

Natural Product-Inspired Pyranonaphthoquinone Inhibitors of Indoleamine 2,3-Dioxygenase-1 (IDO-1)*
David J. A. Bridewell A , Jonathan Sperry B , Jason R. Smith C , Priambudi Kosim-Satyaputra C , Lai-Ming Ching A , Joanne F. Jamie C and Margaret A. Brimble B DA Auckland Cancer Society Research Centre, Faculty of Medical and Health Sciences, The University of Auckland, Auckland Central 1010, New Zealand.
B School of Chemical Sciences, The University of Auckland, Auckland Central 1010, New Zealand.
C Department of Chemistry and Biomolecular Sciences, Macquarie University, Sydney, NSW 2109, Australia.
D Corresponding author. Email: m.brimble@auckland.ac.nz
Australian Journal of Chemistry 66(1) 40-49 https://doi.org/10.1071/CH12393
Submitted: 23 August 2012 Accepted: 29 October 2012 Published: 12 November 2012
Abstract
A series of pyranonaphthoquinone derivatives possessing structural features present in both natural products annulin B and exiguamine A have been shown to exhibit low micromolar inhibition of indoleamine 2,3-dioxygenase-1 (IDO-1). These inhibitors retain activity against the enzyme in a cellular context with an approximate one-log loss of dose potency against IDO-1 in cells. One particular analogue, triazole 8 shows good inhibition of IDO-1 along with little loss of cell viability at low drug concentrations. These results have extended the naphthoquinone series of novel IDO-1 inhibitors based on lead compounds from nature.
Introduction
Indoleamine 2,3-dioxygenase-1 (IDO-1; EC 1.13.11.42) was first discovered in the 1960s[1,2] and was subsequently shown to catalyse the first and rate-limiting step of the catabolism of the essential amino acid l-tryptophan to N-formyl kynurenine in the kynurenine pathway.[3]
The expression of IDO-1 is upregulated in response to IFN-γ,† typically at sites of inflammation, and in those environments where tryptophan levels have been depleted by IDO-1, T-lymphocytes undergo G1 cell-cycle arrest and apoptosis leading to immunosuppression.[4,5] Moreover, in mammals the elevated levels of IDO-1 present in the placenta is thought to provide immune protection to the developing fetus by decreasing the local tryptophan availability.[6]
The overexpression of IDO-1 has now been identified in a range of human pathologies including Alzheimer’s disease, asthma, depression, age-related cataract formation, and HIV-encephalitis.[7–11] It has also been found to be upregulated in a wide range of human tumours and has recently emerged as a possible explanation for the immune escape exhibited by cancer cells, in a mechanism similar to that found in fetal tissue. This hypothesis, together with its expression in such a large number of human diseases has firmly established IDO-1 as an appealing drug target.[12–18] Furthermore, Zheng and co-workers have shown that murine melanoma cells, stably transformed with an IDO-1-silencing construct, exhibited a reduced capacity to form tumours compared with the unmodified parental melanoma cells when inoculated into recipient mice.[19] Moreover, tumours developing from the wild-type parental cells regressed when injected intra-tumourally with an anti-IDO siRNA.‡
The clinical potential of IDO-1 as a target for cancer treatment has been further exemplified by the observation that the modest IDO-1 inhibitors 1-methyltryptophan (1-MT),[20] 5-bromobrassinin,[21] and menadione[22] while failing to regress murine tumours when used as a monotherapy, nevertheless halted the progression of established tumours in vivo when used in combination with the anti-mitotic drug paclitaxel. These observations are particularly noteworthy as paclitaxel was ineffective when used alone in these studies.[20–22]
Although the IDO-1 inhibitors 2,5-dihydro-l-phenylalanine,[23] 1-methyltryptophan,[24] 4-phenylimidazole,[25] and β-carbolines such as norharman[26,27] have been known for decades, it is only since the emergence of IDO-1 as a novel target for the treatment of cancer that extensive studies towards developing inhibitors of this enzyme have emerged.[13,17] An investigation concerning the inhibitory properties of 4-phenylimidazole (4-PI)[25] has witnessed the crystal structure of IDO-1 being determined,[28] along with the production of nanomolar inhibitors of IDO-1 based on the 4-PI scaffold.[29]
Numerous synthetic IDO-1 inhibitors with novel pharmacophores have emerged, including several based on the indole heterocycle,[30–33] S-benzyl isothioureas,[34] 4-amino-1,2,5-oxadiazole-3-carboximidamides,[35,36] the imidodicarbonimidic diamide NSC 401366,[37] ebselen,[38] and several triazole-based IDO inhibitors.[39] Potent inhibitors have also been discovered using a combination of docking-based pharmacophore model development and fragment-based drug design (FBDD),[40a] along with the identification of potential inhibitors by virtual screening[40b] (Fig. 1).
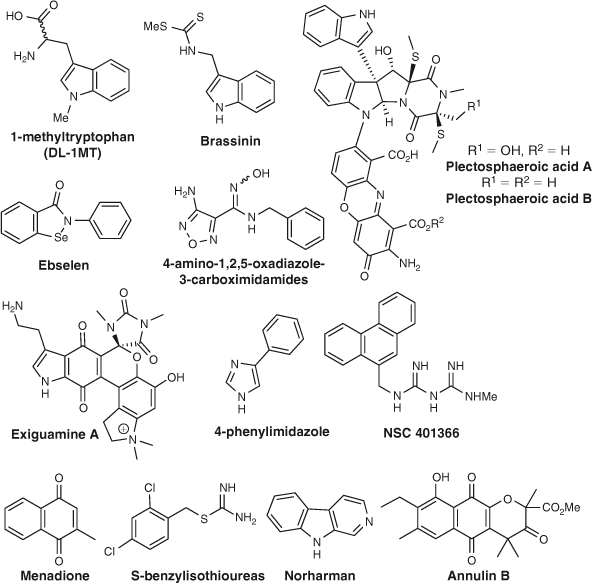
|
Several natural products have also been isolated that show varying levels of inhibition of IDO-1, providing new pharmacophores for the development of potential anti-cancer agents based on those found in nature. Examples include brassinin,[21,41] exiguamine A,[42–44] plectosphaeroic acids A and B,[45] and annulin B[22,46,47] (Fig. 1).
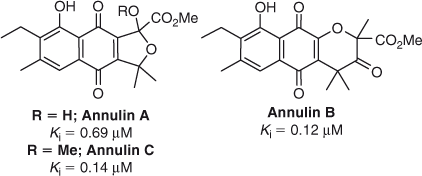
The pyranonaphthoquinones, annulin A and B, were first isolated in 1986 from the marine hydroid Garveia annulata,[47] with annulin C discovered from the same source sometime later[46] (Fig. 2). Annulin B was subsequently shown to be a potent inhibitor of IDO-1 (Ki = 0.12 µM),[46] inspiring the synthesis of a series of pyranonaphthoquinone inhibitors based on the annulin pyranonaphthoquinone core structure.[22] The synthetic pyranonaphthoquinones tested were amongst the most potent IDO-1 inhibitors recorded to date using an isolated enzyme assay (Ki = 61–70 nM), with a select few also causing minimal impact on cell viability at 100 µM after 24 h.[22]
It is well established that various naphthoquinones display potent activity as IDO-1 inhibitors,[22,42–44,46,47] suggesting this moiety to be a key pharmacophore and a likely indole mimetic.[22] As the pyran containing natural products annulin B[46,47] and exiguamine A[42,43] are both potent IDO-1 inhibitors, we planned on testing a series of pyranonaphthoquinone derivatives possessing both the pyran and naphthoquinone structural features of these natural products, thus extending the naphthoquinone series of novel IDO-1 inhibitors based on lead compounds from nature (Scheme 1). Our research group has a long-standing interest in the synthesis of natural products possessing a pyranonaphthoquinone moiety,[48] along with their chemical modification in order to improve their respective bioactivities.[49,50] Thus, the potent IDO-1 inhibitory properties displayed by annulin B and exiguamine A, combined with our ongoing interest regarding the synthesis and biological properties of pyranonaphthoquinone derivatives led us to initiate the testing of pyranonaphthoquinones bearing the core structure 1, which displays a combination of the structural features present in both annulin B and exiguamine A (Scheme 1).
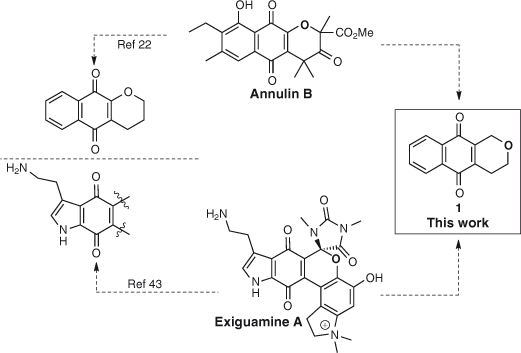
|
Results and Discussion
The pyranonaphthoquinone derivatives tested herein are shown in Fig. 3 and comprise a selection of natural products and synthetic analogues available in our laboratory.[50–56]

|
Activity of Pyranonaphthoquinone Derivatives against Purified Recombinant Human IDO
The pyranonaphthoquinone derivatives were assayed against recombinant human IDO-1 using a methylene blue/ascorbic acid artificial reducing system to maintain IDO-1 in the active ferrous (Fe2+) form.[57] This is the most commonly used in vitro assay system for IDO-1 and has been employed for assessing many IDO-1 inhibitors, including naphthoquinones. Gratifyingly, all the pyranonaphthoquinone derivatives tested were found to inhibit the catalytic activity of IDO-1 in the 1 h enzyme assay in a dose dependent manner, so much so that by ~50 μM all the compounds were inhibiting enzyme activity by over 80 % (Fig. 4). The compounds also showed good IC50 activity in the low micromolar range (Table 1), with the most active compounds, 5 and 6 displaying submicromolar activity (IC50 = 0.8 µM and 0.9 µM respectively). The deoxyeleutherin analogue 3 also showed good inhibition (IC50 = 1.0 µM), considerably more so than the natural product 2 itself (IC50 = 8.0 µM). The natural product ventiloquinone L 4, the dimeric pyranonaphthoquinone 7, and the triazole analogues 8, 10, and 11 were less potent, but still displayed good inhibition (IC50 = 5–6 µM). The least active compound, the benzotriazole analogue 9, still displayed a respectable IC50 of 14.1 µM (Table 1). The IC50 values of the most active compounds (5 and 6) was found to be the same order of magnitude, but slightly less potent, than that measured for the known IDO-1 inhibitors 1,4-naphthoquinone (0.59 μM) and menadione (0.64 μM) (data not shown), which contain the simpler more ligand efficient scaffold.
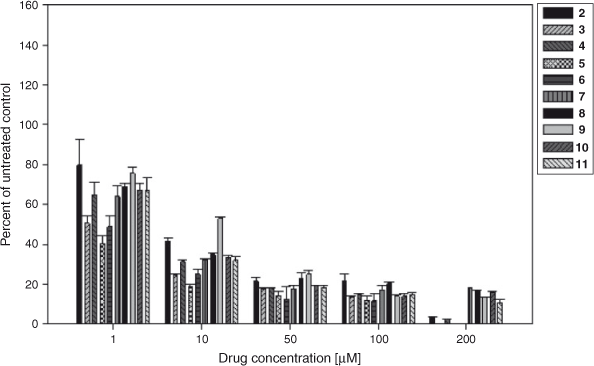
|

|
Of all the pyranonaphthoquinones tested, it appears that the carboxylic acid side-chain is necessary to obtain the submicromolar activity, exemplified by the increased potency exhibited by 5 and 6 compared with all the remaining methyl and triazole substituted analogues tested. As the racemic pyranonaphthoquinone 5 was the most active, we cannot unequivocally confirm at this stage which enantiomer is responsible for the IDO-inhibitory activity, especially considering all the other compounds were tested as single cis-enantiomers as depicted in Fig. 3.
As quinones are oxidizing agents it is possible that the pyranonapthoquinones may inhibit IDO-1 non-selectively by acting as non-specific oxidants of the enzyme. However, Kumar et al.[22] also considered this possibility and they could not find any correlation between the oxidation potential of various quinones and their ability to inhibit IDO-1, thereby arguing against non-specific oxidation of IDO-1 as the primary inhibitory mechanism of the pyranonaphthoquinone class of compounds.[22]
Cytotoxicity of Pyranonaphthoquinone Derivatives against Murine and Human Cancer Cell Lines
As all the pyranonaphthoquinones were reasonably potent inhibitors of the purified enzyme, we next set out to examine the cytotoxicity of pyranonaphthoquinones 2–11 against mammalian cells. An initial examination of these compounds had shown that they could act as topoisomerase II-targeting agents and were thus potentially cytotoxic.[49] Using IC50 assays, we measured the growth inhibitory effects of these compounds against the murine Lewis lung carcinoma cell line LLTC, and LLTC-IDO cells, which have been stably transfected to express human recombinant IDO-1. As a representative human tumour cell line we also examined their activity against the human non-small-cell lung carcinoma cell line, H460.
The results showed that only 5 inhibited cell growth against the murine line LLTC and LLTC-IDO by more than 50 %, producing an IC50 of 1.2 and 2.4 µM respectively (Table 1). The remaining pyranonaphthoquinones did not inhibit cell growth by 50 % even at the highest concentration tested in the assay (6 µM). Moreover, at this concentration the effect on cell growth against the murine lines was minimal (data not shown) suggesting an absence of drug-induced cytotoxicity in these cells. In contrast, the series demonstrated greater activity against the human H460 cells with 4, 5, 7, 9, and 10 inhibiting cell growth by more than 50 %, yielding IC50 values between 4.7 and 6.8 µM. The remaining five compounds 2, 3, 6, 8, and 11 failed to inhibit H460 cell growth by 50 % even at the highest concentration tested (9 µM) against this line (Table 1). The reasons for the increased potency against H460 cells are not clear, but it may possibly reflect altered interactions between human topoisomerase II (the cytotoxic target) and the pyranonaphthoquinones in this line.[49] Nevertheless, the series exhibited much less dose potency against H460 cells than clinically used topoisomerase II-poisons, which typically have IC50 values against this line in the low nanomolar range.[58,59]
Activity of Pyranonaphthoquinone Derivatives against the Catalytic Activity of Human IDO in LLTC-IDO Cells
Next we examined the series’ ability to inhibit the catalytic activity of IDO-1 in a cellular context by determining the extent to which each compound inhibited the production of the l-tryptophan catabolite, l-kynurenine, in the human IDO-1 overexpressing LLTC-IDO cells. The results showed that there was a dose dependent decrease in l-kynurenine production in the drug treated cells; however the potency of the effect was highly compound dependent (Fig. 5a). For example 5 was the most potent compound, inhibiting l-kynurenine production in LLTC-IDO cells by over 90 % at 11.3 µM. In contrast, 6 was less potent showing only a modest inhibition of l-kynurenine production at drug concentrations up to 1.3 μM, and only 40 % inhibition at 11.3 μM. As 5 is cytotoxic to LLTC-IDO cells (Table 1), it was perhaps not surprising to observe a lack of l-kynurenine in the drug treated cells as this was likely due to drug induced cell death.
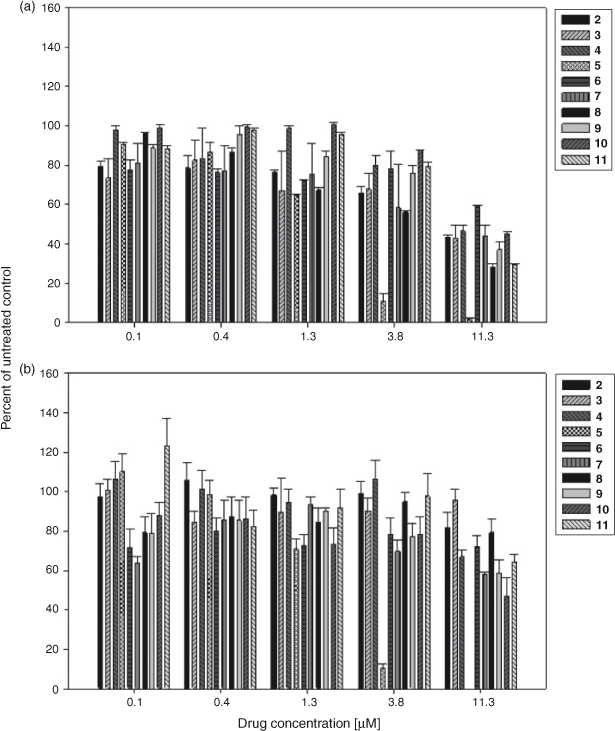
|
Nevertheless, to determine whether the decrease in l-kynurenine production observed in the pyranonaphthoquinone treated cells was in fact due to inhibiting the actions of the enzyme and not as a consequence of drug induced cell death, a MTT assay was performed on the series to establish whether the cells remained metabolically active after a 24 h exposure to the drug. As expected, metabolic activity was rapidly lost in the 5 treated cells at concentrations above 1.3 µM indicating cell death, and at 11.3 µM none of the drug treated cells remained viable (Fig. 5b). However, for the other compounds in the series, while some decrease in metabolic activity with increasing drug concentration was observed, particularly at the highest concentration tested (11.3 µM), overall the loss of activity was variable and compound dependent. For example, at the highest concentration tested, 3 showed little reduction in the metabolism of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (cell viability) following a 24 h drug exposure, whereas compound 10 showed a 50 % loss (Fig. 5b).
To determine the overall relationship between cell viability and the inhibition of l-kynurenine production for this series, linear regression analysis was performed on the results. This indicated that an explanation for the lack of l-kynurenine production in the drug treated cells based solely on a mechanism of drug induced cell death is unlikely, as there was not a strong positive correlation between the two (r2 0.46, Fig. 6a). However, when the dependency between cell viability and inhibition of l-kynurenine production for individual compounds in the series was analysed separately this was found to be highly compound specific, ranging from a strong positive correlation for 5 (r2 0.99) through to no relationship at all for 8 (r2 0.00005, Fig. 6b). These results would therefore suggest that with the exception of compound 5, which is cytotoxic to cells, the pyranonaphthoquinones are likely to be inhibiting IDO-1 directly inside the cells rather than mediating their effects solely by inducing cell death, with efficacy of IDO-1 inhibition varying across the series. This differential in efficacy may in part reflect the ability of each compound to enter the cells, although further work will be needed to fully establish this. Nevertheless these results highlight the importance of screening all potential pyranonaphthoquinone IDO-1 inhibitors for cytotoxicity.
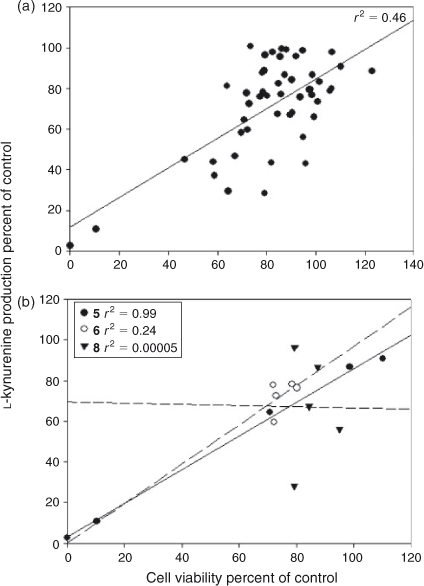
|
On the basis of these results, it would appear that 8 has the most favourable profile of the pyranonaphthoquinone derivatives as a potential IDO-1 inhibitor, showing little cellular cytotoxicity and loss of cell viability in drug treated cells at low drug concentrations, but nevertheless inhibiting l-kynurenine production by over 60 %.
In conclusion, we have reported a series of pyranonaphthoquinone derivatives that are low micromolar IDO-1 enzyme inhibitors that retain activity against the enzyme in a cellular context, exhibiting an approximately one-log loss of dose potency against IDO in cells. Although some in the series showed varying degrees of cytotoxicity against murine LLTC cells and human non-small-cell lung carcinoma cells, the relationship between inhibition of IDO-1 activity and cell death was found to be compound specific with the triazole analogue 8 showing little cytotoxicity but good IDO-1 inhibitory activity. Further work towards the development of IDO-1 inhibitors based on the novel scaffold 1, including identification of the most active enantiomer, is currently in progress.
Experimental
Expression of 6His-IDO
Expression of recombinant human 6His-IDO in the E. coli strain EC 538 (pREP4, pQE9-IDO) and its purification was conducted following the method of Austin et al.[57] Competent bacteria were prepared, transformed, and screened for isopropylthio-β-galactoside (IPTG) inducible IDO-1 expression using standard methods (data not shown). Kinetic analysis of the purified enzyme yielded a Km of 25.3 µM.
Enzyme Assay
The method of Austin and co-workers was used to measure the inhibitory activity of the pyranonaphthoquinones against purified recombinant human IDO-1.[57] Experiments were performed in duplicate. Reactions were assembled on ice in a 96-well plate and each reaction (final volume 200 µL) contained 50 mM potassium phosphate buffer (pH 6.5), 20 mM ascorbic acid (neutralised with 1 N NaOH), 100 µg mL–1 catalase, 10 µM methylene blue, 200 µM l-tryptophan, purified IDO-1 (20 µL, 29.3 IU IDO), and various concentrations of inhibitor (0, 1, 10, 50, 100, 200 μM). At the highest drug concentration tested, the DMSO concentration is 2.5 %. The plates were incubated for 1 h at 37°C before the reaction was terminated by the addition of trichloroacetic acid (TCA) (final concentration 5 %) followed by heating at 65°C for 15 min. Reaction products (125 µL/well) were transferred to a fresh 96-well plate containing p-dimethylaminobenzaldehyde (p-DAB) in acetic acid (125 µL/well) and the absorbance read at 480 nM in a ELx 808 plate reader (BioTek Instruments Inc.). The assay was performed at 200 µM tryptophan, which is at levels that do not cause substrate inhibition. This has been checked in this assay system however, recent reports suggest that substrate inhibition by L-Trp on IDO1 is already significant at concentrations of 200 µM.[60]
Cell Lines
The murine Lewis Lung Carcinoma (LLTC) used in this study has been described.[61] The human H460 non-small-cell lung carcinoma cell line used in this study was purchased from the American Type Culture Collection (ATCC). Each line was cultured in α-modified minimal essential medium supplemented with 10 %, or 5 % respectively, fetal calf serum. Both lines were passaged weekly using trypsin (0.07 % w/v) in citrate-buffered saline. LLTC-IDO cells were transfected with an Ultimate ORF clone purchased from Invitrogen (Carlsbad, CA, USA) that contained human IDO-1 that was subsequently cloned into a cytomegalovirus (CMV) promoter containing vector to drive constitutive expression of the gene using the gateway Cloning System (Invitrogen, Carlsbad, CA, USA). Positive IDO-1 expressing LLTC cells were identified, following limiting dilution and puromycin selection, by both Western Blotting and the enzyme assay, and positive lines were bulked up and frozen in liquid nitrogen for long-term storage.
IC50 Growth Inhibition Assays
LLTC-IDO or H460 cells in exponential phase growth were added to the wells of a 96-well plate (1000 cells/well) and were allowed to continue growth overnight. The next morning, the various pyranonaphthoquinone derivatives were added and serially diluted across the plate. The plates were then incubated for 4 days at 37°C in 5 % CO2 in air. Growth inhibition was assessed by sulforhodamine B staining. Each plate was done in duplicate. IC50 values were determined from the regression line relating the percentage inhibition to logarithmic drug concentration.
Activity of Pyranonaphthoquinone Derivatives against l-Kynurenine Production in LLTC-IDO Cells
LLTC-IDO cells in exponential phase growth were harvested and added (4 × 104 cells/well; 50 µL volume) to the wells of a 96-well plate in which the pyranonaphthoquinone derivatives had already been added and serially diluted across the plate (final volume per well 200 µL). The plates were then incubated for 24 h at 37°C in 5 % CO2 in air. At the end of this time the supernatant (150 μL) was harvested and transferred to a fresh 96-well plate to which TCA (final concentration 5 %) was added and the plate was incubated at 65°C for 15 min. The plate was then centrifuged (1000 g, 5 min) and 125 μL of the clarified supernatant was then transferred to a new 96-well plate that contained p-DAB in acetic acid (125 µL/well) and the absorbance was read at 480 nM referenced against an l-kynurenine standard curve as above. Each plate was done in duplicate.
MTT Cell Viability Assay
LLTC-IDO cells in exponential phase growth were harvested and added (4 × 104 cells/well; 50 µL volume) to the wells of a 96-well plate in which the pyranonaphthoquinone derivatives had already been added and serially diluted across the plate (final volume per well 200 µL). The plates were then incubated for 24 h at 37°C in 5 % CO2 in air. At the end of this time MTT (5 mg mL–1; 20 µL) was added to each plate and the plates incubated for 1 h. The supernatant was then carefully removed from each well and DMSO (150 µL) was added and the plate shaken vigorously to solubilise the MTT. The absorbance was read at 570 nm on an ELx 808 plate reader (BioTek Instruments Inc., Winooski, VT, USA). Each plate was done in duplicate and cell viability as a percentage was assessed with respect to the non-drug treated controls.
Supplementary Material
Full experimental details and purity data of compounds 2–11 can be found on the Journal’s website.
Acknowledgements
This work was supported by the Genesis Oncology Trust, the Auckland Medical Research Foundation, and the Auckland Cancer Society. Dr David Rennison is thanked for HPLC assistance.
References
[1] K. Higuchi, S. Kuno, O. Hayaishi, Fed. Proc. 1963, 22, 243.(abstr.)[2] K. Higuchi, O. Hayaishi, Arch. Biochem. Biophys. 1967, 120, 397.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaF2sXktVyju7w%3D&md5=cc86740cee84e232c1481e4e7157e7f7CAS |
[3] T. Shimizu, S. Nomiyama, F. Hirata, O. Hayaishi, J. Biol. Chem. 1978, 253, 4700.
| 1:CAS:528:DyaE1cXkvVKgtrc%3D&md5=3f8572999185e688d734220090af5536CAS |
[4] O. Takikawa, R. Yoshida, R. Kido, O. Hayaishi, J. Biol. Chem. 1986, 261, 3648.
| 1:CAS:528:DyaL28XitVCnsrs%3D&md5=73ea2821462142b1d97202d52a21531aCAS |
[5] D. H. Munn, E. Shafizadeh, J. T. Attwood, I. Bondarev, A. Pashine, A. L. Mellor, J. Exp. Med. 1999, 189, 1363.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXivFGku7w%3D&md5=364d820b7d974dfe48f79d0623fc79b8CAS |
[6] D. H. Munn, M. Zhou, J. T. Attwood, Science 1998, 281, 1191.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXls1Cqu7c%3D&md5=88f8bd1396afd01fbb8bd513158001f3CAS |
[7] G. J. Guillemin, B. J. Brew, C. E. Noonan, O. Takikawa, K. M. Cullen, Neuropathol. Appl. Neurobiol. 2005, 31, 395.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXpslWisr8%3D&md5=2bc57ea5eeabd3477b37744117a59256CAS |
[8] T. Hayashi, L. Beck, C. Rossetto, X. Gong, O. Takikawa, K. Takabayashi, D. H. Broide, D. A. Carson, E. Raz, J. Clin. Invest. 2004, 114, 270.
| 1:CAS:528:DC%2BD2cXlvFWmsbs%3D&md5=8a4bb449f2121efefc5f516a0c83e070CAS |
[9] R. Potula, L. Poluektova, B. Knipe, J. Chrastil, D. Heilman, H. Dou, O. Takikawa, D. H. Munn, H. E. Gendelman, P. Persidsky, Blood 2005, 106, 2382.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXhtVKqt7zJ&md5=4c68dd5484777317e9476bea778bf869CAS |
[10] O. Takikawa, R. J. Truscott, M. Fukao, S. Miwa, Adv. Exp. Med. Biol. 2003, 527, 277.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXktlOjtrw%3D&md5=409f1d1a9a8f9b2edb7a6642e12659bcCAS |
[11] M. C. Wichers, M. J. Maes, J. Psychiatry Neurosci. 2004, 29, 11.
[12] A. J. Muller, G. C. Prendergast, Cancer Res. 2005, 65, 8065.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXhtVShsb3E&md5=ed7a4d26147122e93864ac681723972bCAS |
[13] A. J. Muller, W. P. Malachowski, G. C. Prendergast, Expert Opin. Ther. Targets 2005, 9, 831.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXmvFels7k%3D&md5=17563065d2397960ccff3072351fa95dCAS |
[14] C. Uyttenhove, L. Pilotte, I. Theate, V. Stroobant, D. Colau, N. Parmentier, T. Boon, B. van den Eynde, Nat. Med. 2003, 9, 1269.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXns1Clu7c%3D&md5=32b2544ce11dbf4ec0e5dd596134b217CAS |
[15] J. B. Katz, A. J. Muller, G. C. Prendergast, Immunol. Rev. 2008, 222, 206.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXlsFejt70%3D&md5=9bd871c57c23a127d8d40cc2035e0a0fCAS |
[16] G. C. Prendergast, Oncogene 2008, 27, 3889.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXns1OnsrY%3D&md5=16419bf7b822ccd87fb8b51de27a274cCAS |
[17] A. J. Muller, P. A. Scherle, Natl. Rev. 2006, 6, 613.
| 1:CAS:528:DC%2BD28XntFGlu7s%3D&md5=0e8701d8409f2dd6620116e60272ecb1CAS |
[18] A. Macchiarulo, E. Camiaoni, R. Nuti, R. Pellicciari, Amino Acids 2009, 37, 219.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXotVKju7s%3D&md5=83c0f06efde73e6867bcb7f7d33c02f1CAS |
[19] X. Zheng, J. Koropatnick, M. Li, X. Zhang, F. Ling, X. Ren, X. Hao, H. Sun, C. Vladau, J. A. Franek, B. Feng, B. L. Urquhart, R. Zhong, D. J. Freeman, B. Garcia, W. P. Min, J. Immunol. 2006, 177, 5639.
| 1:CAS:528:DC%2BD28XhtVans7%2FM&md5=f1f0808271e863b29ecb0bd6bb5eb126CAS |
[20] A. J. Muller, J. B. Duhadaway, P. S. Donover, E. Sutanto-Ward, G. C. Prendergast, Nat. Med. 2005, 11, 312.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXhvVSru7c%3D&md5=7848d37526f640e1881cb4b17604ac55CAS |
[21] T. Banerjee, J. B. Duhadaway, P. Gaspari, E. Sutanto-Ward, D. H. Munn, A. L. Mellor, W. P. Malachowski, G. C. Prendergast, A. J. Muller, Oncogene 2008, 27, 2851.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXltlSgsr0%3D&md5=a35e1414025f40ace8d4b1beffb67f07CAS |
[22] S. Kumar, W. P. Malachowski, J. B. DuHadaway, J. M. LaLonde, P. J. Carroll, D. Jaller, R. Metz, G. C. Prendergast, A. J. Muller, J. Med. Chem. 2008, 51, 1706.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXislKqtL4%3D&md5=3c324c27f3d3e25d8fbc4e318ab3d573CAS |
[23] Y. Watanabe, F. Motokazu, H. Osamu, T. Tomio, U. Hamao, Biochem. Biophys. Res. Commun. 1978, 85, 273.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE1MXltVCrug%3D%3D&md5=73c3d543d9a61246343303f5ee7d16bbCAS |
[24] S. G. Cady, M. Sono, Arch. Biochem. Biophys. 1991, 291, 326.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK38XhtVWmu78%3D&md5=d4df4f6d198c972a51b88ad0dedab0ebCAS |
[25] M. Sono, S. G. Cady, Biochemistry 1989, 28, 5392.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL1MXktF2qtLg%3D&md5=681786fc2063386ac731f0082829ee75CAS |
[26] N. Eguchi, Y. Watanabe, K. Kawanishi, Y. Hashimoto, O. Hayaishi, Arch. Biochem. Biophys. 1984, 232, 602.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2cXlsVKltro%3D&md5=f550e03cb72f8243d64340544ecc79e5CAS |
[27] A. C. Peterson, A. J. La Loggia, L. K. Hamaker, R. A. Arend, P. L. Fisette, Y. Ozaki, J. A. Will, R. R. Brown, J. M. Cook, Med. Chem. Res. 1993, 7, 473.
[28] H. Sugimoto, S. Oda, T. Otsuki, T. Hino, T. Yoshida, Y. Shiro, Proc. Natl. Acad. Sci. USA 2006, 103, 2611.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XksF2rsb4%3D&md5=786720ae26e106637fa6974b70149948CAS |
[29] S. Kumar, D. Jaller, B. Patel, J. M. LaLonde, J. B. DuHadaway, W. P. Malachowski, G. C. Prendergast, A. J. Muller, J. Med. Chem. 2008, 51, 4968.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXptVyrtrY%3D&md5=c80bd2c1b95ecf58d85900634769289eCAS |
[30] H. Nakashima, Y. Uto, E. Nakata, H. Nagasawa, K. Ikkyu, N. Hiraoka, K. Nakashima, Y. Sasaki, H. Sugimoto, Y. Shiro, T. Hashimoto, Y. Okamoto, Y. Asakawa, H. Hori, Bioorg. Med. Chem. 2008, 16, 8661.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtFantLjK&md5=88322fc8a26e796cff0018e505cb4759CAS |
[31] H. Nakashima, K. Ikkyu, K. Nakashima, K. Sano, Y. Uto, E. Nakata, H. Nagasawa, H. Sugimoto, Y. Shiro, Y. Nakagawa, H. Hori, Adv. Exp. Med. Biol. 2010, 662, 415.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhs1Wmsr%2FL&md5=c7368e79cb5ebbf3fdb76189f8b6023fCAS |
[32] E. Dolušić, P. Larrieu, S. Blanc, F. Sapunaric, B. Norberg, L. Moineaux, D. Colette, V. Stroobant, L. Pilotte, D. Colau, T. Ferain, G. Fraser, M. Galeni, J.-M. Frère, B. Masereel, B. van den Eynde, J. Wouters, R. Frédérick, Bioorg. Med. Chem. 2011, 19, 1550.
| Crossref | GoogleScholarGoogle Scholar |
[33] E. Dolušić, P. Larrieu, S. Blanc, F. Sapunaric, J. Pouyez, L. Moineaux, D. Colette, V. Stroobant, L. Pilotte, D. Colau, T. Ferain, G. Fraser, M. Galleni, J.-M. Frere, B. Masereel, B. van den Eynde, J. Wouters, R. Frederick, Eur. J. Med. Chem. 2011, 46, 3058.
| Crossref | GoogleScholarGoogle Scholar |
[34] K. Matsuno, K. Takai, Y. Isaka, Y. Unno, M. Sato, O. Takikawa, A. Asai, Bioorg. Med. Chem. Lett. 2010, 20, 5126.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtVehsbfE&md5=14903238dedada29abe24aa8a309e051CAS |
[35] E. W. Yue, B. Douty, B. Wayland, M. Bower, X. Liu, L. Leffet, Q. Wang, K. J. Bowman, M. J. Hansbury, C. Liu, M. Wei, Y. Li, R. Wynn, T. C. Burn, H. K. Koblish, J. S. Fridman, B. Metcalf, P. A. Scherle, A. P. Combs, J. Med. Chem. 2009, 52, 7364.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXntVSksb8%3D&md5=a911bbb5810a2486bd7a0c0720946ec1CAS |
[36] H. K. Koblish, M. J. Hansbury, K. J. Bowman, G. Yang, C. L. Neilan, P. J. Haley, T. C. Burn, P. Waeltz, R. B. Sparks, E. W. Yue, A. P. Combs, P. A. Scherle, K. Vaddi, J. S. Fridman, Mol. Cancer Ther. 2010, 9, 489.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhslersrY%3D&md5=ce08f652df240ebbe736829152223913CAS |
[37] E. Vottero, A. Balgi, K. Woods, S. Tugendreich, T. Melese, R. J. Andersen, A. G. Mauk, M. Roberge, Biotechnol. J. 2006, 1, 282.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XmtlKqtbs%3D&md5=8acad6aba14afc14e95d1002a54e2f6fCAS |
[38] A. C. Terentis, M. Freewan, T. S. Sempertegui Plaza, M. J. Raftery, R. Stocker, S. R. Thomas, Biochemistry 2010, 49, 591.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhs1Smsb3K&md5=c3e9843104d62a2ad3fda46f7e2fe626CAS |
[39] (a) Q. Huang, M. Zheng, S. Yang, C. Kuang, C. Yu, Q. Yang, Eur. J. Med. Chem. 2011, 46, 5680.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtlKkurvL&md5=4fb6b8f81e0fd57d6a08721e992ac33dCAS |
(b) F. U. Röhrig, S. R. Majjigapu, A. Grosdidier, S. Bron, V. Stroobant, L. Pilotte, D. Colau, P. Vogel, B. J. Van den Eynde, V. Zoete, O. Michielin, J. Med. Chem. 2012, 55, 5270.
| Crossref | GoogleScholarGoogle Scholar |
[40] (a) U. F. Röhrig, L. Awad, A. Grosdidier, P. Larrieu, V. Stroobant, D. Colau, V. Cerundolo, A. J. Simpson, P. Vogel, B. J. van den Eynde, V. Zoete, O. Michielin, J. Med. Chem. 2010, 53, 1172.
| Crossref | GoogleScholarGoogle Scholar |
(b) S. John, S. Thangapandian, S. Sakkiah, K. W. Lee, Eur. J. Med. Chem. 2010, 45, 4004.
| Crossref | GoogleScholarGoogle Scholar |
[41] P. Gaspari, T. Banerjee, W. P. Malachowski, A. J. Muller, G. C. Prendergast, J. DuHadaway, S. Bennett, A. M. Donovan, J. Med. Chem. 2006, 49, 684.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXhtlWmurfN&md5=340e20037e6f2099c9823749c08960c2CAS |
[42] H. C. Brastianos, E. Vottero, B. O. Patrick, R. Van Soest, T. Matainaho, A. G. Mauk, R. J. Andersen, J. Am. Chem. Soc. 2006, 128, 16046.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XhtlCgsrbO&md5=fb105dd3a280b47ed2a1665a6d02e2adCAS |
[43] G. Carr, M. K. Chung, A. G. Mauk, R. J. Andersen, J. Med. Chem. 2008, 51, 2634.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXktlCit7Y%3D&md5=8107907174856adf989fb79b86339736CAS |
[44] M. Volgraf, J.-P. Lumb, H. C. Brastianos, G. Carr, M. K. W. Chung, M. Muenzel, A. G. Mauk, R. J. Andersen, D. Trauner, Nat. Chem. Biol. 2008, 4, 535.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXpvFGrtLo%3D&md5=2a3b1bb99032b55341ff92edb44284a5CAS |
[45] G. Carr, W. Tay, H. Bottriell, S. K. Andersen, G. A. Mauk, R. J. Andersen, Org. Lett. 2009, 11, 2996.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXnsVOmtLw%3D&md5=1c2d5b327a5fb1facb5c58e31916729dCAS |
[46] A. Pereira, E. Vottero, M. Roberge, A. G. Mauk, R. J. Andersen, J. Nat. Prod. 2006, 69, 1496.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XhtVShs7nI&md5=a2bcd274634c53cfe179a733ed4f9c62CAS |
[47] E. Fahy, R. J. Andersen, C. Xu, J. Clardy, J. Org. Chem. 1986, 51, 5145.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2sXjtVGn&md5=25abe26991ec0177cccd3a3503e31a57CAS |
[48] J. Sperry, P. Bachu, M. A. Brimble, Nat. Prod. Rep. 2008, 25, 376.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXkt1Sms7g%3D&md5=cfff6e81b84af3f63fcbd7ec8ba9971eCAS |
[49] J. Sperry, I. Lorenzo-Castrillejo, M. A. Brimble, F. Machín, Bioorg. Med. Chem. 2009, 17, 7131.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXht1ais7vL&md5=d147a1617af3b56d16d9ea2994748791CAS |
[50] K. Rathwell, J. Sperry, M. A. Brimble, Tetrahedron 2010, 66, 4002.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXmtFyrtr8%3D&md5=196d17c5d653e6c7bdc14c738ef3b512CAS |
[51] J. S. Gibson, O. Andrey, M. A. Brimble, Synthesis 2007, 2611.
| 1:CAS:528:DC%2BD2sXhtVKls7%2FN&md5=f47ba938779647f7a664b589225f2d51CAS |
[52] P. Bachu, J. Sperry, M. A. Brimble, Tetrahedron 2008, 64, 4827.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXlvVWgsL4%3D&md5=0fb097f6fd6149eec56579afaa92d0dcCAS |
[53] J. Sperry, J. J. P. Sejberg, F. M. Stiemke, M. A. Brimble, Org. Biomol. Chem. 2009, 7, 2599.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXnvV2nsbY%3D&md5=03795d8685d46f06ad1e2c282b671cc0CAS |
[54] P. Bachu, J. Sperry, M. A. Brimble, Tetrahedron 2008, 64, 3343.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXis1Clsro%3D&md5=562758c8dda50157afdc138df47a2c57CAS |
[55] M. A. Brimble, P. Bachu, J. Sperry, Synthesis 2007, 2887.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXht1KqsL7F&md5=3f4c3e2d06377d1c35bb87e945b43656CAS |
[56] J. Sperry, J. S. Gibson, J. J. P. Sejberg, M. A. Brimble, Org. Biomol. Chem. 2008, 6, 4261.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtlWjt7nM&md5=faaac8c4b17fdb7a3e0275ecc28d8b08CAS |
[57] C. J. Austin, J. Mizdrak, A. Matin, N. Sirijovski, P. Kosim-Satyaputra, R. D. Willows, T. H. Roberts, R. J. Truscott, G. Polekhina, M. W. Parker, J. F. Jamie, Protein Expr. Purif. 2004, 37, 392.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXnsFGgtL4%3D&md5=9b9f6cd64f1e228ae190413599e09190CAS |
[58] D. J. Bridewell, G. J. Finlay, B. C. Baguley, Oncol. Res. 1997, 9, 535.
| 1:CAS:528:DyaK1cXisVCntLk%3D&md5=797b28afcd1682a4038f21e27d299392CAS |
[59] D. J. Bridewell, A. C. Porter, G. J. Finlay, B. C. Baguley, Cancer Chemother. Pharmacol. 2008, 62, 753.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXht1CjtL3N&md5=6828b659a1f1ee4bfd39341f371e36efCAS |
[60] I. Efimov, J. Basran, X. Sun, N. Chauhan, S. K. Chapman, C. G. Mowat, E. L. Raven, J. Am. Chem. Soc. 2012, 134, 3034.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xhs1ehsLk%3D&md5=e7cc6399e3f8764f23bfde6180f1bf11CAS |
[61] A. Haldane, K. M. Holdaway, G. J. Finlay, B. C. Baguley, Cancer Chemother. Pharmacol. 1993, 32, 463.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2cXhsFeisb4%3D&md5=29bd4883539aabe9d6f09e467286a785CAS |
* We are particularly grateful to Dr Kosim-Satyaputra’s expert advice on the growth and purification of the IDO that made this work possible. Sadly, Boeddi passed away before this manuscript could be submitted for publication.
† IFN = interferon.
‡ siRNA = small interfering RNA.