then primary, and this will dictate structure. This argument was still being used in radical systems until the 1990s.
Few Australian laboratories were interested in the detailed chemistry of radical polymerization; for example at the Royal Australian Chemical Institute (RACI) Symposium on Polymers held in 1957 there were two papers on the synthetic chemistry of polymers and 28 on the physical properties of polymers. Often the structure of the polymers produced by radical chemistry was represented as shown in Fig. 1. The end groups and any H–H groups were considered irrelevant (0.1 % of the structure).[3]
|
However, there were several theoretical and practical observations that this model did not satisfy. Examples are:
-
In practical systems where the polymer is exposed to heat and/or UV light (i.e. paint) the initiator chosen for the synthesis had a significant influence on the stability of the resultant polymer.
-
Poly(methyl methacrylate) (PMMA) synthesized by ionic methods was more stable than the (apparently) same polymer made by radical polymerization. Similar observations applied to poly(vinyl chloride) (PVC).
-
In simple organic radicals, and in some polymerizations (e.g. cyclo-polymerization), the concept of stereo-electronic effects was shown to be important and kinetically preferred structures were formed,[4] and
-
There was a major contradiction in the teaching of radical chemistry. On the one hand radicals were described as highly reactive, neutral species that attack indiscriminately, yet in polymerizations they were believed to give a regular H–T linkage with very specific selectivity.
The above observations led us to a project to study defect groups in polymers produced by radical chemistry. Defect groups are defined as any monomer residue in a different environment (structural) to the major repeat units.[5] These included the initiator end-group (1 in Fig. 2), the end-groups resulting from termination by disproportionation (2 and 3 in Fig. 2) or combination (4 in Fig. 2), as well as any other H–H linkages (4 in Fig. 2).
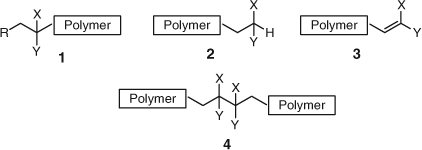
|
Defect Groups
Obvious differences between PMMA synthesized by ionic and radical chemistries are the H–H and unsaturated disproportionation structures observed in the free radical system as shown in Fig. 2. Therefore a model for the H–H component was synthesized and a macromonomer equivalent to the disproportionation molecule was made by chain transfer methods using cobalt tetraphenylporphyrin (CoTpp) as shown in Scheme 2.[6]
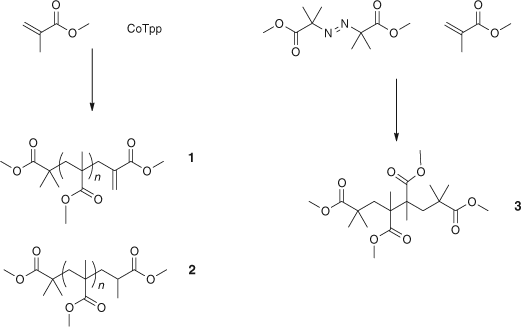
|
Thermal stability studies showed that the stability increased from H–H linkages (3): 195°C least stable, and then the unsaturated macromonomer (1): 225°C, and finally the saturated oligomers and polymers (2): 300°C.[6] These studies helped explain the different thermal stability of ionic and radical PMMA.
Given the mechanism for radical polymerization in Scheme 1 the question arises what is the structure of PMMA at high conversions? That is, in commercial PMMA does the unsaturated disproportionation macromonomer copolymerize to yield a branched structure? Our studies showed that the macromonomer added a cyanoisopropyl radical, it did not homopolymerize, and importantly, the molecular weight of PMMA formed was lowered significantly by the macromonomer.[7] This was explained by what in retrospect was an early version of RAFT, that is a facile β-scission of the adduct as shown in Scheme 3.
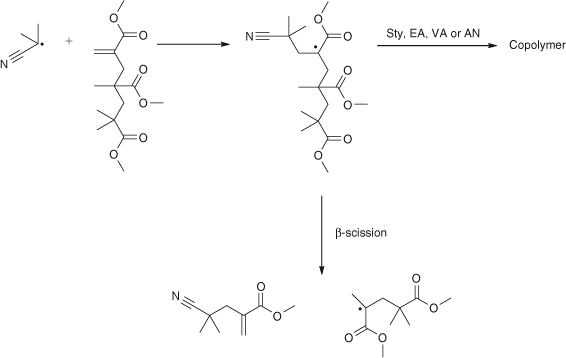
|
However, the incorporation of PMMA macromonomer was much higher in copolymerizations with ethyl acrylate (EA), vinyl acetate (VAc), styrene (Sty), or acrylonitrile (AN).[7] This discovery is discussed more fully in the section on the exciting development of what is termed RAFT (see later section on RAFT).
Nitroxides as Trapping Agents
Two other observations from our studies on commercially important polymer systems were significant in leading to our detailed study of radical polymerization. First was the cyclopolymerization of 1,6-dienes, such as N-alkyl diallylamines. We were able to show that the kinetically preferred 5-membered ring was formed as shown in Scheme 4.[8,9]
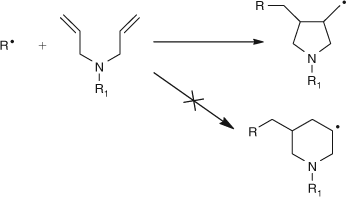
|
Thus in conventional radical polymerization we were firmly of the view that, for example, the regular H–T structure was the result of easier (faster) addition to the tail, not the stability of the radical formed. That is, kinetic (rate) factors were of greater significance than thermodynamic stability.
Likewise further impetus for a detailed study of the initiation step came from an unexpected source. When the clay mineral kaolinite is added to organic systems it gives high viscosities. Treatment of the kaolinite with oleic acid greatly reduces the viscosity (from 48 to 3 Pa in paraffin oil[10]) and also offers the possibility of the bound oleic acid entering into the polymer matrix in, e.g. cured rubber or unsaturated polyesters (the so called graded seal effect observed with carbon black).[10] These oleic acid modified clays were exciting to manufacture because some batches ignited spontaneously in the storeroom; however, industry solved this problem by using an extended drying step. Unravelling the chemistry behind this process was an important step in the study of initiation and is detailed here because of its historical importance in leading to LRP and its illustration of the virtue of understanding the underlying chemistry as distinct from only solving the practical problem. We predicted some radical chain process was responsible for this spontaneous combustion.
Studies on clay/oleic acid chemistry are difficult experimentally and a model system was set up based on the following logic. Oleic acid is known to oxidize at the methylenes adjacent to the double bond to form a hydroperoxide so tertiary butyl hydroperoxide was chosen to model this hypothesized first step. Clay was known to be strongly acidic when dried hence sulfuric acid was used.[11] It was also known that acids decomposed hydroperoxides by an ionic mechanism[12] hence this would compete with any radical process. Therefore methyl methacrylate (MMA) was used as a solvent (trap); the idea being that each radical produced would be reflected in a PMMA molecule, but ionic intermediates would not attack the MMA, i.e. one radical would produce a detectable amount of PMMA.
PMMA was in fact formed and in a very careful product balance and analysis of the PMMA we concluded that[13,14]:
-
Approximately 30 % of t-butyl hydroperoxide underwent an acid induced radical decomposition. This was an important discovery for many reasons including the safe disposal of peroxides.
-
Of the t-butoxy radicals that were generated only 60 % added to the MMA; the remainder formed t-butanol by abstraction.
However, we did not know the precise points of attack of the t-butoxy on the MMA and therefore needed an effective trapping agent to isolate the initial reactions.
Nitroxide Trapping
Ideally the trapping agent would stop the chain after only one or two units had added. Diphenyl picrylhydrazyl (DPPH) had previously been used to trap radicals but we had shown that the use of this was complicated and not definitive.[15] Of the variety of trapping agents and techniques available, such as spin traps, transition metal ions, and nitroxides, we chose the latter for several reasons; nitroxides combine at near diffusion rates with carbon centred radicals to give alkoxyamines which can be separated and identified by conventional organic methods, and nitroxides do not react with oxygen centred radicals, i.e. from our initiating species.
Our first radical trapping experiments were conducted with the readily available nitroxide 2,2,6,6,-tetramethylpiperidinyl-1-oxy (TEMPO, 1 in Fig. 3),[16] but for more detailed work we synthesized the isoindoline nitroxide (1,1,3,3-tetramethylisoindoline-2-oxyl, 2 in Fig. 3) with a chromophore to aid in identification and separation.[17]
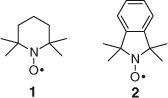
|
Over the years a vast array of other nitroxides has been evaluated. Details of this work are reported in numerous reviews,[18–20] so here we will report on some of our more significant findings and some of their implications.
Effect of Initiator
It is informative to compare two common initiating radicals: t-butoxy and benzoyloxy, and their reactions with two common monomers, Sty and MMA. These studies used either of the two nitroxides shown in Fig. 3.
t-Butoxy
The initiation of MMA with t-butoxy radicals is shown in Scheme 5.[21]
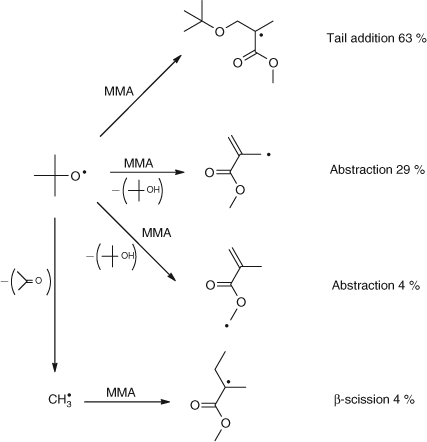
|
This was a seminal study and provided important data including:
-
The significant hydrogen abstraction from the α-methyl group (29 %).
-
Abstraction from the methyl group of the ester. Presumably this could also occur from the polymer.
-
The β-scission of the t-butoxy to give acetone and a methyl radical and hence product (4). Since the rate constant for this unimolecular fragmentation was known we had a ‘radical clock’ to calibrate the absolute rate constant of the reaction pathways.
In contrast the reaction scheme for the initiation of Sty with t-butoxy radicals is essentially clean, and is shown in Scheme 6.[3]
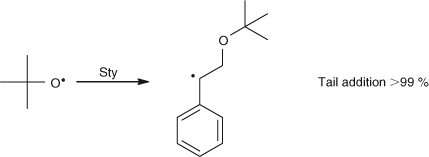
|
Benzoyl Peroxide
The complex reaction of t-butoxy with MMA contrasts with that of benzoyl peroxide which gave essentially only tail addition as shown in Scheme 7.[3]
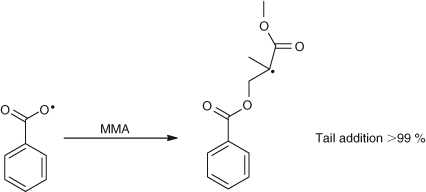
|
However, the reaction scheme for the initiation of styrene with benzoyloxy radicals is shown in Scheme 8 and gives a range of products.[22,23]
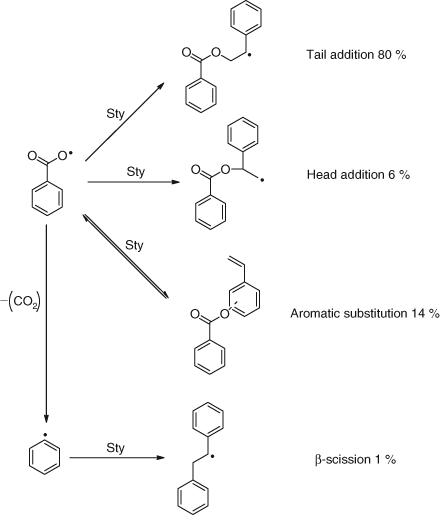
|
Several products in Scheme 8 require comment[22]:
-
The head addition to give the non-resonance stabilized radical. This is further support for kinetic control since thermodynamic considerations would strongly favour the resonance stabilized tail addition. Furthermore we have shown that the secondary benzoate formed by tail addition is thermally less stable than the primary benzoate.[24]
-
The large amount of aromatic substitution. This is reversible but can be trapped with efficient scavengers such as nitroxides.[22]
-
Again we have a radical clock with the formation of phenyl radicals.
These studies clearly showed the need to match the initiating radical to the monomer. There is no one best system.
Other Initiators
Since our original studies numerous other systems have been studied using nitroxide trapping and here we give two examples:
-
Hydroxyl radicals: We generated these in organic media by the use of 2-(t-butylazo)prop-2-yl hydroperoxide.[25] In summary hydroxyl radicals give more head addition to methyl acrylate (17 %) than t-butoxyl (2 %) but less ester abstraction.[3]
-
Cumyloxy radicals: These behave in a similar manner to t-butoxyl but undergo much greater β-scission in MMA.[26]
For more detailed analysis of other radicals refer to Moad and Solomon.[3]
Effect of Monomer
The value of nitroxide trapping is well exemplified in studies involving monomers with multiple points of attack, particularly alkyl methacrylates, and results are indicated in Fig. 4 for t-butoxy radicals.[21] Likewise with allyl methacrylate the attack is also shown in Fig. 4.[27]
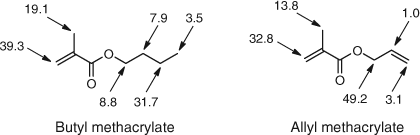
|
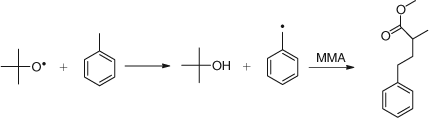
Effect of Solvents
The ability, particularly, of the t-butoxy radical to abstract hydrogen is reflected in the choice of solvent for the polymerization. For example in toluene as solvent, at high conversion, most initiation is by way of solvent derived radicals with the scheme shown in Scheme 9.[28,29]
|
This offers the possibility of end group functionalization by the use of functional solvents.[28]
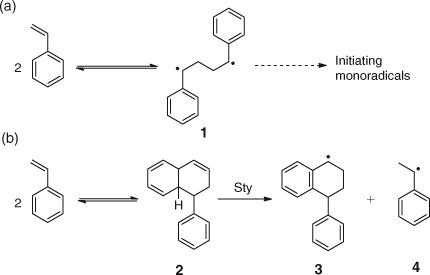
Thermal Polymerization of Styrene
The thermal polymerization of monomers has long been an area of controversy and it is often argued that trace impurities (oxygen) or the type of reaction vessel used could be a factor. However, in the case of Sty there is more acceptance that highly purified monomer can undergo spontaneous polymerization at a rate of ~2 % per hour at around 100°C.[3] The two most widely considered mechanisms are the biradical scheme proposed by Flory, (a) in Scheme 10,[30] and the Mayo molecular assisted homolysis (MAH) mechanism, (b) in Scheme 10.[31]
|
The use of nitroxides results in a much higher rate of radical formation suggesting the nitroxide is interacting with an intermediate such as the Diels–Alder dimer of Sty. This is indirect evidence in support of the Mayo MAH mechanism. Conclusive proof of the MAH mechanism came from the use of FeCl3/DMF as a retarder when both 1-phenyl-1,2,3,4-tetrahydronaphthalenyl and phenylethyl end groups were identified.[32] This confirms that radicals 3 and 4 in Scheme 10 had initiated polymerization and hence the Mayo mechanism is correct.
In summary, the initiation studies provided a sound basis for a scientific selection of the solvent, initiator, and reaction conditions for the polymerization of a given monomer.
Initiator Efficiency
As part of our study on defect groups and radical polymerization we considered the efficiency of traditional radical initiators. Here we will exemplify this work by reference to AIBN.
Initiator efficiency (f) is defined by Eqn 1 where n is the number of moles of radicals generated per mole of initiator
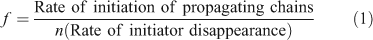
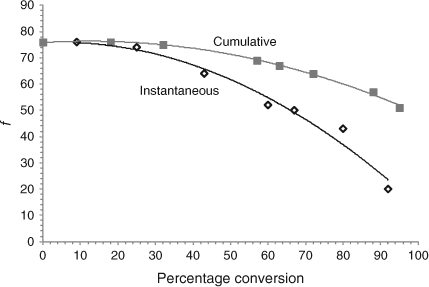
Initiator efficiency is typically measured at zero or low conversions. As the polymerization progresses the viscosity of the medium increases and monomer concentration decreases. Fig. 5 shows the ‘instantaneous’ f, for azoisobutyronitrile (AIBN) initiated Sty, reducing over the course of the reaction from 76 % at low conversion to <20 % at 90–95 % conversion.[33]
|
Loss of useful radicals via the cage reaction is influenced by factors such as viscosity and the proportion of ‘useful’ radicals is in the range 50–70 %. In the case of AIBN this loss, shown in Scheme 11, leads to the combination product tetramethylsuccinonitrile (1 in Scheme 11) which is claimed to be toxic.[34,35] The other disproportionation product, methacrylonitrile (3 in Scheme 11), is a potential comonomer.[36,37] Furthermore, the formation of the ketenimine (2 in Scheme 11) can complicate the analysis.
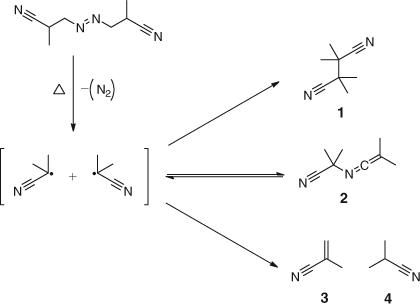
|
Termination in Copolymerizations
Given the influence of macromonomers and other defect structures on polymer properties it is worth discussing an example of copolymerization and the likely end groups that can arise during the termination step. Here we consider a copolymer of Sty and MMA. We used computer simulation to show that during copolymerization there is a greater concentration of styryl ended chains and termination is predominately by Sty–Sty coupling. We also used the appropriate azo compounds and decomposed these in the presence of nitroxides so as to isolate the cage reaction. In this way we could study the cage products. Styrene is generally considered to terminate predominately by combination and MMA by both combination and disproportionation. Of importance is the relative concentrations of each terminal radical and these in turn are determined by reactivity ratios and propagation rate constants.[38]
We showed that in methacylates the size of the ester group has an influence on termination. In the copolymerization of butyl methacrylate and MMA, there is a small preference for the transfer of hydrogen from the radical of the model for the butyl ester of methacrylate to the methyl ester as shown in Scheme 12.[39]
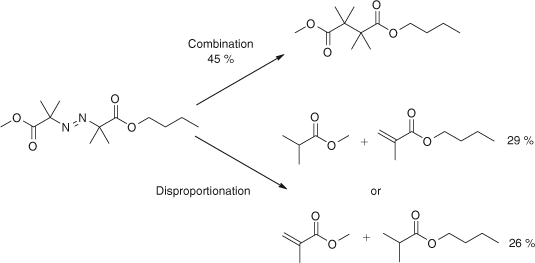
|
Nitroxide Mediated Polymerization (NMP)
Several observations were made from the trapping work with nitroxides which were to lead to NMP:
-
During the isolation and separation of trapped alkoxyamines the characteristic purple colour of the nitroxide was sometimes observed. We avoided this by using low temperatures and other techniques.[40]
-
The cis–trans ratio of Sty dimers changed on heating as shown in Scheme 13.[41]
-
Allylic alkoxyamines from hydrocarbons reverted to equilibrium mixtures as shown in Scheme 14.[42]
-
In several systems we observed molecules with more than one monomer residue.[22,26] Whereas we did not favour the mechanism for their formation as being a reversing of the trapped alkoxyamine with one monomer residue followed by insertion of a second monomer unit, we certainly thought about this possibility. We considered these dimer products formed by the slow trapping of the first formed radical.
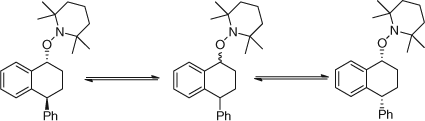
|
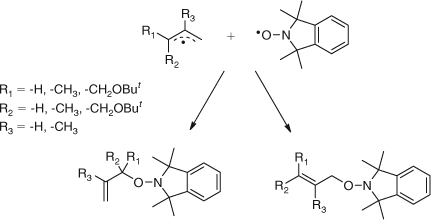
|
These observations suggested that the alkoxyamines formed could be heat labile, with the equilibrium shown in Scheme 15, and this offered the possibility of a controlled growth and the synthesis of structures not previously possible by radical chemistry.
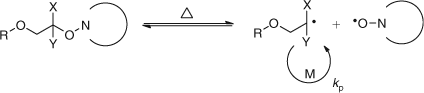
|
The significance of this chemistry was recognized early and we explored the many options that were possible. For commercial reasons we were not permitted to publish this work and a detailed patent was filed in 1984 with the front page shown in Fig. 6.[43]

|
There were two publications, a communication in 1987[44] and a conference paper in 1991.[45] We did publish a theoretical paper that showed that NMP could, in principle, provide narrow polydispersity polymers.[46] The patent used the terms ‘living’, ‘controlled-growth free radical polymerization’, and ‘step growth’ and was a milestone in the renaissance of radical polymerization; the equivalent of the four minute mile.
The 1984 patent was extensive and, in contrast to many patents, contained quite detailed experimental procedures. In fact the patent was essentially a scientific paper. In view of the extensive literature on NMP and some of the claims made it is informative to summarize the disclosures of the 1984 patent since we have published only some of the details in scientific journals.[47,48]
New Initiators
The patent details the preparation of three types of new initiators:
-
The alkoxyamine of an initiator such as AIBN and a nitroxide ((a) in Scheme 16),
-
The alkoxyamine formed from an initiator, a monomer, and a nitroxide ((b) in Scheme 16), and,
-
Alkoxyamines prepared from functional initiators such as 4,4′-azobis(4-cyano-n-pentanol) (compounds 3 and 4 in Scheme 16).
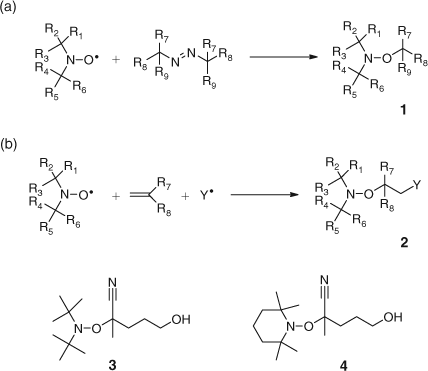
|
In preparing these initiators we were guided by the results of our previous trapping work. For example in preparing the alkoxyamine from styrene we used t-butoxy, not benzoyl peroxide. More recently these alkoxyamines have been described as ‘unimers’. This term relates to the fact that there is only one monomer unit in the compound. Of course we reported that any of the above could be prepared in situ using the free nitroxide. However, in the work we reported we purified and characterized each alkoxyamine since our previous nitroxide trapping work had shown that few of the initiating steps were clean.
Nitroxide Structure
A broad range of nitroxides was disclosed. Of great practical and theoretical significance was the reporting of sterically crowded nitroxides of the general formula shown as 1 in Fig. 7. Initially we used TEMPO and 1,1,3,3-tetramethylisoindoline-2-oxyl (shown in Fig. 3).[21] In the patent we further disclosed the synthesis and use of 1,1,3,3-tetraethylisoindolin-2-yloxyl (2 in Fig. 7), 1,1,3,3-tetra-n-propylisoindolin-2-yloxy (3 in Fig. 7), 2,6-dimethyl-2,6-di-n-propylpiperidin-1-yloxyl (4 in Fig. 7), and di-t-butyl nitroxide (5 in Fig. 7).
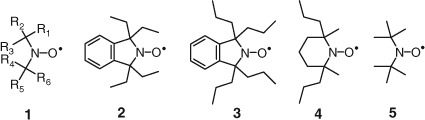
|
Monomers Studied
The monomers disclosed in the patent were: Sty, α-methyl styrene, AN, methacrylonitrile, methyl acrylate, EA, MMA, isobutyl methacrylate and VAc. The patent also used polybutadiene and poly(isobutyl methacrylates).
Molecular Weight and Molecular Weight Distribution
The patent covered molecular weights ranging from 946 to 60000 Da. Most of the examples in the patent had a broad molecular weight distribution (MWD or Mw/Mn) although one example with PMMA did have a MWD of 1.15. A few years later we realised that in principle NMP was capable of yielding narrow MWD and we published a theoretical paper on what is now known as the persistent radical effect.[46]
Polymer Structure
Homopolymers, block copolymers (di- and tri-), as well as graft copolymers were described. Later we were to use NMP to produce microgels.[49] Various homopolymers (1 and 2 in Fig. 8), block copolymers 3 and 4, and a graft copolymer 5 reported in the patent are shown in Fig. 8.
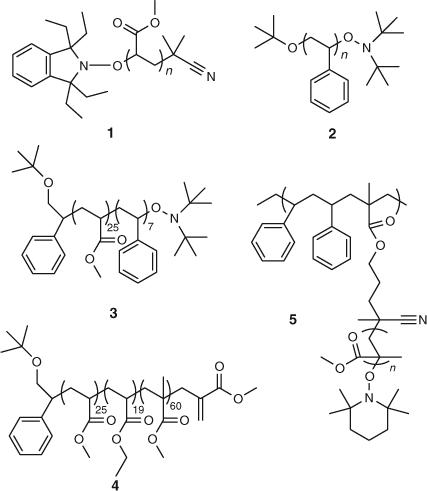
|
Macromonomers
These were formed by elimination of the hydroxylamine from an oligomer. Scheme 17 shows the removal of the hydroxylamine to form macromonomer 1, while the structures of two other macromonomers are also shown.
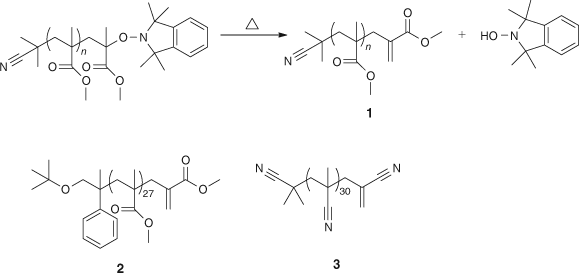
|
Reactions of Aminoxyl End Groups
Removal of the aminoxyl end group is desirable in most commercial applications and two methods were reported (as well as the elimination of the hydroxylamine to form macromonomers) and are shown in Scheme 18. Part (a) shows the conversion of the aminoxyl end group into a hydroxyl end group by zinc-acetic acid reduction while part (b) shows the replacement of the aminoxy end group by a hydrogen through the use of a thiol.
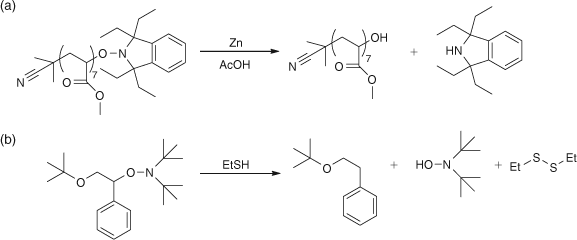
|
Solvent Effects and Stability of Alkoxyamine
Most alkoxyamines were characterized by purification and then NMR spectroscopy and a half-life stability measured by the loss followed by HPLC when heated in ethyl acetate in the presence of a 10–20 fold excess of another nitroxide.[48] Typical results are shown in Fig. 9.
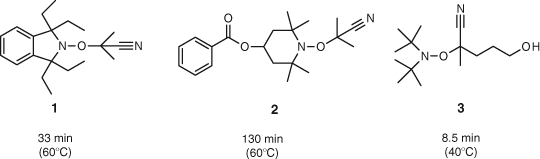
|
Note the wide range of stabilities and the clear effect of the steric hindrance on the stability. Also a change in solvent had a significant effect on half-life as can be seen in Table 1 for alkoxyamine 1 in Fig. 9.

|
Reversible Addition–Fragmentation Chain Transfer Polymerization (RAFT)
We have previously mentioned the β-scission of the radicals derived from addition of radicals to certain macromonomers (Scheme 3). The following section describes how this work led to the development of small-molecule addition–fragmentation chain transfer agents (AFCTAs) and ultimately to the discovery of RAFT.[50]
Macromonomers as AFCTAs
The concept of chain transfer by a mechanism involving addition–fragmentation arose from observations during a study on the chemistry of MMA macromonomers (Fig. 10).
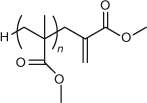
|
A detailed study of cyanoisopropyl radical addition to MMA trimer (Fig. 10 when n = 2) revealed that the favoured reaction of the adduct radical was β-scission.[7] The major products from this pathway, which are shown in Scheme 3, are a propagating radical and a new alkene terminated oligomer, i.e. a new macromonomer. The new propagating radical terminates by disproportionation to yield MMA dimer, (Fig. 10 when n = 1) or by combination with itself or with the cyanoisopropyl radical.
In an extension to this work, we investigated the copolymerization of methacrylate based macromonomers with acrylates, methacrylates, Sty, AN, VAc, and acrylamide. It was observed that macromonomers copolymerize with the less sterically hindered monomers (e.g. acrylates, Sty, AN, acrylamide) to afford graft copolymers.[7,51] In contrast, little[7] or no[51] copolymerization was observed with the more sterically hindered methacrylates (e.g. see Table 2).

|
Based on the above observations, we proposed a general mechanism to explain the results of copolymerization of methacrylate macromonomers with various monomers (Scheme 19).[51]
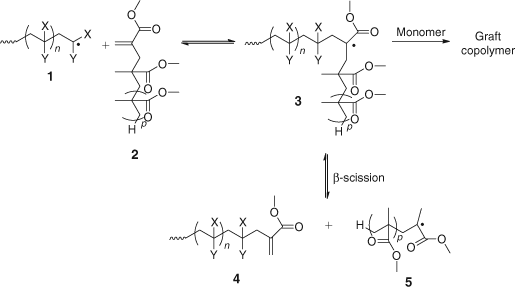
|
The addition of propagating radical 1 to methacrylate macromonomer 2 in Scheme 19 is expected to occur readily to give adduct radical 3.[7,51] The adduct radical 3 can react by one of three different pathways: (a) it can add to monomer which would result in the macromonomer becoming incorporated into the polymer backbone (yielding a graft copolymer), (b) it can revert to the starting species in which case the propagating radical 1 can continue to grow by further addition of monomer, or (c) it can fragment, by β-scission, and in so doing generates a polymeric re-initiating radical 5 and a new alkene terminated oligomer 4 whose backbone is based on the propagating radical 1. The relative rates of these pathways depend on the monomer used. For alkyl acrylates (1; X = H, Y = CO2R), fragmentation of the adduct radical 3 to 4 and 5 is likely to be more favourable than fragmentation by the reverse reaction to 1 and 2. This is because the former route gives rise to a more stable tertiary radical 5 compared with the secondary radical of the alkyl acrylate propagating species 1.
However, propagation of the adduct radical 3 (X = H, Y = CO2R) in Scheme 19 by reaction with monomer is also favourable presumably because the alkyl acrylate monomer is relatively unhindered and so can be attacked by the bulky radical 3. This is evident from the results in Table 2 which show that for EA, incorporation of MMA macromonomer is high. In contrast, there is little incorporation of macromonomer in copolymerizations with MMA (i.e. propagating radical 1; X = Me, Y = CO2CH3).[7]
In a follow up study, ethyl methacrylate (EMA) was polymerized in the presence of MMA dimer or trimer (2, P = 0 or P = 1, respectively). A 1H NMR analysis of the product revealed that only one macromonomer unit per chain was incorporated and that this resided at the chain ends, in agreement with the mechanism in Scheme 19. The lack of macromonomer copolymerization with MMA suggests that propagation of adduct radical 3 is severely limited.[51] Indeed, the large van der Waals interactions between a bulky radical 3 approaching a hindered methacrylate monomer is likely to be the reason for propagation of radical 3 to be unfavourable. Ultimately, the favoured route and the path forward is through fragmentation by β-scission. In the case of a propagating radical derived from styrene (1; X = H, Y = Ph), the situation is between the methacrylate and acrylate examples with a higher level of macromonomer incorporation than methacrylates but lower than the acrylates.
It is evident from the discussion above that methacrylate macromonomers act as chain transfer agents in free radical polymerization by a mechanism of addition–fragmentation and in so doing install an olefinic group at the end of the polymer chains. These products are themselves macromonomers that can react further as the polymerization progresses. For polymerization of acrylates in the presence of macromonomers the newly formed macromonomers will be branched because of concurrent copolymerization. The continued copolymerization of these would give rise to highly branched block copolymer structures. The polymerization of methacrylates in the presence of methacrylate macromonomers will yield new methacrylate macromonomers that will in turn participate in the addition–fragmentation process. This ‘recycling’ is the genesis of reversible addition–fragmentation chain transfer discussed in a later section.
Small Molecule AFCTA
The utility of the addition–fragmentation chain transfer was further developed by designing three classes of simple organic molecules with a general structure shown in Fig. 11.[52]
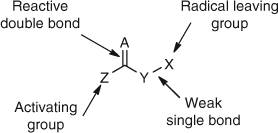
|
These classes are represented by allylic compounds (Fig. 11, A = Y = CH2), which include allylic sulfides (X = SR),[52–54] allylic peroxides (X = OOR),[55,56] allylic bromides (X = Br),[52,57] allylic sulfones (X = S(O)R),[52,57] allylic phosphonates (X = P(O)(OR)2),[52,57] and allylic stannanes (X = SnR).[52,57] The other two classes are vinyl ethers (Fig. 11, A = CH2, Y = O)[52,58,59] and thiocarbonyl compounds (Fig. 11, A = S, Y = O).[60,61] The group X is a good homolytic leaving group and Z can be varied to give an optimum chain transfer constant (Ctr).
Addition–fragmentation chain transfer is a versatile process being applicable to a range of monomers (Sty, (meth)acrylates, vinyls) and having the flexibility to introduce functionality at the α and/or ω terminal ends.[62] The general mechanism of the addition–fragmentation chain transfer process is outlined in Scheme 20 and gives rise to polymers as shown. The molecular weights of the polymers are controlled by the amounts of chain transfer agent added, the rates of polymerization are essentially identical to those in the absence of transfer agent and the polydispersities are similar to those of conventional polymerizations (1.5–2.0).
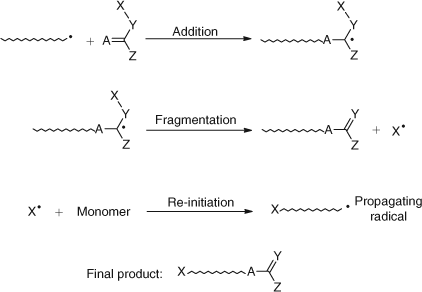
|
The incorporation of appropriate functionality in the X and Z groups (Scheme 20) will result in mono and/or di end functional oligomers.
The concept of carbon–carbon bond formation by radical addition–fragmentation has its origins in the original work by Lewis et al. who reported the allylation of aldehydes utilizing radical addition–fragmentation.[63] Sometime later, the major efforts of Keck et al.[64] Baldwin et al.,[65] and Barton and Crich[66] all helped to establish radical addition–fragmentation as a useful tool for C–C bond formation. Although the addition–fragmentation chain transfer process has its roots in these original works, there is an added dimension when applying the basic concepts of radical addition–fragmentation to free radical polymerization. In order for chain transfer by radical addition–fragmentation to operate effectively, the process must compete with propagation. To achieve this, the chain transfer agent must have: (a) a double bond whose reactivity towards propagating radicals is similar to that of the polymerizing monomer, (b) fast fragmentation of the resulting adduct radical, and (c) efficient re-initiation of polymerization by the expelled radical.[62,67]
This section will discuss how we employed simple AFCTAs to control molecular weight and to prepare end functional polymers.
Allylic Class of AFCTAs
The allylic class of AFCTAs, represented in Fig. 12, encompasses a group of compounds which include the allylic sulfides, allylic peroxides, allylic bromides, allylic sulfones, allylic phosphonates, and allylic stannanes.
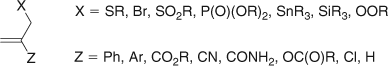
|
The allylic class of AFCTAs yield macromonomers as final products by the mechanism shown in Scheme 21.
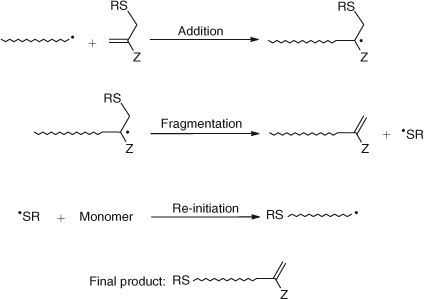
|
The majority of compounds in the allylic class have been reported in a 1988 patent.[52] A summary of the different types of allylic compounds used as AFCTAs, and hence the macromonomers formed (Scheme 21), is given in Table 3.
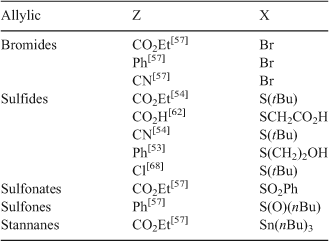
|
As is evident from Table 4, the allylic sulfides (Fig. 12, X = SR) are attractive AFCTAs. First, their Ctr are in the range of greatest utility (an ideal Ctr is 1.0) for controlling molecular weight in batch polymerizations.[69] Second, they can be used to prepare a range of macromonomers based on methacrylates, Sty, and acrylates, and third, they can provide both mono- and di- end functional polymers when appropriate functionality is introduced in Z and R groups.
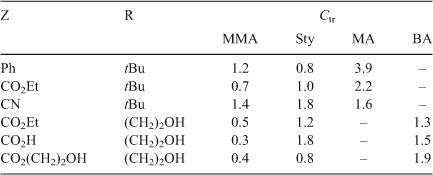
|
For example, according to Scheme 21, utilizing a dihydroxy functional AFCTA (last entry, Table 4) will lead to a dihydroxy end functional macromonomer (Fig. 13).
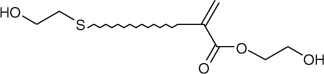
|
The other compounds in the allylic class of AFCTA (e.g. allylic bromides, allylic sulfones, allylic phosphonates, and allylic stannanes) are also useful chain transfer agents in controlling molecular weight in polymerizations as judged by their Ctr (Table 5).
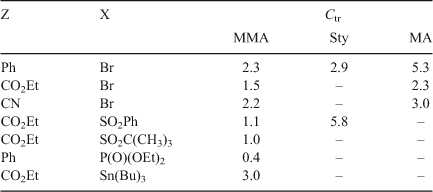
|
The allylic peroxides (Fig. 12, X = OOR) fall under the allylic class of AFCTAs, but yield oligomers with an oxirane end group rather than a 1,1-disubstituted alkene end group as for the general allylic class.
The mechanism by which the oxirane end group is obtained is depicted in Scheme 22 and is largely similar to the mechanism of action of allylic AFCTAs. A propagating radical adds to the allylic peroxides giving an intermediate adduct radical. This radical undergoes a 1,3-SHi intra-molecular homolytic substitution resulting in the cleavage of the weak peroxy bond. In contrast to the general allylic class of AFCTA which fragment by β-scission, fragmentation for the allylic peroxides proceeds by γ-scission; this results in the formation of an epoxide and the expulsion of an alkoxyl radical (Scheme 22). The epoxide end group structure has been verified by 1H NMR analysis.[62]
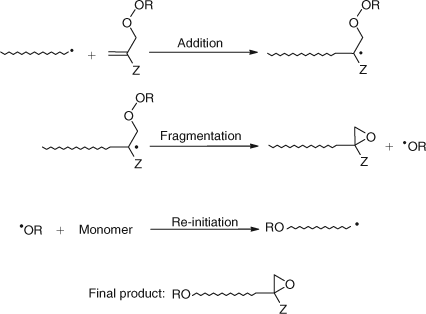
|
The Ctr of allylic peroxides determined in MMA, Sty, and methyl acrylate (Table 6) show that these compounds are effective chain transfer agents. The Ctr of allylic peroxides is several orders of magnitude higher than simple dialkyl peroxides (compare with di-t-butyl peroxide; Ctr of 0.00023–0.0013 and di-isopropyl peroxide; Ctr of 0.0003) which suggested that chain transfer with the allylic peroxides is not due to direct attack at the peroxy bond.
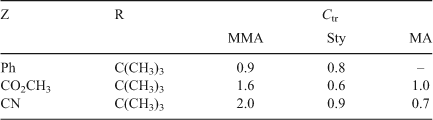
|
Vinyl Ether Class of AFCTAs
A range of compounds in the vinyl ether class, as shown in Fig. 14, are effective as AFCTAs.[58,59]
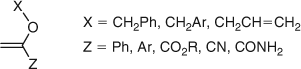
|
The mechanism of addition–fragmentation chain transfer follows the general route discussed for allylic compounds. The vinyl ether AFCTAs yield oligomers with a ketone end group structure as shown in Scheme 23. In this case, the formation of the stable C–O double bond is an added driving force for the fragmentation process.
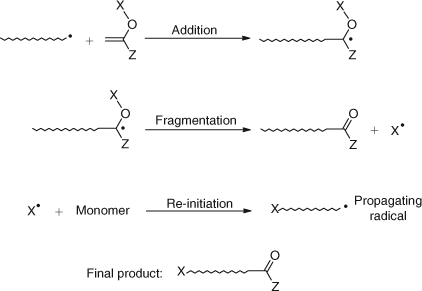
|
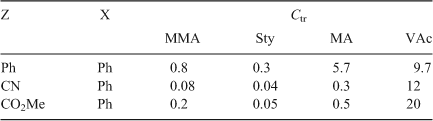
Several compounds in the vinyl ether class have Ctrs that approach the ideal value of 1.0 (Table 7).
|
Thiocarbonyl Class of AFCTAs
The use of thiohydroxamic esters for the generation of alkyl radicals in organic chemistry is well known through the work of Barton and coworkers.[70] The thiocarbonyl class of AFCTAs was broadened when thionoesters, represented in Fig. 15, were reported to undergo chain transfer by the addition–fragmentation process.[62]
|
The use of thionoesters as AFCTAs produced oligomers with a thioloester end group, and is outlined in Scheme 24.
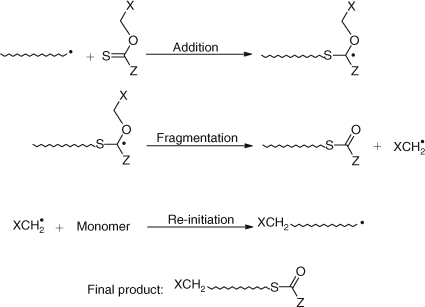
|
Examples of thionoester AFCTAs are listed in Table 8, along with their Ctr in Sty, methyl acrylate, and VAc polymerizations.[62]
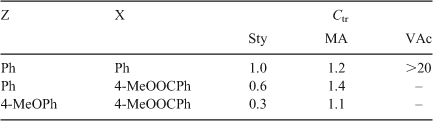
|
The RAFT Process
In an extension of the addition–fragmentation concept, methacrylate based macromonomers have been shown to impart ‘living’ behaviour to free radical polymerizations. To demonstrate this feature, narrow polydispersity block copolymers, based on methacrylates, were prepared under feed conditions.[68,71,72] The mechanism was envisaged to operate by RAFT and represented a new process for achieving LRP. Subsequently, it was found that simple organic compounds possessing the thiocarbonylthio moiety (S=C–S) were much more effective and versatile in inducing ‘livingness’ by RAFT.[73–77] For convenience the terms ‘RAFT process’ and ‘RAFT agents’ were coined.[75] It should be noted here that researchers at Rhodia in France simultaneously and independently observed that xanthates provide living characteristics to the polymerization of certain monomers. They labelled their mechanistically identical process: macromolecular design by the interchange of xanthates (MADIX).
Representative examples of thiocarbonylthio RAFT agents are shown in Fig. 16.
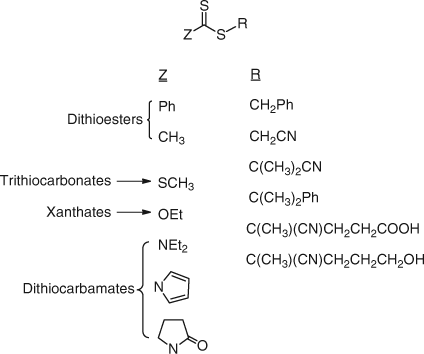
|
There are four common classes of thiocarbonylthio RAFT agents, depending on the nature of the Z group; (a) dithioesters (Z = aryl or alkyl), (b) trithiocarbonates (Z = substituted sulfur), (c) dithiocarbonates (xanthates) (Z = substituted oxygen), and (d) dithiocarbamates (Z = substituted nitrogen).
RAFT polymerization is performed by adding a chosen quantity of an appropriate RAFT agent to a conventional free radical polymerization mixture and yields polymers of predetermined chain length and narrow polydispersity. Polydispersity indices of less than 1.1 can be usually achieved under optimal conditions. The RAFT process offers the same versatility and convenience as conventional free radical polymerization. It is applicable to the same range of monomers (e.g. (meth)acrylates, Sty, acrylamides, vinyls), solvents, functional groups (e.g. OH, CO2H, NR2, NCO) and reaction conditions (e.g. bulk, solution, suspension, and emulsion).
In emulsion polymerizations, RAFT introduces some additional challenges related to the difficulty in the transport of the RAFT agent from the organic layer through the aqueous phase and into the growing polymer particle. We have discussed these challenges in our contributions to this topic[78–80] and in our comprehensive reviews of the RAFT process.[81–83]
RAFT polymerization can be initiated by any source of free radicals including UV irradiation[84] and gamma irradiation[85] and can be performed over a wide range of temperatures, including room temperature.[86] We have also shown that it can be carried out successfully in a continuous flow microreactor[87] or performed in the presence of Lewis Acids to modify the stereochemistry of certain polymers.[88,89]
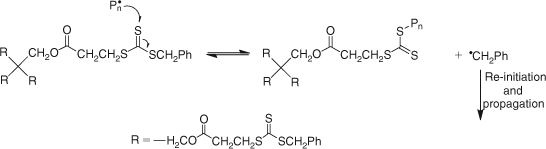
The mechanism of RAFT polymerization with the thiocarbonylthio-based RAFT agents involves a series of addition–fragmentation steps as depicted in Scheme 25.
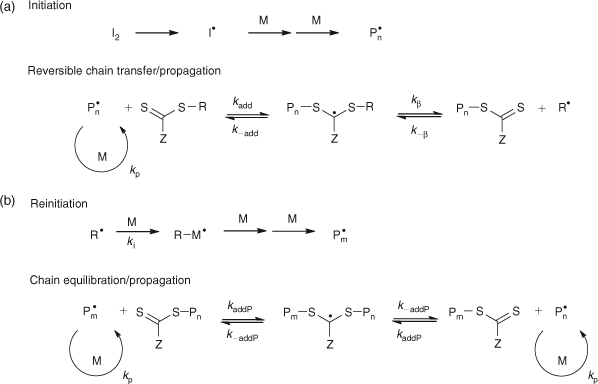
|
As for conventional free radical polymerization, initiation by decomposition of an initiator leads to the formation of propagating chains. In the early stages, addition of a propagating radical (Pn•) to the RAFT agent (S = C(Z)SR) followed by fragmentation of the intermediate radical gives rise to a polymeric RAFT agent and a new radical (R•). The radical R• re-initiates polymerization by reaction with monomer to form a new propagating radical (Pm•). In the presence of monomer, the equilibrium between the active propagating species (Pn• and Pm•) with the dormant polymeric RAFT compound provides an equal probability for all the chains to grow. This feature of the RAFT process leads to the production of polymers with narrow molecular weight distributions, as exemplified in Fig. 17.
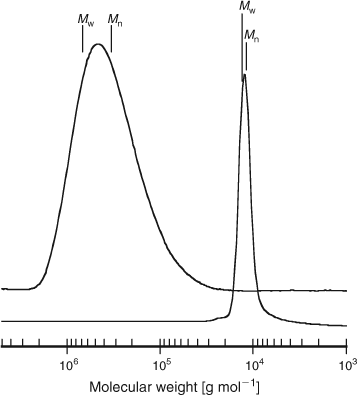
|
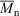 324000,
324000, 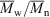 1.74, 72 % conversion) and a similar polymerization in the presence of cumyl dithiobenzoate (0.0029 M) (
1.74, 72 % conversion) and a similar polymerization in the presence of cumyl dithiobenzoate (0.0029 M) (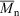 14400,
14400, 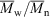 1.04, 55 % conversion).[
1.04, 55 % conversion).[When the polymerization is complete, the great majority of the chains contain the thiocarbonylthio moiety as the end group (Scheme 25) which has been identified by 1H NMR and UV-vis spectroscopy[75] in addition to matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF/MS).[91] Further evidence for the proposed mechanism was provided by the identification of the intermediate thioketal radical ((a) and/or (b), Scheme 25) by ESR spectroscopy.[92]
In order for the RAFT process to function effectively there are certain aspects of the polymerization conditions that require consideration. The most critical considerations are choice of the RAFT agent and an appropriate rate of initiation.
The RAFT agent must be chosen such that its chain transfer activity is appropriate to the monomer(s) to be polymerized. The electronic properties of the Z group and the stereoelectronic properties of the R group determine the chain transfer activity of the RAFT agents (Fig. 16). In general, many RAFT agents behave as ideal chain transfer agents as judged by the rates of polymerization being similar to rates of polymerization without the RAFT agent (within 20 %). However, inappropriate choice of the RAFT agent can lead to significant retardation, particularly when high concentrations are employed to prepare low molecular weight polymers. The retardation phenomenon has been attributed to several factors which have been explained in terms of the mechanism of the RAFT process (Scheme 25)[90]: (a) slow fragmentation of the adduct radical (A) formed from addition to the original RAFT agent, (b) slow re-initiation by the expelled radical R•, (c) preference for the expelled radical R• to add to the RAFT agent rather than to monomer, (d) slow fragmentation of the adduct radical (B) formed from addition to the polymeric RAFT agent, (e) preference for the propagating radical (Pn• and Pm•) to add to the RAFT agent rather than to monomer. The observed retardation phenomenon has been addressed by the appropriate choice of Z and R groups. For example, in styrene polymerization with cumyl dithiobenzoate (1a in Fig. 18) retardation was observed as an inhibition period accompanied by slow consumption of the RAFT agent. Since the cumyl radical is expected to be a good leaving group, the observed inhibition was attributed to (b) and/or (c) above. The retardation in Sty polymerization was averted by using a RAFT agent with cyanoisopropyl as the radical leaving group (1b in Fig. 18).[90] Similarly, retardation was observed in the polymerization of n-butyl acrylate with cumyl dithiobenzoate (1a in Fig. 18) or benzyl dithiobenzoate (1c in Fig. 18), even though the original RAFT agent was rapidly consumed. The observed retardation was attributed to the polymeric RAFT agent (i.e. (d) and/or (e) above). The retardation was alleviated by using benzyl dithioacetate (2 in Fig. 18) as this RAFT agent is less active implying that addition to the C=S bond is less favoured and hence occurs at a slower rate but the resultant adduct radical is less stable thus favouring fragmentation (refer to Scheme 25). A possible side reaction in RAFT polymerization is termination of propagating radicals by radicals (A) and (B) Scheme 25.[93]
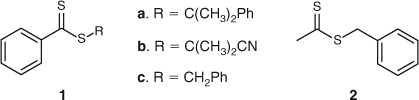
|
The Z group in the RAFT agent influences the reactivity of the double bond. As such the Z group must be chosen so that it activates the double bond towards radical addition but at the same time does not provide too great a stabilizing influence on the adduct radical (as this will contribute to slow fragmentation and hence retardation) (see Scheme 25). Similarly, the R group must be chosen such that it is a good radical leaving group relative to the radical of the propagating species.[75,94] The leaving group R• should also preferentially add to monomer.
In contrast to the chain transfer constants for macromonomers which range from 0.01 to a theoretical maximum of 0.5, the transfer constants of various RAFT agents have been found to span more than five orders of magnitude. The transfer constants have been measured in the range of 0.01 to above 1000 depending on the nature of Z, R, and type of monomer.[90,94–96] It has been reported that to obtain narrow polydispersity polymers (Mw/Mn < 1.5) in a batch process with degenerative chain transfer (e.g. RAFT process) the Ctr of the transfer agent should be greater than two.[97] However, it has been shown that this limitation can be overcome by the use of a monomer feed polymerization process to reduce the rate of propagation and thereby produce narrow polydispersity polymers from a less active CTA.[68,98]
The effect of various Z groups on the chain transfer activity of RAFT agents was discussed in a recent report. By keeping the R group constant, a direct correlation between the chain transfer activity and the reactivity of the C=S bond was observed. The change in the reactivity of the C=S bond was related to the heats of reaction for C=S addition and the LUMO energies.[95] For the RAFT polymerization of Sty, the chain transfer constants were found to decrease in the series where Z is: aryl (Ph) >> alkyl (CH3) ~alkylthio (SCH2Ph, SCH3) ~N-pyrrolo >> N-lactam > aryloxy (OC6H5) > alkoxy >> dialkylamino. Some examples of chain transfer constants are given in Table 9.

|
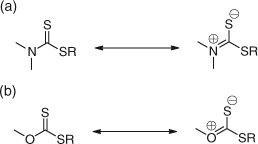
As is evident from Table 9, the chain transfer activity of dithiocarbamates and xanthates is generally low but also dependent on the nature of the substituents on nitrogen and oxygen. For dithiocarbamates, the Ctr increases when the N substituent is: N,N-dialkyl > N-lactam > N-pyrrolo. The low activity of the N,N-dialkyl dithiocarbamates and xanthates has been explained in terms of the contributions from the zwitterionic resonance structures (Scheme 26).[93,99]
|
The conjugation of the lone pair of electrons (on the nitrogen or oxygen) with the C=S double bond reduces the double bond character, thus raising the LUMO and HOMO energies and making radical addition less favourable.[95] The implications for the less reactive propagating radicals derived from Sty and MMA is that the use of N,N-dialkyl and N-phenyl, N-alkyl dithiocarbamates are ineffective as RAFT agents giving no significant molecular weight control. For more reactive propagating radicals, for example, acrylyl, these RAFT agents are mildly effective giving molecular weight control but generally broad polydispersities. In the case of highly reactive propagating radicals, for example, derived from vinyl esters, these RAFT agents give good molecular weight control and low polydispersity indices. The xanthates are only mildly effective as RAFT agents for acrylates, styrenes, and acrylamides giving some molecular weight control but broad polydispersities. This limitation has been overcome by attaching electron withdrawing groups (both inductive and mesomeric) to the nitrogen and oxygen centres of dithiocarbamates and xanthates, respectively, thus reducing the conjugation of the lone pair of electrons with the C=S bond.[74,93,99] This results in an increase in the Ctr value, with the extent of increase depending on the extent to which the lone pair of electrons are removed from conjugation with the C=S bond (Table 9).
In a parallel study the Z group was kept constant and the effect of the R group on the chain transfer activity of RAFT agents was investigated.[94] It was reasoned that the Ctr should reflect the effect of the R group on the partitioning of the intermediate adduct radical between starting material and product (see Scheme 25). In the RAFT polymerization of MMA with dithiobenzoates (S=C(Ph)SR), the effectiveness of the RAFT agent (i.e. the leaving group ability of R) decreases in the order: C(Alkyl)2CN ~C(Alkyl)2Ph > C(CH3)2C(=O)OEt >C(CH3)2C(=O)NH(alkyl) > C(CH3)2CH2C(CH3)3 ~CH(CH3)Ph > C(CH3)3 ~CH2Ph. In reality, only the first two groups are effective in preparing PMMA of narrow polydispersity (Mw/Mn ≤ 1.1) and predetermined molecular weight. Some examples of Ctr are listed in Table 10. These indicate that both steric and polar factors determine the leaving group ability of R. As discussed above, when choosing an appropriate R group in the RAFT agent the polar and steric factors must be balanced with the requirement that R• must be efficient in re-initiating polymerization.

|
The Ctr of polymeric RAFT agents have much higher values[96] (Table 11), possibly reflecting the increased leaving group ability of the bulkier polymeric group compared with the R groups of the RAFT agents in Table 9 and Table 10.

|
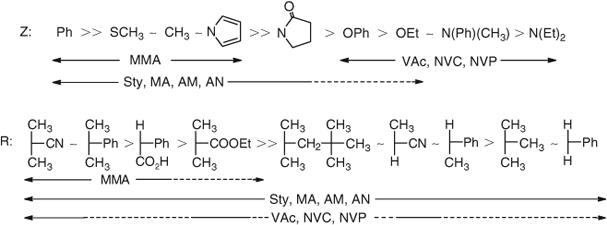
On the basis of these studies we have been able to provide general guidance on how to select the appropriate thiocarbonylthio RAFT agent for a particular monomer.[81,82,89] This is shown in Fig. 19.
|
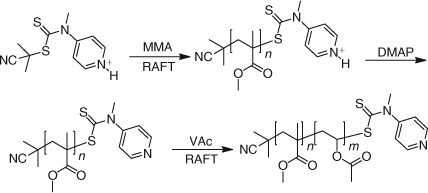
We have found that RAFT agents such as dithioesters or trithiocarbonates suitable for controlling the polymerization of ‘more-activated’ monomers (MAMs) e.g. MMA, Sty, MA, inhibit or retard the polymerizations of ‘less activated’ monomers (LAMs) e.g. VAc, N-vinylpyrrolidone (NVP), N-vinylcarbazole (NVC), while RAFT agents suitable for controlling the polymerizations of LAMs such as N,N-dialkyl- or N-alkyl-N-aryl dithiocarbamates and xanthates tend to be ineffective with MAMs. We have solved this problem by designing a new class of RAFT agents that can be switched by protonation/deprotonation to achieve good control over polymerization of both MAMs and LAMs and hence offer a route to polyMAM-block-polyLAM.[100–102] N-(4-Pyridinyl)-N-methyldithiocarbamates are effective with LAMs and in the presence of a strong acid, the protonated form of the N-(4-pyridinyl)-N-methyldithiocarbamates provide excellent control over the polymerization of MAMs. The process is illustrated in Scheme 27 for the preparation of PMMA-b-PVAc. Thus in the first step the protonated RAFT agent (formed by adding 4-toluenesulfonic acid) is used to control the polymerization of MMA to form PMMA. This macroRAFT agent is then neutralized in situ by adding a stoichiometric amount of N,N-dimethylaminopyridine (DMAP). RAFT polymerization of VAc then provided the desired block copolymer.
|
RAFT Polymerization of Methacrylates
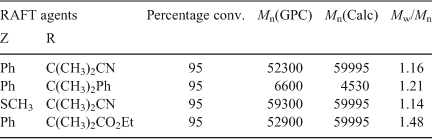
The methacrylyl propagating radical is a sterically bulky radical of moderate reactivity. In choosing the requirements for a RAFT agent, group Z could be aryl, alkyl, or S(alkyl) as these groups are sufficiently radical stabilizing to allow an appropriate rate of addition of the methacrylyl propagating radical to the C=S bond. The R group should be a better leaving group than the methacrylyl radical, hence R• should also be sterically bulky (i.e. tertiary) and possess a radical stabilizing functionality. As discussed above, RAFT agents containing C(alkyl)2CN and C(alkyl)2Ph as the R group are effective in preparing PMMA of narrow polydispersity and predetermined molecular weight.[93,94] Some examples of PMMA prepared with RAFT agents meeting these criteria is shown in Table 12.
|
Interestingly, when R = C(CH3)2CO2Et, we obtained good molecular weight control but the polydispersity index was only 1.48 (last entry, Table 12). This indicates that this RAFT agent has relatively poor chain transfer activity which is likely to be a reflection of the poorer leaving group ability of ethyl isobutyrate compared with, for example, cyanoisopropyl or cumyl (first and second entries respectively in Table 12). Since the steric bulk between the three groups is likely to be similar, it may reflect the lower radical stabilizing ability if the CO2Et group compared with CN or Ph.
The utility of the RAFT process can be illustrated further by the following example of the RAFT polymerization of MMA. A series of MMA polymerizations was carried out at 90°C with 1,1′-azobis(1-cyclohexanenitrile) initiator, and using a ~60-fold range of concentrations of S-dodecyl S-(2-cyano-4-carboxy)but-2-yl trithiocarbonate.[103] The molecular weights, ranging from 2600 to 125000, agree with expectation based on the concentrations of RAFT agent and initiator used (Table 13). All samples have narrow molecular weight distributions (Mw/Mn < 1.2).

|
RAFT Polymerization of Styrenes
For the RAFT polymerization of Sty there is more freedom in the choice of the RAFT agent compared with the RAFT polymerization of methacrylates. This is particularly true in the choice of R groups. The list of RAFT agents producing narrow polydispersity PS also includes groups where R = CH2Ph (Table 14). The greater flexibility in effective R groups is due to the less bulky nature and lower propagation rate of the polystyryl radical compared with the polymethacrylyl radical. Hence the steric and electronic stabilization parameters are not as demanding as for methacrylate polymerization.

|
For reasons discussed previously, the low Ctr of N,N-dialkyl dithiocarbamates (Table 9) makes these ineffective agents for the RAFT polymerization of Sty. There is no control in molecular weight or narrowing of polydispersity in styrene polymerization when Z = N(Et)2 and R = CH2Ph (Table 14) This is an indication that the styryl propagating radical is not adding to the RAFT agent due to the reduced reactivity of the C=S bond arising from conjugation with the nitrogen lone pair of electrons. When the extent of this conjugation is reduced, the RAFT agent is effective in styrene polymerization. This is exemplified in the last entry in Table 14.
RAFT Polymerization of Acrylates
The acrylyl propagating radicals have relatively low steric bulk and high reactivity. These characteristics are ideal for addition to the C=S bond and for expulsion of the R group in RAFT agents. Consequently, there is a wider choice in both the Z and R groups available for the RAFT polymerization of acrylates. This is evident from the examples presented in Table 15.

|
The xanthates are only mildly effective RAFT agents in acrylate polymerizations, providing good molecular weight control but polydispersity remains relatively high (last entry, Table 15).
RAFT Polymerization of Acrylamides
As for the acrylyl propagating radical, the acrylamidyl propagating radical possesses relatively low steric bulk and high reactivity. Consequently, a similarly wide range of RAFT agents would be useful in the RAFT polymerization of acrylamides. We have reported some examples of the RAFT polymerization of N,N-dimethylacrylamide (DMA) and N-isopropylacrylamide (NIPAM) (Table 16).[93,104]

|
RAFT Polymerization of Vinyl Esters
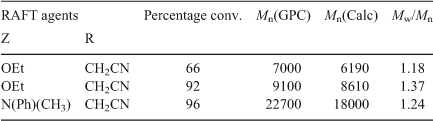
The VAc propagating radical has quite different reactivity characteristics compared with (meth)acrylate and Sty derived propagating radicals. For example, the VAc propagating radical has relatively little steric bulk and the radical is poorly stabilized making it highly reactive and, consequently, a very poor homolytic leaving group. This marked change in reactivity has severe consequences in the choice of RAFT agents for vinyl ester polymerization. The results in Table 17 show that the RAFT polymerization of VAc is only effective with xanthates (Z = OR) and dithiocarbamates (Z = NR2).[74] We have shown that the polymerization of VAc is completely inhibited in the presence of dithioesters, trithiocarbonates, and aromatic dithiocarbamates, i.e. the RAFT agents which are preferred for RAFT polymerization of (meth)acrylates, styrenes and acrylamides.[93] The inhibition observed with these RAFT agents has been explained by a slow fragmentation of the intermediate radical (A) (Scheme 25). The slow fragmentation arises from the poor homolytic leaving ability of the VAc radical, i.e. the intermediate radical is more stable than the products arising from fragmentation.
|
Random and Gradient Copolymers
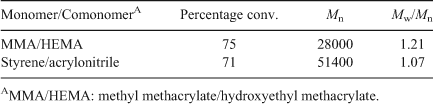
We have used analysis by 1H NMR spectroscopy to show that the RAFT process does not alter the composition of copolymers in random copolymerizations when compared with copolymerizations without the RAFT agent.[77] Two examples of random copolymers are shown in Table 18.
|
We have also reported the preparation of a BA/MMA gradient copolymer by the RAFT process.[77] The results (Table 19) showed that using cumyl dithiobenzoate, S=C(Ph)SC(CH3)2Ph, as the RAFT agent gave a narrow polydispersity gradient copolymer, rich in MMA at one end and rich in BA at the other end.

|
Diblock and Triblock Copolymers
There are numerous examples utilizing the living nature of the RAFT process to prepare various AB, ABA, and ABC blocks.[73,76,93,99] Some examples of hard–soft, hydrophilic–hydrophobic, and other AB diblocks are listed in Table 20.

|
The last entry in Table 20 illustrates an alternative strategy for block synthesis – linking a preformed polymer, e.g. poly(ethylene oxide), to a functional RAFT agent and chain extending using the RAFT process.
A consideration in block synthesis with the RAFT process is that in order to obtain a narrow polydispersity block copolymer the leaving group ability of the propagating radical of the A block is greater than or at least comparable to the leaving group ability of the propagating radical of the B block.
ABC triblocks can be readily prepared by chain extending a preformed AB diblock. We have illustrated this by adding t-butyl methacrylate to poly(benzyl methacrylate-block-dimethylaminoethyl methacrylate) (Mn = 3500; Mw/Mn = 1.06) to give poly(benzyl methacrylate-block-dimethylaminoethyl methacrylate-block-t-butyl methacrylate) (Mn = 8250; Mw/Mn = 1.12).[76]
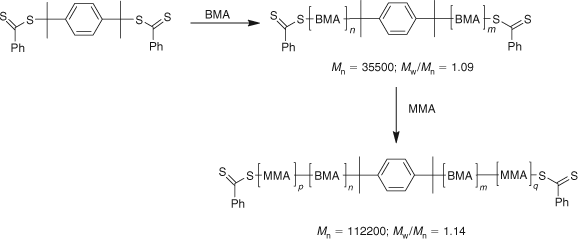
We have also prepared ABA triblocks by utilizing a difunctional RAFT agent and this is exemplified by the preparation of poly(methyl methacrylate-block-butyl methacrylate-block-methyl methacrylate) (Mn = 112200; Mw/Mn = 1.14) (Scheme 28).[76]
|
The advantage of this strategy is that ABA triblocks can be prepared in two steps. An alternative to this two-step ABA triblock strategy is the use of symmetrical trithiocarbonates as RAFT agents.[74] We have reported several examples to illustrate the utility of this approach which, in general, follows the procedure outlined in Scheme 29.[105]
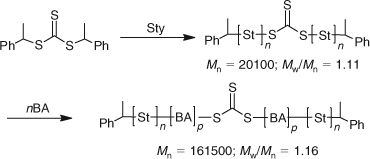
|
Star Polymers
Star polymers can be readily prepared by utilizing RAFT agents that contain multiple thiocarbonylthio moieties.[76,106,107] We have reported two distinct strategies for accessing star polymers using two classes of RAFT agents. In the first class, the representative structures depicted in Fig. 20, show multi-functional dithioester and trithiocarbonate RAFT agents which are designed to have the arms grow from the core (i.e. the propagating chains are attached to the core). Examples whereby these agents are used to give 4-arm and 6-arm star PS polymers have been reported.[76,106]
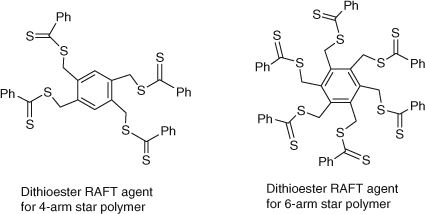
|
An unavoidable consequence with this class of RAFT agent is that since the propagating chains are attached to the core, the inevitable termination coupling reactions of radicals lead to small amounts of star–star coupled products. These coupled products have been observed by GPC as peaks with two times the molecular weight of the main peak of the star polymer.[93,106]
In the second class of RAFT agents, the trithiocarbonate shown in Fig. 21 has been designed to have the propagating chains grow away from the core (i.e. detached from the core).
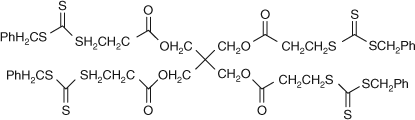
|
Both these approaches provide 4 and 6-arm star polymers with good molecular weight control and low polydispersity.[76,93,106] For example, the trithiocarbonate RAFT agent in Fig. 21 was used to prepare 4-arm PS (Mn(GPC) = 63900; Mn(Calc) = 78700; Mw/Mn = 1.08, 63 % conversion).[106] There is a distinct advantage in the use of RAFT agents of the type shown in Fig. 21 (i.e. where propagating chains grow detached from the core) because complications arising from star–star coupling reactions are avoided. This is highlighted in Scheme 30. Since the propagating chains are detached from the core, the core remains in the ‘dormant’ form and any radical–radical coupling reactions will yield linear polymers which are produced in lower amounts than if star–star coupling occurred. These linear termination products have been observed by GPC as peaks with approximately half the molecular weight of the star polymer.[31,106]
|
End-Group Removal and/or Transformation
The RAFT process yields thiocarbonylthio terminated polymers. This end-group is what imparts ‘living’ character to the polymer chains but it also means that the polymers are coloured. The colour ranges from deep red to pale yellow depending on the absorption spectrum of the particular thiocarbonylthio chromophore. Over the years there have been strong incentives to develop effective methods to modify the thiocarbonylthio end groups, not only to remove the colour but also to convert them into more useful functionalities. A variety of processes have now been reported for this purpose including reactions with nucleophiles, radical-induced reductions, thermolysis, electrocyclic reactions, and ‘click’ processes as outlined in our recent review.[108] We have shown that the thiocarbonylthio end group can be reduced to a hydrogen end-group with a source of radicals and a hydrogen atom donor,[109] decolourized with a mix of azo and peroxide initiators,[110] or converted into colourless cyclic structures by cycloaddition with diazomethane.[111]
Conclusion
Radical trapping with nitroxides has provided a detailed picture of the initiation of radical polymerization by oxygen-centred radicals and has shown many unexpected results. With t-butoxy radicals, for example, we have observed a great deal of hydrogen abstraction from monomers and/or solvents. In some cases this pathway exceeded the expected pathway of radical addition to the double bond. These studies, together with a better understanding of the way polymer chains terminate, has helped us to explain why polymers prepared with different initiators can have different properties. The trapping of radicals with ferric chloride allowed us to confirm the Mayo mechanism for the thermal polymerization of Sty. This unusual mechanism for the formation of radicals by a ‘molecule-assisted homolysis’ had been in dispute for more than 20 years. Observations during the trapping of radicals with nitroxides led us to conclude that the formation of alkoxyamines by the combination of carbon-centred radicals with nitroxides was in fact reversible, especially at more elevated temperatures. This suggested that polymer chains could be grown in a controlled and stepwise fashion by the insertion of monomer between the carbon-centred radical and the nitroxide and that the process could be controlled by varying the temperature of the reaction. This ‘living-radical’ polymerization process is now known as nitroxide-mediated polymerization or NMP. We showed that it is applicable to the preparation of many ‘living’ polymers and that the temperature at which it can be conducted is inversely proportional to the steric bulk of the nitroxide moiety.
When we applied NMP to α-methyl monomers, such as MMA, we encountered a disproportionation reaction between the propagating radical and the nitroxide which led to the formation of omega-unsaturated polymers/oligomers. We then demonstrated that these macromonomers can be used as prepolymers in copolymerization to prepare graft copolymers and, depending on the monomers employed, can also act as useful chain transfer agents to regulate the molecular weight of polymers and under certain conditions make low polydispersity block copolymers. We showed that the chain transfer process for these macromonomers took place by a mechanism of radical addition–fragmentation and were able to extrapolate to the use of simple organic compounds for the same purpose. These included allylic compounds (such as allylic sulfides, peroxides, bromides, sulfones, etc), vinyl ethers and thiocarbonyl compounds. When we investigated thiocarbonylthio compounds (e.g. dithioesters) as chain transfer agents we observed something special; the polymer produced had a very narrow molecular weight distribution. We concluded that this was a case of reversible addition–fragmentation chain transfer and labelled it the RAFT process. Subsequently we showed that it offers the same convenience and versatility as conventional free radical polymerization since it is applicable to the same range of monomers, initiators, solvents, functional groups, and reaction conditions. The polymerization is simply performed by adding a quantity of RAFT agent (dithioester, trithiocarbonate, dithiocarbamate, or xanthate) to conventional free radical polymerizations and yields thiocarbonylthio terminated polymers of predetermined molecular weight and low polydispersity indices (less than 1.1 under optimal conditions). These polymers can be chain extended to give a variety of block, gradient, and star copolymers and the thiocarbonylthio end group can be converted into many other useful functional groups.
Acknowledgements
The authors acknowledge the expert contributions of their many collaborators, listed as coauthors in the References section, and especially thank Dr Emma Prime and Dr Ming Chen for their help with this manuscript.
References
[1] M. Szwarc, Advances in Polymer Science, Vol. 49: Living Polymers and Mechanisms of Anionic Polymerization 1983 (Springer-Verlag: Berlin).[2] F. R. Mayo, C. Walling, Chem. Rev. 1940, 27, 351.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaH3MXis1el&md5=849adb7d2d1f3bad76d1b28482bd2918CAS |
[3] G. Moad, D. H. Solomon, The Chemistry of Radical Polymerization 2006, 2nd edition (Elsevier Ltd: Oxford).
[4] A. L. J. Beckwith, G. Moad, J. Chem. Soc., Perkin Trans. 2 1980, 1083.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL3cXls1Ohtrk%3D&md5=af1896eef2c2d515d9a6367da23e5bcfCAS |
[5] D. H. Solomon, P. Cacioli, G. Moad, Pure Appl. Chem. 1985, 57, 985.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2MXltVGntbY%3D&md5=27ceb7f3b3c8a7c0ee6988eaeff80742CAS |
[6] P. Cacioli, G. Moad, E. Rizzardo, A. K. Serelis, D. H. Solomon, Polym. Bull. 1984, 11, 325.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2cXktVOktL0%3D&md5=706f91c317a65ba930fe59a31c500f0fCAS |
[7] P. Cacioli, D. G. Hawthorne, R. L. Laslett, E. Rizzardo, D. H. Solomon, J. Macromol. Sci. Chem. 1986, A23, 839.
| 1:CAS:528:DyaL28XhvVKqurw%3D&md5=c4d66eddd668aa9a63ae66c8450c036cCAS |
[8] D. H. Solomon, J. Macromol. Sci. Chem. 1975, A9, 97.
| 1:CAS:528:DyaE2MXlvVWgsQ%3D%3D&md5=1b6e4719e06feee8030faa3608a0d288CAS |
[9] A. L. J. Beckwith, A. K. Ong, D. H. Solomon, J. Macromol. Sci. Chem. 1975, A9, 125.
| 1:CAS:528:DyaE2MXlvVaisw%3D%3D&md5=b71190c37e6f45f25e924c4518707c2cCAS |
[10] D. H. Solomon, Chem. Aust. 1982, 49, 192.
| 1:CAS:528:DyaL38XitFylsr8%3D&md5=0d81b84419729d6930556aa1c23ed713CAS |
[11] D. H. Solomon, Chemistry of Pigments and Fillers 1983 (Wiley: New York, NY).
[12] R. Hiatt, Organic Peroxides 1971, Volume 3 (Wiley-Interscience: New York, NY).
[13] E. Rizzardo, D. H. Solomon, J. Macromol. Sci. Chem. 1980, A14, 33.
[14] E. Rizzardo, D. H. Solomon, J. Macromol. Sci. Chem. 1979, A13, 1005.
| 1:CAS:528:DyaE1MXlslGqs7Y%3D&md5=bc227a1f21b74f9f6dbb5d318f44a516CAS |
[15] D. G. Hawthorn, D. H. Solomon, J. Macromol. Sci. Chem. 1972, 6, 661.
| Crossref | GoogleScholarGoogle Scholar |
[16] E. Rizzardo, D. H. Solomon, Polym. Bull. 1979, 1, 529.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL3cXpsVOgsw%3D%3D&md5=bc32a52a7e9af38e5d36d27dbeface24CAS |
[17] P. G. Griffiths, G. Moad, E. Rizzardo, D. H. Solomon, Aust. J. Chem. 1983, 36, 397.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL3sXkt1ahtL8%3D&md5=a8da3bf07669e9a8116f59cc11e433dbCAS |
[18] C. J. Hawker, A. W. Bosman, E. Harth, Chem. Rev. 2001, 101, 3661.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXnslaqsrc%3D&md5=f526c4187293bb403eb06f2cb420f495CAS |
[19] A. Studer, T. Schulte, Chem. Rec. 2005, 5, 27.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXksVagsr8%3D&md5=89b6d955fd2294b0b512215e9551b7f2CAS |
[20] C. J. Hawker, in Handbook of Radical Polymerization 2002, p. 463 (Eds T. P. Davis, K. Matyjaszewski) (John Wiley & Sons: New York, NY).
[21] P. G. Griffiths, E. Rizzardo, D. H. Solomon, J. Macromol. Sci. Chem. 1982, A17, 45.
| 1:CAS:528:DyaL38XitVGjtw%3D%3D&md5=e2d8576a354cd688e34951fc564cf33bCAS |
[22] G. Moad, E. Rizzardo, D. H. Solomon, Macromolecules 1982, 15, 909.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL38XktVKrtrw%3D&md5=df471c1dc9789f34d5da6c1b603b9cc0CAS |
[23] G. Moad, D. H. Solomon, S. R. Johns, R. I. Willing, Macromolecules 1982, 15, 1188.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL38Xksleju7s%3D&md5=7c55bef4d268a0f4f1d9606e36d00726CAS |
[24] G. Moad, D. H. Solomon, R. I. Willing, Macromolecules 1988, 21, 855.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL1cXhtFeju74%3D&md5=ef533f6bdb34f3bb9eb53549cb62ec9aCAS |
[25] R. D. Grant, E. Rizzardo, D. H. Solomon, J. Chem. Soc. Chem. Commun. 1984, 867.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2MXlt12n&md5=06f090dcf3676ed13e1a1bd471e28ae5CAS |
[26] E. Rizzardo, A. K. Serelis, D. H. Solomon, Aust. J. Chem. 1982, 35, 2013.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL3sXhslWjsw%3D%3D&md5=5eefe0e74b63d06ce9a90ff03f9e8163CAS |
[27] W. K. Busfield, I. D. Jenkins, S. H. Thang, E. Rizzardo, D. H. Solomon, Aust. J. Chem. 1985, 38, 689.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2MXktlOkurY%3D&md5=877a95c6ba438c47f7cd97dce332a400CAS |
[28] R. D. Grant, P. G. Griffiths, G. Moad, E. Rizzardo, D. H. Solomon, Aust. J. Chem. 1983, 36, 2447.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2cXhtl2ltLk%3D&md5=cd1a286ab3067b7809dd4af3512793eeCAS |
[29] D. Bednarek, G. Moad, E. Rizzardo, D. H. Solomon, Macromolecules 1988, 21, 1522.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL1cXitVGit7g%3D&md5=c4a5b8048ec2423128eebe431824a1f7CAS |
[30] P. J. Flory, J. Am. Chem. Soc. 1937, 59, 241.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaA2sXhsl2jtg%3D%3D&md5=a9132380d9adad9e6675aed0ad682b50CAS |
[31] F. R. Mayo, Polym. Prepr. 1961, 2, 55.
[32] Y. K. Chong, E. Rizzardo, D. H. Solomon, J. Am. Chem. Soc. 1983, 105, 7761.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2cXnt1ei&md5=de87302480351f1ad6b599e827c3d0dbCAS |
[33] D. H. Solomon, G. Moad, Makromol. Chem. Macromol. Symp. 1987, 10–11, 109.
[34] H. Ishiwata, T. Inoue, K. Yoshihira, J. Chromatogr. A 1986, 370, 275.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2sXmsF2itw%3D%3D&md5=757f7cdc6b0c266b57998549555f203cCAS |
[35] P. J. Fordham, J. W. Gramshaw, L. Castle, Food Addit. Contam. 2001, 18, 461.
| 1:CAS:528:DC%2BD3MXjs1aiu7s%3D&md5=03bb4fbb30101b36586f1431be1006dcCAS |
[36] G. Moad, D. H. Solomon, S. R. Johns, R. I. Willing, Macromolecules 1984, 17, 1094.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2cXhvFagt7Y%3D&md5=a5ea8169c62f77cb38a55e42261118fbCAS |
[37] W. H. Starnes, I. M. Plitz, F. C. Schilling, G. M. Villacorta, G. S. Park, A. H. Saremi, Macromolecules 1984, 17, 2507.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2MXntFGg&md5=c82506bfbebe7cdaf901e9ad83454fc0CAS |
[38] G. Moad, A. K. Serelis, D. H. Solomon, T. H. Spurling, Polym. Commun. (Guildf.) 1984, 25, 240.
| 1:CAS:528:DyaL2cXltlWitbs%3D&md5=6dc11e0e7dc940e8077eb231ea0a7f3dCAS |
[39] D. P. Kelly, A. K. Serelis, D. H. Solomon, P. E. Thompson, Aust. J. Chem. 1987, 40, 1631.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL1cXksFCgsr4%3D&md5=16f0587d0fee218bb08d934751efa078CAS |
[40] D. H. Solomon, unpublished results.
[41] G. Moad, D. H. Solomon, unpublished results.
[42] M. J. Cuthbertson, E. Rizzardo, D. H. Solomon, Aust. J. Chem. 1983, 36, 1957.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2cXhtlyhu7g%3D&md5=7e598d634b79593c7891710d84894d05CAS |
[43] D. H. Solomon, E. Rizzardo, P. Cacioli, US patent 4,581,429, 1986.
[44] E. Rizzardo, Chem. Aust. 1987, 54, 32.
[45] E. Rizzardo, Y. K. Chong, 2nd Pacific Polymer Conference, Preprints 1991, p. 26 (Pacific Polymer Federation: Tokyo).
[46] C. H. J. Johnson, G. Moad, D. H. Solomon, T. H. Spurling, D. J. Vearing, Aust. J. Chem. 1990, 43, 1215.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3cXkvFyjurk%3D&md5=cf651435071649d3c89d9379e402d3b1CAS |
[47] D. H. Solomon, J. Polym. Sci., Part A: Polym. Chem. 2005, 43, 5748.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXht1GrtbfL&md5=574c5adce268b32c13c5456cb75fd6b5CAS |
[48] G. Moad, E. Rizzardo, Macromolecules 1995, 28, 8722.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXpsFWqu7o%3D&md5=83371dddf357140a147e9321280d51ddCAS |
[49] S. Abrol, P. A. Kambouris, M. G. Looney, D. H. Solomon, Macromol. Rapid Commun. 1997, 18, 755.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXmtF2nsbo%3D&md5=15093a8a7829c09c24ba058766f7f083CAS |
[50] G. Moad, E. Rizzardo, S. H. Thang, Polymer (Guildf.) 2008, 49, 1079.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXitlels7c%3D&md5=03552fa69703a325d5ca3be6a3b68ce2CAS |
[51] E. Rizzardo, D. S. Harrison, R. L. Laslett, G. F. Meijs, T. C. Morton, S. H. Thang, in Progress in Pacific Polymer Science, Proceedings of the Pacific Polymer Conference, 1st 1991, p. 77 (Eds B. C. Anderson, Y. Imanishi) (Springer-Verlag: Berlin).
[52] E. Rizzardo, G. Meijs, S. H. Thang, G. F. Meijs, S. Thang, WO8804304–A1, 1988.
[53] G. F. Meijs, T. C. Morton, E. Rizzardo, S. H. Thang, Macromolecules 1991, 24, 3689.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3MXisVOrtrc%3D&md5=aa2c9d970133137b8136dcb1a69e6c43CAS |
[54] G. F. Meijs, E. Rizzardo, S. H. Thang, Macromolecules 1988, 21, 3122.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL1cXlslWjs7o%3D&md5=241f8c7e0a1bed083438594b0de6f8deCAS |
[55] E. Rizzardo, G. F. Meijs, S. Thang, S. H. Thang, WO9106535–A1, 1990.
[56] G. F. Meijs, E. Rizzardo, S. H. Thang, Polym. Prepr. 1992, 33, 893.
| 1:CAS:528:DyaK2cXhvFams70%3D&md5=8045236fe7fbd3206779c11e6ec74fcaCAS |
[57] G. F. Meijs, E. Rizzardo, S. H. Thang, Polym. Bull. 1990, 24, 501.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3MXot1Kjsw%3D%3D&md5=9a30e63cf64396fdadd17831ec9864bdCAS |
[58] G. F. Meijs, E. Rizzardo, Makromol. Chem., Rapid. Commun. 1988, 9, 547.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL1cXmtFWmtr0%3D&md5=f7bc9dcb387a29472deceec8978c3c53CAS |
[59] G. F. Meijs, E. Rizzardo, Makromol. Chem. 1990, 191, 1545.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3cXltlaru78%3D&md5=1660a944410455ed34b56ffb3ace47c9CAS |
[60] G. F. Meijs, E. Rizzardo, Polym. Bull. 1991, 26, 291.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3MXlvVCjs78%3D&md5=06b2d062a26bbb6314eb6efbe7f63da2CAS |
[61] G. F. Meijs, E. Rizzardo, T. P. T. Le, Y. C. Chen, Makromol. Chem. 1992, 193, 369.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK38XitFGjsrY%3D&md5=dc099785dabc64766907b0643d83ac70CAS |
[62] E. Rizzardo, G. F. Meijs, S. H. Thang, Macromol. Symp. 1995, 98, 101.
| Crossref | GoogleScholarGoogle Scholar |
[63] S. N. Lewis, J. J. Miller, S. Winstein, J. Org. Chem. 1972, 37, 1478.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE38XktFyqsrc%3D&md5=fc7215133d4fa0db8c51f5902321822aCAS |
[64] G. E. Keck, A. M. Tafesh, J. Org. Chem. 1989, 54, 5845.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3cXitFahsw%3D%3D&md5=c48ede67eb59d920aa21df699d4c2363CAS |
[65] R. M. Adlington, J. E. Baldwin, A. Basak, R. P. Kozyrod, J. Chem. Soc. Chem. Commun. 1983, 944.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2cXhtl2msr8%3D&md5=edbc737b1b50150e88df3361070b7d36CAS |
[66] D. H. R. Barton, D. Crich, J. Chem. Soc., Perkin Trans. 1 1986, 1613.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2sXhvFGmsbc%3D&md5=faaacffc8745c3f3eb979e9bd90ed02fCAS |
[67] E. Rizzardo, Y. K. Chong, R. A. Evans, G. Moad, S. H. Thang, Macromol. Symp. 1996, 111, 1.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXktFaqsw%3D%3D&md5=ba33bf43d7fdf316391f13d1e5a63030CAS |
[68] J. Krstina, C. L. Moad, G. Moad, E. Rizzardo, C. T. Berge, M. Fryd, Macromol. Symp. 1996, 111, 13.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXktFaqsA%3D%3D&md5=176190d7eeeb2d11a3df5609ef8f4549CAS |
[69] T. Corner, Advances in Polymer Science 1984, 62, 95.(Springer: Berlin)
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2MXks1altA%3D%3D&md5=d976a54bca2f03877facd90cf4295f26CAS |
[70] D. H. R. Barton, D. Crich, G. Kretzschmar, J. Chem. Soc., Perkin Trans. 1986, 1, 39.
| Crossref | GoogleScholarGoogle Scholar |
[71] J. Krstina, G. Moad, E. Rizzardo, C. L. Winzor, C. T. Berge, M. Fryd, Macromolecules 1995, 28, 5381.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXmsFejt74%3D&md5=de37f71003577308e13a7950ff4ac560CAS |
[72] G. Moad, C. L. Moad, J. Krstina, E. Rizzardo, C. T. Berge, T. R. Darling, 1995–US14428 9615157, 1996.
[73] T. P. Le, G. Moad, E. Rizzardo, S. H. Thang, 1997–US12540 9801478, 1998.
[74] J. Chiefair, R. T. Mayadunne, G. Moad, E. Rizzardo, S. H. Thang, 1998–US26428 9931144, 1999.
[75] J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo, S. H. Thang, Macromolecules 1998, 31, 5559.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXkvF2gs7k%3D&md5=7b317daf025894dcec376159155980ccCAS |
[76] Y. K. Chong, T. P. T. Le, G. Moad, E. Rizzardo, S. H. Thang, Macromolecules 1999, 32, 2071.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXht1art7k%3D&md5=572bd1689174c866b88fb2929cb7571dCAS |
[77] E. Rizzardo, J. Chiefari, B. Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, S. H. Thang, Macromol. Symp. 1999, 143, 291.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXnt1Sitb8%3D&md5=9bb03833d28f73094ea7e566f5d8d023CAS |
[78] S. W. Prescott, M. J. Ballard, E. Rizzardo, R. G. Gilbert, Aust. J. Chem. 2002, 55, 415.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XotFSju7Y%3D&md5=121e903760bdcfc7a2c916104efb7c25CAS |
[79] S. W. Prescott, M. J. Ballard, E. Rizzardo, R. G. Gilbert, Macromolecules 2002, 35, 5417.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XktFKrtLg%3D&md5=d66a2b44965c052bdcfde8fb5486d8c0CAS |
[80] S. W. Prescott, M. J. Ballard, E. Rizzardo, R. G. Gilbert, Macromolecules 2005, 38, 4901.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXjsVGhu7w%3D&md5=3f013c0a3b140b5bae0d95fd43c6ce9cCAS |
[81] G. Moad, E. Rizzardo, S. H. Thang, Aust. J. Chem. 2005, 58, 379.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXltFymsL4%3D&md5=0464d702bb4d20d3f965209912e5cd9bCAS |
[82] G. Moad, E. Rizzardo, S. H. Thang, Aust. J. Chem. 2006, 59, 669.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XhtFeqsr%2FM&md5=dbce39d0fb32872e6e9f9a55bde54681CAS |
[83] G. Moad, E. Rizzardo, S. H. Thang, Aust. J. Chem. 2009, 62, 1402.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVers7bN&md5=fb2b73394696eb457b81af596f1722b8CAS |
[84] J. F. Quinn, L. Barner, C. Barner-Kowollik, E. Rizzardo, T. P. Davis, Macromolecules 2002, 35, 7620.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XmsVCls74%3D&md5=8973b9c26938c52a47f86d84ba9e8ec5CAS |
[85] J. F. Quinn, L. Barner, E. Rizzardo, T. P. Davis, J. Polym. Sci., Part A: Polym. Chem. 2002, 40, 19.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XlsVWj&md5=6b056769368bc5299a959bbef95cfc67CAS |
[86] J. F. Quinn, E. Rizzardo, T. P. Davis, Chem. Commun. (Camb.) 2001, 1044.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXjs1Grt74%3D&md5=dfae1c42132d0cfd2c1bf3d616cd83aeCAS |
[87] C. H. Hornung, C. Guerrero-Sanchez, M. Brasholz, S. Saubern, J. Chiefari, G. Moad, E. Rizzardo, S. H. Thang, Org. Process Res. Dev. 2011, 15, 593.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXivFygs7w%3D&md5=15ca10d5c3401d3e38eac3e61c9cc890CAS |
[88] Y. K. Chong, G. Moad, E. Rizzardo, M. A. Skidmore, S. H. Thang, Macromolecules 2007, 40, 9262.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhtlGntrvO&md5=44d9bfa6bb01cb1e593b26decfff424cCAS |
[89] E. Rizzardo, M. Chen, B. Chong, G. Moad, M. Skidmore, S. H. Thang, Macromol. Symp. 2007, 248, 104.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXktVSmsb4%3D&md5=dce0f51ab9dae7167261b6d8b47d8d33CAS |
[90] G. Moad, J. Chiefari, Y. K. Chong, J. Krstina, R. T. A. Mayadunne, A. Postma, E. Rizzardo, S. H. Thang, Polym. Int. 2000, 49, 993.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXmtFemsbs%3D&md5=a1f943c51966d6dc4e7e06baa69f7b09CAS |
[91] M. Chen, K. P. Ghiggino, A. W. H. Mau, E. Rizzardo, S. H. Thang, G. J. Wilson, Chem. Commun. (Camb.) 2002, 19, 2276.
| Crossref | GoogleScholarGoogle Scholar |
[92] D. G. Hawthorne, G. Moad, E. Rizzardo, S. H. Thang, Macromolecules 1999, 32, 5457.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXksFSnt74%3D&md5=8654a78c6068830d7dc67fa3c416e56bCAS |
[93] E. Rizzardo, J. Chiefari, R. T. A. Mayadunne, G. Moad, S. H. Thang, ACS Symposium Series 2000, 768, 278.(American Chemical Society: Washington, DC)
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXmtFSjtr0%3D&md5=4f86b1f0463f9994b174cf39dabb2575CAS |
[94] Y. K. Chong, J. Krstina, T. P. T. Le, G. Moad, A. Postma, E. Rizzardo, S. H. Thang, Macromolecules 2003, 36, 2256.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXitVemuro%3D&md5=be1b7c9c3c7f9a31c2251b589d41e6e5CAS |
[95] J. Chiefari, R. T. A. Mayadunne, C. L. Moad, G. Moad, E. Rizzardo, A. Postma, M. A. Skidmore, S. H. Thang, Macromolecules 2003, 36, 2273.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXitVemurs%3D&md5=177a2137dc017ac41edc8c1bbc67173aCAS |
[96] A. Goto, K. Sato, Y. Tsujii, T. Fukuda, G. Moad, E. Rizzardo, S. H. Thang, Macromolecules 2001, 34, 402.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXhtVajtg%3D%3D&md5=4f36d4d9470931c90ab263c0830eb6d5CAS |
[97] G. Litvinenko, A. H. E. Müller, Macromolecules 1997, 30, 1253.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXhtF2is7k%3D&md5=3819bb4547fd2f7b64295fec08c3fdbbCAS |
[98] G. Moad, A. G. Anderson, F. Ercole, C. H. J. Johnson, J. Krstina, C. L. Moad, E. Rizzardo, T. H. Spurling, S. H. Thang, ACS Symposium Series 1998, 685, 332.(American Chemical Society: Washington, DC)
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXhtFWrurY%3D&md5=a9fd42c1c3d948831ab2560455c3beb2CAS |
[99] R. T. A. Mayadunne, E. Rizzardo, J. Chiefari, Y. K. Chong, G. Moad, S. H. Thang, Macromolecules 1999, 32, 6977.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXmtVOmt78%3D&md5=e1e43b0941409cdd64bc63d773f145b8CAS |
[100] M. Benaglia, J. Chiefari, Y. K. Chong, G. Moad, E. Rizzardo, S. H. Thang, J. Am. Chem. Soc. 2009, 131, 6914.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXltFOlsr4%3D&md5=b20f45aece9d3de513ad7f0d683d45e4CAS |
[101] M. Benaglia, M. Chen, Y. K. Chong, G. Moad, E. Rizzardo, S. H. Thang, Macromolecules 2009, 42, 9384.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVWhtr3N&md5=337ba2180bc3d4971308680740ad186aCAS |
[102] D. J. Keddie, C. Guerrero-Sanchez, G. Moad, E. Rizzardo, S. H. Thang, Macromolecules 2011, 44, 6738.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtVags7nK&md5=daabb09aa1e88a4e12a702f889ceb83bCAS |
[103] G. Moad, Y. K. Chong, A. Postma, E. Rizzardo, S. H. Thang, Polymer (Guildf.) 2005, 46, 8458.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXpt1WksLc%3D&md5=57f3e048f17ac2ad0d51e90f1b4cc742CAS |
[104] F. Ganachaud, M. J. Monteiro, R. G. Gilbert, M.-A. Dourges, S. H. Thang, E. Rizzardo, Macromolecules 2000, 33, 6738.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXlsVems7g%3D&md5=32adeb983a54fceb3870e8ff7e3e23d1CAS |
[105] R. T. A. Mayadunne, E. Rizzardo, J. Chiefari, J. Krstina, G. Moad, A. Postma, S. H. Thang, Macromolecules 2000, 33, 243.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXhtlWgtA%3D%3D&md5=60f9db1847a9b480d723c42262db848cCAS |
[106] R. T. A. Mayadunne, J. Jeffery, G. Moad, E. Rizzardo, Macromolecules 2003, 36, 1505.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXhtFCgt7w%3D&md5=5256aa7f0f03ec70406c7147248c62dcCAS |
[107] M. Chen, K. P. Ghiggino, A. Launikonis, A. W. H. Mau, E. Rizzardo, W. H. F. Sasse, S. H. Thang, G. J. Wilson, J. Mater. Chem. 2003, 13, 2696.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXotlyqsbc%3D&md5=bed121e4cae2f17eab7dc9dff118f49dCAS |
[108] G. Moad, E. Rizzardo, S. H. Thang, Polym. Int. 2011, 60, 9.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhs1Slt7rK&md5=28dd877fd4b6adc5e5acd0b406f84e41CAS |
[109] Y. K. Chong, G. Moad, E. Rizzardo, S. H. Thang, Macromolecules 2007, 40, 4446.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXlsVKisb0%3D&md5=966471da00ec76ebc2948d026416c2ccCAS |
[110] M. Chen, G. Moad, E. Rizzardo, J. Polym. Sci., Part A: Polym. Chem. 2009, 47, 6704.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtlGjurzE&md5=aea33b9b58bc39553c81ed74cc6ed7a6CAS |
[111] M. Chen, G. Moad, E. Rizzardo, Aust. J. Chem. 2011, 64, 433.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXkvVCrs7w%3D&md5=456830fbbf54ddeefe77b02863511ffeCAS |
* Ezio Rizzardo and David Solomon shared the 2011 Prime Minister’s Prize for Science for their role in revolutionizing polymer science through their discovery and development of living radical polymerization (LRP). Nitroxide mediated polymerization (NMP) and reversible addition–fragmentation chain transfer (RAFT) polymerization put Australian polymer science at the forefront of research into free radical polymerization. This and related work was largely responsible for the transformation of free radical polymerization from a mature science in the 1960–1970s to the great activity we see today.