

Transition Metal-Promoted Arylation: An Emerging Strategy for Protein Bioconjugation*
Lara R. Malins AA Department of Chemistry, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA 92037, USA. Email: malins@scripps.edu
Australian Journal of Chemistry 69(12) 1360-1364 https://doi.org/10.1071/CH16416
Submitted: 18 July 2016 Accepted: 12 September 2016 Published: 10 October 2016
Abstract
Transition metal-mediated arylation chemistry is emerging as a powerful tool for the selective modification of native peptides and proteins, providing new opportunities in the field of bioconjugation. This highlight paper will summarize recent methodologies for the regio- and chemoselective arylation of select proteinogenic side chains and backbone amide N–H bonds within unprotected peptides and proteins. The importance of the metal–ligand complex in achieving tunable selectivity and the inherent benefits of arylation as a mode of covalent protein modification will be highlighted.
Introduction
Strategies for the selective modification of peptides and proteins[1] are crucial for the design of protein-based therapies and for furthering our understanding of protein structure and function. The ability to form predictable and stable covalent linkages between complex biomolecules and target functional groups drives development in bioconjugation chemistry and the exploration of promising new classes of therapeutics including antibody–drug conjugates[2] and stapled peptides.[3] Although nature is readily able to modify complex peptides and proteins using highly specific enzyme-catalyzed transformations, termed post-translational modifications (PTMs),[4] duplicating the mild reaction conditions (physiological pH, aqueous media, and ambient temperature) and the exquisite chemo- and regioselectivity of enzymatic transformations in the laboratory is an on-going challenge.[5]
The majority of classical methods for bioconjugation rely on the modification of select proteinogenic amino acid residues, generally exploiting the native nucleophilicity of lysine (Lys), tyrosine (Tyr), and cysteine (Cys) side chain functionalities.[1b,6] Unnatural amino acids, incorporated using chemical synthesis or biological expression, have enabled the introduction of strategic non-native functional handles (e.g. azides, alkenes, alkynes, ketones, aryl halides) into peptide and protein precursors, allowing bioorthgonal and site-selective modifications.[7] Many modern bioconjugation methods have exploited these functional handles through the application of transition metal catalysis,[1b] as in the venerable CuI-catalyzed azide–alkyne cycloaddition (CuAAC) or ‘click’ reaction[8] or metal-mediated olefin cross-metathesis.[9] Despite these prominent examples, transition metal-mediated reactions are still underutilized as a means of modifying complex biomolecules, particularly native substrates bearing no bioorthogonal handles. This is in part due to the stringent demands for protein modification (physiological pH, aqueous media, low substrate concentrations, and ambient temperatures), which are often perceived to be incompatible with most organometallic reagents and reaction protocols.
Early successes in the selective transition metal-promoted functionalization of native peptides and proteins include the reductive amination of Lys residues,[10] application of pi-allylpalladium complexes for Tyr modification,[11] and pioneering work on the use of rhodium carbenoids for the selective modification of tryptophan (Trp).[12] Contributions to the general area of metal-mediated protein modifications have been thoroughly and exquisitely reviewed.[1b,13d] This concise review will focus instead on a burgeoning trend in bioconjugation towards the development of metal-mediated strategies for the chemoselective arylation of native amino acids within peptides and proteins (Fig. 1). Such metal-promoted arylations offer several attractive features for protein bioconjugation. This review will highlight the functional advantages of these emerging technologies by outlining the most recent advances (published in 2015 and 2016) in transition metal-mediated arylations for the modification of native proteins. As such, promising metal-free arylation strategies for bioconjugation and stapling using SNAr chemistry[14] and metal-mediated arylation strategies employing non-proteinogenic or protected amino acids are not included in this focussed overview. A highlight of new strategies for side chain arylation (Fig. 1a) and directed backbone arylation (Fig. 1b) will provide a snapshot of the emerging interest and practical utility of metal-mediated arylations for bioconjugation.
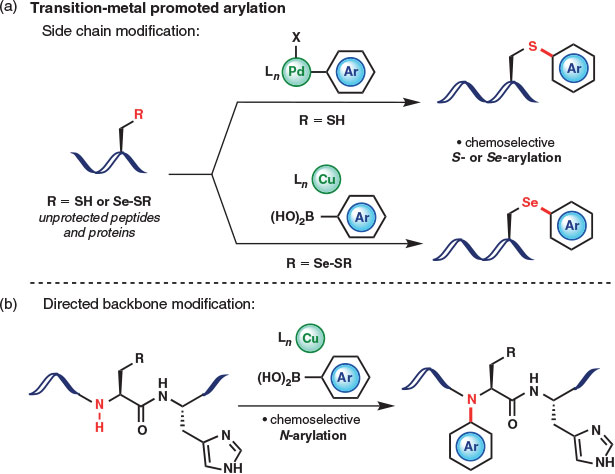
|
Advantages of Metal-Mediated Arylation
To understand the current trend towards metal-mediated arylation as a tool for covalent protein modification, it is prudent to evaluate the strategic advantages offered by the aryl motif as a functionalizable unit for bioconjugation. First, the introduction of an aryl bridge onto native proteins diversifies the realm of ‘linker space’ currently accessible using conventional bioconjugation methods, which often utilize alkylation or acylation chemistry. Altering the spacing and rigidity of the linker unit has important implications for the physicochemical properties and in vivo efficacy of bioconjugates, including stapled peptides.[3] Second, the ability to introduce diverse aryl ring substituents facilitates the modulation of bioconjugate properties such as stability and solubility. Finally, the introduction of aryl motifs using transition metal complexes offers additional strategic advantages. Careful optimization of the metal, ligand, and reaction additives offers vast potential for tunable functional group selectivity and mild reaction conditions. These features are crucial for the effective modification of native peptides and proteins, which are rich in functionality and in which chemo- and regioselectivity are not provided by the introduction of a non-native, bioorthgonal handle. The following sections will therefore emphasize the crucial role of the metal and ligand complex in achieving highly selective metal-mediated side chain and backbone arylation reactions.
Side Chain Arylation
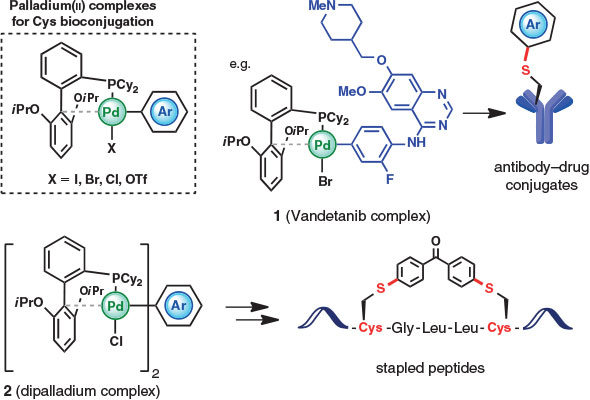
The rapid and selective metal-promoted arylation of Cys side chains within unprotected peptides and proteins was first reported in 2015 by Buchwald, Pentelute, and coworkers through the preparation of bench-stable aryl palladium(ii) complexes (Fig. 2) capable of highly efficient aryl transfer to cysteine residues.[15] The organopalladium species were readily formed by oxidative addition of an aryl halide or triflate and the judicious choice of a ligand (2-dicyclohexylphosphino-2′,6′-diisopropoxybiphenyl, RuPhos), which provided a stable yet reactive palladium species that could effectively facilitate C–S reductive elimination to afford diverse S-aryl cysteine derivatives at room temperature in aqueous media (pH 5.5–8.5) and at high dilution (low micromolar concentrations). Although pi-allylpalladium complexes have been previously employed for the modification of Tyr residues,[11] the arylpalladium species employed by Buchwald and coworkers[15] are less electrophilic, which facilitates their selectivity for cysteine. With a model peptide, quantitative conversion to the Cys-labelled products was achieved with a variety of different arylpalladium reagents, including fluorescent tags, bioconjugation linkers, affinity labels, and therapeutics. The method was also extended to the modification of Cys residues within proteins and was successfully employed in the preparation of an antibody–drug conjugate linking the cancer drug vandetanib (using arylpalladium reagent 1, Fig. 2) to trastuzumab, a cysteine-containing monoclonal antibody targeting the breast cancer protein HER2. By employing a reagent with two electrophilic palladium centres (e.g. 2), the authors were also able to prepare a stapled peptide containing a novel diaryl linker unit. Importantly, the arylated peptide adducts prepared in this study were highly stable to acid, base, and exogenous thiols, unlike bioconjugates formed using traditional reagents such as N-ethyl maleimide and 2-bromoacetamide. Modification of substituents on the aryl ring also enabled tuning of the oxidative stability of the S-aryl moiety.[15]
Another powerful approach to palladium-mediated Cys arylation was recently reported in 2016 by Davis and coworkers.[16] This method exploits the endogenous metal binding motifs often displayed in the active sites of metal-dependent enzymes to direct regioselective palladium-catalyzed S-arylation in the presence of multiple Cys residues. The authors hypothesized that a native binding site might serve to guide a reactive metal complex, which could then covalently modify a proximal reactive Cys residue (Fig. 3). The proposed selective arylation was interrogated using mannosylglycerate synthase (MGS), a class A glycosyltransferase and metal-dependent enzyme bearing an aspartate metal-binding motif (Asp100Ala101Asp102), as a model. Employing an aryl iodide and a Pd(OAc)2 complex with the disodium salt of N,N-dimethyl-2-amino-4,6-dihydroxypyrimidine (DM-ADHP) as a precatalyst in aqueous medium, the authors were able to form in situ an activated arylpalladium(ii) species capable of selective S-arylation of Cys233, which is proximal to the MGS metal-binding motif. The remaining Cys residues were unmodified, despite reacting readily in a control experiment employing non-directed alkylation chemistry. Alanine (Ala)-scanning mutagenesis also established a role for the metal-binding motif and an accessory ion-engagement site (Thr139) in guiding the reactive palladium complex, although residual directing ability was observed on replacement of these residues with Ala. The optimized catalyst and ligand complex was crucial for reaction success, with alternative precatalysts returning only unmodified protein. Interestingly, elevated temperatures (65°C) were also required, suggesting that the arylpalladium(ii) species formed in situ during these transformations is less reactive than the isolated palladium complexes employed for the room temperature arylations reported by Buchwald and coworkers.[15] These results highlight a primary benefit of using transition metals for protein modification – judicious choice of ligands allows precise tailoring of both reactivity and selectivity.
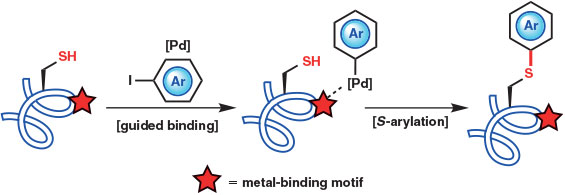
Davis and coworkers went on to demonstrate selective palladium-mediated S-arylation of MGS using a broad range of substituted aryl iodides, including isotope labels, carbohydrates, and functional handles for bioconjugation.[16] Given the proximity of most metal-binding sites (and therefore the Cys residues modified by the guided S-arylation protocol) to the enzyme active site, the authors also evaluated select S-aryl constructs as covalent modulators of MGS enzymic activity. These studies provided proof of concept for the design of metal-promoted, active site-directed covalent inhibition of enzyme targets. Finally, guided S-arylation using a biotin aryl iodide was also used to label and identify metal-dependent proteins within complex mixtures (e.g. cell lysates). As a general tool, proteomic analysis of S-arylation sites can provide valuable insight into the environment surrounding metal-binding motifs.[16]
Transition metal-mediated arylations of Cys, including the two described above, rely on the nucleophilicity of the side chain thiol at physiological pH. Selenocysteine (Sec), the selenium analogue of Cys and so-called 21st proteinogenic amino acid,[17] also displays potent nucleophilicity at physiological pH, at which its acidity (pKa 5.47)[18] renders it primarily in the anionic selenolate form. Despite these properties and the importance of Sec in several essential proteins,[19] difficulties in functionalizing and handling Sec residues limit their applicability in bioconjugation chemistry. In particular, Sec rapidly oxidizes to the corresponding diselenide as a result of its low redox potential.[20] Reactions employing electrophilic bioconjugation reagents therefore require oxygen-free conditions and an exogenous reducing agent to generate the nucleophilic selenol in situ.
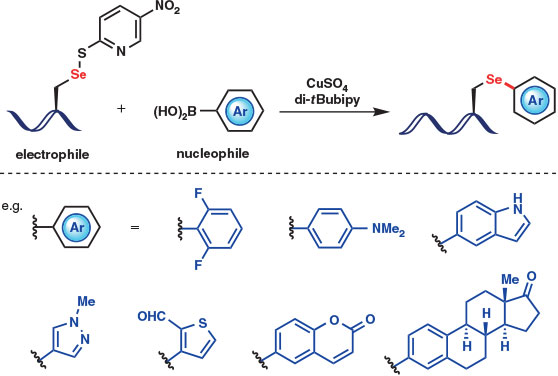
To overcome this limitation, Buchwald, Pentelute, and coworkers reported in 2015 a robust umpolung approach to the selective arylation of Sec that relies on the electrophilicity of oxidized Sec in a copper-promoted reaction with nucleophilic arylboronic acids (Fig. 4).[21] Employing Sec residues masked as the corresponding selenyl sulfides with 2-thiol-5-nitropyridine (TNP), selective and high-yielding Se-arylation within unprotected peptides was achieved using CuSO4 and the optimal ligand 4,4′-di-tert-butyl-2,2′-bipyridine (di-tBubipy) in aqueous buffer at 37°C. The desired reaction did not proceed in the absence of copper or without a ligand, and control studies employing a free Cys thiol or a TNP–Cys disulfide resulted in low conversion to the S-arylated product, highlighting the superior reactivity of Sec. A broad range of arylboronic acids was employed, including heterocycles, fluorescent tags, and drug molecules (Fig. 4). Interestingly, the Se-aryl constructs formed were shown to be more stable to oxidative conditions than an analogous benzylated Sec derivative, which readily formed dehydroalanine (Dha) as the oxidation–elimination product. The rate of Dha formation for Se-aryl adducts was highly dependent on the electron density of the aryl ring, enabling the tunable stability of arylated Sec adducts formed using the copper-mediated arylation protocol.[21]
Backbone Arylation
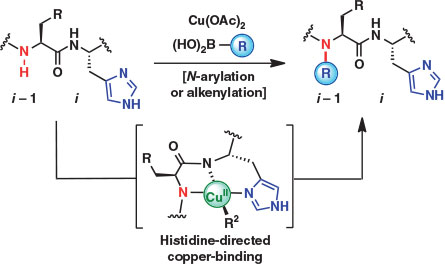
A conceptually unique approach to protein modification that combines the notion of directed functionalization with copper-mediated arylation chemistry was described in 2016 by Ball and coworkers.[22] The authors of this study report a method for the site-selective arylation and alkenylation of backbone N–H bonds directed by a proximal histidine (His) residue (Fig. 5). Although backbone modifications have profound impacts on folding, function, and stability,[23] methods for direct backbone amide functionalization are far less common than side chain modifications, likely as a result of inherent issues with chemo- and regioselectivity in the polyamide-based structures of native peptides and proteins. The Cu(OAc)2-mediated, His-directed oxidative coupling of boronic acids described by Ball and coworkers provides a method for exclusive backbone N–H modification at the i – 1 position (one residue before His, Fig. 5). The remarkable selectivity observed is thought to be attributed to the binding of copper to the His imidazole side chain and coordination to the deprotonated i and i – 1 backbone nitrogen atoms (Fig. 5). The scope and selectivity of the method were evaluated on several model peptides, with His-directed site-selective modification accomplished using several substituted aryl and alkenylboronates in aqueous media and at ambient temperature. In some cases, alkenylboronates resulted in higher conversions to the backbone-modified products, a result attributed to the increased steric demands associated with arylboronate reagents. Finally, the method was shown to be applicable to the selective modification of the protein lysozyme, which contains one His residue (His15) and was singly modified in the presence of copper(ii) and trifluoroborate salts bearing handles for further bioconjugation (azide, alkyne, and desthiobiotin).[22] Such discrete backbone modification of native proteins promoted by transition metal complexes opens exciting new opportunities to study the effects of N-substitution on protein structure and function.
Conclusions
The work summarized in this highlight illustrates a strong trend towards the use of transition metal-mediated chemistry, specifically site-selective arylation, for the modification of native peptides and proteins. With appropriately tuned catalyst and ligand complexes, regio- and chemoselective covalent modifications have been accomplished under mild conditions and in aqueous media for both side chain and backbone functionalization. The arylated peptide and protein adducts accessible using metal-mediated transformations provide access to diverse areas of chemical space and new modes of bioconjugation from native, proteinogenic amino acids. The ability to modulate conjugate properties, including stability, by introducing aryl substituents also provides the opportunity for exquisite control of physicochemical properties. One current limitation of these otherwise powerful methods is the reliance on large excesses of metal–ligand complex and aryl substrate (typically at least 10 equivalents each for the modification of protein substrates), which might hamper the use of precious aryl moieties. In some cases, residual metal species may also complicate protein purification. Nevertheless, the compelling advantages offered by metal-mediated protein arylation chemistry outweigh its limitations and will likely drive the rapid adoption of such methodologies. Future studies that further expand the toolbox of covalent protein modification strategies will undoubtedly emerge in due course.
* The author is the recipient of the 2015 RACI Cornforth Medal.
References
[1] (a) C. D. Spicer, B. G. Davis, Nat. Commun. 2014, 4740.| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXksVeksb4%3D&md5=cc74f3b2512ff075852d437738a0c2efCAS | 25190082PubMed |
(b) O. Boutureira, G. J. Bernardes, Chem. Rev. 2015, 115, 2174.
| Crossref | GoogleScholarGoogle Scholar |
[2] R. V. J. Chari, M. L. Miller, W. C. Widdison, Angew. Chem. Int. Ed. 2014, 53, 3796.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXjtVGmsr0%3D&md5=6189adad9463d03421d6df26cd02bf1bCAS |
[3] Y. H. Lau, P. de Andrade, Y. Wu, D. R. Spring, Chem. Soc. Rev. 2015, 44, 91.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXhsV2mtb%2FL&md5=ceeb62c0c4c96ac750ea9a37bf80e00bCAS | 25199043PubMed |
[4] (a) C. Walsh, Post-Translational Modification of Proteins: Expanding Nature’s Inventory 2006 (Roberts and Co. Publishers: Englewood, CO).
(b) C. T. Walsh, S. Garneau-Tsodikova, G. J. Gatto, Angew. Chem. Int. Ed. 2005, 44, 7342.
| Crossref | GoogleScholarGoogle Scholar |
[5] N. Stephanopoulos, M. B. Francis, Nat. Chem. Biol. 2011, 7, 876.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsVOisbbP&md5=429f41e33de8af0251aa81269b7b7ad1CAS | 22086289PubMed |
[6] (a) G. T. Hermanson, Bioconjugate Techniques (3rd edn) 2013 (Academic Press: Boston, MA).
(b) G. J. L. Bernardes, J. M. Chalker, B. G. Davis, in Ideas in Chemistry and Molecular Sciences (Ed. B. Pignataro) 2010, pp. 59–91 (Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim).
[7] (a) K. Lang, J. W. Chin, Chem. Rev. 2014, 114, 4764.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXkslamtrY%3D&md5=769f3507405299b28fe5c2086605d592CAS | 24655057PubMed |
(b) W. H. Zhang, G. Otting, C. J. Jackson, Curr. Opin. Struct. Biol. 2013, 23, 581.
| Crossref | GoogleScholarGoogle Scholar |
(c) E. M. Sletten, C. R. Bertozzi, Angew. Chem. Int. Ed. 2009, 48, 6974.
| Crossref | GoogleScholarGoogle Scholar |
(d) C. H. Kim, J. Y. Axup, P. G. Schultz, Curr. Opin. Chem. Biol. 2013, 17, 412.
| Crossref | GoogleScholarGoogle Scholar |
[8] V. V. Rostovtsev, L. G. Green, V. V. Fokin, K. B. Sharpless, Angew. Chem. Int. Ed. 2002, 41, 2596.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38Xls1Ohsr4%3D&md5=fe9f6b13c78335a5d346bc92864dea5dCAS |
[9] Y. A. Lin, J. M. Chalker, B. G. Davis, ChemBioChem 2009, 10, 959.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXltVOlt7g%3D&md5=2ab29a3375d63cf0426011ac4a4b5226CAS | 19343741PubMed |
[10] J. M. McFarland, M. B. Francis, J. Am. Chem. Soc. 2005, 127, 13490.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXpslequrk%3D&md5=d6f8a2a0a81c361f51a8edf1acff0d69CAS | 16190700PubMed |
[11] S. D. Tilley, M. B. Francis, J. Am. Chem. Soc. 2006, 128, 1080.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XitVymsw%3D%3D&md5=589aa5537e4affc148afbe1872103679CAS | 16433516PubMed |
[12] J. M. Antos, M. B. Francis, J. Am. Chem. Soc. 2004, 126, 10256.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXmtVOjtrc%3D&md5=d48de9fb95ac241874a507bf9df55f78CAS | 15315433PubMed |
[13] (a) J. M. Antos, M. B. Francis, Curr. Opin. Chem. Biol. 2006, 10, 253.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XltlKkt7c%3D&md5=c37c18dd4ffee417a7b9b6a13ab027d3CAS | 16698310PubMed |
(b) J. H. van Maarseveen, J. N. Reek, J. W. Back, Angew. Chem. Int. Ed. 2006, 45, 1841.
| Crossref | GoogleScholarGoogle Scholar |
(c) M. Yang, J. Li, P. R. Chen, Chem. Soc. Rev. 2014, 43, 6511.
| Crossref | GoogleScholarGoogle Scholar |
(d) A. F. Noisier, M. A. Brimble, Chem. Rev. 2014, 114, 8775.
| Crossref | GoogleScholarGoogle Scholar |
[14] (a) A. M. Spokoyny, Y. Zou, J. J. Ling, H. Yu, Y. S. Lin, B. L. Pentelute, J. Am. Chem. Soc. 2013, 135, 5946.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXlsV2gtrc%3D&md5=2266a9d99a353bd9cb567db56a5a24bcCAS | 23560559PubMed |
(b) S. P. Brown, A. B. Smith, J. Am. Chem. Soc. 2015, 137, 4034.
| Crossref | GoogleScholarGoogle Scholar |
(c) G. Lautrette, F. Touti, H. G. Lee, P. Dai, B. L. Pentelute, J. Am. Chem. Soc. 2016, 138, 8340.
| Crossref | GoogleScholarGoogle Scholar |
(d) C. Zhang, M. Welborn, T. Zhu, N. J. Yang, M. S. Santos, T. Van Voorhis, B. L. Pentelute, Nat. Chem. 2016, 8, 120.
| Crossref | GoogleScholarGoogle Scholar |
(e) S. Kalhor-Monfared, M. R. Jafari, J. T. Patterson, P. I. Kitov, J. J. Dwyer, J. M. Nuss, R. Derda, Chem. Sci. 2016, 7, 3785.
| Crossref | GoogleScholarGoogle Scholar |
[15] E. V. Vinogradova, C. Zhang, A. M. Spokoyny, B. L. Pentelute, S. L. Buchwald, Nature 2015, 526, 687.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXhslCnu7zK&md5=309e6582a882d9248aaeb2be85f6450cCAS | 26511579PubMed |
[16] J. Willwacher, R. Raj, S. Mohammed, B. G. Davis, J. Am. Chem. Soc. 2016, 138, 8678.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC28XhtVGjtLrF&md5=f955cfa383491a169a347f8978bb1069CAS | 27336299PubMed |
[17] A. Bock, K. Forchhammer, J. Heider, W. Leinfelder, G. Sawers, B. Veprek, F. Zinoni, Mol. Microbiol. 1991, 5, 515.
| Crossref | GoogleScholarGoogle Scholar | 1:STN:280:DyaK3M3mtVCgtw%3D%3D&md5=e7131a81cedc2ce49cdb6af9652b31b1CAS | 1828528PubMed |
[18] B. J. Byun, Y. K. Kang, Biopolymers 2011, 95, 345.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXitlKrtrc%3D&md5=8248b9194895a247178ebab1b0323f76CAS | 21213257PubMed |
[19] (a) L. Johansson, G. Gafvelin, E. S. J. Arner, Biochim. Biophys. Acta, Gen. Subj. 2005, 1726, 1.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXhtFKns7fE&md5=47b9445d6fd17d720520d5cc1adfccb2CAS |
(b) J. Lu, A. Holmgren, J. Biol. Chem. 2009, 284, 723.
| Crossref | GoogleScholarGoogle Scholar |
[20] (a) L. R. Malins, N. J. Mitchell, R. J. Payne, J. Pept. Sci. 2014, 20, 64.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXhvVGgtLzP&md5=20a1aff93d0c22a6ad44b5b4a318a7acCAS | 24285588PubMed |
(b) D. Besse, F. Siedler, T. Diercks, H. Kessler, L. Moroder, Angew. Chem. Int. Ed. Engl. 1997, 36, 883.
| Crossref | GoogleScholarGoogle Scholar |
(c) T. Nauser, S. Dockheer, R. Kissner, W. H. Koppenol, Biochemistry 2006, 45, 6038.
| Crossref | GoogleScholarGoogle Scholar |
[21] D. T. Cohen, C. Zhang, B. L. Pentelute, S. L. Buchwald, J. Am. Chem. Soc. 2015, 137, 9784.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXht1KmsrnK&md5=26a04177710e07a219becda80b2ec853CAS | 26225900PubMed |
[22] J. Ohata, M. B. Minus, M. E. Abernathy, Z. T. Ball, J. Am. Chem. Soc. 2016, 138, 7472.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC28XptVKrur4%3D&md5=f9cb4a6277974c655b0f39696eb2d008CAS | 27249339PubMed |
[23] (a) U. Kazmaier, J. Deska, Curr. Org. Chem. 2008, 12, 355.
| Crossref | GoogleScholarGoogle Scholar |
(b) J. Chatterjee, F. Rechenmacher, H. Kessler, Angew. Chem. Int. Ed. 2013, 52, 254.
| Crossref | GoogleScholarGoogle Scholar |