

Methanal Extrusion in ipso-Substitution Reactions of Hydroxymethylindoles*
Jeremy C. Dobrowolski A , Kittiya Somphol A , Mardi Santoso A , Hung Duong A , Christopher R. Gardner A , Naresh Kumar A and David StC. Black A BA School of Chemistry, The University of New South Wales, UNSW Australia, Sydney, NSW 2052, Australia.
B Corresponding author. Email: d.black@unsw.edu.au
Australian Journal of Chemistry 70(11) 1188-1195 https://doi.org/10.1071/CH17257
Submitted: 12 May 2017 Accepted: 2 August 2017 Published: 29 August 2017
Abstract
A range of 3-hydroxymethylindoles undergo acid-catalysed reactions involving ipso-electrophilic substitution with the extrusion of methanal and the formation of diindolylmethane moieties. Both inter- and intramolecular processes lead to macrocyclic compounds 10 and 14.
Introduction
Hydroxymethylindoles undergo a variety of acid-catalysed reactions leading to cyclic indolylmethylene oligomers. For instance, 2- and 7-hydroxymethyl-4,6-dimethoxyindoles are converted into calix-3- and calix-4-indoles.[1,2] However, when the indole nitrogen atom is substituted, the products of similar reactions are indolocyclotriveratrylenes. Reaction of 3-hydroxymethyl-1-methylindole[3–5] with a catalytic amount of p-toluenesulfonic acid in dichloromethane gives a simple indolocyclotriveratrylene.[6] Similar but more complex cyclotriveratrylenes can also be generated from other N-substituted hydroxymethylindoles and also those derived from activated indoles.[7] Given the formation of indolocyclotriveratrylenes 2 from N-substituted hydroxymethylindoles 1, it was considered that the reactions of bis-hydroxymethylindole systems 3 containing two indole rings joined by linkers through their nitrogen atoms might lead to the formation of cylindrical structures 4 capable of selectively attracting small molecules (Scheme 1). The construction and mechanistic investigation for synthesis of these precursor systems are now described.
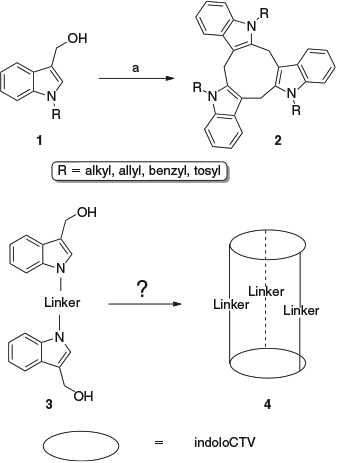
|
Results and Discussion
Synthesis of 3,3′-Bishydroxymethylindoles
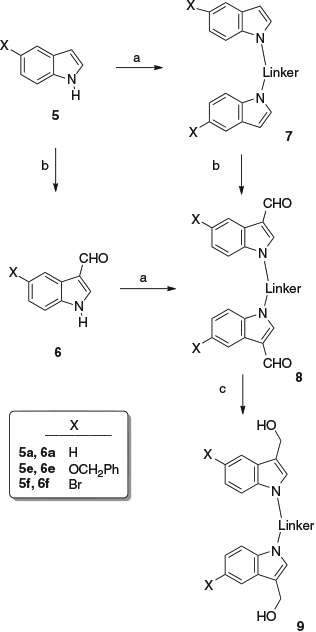
In principle, these compounds can be prepared using two simple routes (Scheme 2).
|
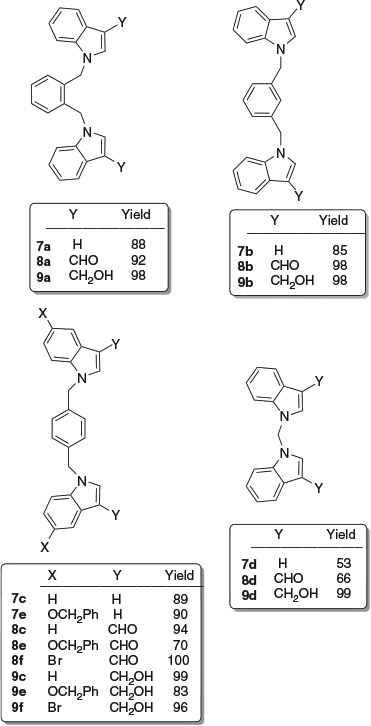
The indoles 5 can first be linked by an alkylation reaction with a dibromo-substituted linker, and then the resulting compounds 7 can be formylated at the 3- and 3′-positions (usually using the Vilsmeier reagent), resulting in biscarbaldehydes 8, which can be reduced to give the bishydroxymethyl compounds 9. Alternatively, the biscarbaldehydes 8 can be formed by linkage of two indole-3-carbaldehydes 6. A combination of these two processes was employed for the synthesis of the dialdehydes 8. The chosen linkers were o-, m-, and p-di(bromomethyl)benzenes and diiodomethane, providing compounds 7–9 (Fig. 1). In this way, compounds 7a–e were prepared, compounds 7a–d being already known,[8–10] and compound 7e being new. The dialdehydes 8a–f were usually but not always prepared from the indole-3-carbaldehydes 6 (see Experimental). Compounds 8a–c have previously been reported,[11,12] whereas compounds 8d–f are new. Compound 7e was prepared from 5-benzyloxyindole[13] and p-di(bromomethyl)benzene, and formylation gave the dialdehyde 8e. The biscarbaldehyde 8d was prepared from indole-3-carbaldehyde 6a and diiodomethane, whereas the related compound 8f was obtained by reaction of 5-bromoindole-3-carbaldehyde 6f[14] with p-di(bromomethyl)benzene. The bishydroxymethylindoles 9a–f were formed by reduction of the dicarbaldehydes 8a–f with sodium borohydride.
|
Acid-Catalysed Reactions of 3,3′-Bishydroxymethylindoles
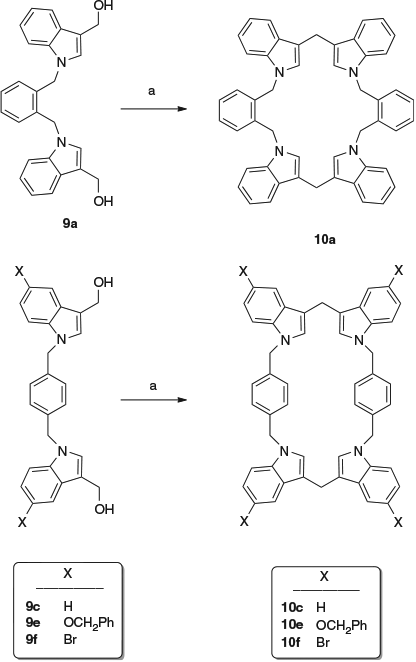
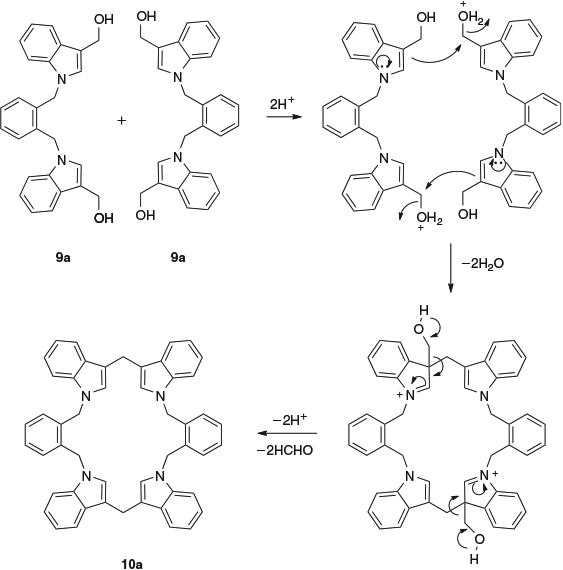
The plan outlined in the introduction, namely to generate two cyclotriveratrylene rings joined by the linkers, was unsuccessful. No indication of such products was observed. However, clean reactions occurred in the case of compounds 9a, 9c, 9e, and 9f, whereas reactions of compounds 9b and 9d gave complex mixtures. None of the isolated reaction products were the result of oligomerisation of indolylmethylene moieties; instead, simple dimerisations occurred to give interesting macrocyclic compounds, as the result of ipso-electrophilic aromatic substitution with the loss of methanal. Bis-hydroxymethylindole 9a gave the 22-membered macrocylic ring compound 10a in 62 % yield, and the bis-hydroxymethylindoles 9c, 9e, and 9f gave the 26-membered macrocyclic ring compounds 10c, 10e, and 10f in yields of 60, 60 and 50 % respectively (Scheme 3).
|
The macrocycle 10a was too insoluble for NMR data to be measured, but its molecular weight determination by matrix-assisted laser desorption–ionization (MALDI) mass spectrometry gave a molecular ion at m/z 696.80, consistent with the macrocyclic structure. Macrocycles 10c, 10e, and 10f gave NMR spectra that indicated their structural simplicity. For example, the 13C NMR spectrum of compound 10c showed two methylene carbon signals at 29.8 and 49.3 ppm, six tertiary carbon signals at 109.3, 119.0, 119.4, 121.7, 126.3, and 127.1 ppm, and four quaternary carbon signals at 115.1, 128.2, 136.7, and 137.2 ppm. A proposed mechanism for the formation of compound 10a (as a typical case) is shown in Scheme 4.
|
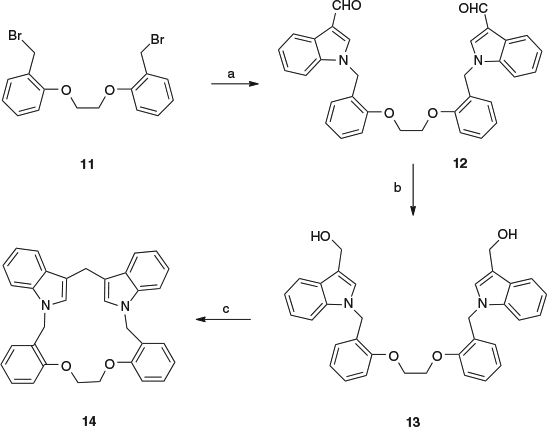
Given the outcome in which the linked 3,3′-bishydroxymethylindoles underwent dimerisation, an example with a longer linker was investigated. The dialdehyde 12 was prepared from indole-3-carbaldehyde and 1,2-bis-(2-bromomethylphenoxy)ethane[15] 11 in 85 % yield. Reduction with sodium borohydride gave the bis-hydroxymethyl analogue 13 in 87 % yield. Treatment of this compound 13 with a catalytic amount of p-toluenesulfonic acid in dichloromethane for 1 h at room temperature yielded the macrocyclic compound 14 in 68 % yield (Scheme 5).
|
In this case, the ipso-substitution reaction is intramolecular and leads to a 17-membered ring, as opposed to the formation of a 34-membered ring from an intermolecular dimerisation. The structure of compound 14 was confirmed by NMR spectroscopy. The 1H NMR spectrum showed three singlet resonances at 4.14, 4.26 and 5.21 ppm, in the ratio of 1 : 2 : 2, corresponding to the three methylene groups in the molecule, namely the 3-indole-methylene, the 1-indole-methylene, and the O-methylene respectively.
The extrusion of methanal in ipso-substitution reactions involving indoles was initially observed by Leete and Marion,[16] and authenticated by Thesing[17] who obtained 3,3′-diindolylmethane in a 74 % yield after shaking an aqueous solution of 3-hydroxymethylindole at 25°C for 20 h. Crucially, methanal in the aqueous filtrate was identified by formation of the dimedone condensation product. This methodology was later extended to other related 3-hydroxymethylindole derivatives by Leete.[18] Some more recent conversions of 3-hydroxymethylindole to 3,3′-diindolylmethane have been reported but involve more complex reaction conditions.[19,20] The reaction methodology involving methanal extrusion has also been reported for activated 3-, 4-, 6-, and 7-hydroxymethylindoles. 3-Hydroxymethyl-4,6-dimethoxy-1-methylindole was converted into the related diindolylmethane in 90 % yield by heating in water.[21] Various 4- and 6-hydroxymethyl-dimethoxyindoles can also be converted into the related diindolylmethanes by treatment with ethanoic acid at room temperature.[21] However, the most common general reactions of this kind involve the treatment of 2,3-disubstituted-7-hydroxymethyl-4,6-dimethoxyindoles with a range of acidic conditions such as ethanoic acid, trifluoroacetic acid, and K-10 clay.[22–25] The essential imperative for these methanal extrusion reactions is that the carbon atom bearing the hydroxymethyl group must be the most nucleophilic site.
Acid-Catalysed Reactions of Indole-Linked Hydroxymethylarenes
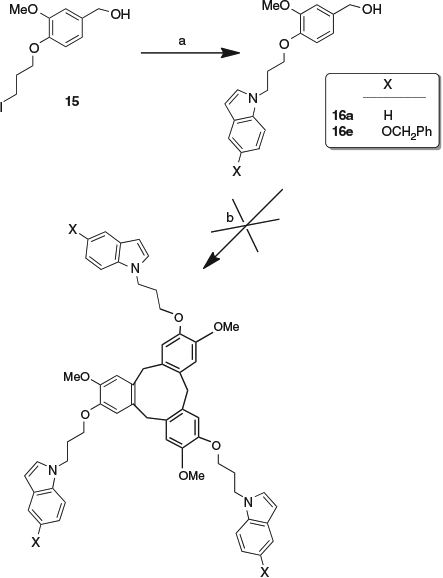
A different stepwise approach to the original aim of constructing structures in which a cyclotriveratrylene and an indolocyclotriveratrylene are linked was briefly investigated. The new compounds 16a and 16e were synthesised in good yield from the known[26] iodo compound 15 by reaction with indole and 5-benzyloxyindole respectively (Scheme 6). The aim was to form a cyclotriveratrylene from the hydroxymethylarene and subsequently formylate the indole in the 3-position and reduce it to the 3-hydroxymethylindole before subjecting that to acidic conditions. The iodo compound 15 had been used as a starting material in the synthesis of cryptophanes, in which two cyclotriveratrylenes were linked together.[26] However, on treatment of compounds 16a and 16e with a range of acidic reagents, the desired cyclotriveratrylenes could not be isolated from the complex reaction mixtures.
|
Conclusion
A new range of 3-hydroxymethylindoles undergo acid-catalysed reactions involving ipso-electrophilic substitution with the extrusion of methanal and the formation of diindolylmethane moieties. Both inter- and intramolecular reactions have been observed, but no cyclotriveratrylenes were formed. Bis-(3-hydroxymethyl)indoles linked by ortho- and para-xylyl moieties undergo intermolecular dimerisation processes to give 22- and 26-membered macrocyclic compounds respectively, whereas the related meta-xylyl linked compounds gave only polymeric mixtures. A compound in which two 3-hydroxymethylindole units are linked by a di-(ortho-xylyl)ethyleneglycol moiety undergoes an intramolecular process to give a 17-membered macrocyclic compound, in preference to an intermolecular dimerisation that would lead to a 34-membered macrocylic compound.
Experimental
General
Melting points were measured using a Mel-Temp melting point apparatus, and are uncorrected. Microanalysis was performed on a Carlo Erba Elemental Analyser EA 1108 at the Campbell Microanalytical Laboratory, University of Otago, New Zealand. 1H and 13C NMR spectra were obtained on a Bruker DPX300 (300 MHz) spectrometer. Mass spectra were recorded on either a Bruker Fourier transform ion cyclotron resonance mass spectrometry (electron ionisation) (FT-ICR MS (EI)) or a Micromass ZQ2000 (electrospray ionisation (ESI)) at UNSW, or a Shimadzu LCMS QP 8000 (ESI) at the University of Otago, New Zealand. Infrared spectra were recorded with a Thermo Nicolet 370 FT-IR spectrometer using KBr discs. Ultraviolet–visible spectra were recorded using a Varian Cary 100 Scan spectrometer. Column chromatography was carried out using Merck 230–400-mesh ASTM silica gel, whereas preparative thin-layer chromatography was performed using Merck silica gel 7730 60GF254.
Synthesis of Novel Bis-Indole
1,4-Di-(5-benzyloxyindol-1-ylmethyl)benzene (7e)
5-Benzyloxyindole 5e (0.22 g, 1 mmol), freshly crushed potassium hydroxide (0.06 g, 1 mmol), and dry dimethyl sulfoxide (20 mL) were stirred together at room temperature for 1 h, then α,α′-dibromo-p-xylene (0.13 g, 0.50 mmol) was added and stirring continued for a further 1 h. Water was added and the resulting precipitate was filtered off, washed with water, and dried. After flash chromatography, the diindolylxylene 7e was obtained as a white solid (0.25 g, 90 %). Mp 179°C. Found: C 82.5, H 6.0, N 5.2; C38H32N2O2.0.25 H2O requires C 82.5, H 5.9, N 5.1 %. vmax (KBr)/cm−1 1618, 1569, 1552, 1483, 1450, 1234, 1148, 1123, 1013, 833, 792, 759, 751, 743, 702, 670. λmax (CH2Cl2)/nm (ϵ/M−1 cm−1) 230 (55900), 276 (22300). δH (300 MHz, [D6]DMSO) 5.04 (4H, s, CH2N), 5.27 (4H, s, OCH2Ph), 6.34 (2H, d, J 2.6, ArH), 6.78 (2H, dd, J 2.3 and 2.6, ArH), 7.06 (4H, s, ArH), 7.11 (2H, d, J 2.3, ArH), 7.24–7.35 (14H, m, ArH). δC (75 MHz, CDCl3) 49.8 (CH2N), 70.8 (OCH2Ph), 101.0, 104.2, 111.2, 112.3, 127.5, 127.9, 128.7, 129.9 (ArCH), 128.0, 129.1, 131.5, 137.8, 138.1, 152.9 (ArC). m/z (ESI+) 571 ([M + Na]+, 100 %).
Synthesis of Diindolylxylene-3,3′-dicarbaldehydes
General Method A
To an ice-cold solution of phosphoryl chloride in dry DMF, a cold solution of the diindolylbenzene in dry DMF was added slowly. The reaction mixture was stirred with ice-cooling for 0.5 h, followed by addition of water, and basifying with 10 % sodium hydroxide solution. The resulting precipitate was filtered off, washed with water until the filtrate was neutral, and dried to yield the diindolylbenzene-3,3′-dicarbaldehyde.
General Method B
A mixture of indole aldehyde, freshly crushed potassium hydroxide, and dry DMSO was stirred at room temperature. After 1 h, the dibromomethylxylene was added and the mixture stirred for a further 1 h. Water was added and the resulting precipitate was filtered off, washed with water, and dried to yield the diindolylbenzene-3,3′-dicarbaldehyde.
Di-(indol-1-yl)methane-3′,3′-dicarbaldehyde (8d)
A mixture of indole-3-carbaldehyde (6a) (0.25 g, 2.00 mmol), freshly crushed potassium hydroxide (0.11 g, 2.00 mmol), and dry DMSO (20 mL) was stirred at room temperature. After 1 h, diiodomethane (0.27 g, 1.00 mmol) was added and the mixture was stirred for a further 1 h. Water was added and the resulting precipitate was filtered off, washed with water, and dried to yield the dialdehyde 8d as a white solid (0.20 g, 66 %). Mp 228–230°C. Found: C 74.4, H 4.7, N 9.2; C19H14N2O2.0.25 H2O requires: C 74.4, H 4.8, N 9.1 %. vmax (KBr)/cm−1 1654, 1612, 1526, 1483, 1454, 1401, 1383, 1371, 1356, 1333, 1319, 1256, 1170, 1128, 784, 746. λmax (CH2Cl2)/nm (ϵ/M−1 cm−1) 240 (34400), 294 (32900). δH (300 MHz, [D6]DMSO) 6.88 (2H, s, CH2), 7.23 (4H, m, H5, H6), 7.87 and 8.07 (4H, 2d, J 8.3, 7.5, H4, H7), 8.67 (2H, s, H2), 9.94 (2H, s, CHO). δC (75 MHz, CDCl3) 57.1 (CH2), 109.3, 120.0, 122.7, 123.8, 125.2, 125.5, 136.2, 136.5 (ArCH and ArC), 184.6 (CHO). m/z (ESI+) 303 ([M + 1]+ (100 %), 287 (10), 275 (10), 174 (10), 158 (95), 146 (10), 130 (10), 118 (10).
1,4-Di-(benzyloxyindol-1-ylmethyl)benzene-3′,3′-dicarbaldehyde (8e)
According to Method A, phosphoryl chloride (0.10 mL, 1.07 mmol), diindolylbenzene 7e (0.26 g, 0.41 mmol), and DMF (7 mL) gave compound 8e as a white solid (0.17 g, 70 %). Mp 210–212°C. Found: C 79.2, H 5.3, N 4.3; C40H32N2O4 requires: C 79.4, H 5.3, N 4.6 %. vmax (KBr)/cm−1 1649, 1619, 1530, 1479, 1467, 1392, 1261, 1228, 1160, 1043, 756. λmax (CH2Cl2)/nm (ϵ/M−1 cm−1) 228 (42100), 256 (54600), 298 (29600). δH (300 MHz, [D6]DMSO) 5.09 (4H, s, CH2N), 5.43 (4H, s, OCH2Ph), 6.93 (2H, dd, J 2.6, 2.3, ArH), 7.23 (4H, s, ArH), 7.29–7.45 (14H, m, ArH), 7.67 (2H, d, J 2.6, ArH), 8.32 (2H, s, ArH), 9.85 (2H, s, CHO). δC (75 MHz, CDCl3) 50.0 (CH2N), 70.1 (OCH2Ph), 104.9, 112.6, 114.2, 127.9, 128.1, 128.7, 141.3 (ArCH), 117.5, 126.0, 132.3, 136.8, 137.7, 155.4 (ArC), 184.9 (CHO). m/z (ESI+) 605 ([M + 1]+ (25 %), 577 (20), 337 (25), 220 (100).
1,4-Di-(bromoindol-1-ylmethyl)benzene-3′,3′-dicarbaldehyde (8f)
According to Method B, the aldehyde 6f (1.12 g, 5.00 mmol), potassium hydroxide (0.28 g, 5.00 mmol), α,α′-dibromo-m-xylene (0.66 g, 2.50 mmol), and DMSO (20 mL) gave the dialdehyde 8f as a yellow solid (1.38 g, 100 %). Mp 282°C (dec.). Found: C 56.3, H 3.5, N 5.0; C26H18Br2N2O2.0.25 H2O requires: C 56.3, H 3.4, N 5.1 %. vmax (KBr)/cm−1 1661, 1532, 1465, 1369, 1368, 1164, 1031. λmax (MeOH)/nm (ϵ/M−1 cm−1) 246 (23300), 301 (26200). δH (300 MHz, [D6]DMSO) 5.46 (4H, s, CH2), 7.22 (4H, s, ArH xylene), 7.34 (2H, dd, J 2.3, 7.5, H6), 7.48 (2H, d, J 9.0, H7), 8.20 (2H, d, J 1.9, H4), 8.40 (2H, s, H2), 9.87 (2H, s, CHO). m/z (ESI+) 552 ([M + 1]+ 81,81Br, 15 %), 550 ([M + 1]+ 79,81Br, 40 %), 548 ([M + 1]+ 79,79Br, 100 %).
Synthesis of 3,3′-Bishydroxymethylindoles
General Method
Sodium borohydride was added to the dialdehyde in a mixture of THF and absolute EtOH and the reaction mixture was stirred at room temperature overnight. The solvent was removed and the residue suspended in 5 % aqueous sodium hydroxide. The resulting precipitate was filtered off, washed with water, and dried to yield the dialcohol.
1,2-Di-(3-hydroxymethylindol-1-ylmethyl)benzene (9a)
The dialdehyde 8a (0.34 g, 0.88 mmol), sodium borohydride (0.33 g, 8.80 mmol), THF (40 mL), and absolute ethanol (20 mL) yielded dialcohol 9a as a yellow solid (0.30 g, 87 %). Product not obtained analytically pure but used directly. Mp 181–182°C. vmax (KBr)/cm−1 3350, 1610, 1465, 1380, 1360, 1320, 1280, 1040, 1010, 980, 750. λmax (MeOH)/nm (ϵ/M−1 cm−1) 221 (82300), 276 (9900), 283 (10600), 293 (9100). δH (300 MHz, [D6]DMSO) 4.86 (4H, s, CH2OH), 5.00 (2H, s, CH2OH), 5.69 (4H, s, CH2N), 6.75 (2H, s, ArH), 7.22–7.49 (6H, m, ArH), 7.52–7.83 (4H, m, ArH), 7.85 (2H, d, J 1.1, ArH). δC (75 MHz, [D6]DMSO) 46.6 (CH2OH), 55.7 (CH2N), 110.4, 119.2, 119.7, 121.8, 126.8, 127.3, 127.4 (ArCH), 116.7, 127.8, 136.0, 136.9 (ArC). m/z (EI) 397 ([M + 1]+ (2 %), 396 M (10), 232 (35), 218 (100), 204 (10), 130 (15), 117 (10), 104 (25), 91 (15), 78 (20).
1,3-Di-(3-hydroxymethylindol-1-ylmethyl)benzene (9b)
The dialdehyde 8b (0.50 g, 1.28 mmol), sodium borohydride (0.48 g, 12.8 mmol), THF (40 mL), and absolute ethanol (20 mL) yielded dialcohol 9b as a yellow solid (0.49 g, 98 %). Product not obtained analytically pure but used directly. Mp 132–133°C. vmax (KBr)/cm−1 3350, 1610, 1540, 1440, 1370, 1340, 1260, 1170, 1120, 1030, 995, 960, 870, 740, 730, 710. λmax (MeOH)/nm (ϵ/M−1 cm−1) 221 (98300), 275 (15500), 283 (15900), 294 (13900). δH (300 MHz, [D6]DMSO) 4.61 (4H, d, J 5.3, CH2OH), 4.76 (2H, t, J 5.3, CH2OH), 5.30 (4H, s, CH2N), 6.96–7.09 (6H, m, ArH), 7.15–7.20 (1H, m, ArH), 7.27–7.36 (6H, m, ArH), 7.57–7.60 (2H, m, ArH). δC (75 MHz, [D6]DMSO) 49.3 (CH2OH), 55.7 (CH2N), 110.4, 119.0, 119.6, 121.7, 126.5, 126.7, 127.5, 129.2 (ArCH), 116.3, 127.2, 136.6, 138.9 (ArC). m/z (EI) 397 ([M + 1]+ (5 %), 396 M (10), 378 (45), 249 (15), 233 (15), 220 (25), 204 (20), 189 (10), 130 (30), 117 (25), 104 (100), 91 (25), 78 (50).
1,4-Di-(3-hydroxymethylindol-1-ylmethyl)benzene (9c)
The dialdehyde 8c (0.24 g, 0.61 mmol), sodium borohydride (0.26 g, 7.0 mmol), THF (40 mL), and absolute ethanol (20 mL) yielded the dialcohol 9c as a pale yellow solid (0.24 g, 99 %). Mp 181–182°C. Found: C 75.1, H 6.3, N 6.6; C26H24N2O2.H2O requires: C 75.3, H 6.3, N 6.8 %. vmax (KBr)/cm−1 3330, 1610, 1545, 1460, 1370, 1330, 1250, 1220, 1160, 1130, 1100, 1050, 1035, 990, 960, 920, 890, 800, 785, 745, 730, 695. λmax (MeOH)/nm (ϵ/M−1 cm−1) 222 (72400), 274 (10400), 284 (10900), 293 (9900). δH (300 MHz, [D6]DMSO) 4.59 (4H, d, J 4.9, CH2OH), 4.76 (2H, t, J 5.3, CH2OH), 5.28 (4H, s, CH2N), 6.96 (2H, t, J 7.2, ArH), 7.04 (2H, t, J 7.3, ArH), 7.11 (4H, s, ArH), 7.30 (2H, s, ArH), 7.36 (2H, d, J 8.3, ArH), 7.56 (2H, d, J 7.9, ArH). δC (75 MHz, [D6]DMSO) 49.7 (CH2N), 57.3 (CH2OH), 109.8, 119.3, 119.8, 122.3, 126.9, 127.4 (ArCH), 115.6, 127.3, 136.8, 136.9 (ArC). m/z (EI) 397 ([M + 1]+ (5 %), 396 M (15), 378 (10), 249 (10), 220 (10), 204 (5), 189 (5), 146 (10), 130 (25), 117 (20), 104 (100), 91 (25).
Di-(3-hydroxymethylindol-1-yl)methane (9d)
The dialdehyde 8d (0.05 g, 0.16 mmol), sodium borohydride (0.06 g, 1.65 mmol), THF (10 mL), and absolute ethanol (10 mL) yielded the dialcohol 9d as a white solid (0.05 g, 99 %). Mp 185–186°C. Found: C 74.0, H 6.0, N 9.1; C19H18N2O2.0.125H2O requires: C 74.0, H 6.0, N 9.1 %. vmax (KBr)/cm−1 3353, 3274, 1617, 1561, 1481, 1467, 1455, 1327, 1315, 1166, 1128, 1100, 999, 979, 743, 734. λmax (CH2Cl2)/nm (ϵ/M−1 cm−1) 230 (50400), 273 (19800). δH (300 MHz, [D6]DMSO) 4.57 (4H, d, J 5.0, CH2OH), 4.80 (2H, t, J 5.0, CH2OH), 6.55 (2H, s, CH2 bridging), 7.02 and 7.16 (4H, 2t, J 8.0, H5, H6), 7.50 (2H, s, H2), 7.54 and 7.78 (4H, 2d, J 8.0, H4, H7). δC (75 MHz, [D6]DMSO) 55.3 (CH2OH), 55.6 (CH2 bridging), 110.6, 117.3, 119.6, 119.7, 122.2, 126.7, 127.6, 136.3 (ArCH and ArC). m/z (ESI) 303 ([M – 3]+ (10 %), 287 (20), 273 (15), 174 (65), 160 (45), 144 (15), 130 (100).
1,4-Di-(5-benzyloxy-3-hydroxymethylindol-1-ylmethyl)benzene (9e)
The dialdehyde 8e (1.10 g, 1.82 mmol), sodium borohydride (0.69 g, 18.2 mmol), THF (40 mL), and absolute ethanol (20 mL) yielded the dialcohol 9e as a white solid (0.92 g, 83 %). Product not obtained analytically pure but used directly. Mp 135–137°C. vmax (KBr)/cm−1 3409, 2923, 2866, 1645, 1620, 1587, 1529, 1484, 1454, 1395, 1263, 1223, 1200, 1028. λmax (CH2Cl2)/nm (ϵ/M−1 cm−1) 227 (58000), 273 (22000). δH (300 MHz, [D6]DMSO) 4.55 (4H, s, CH2OH), 4.69 (2H, bs, CH2OH), 5.04 (4H, s, CH2N), 5.23 (4H, s, OCH2Ph), 6.77 (2H, dd, J 2.6, 2.3, ArH), 7.09 (2H, s, ArH), 7.17 (2H, d, J 2.3 Hz ArH), 7.24–7.45 (14H, m, ArH). δC (75 MHz, [D6]DMSO) 49.1 (CH2N), 55.6 (CH2OH), 70.3 (OCH2Ph), 103.2, 111.1, 112.2, 115.9, 127.6, 127.9, 128.0, 128.7, 132.0, 137.9, 138.1, 152.7 (ArCH and ArC). m/z (ESI) [M + Na]+ 631.2574. C40H36N2NaO4 requires 631.2568.
1,4-Di-(5-bromo-3-hydroxymethylindol-1-ylmethyl)benzene (9f)
The dialdehyde 8f (1.26 g, 2.30 mmol), sodium borohydride (0.86 g, 23.0 mmol), THF (40 mL), and absolute ethanol (20 mL) yielded the dialcohol 9f as a yellow solid (1.22 g, 96 %). Mp 214–215°C. Found: C 56.6, H 4.2, N 4.9; C26H22Br2N2O2 requires: C 56.3, H 4.0, N 5.1 %. vmax (KBr)/cm−1 3424, 2923, 2877, 1469, 1241, 1910, 1038. δH (300 MHz, [D6]DMSO) 4.56 (4H, s, CH2OH), 4.83 (2H, bs, CH2OH), 5.28 (4H, s, CH2N), 7.10 (4H, s, ArH), 7.16 (2H, dd, J 1.7, H6), 7.34–7.37 (4H, m, H2, H7), 7.75 (2H, d, J 1.9, H4), 7.36 (2H, d, J 8.3, ArH), 7.56 (2H, d, J 7.9, ArH). δC (75 MHz, [D6]DMSO) 49.1 (CH2N), 55.4 (CH2OH), 111.8, 112.5, 116.1, 122.0, 124.1, 127.7, 128.7, 129.3, 135.3, 137.6 (ArCH and ArC). m/z (ESI) 553 ([M – 3]+ 81,81Br, 70 %), 551 ([M – 3]+ 79,81Br, 100 %), 548 ([M – 3]+ 79,79Br, 50 %).
10,19,35,44-Tetraazaundecacyclo[42,6,1,13,10,119,26,128,35,04,9,012,17,020,25,029,34,037,42,045,50]tetrapentaconta-1(51),3(52),4,6,8,12,14,16, 20,22,24,26(53),28(54),29,31,33,37,39,41,45,47,49-docosaene (10a)
The dialcohol 9a (0.26 g, 0.66 mmol) in anhydrous acetone (100 mL) was treated with a catalytic amount of p-toluenesulfonic acid monohydrate, and the mixture stirred for 1 h at room temperature. The solvent was removed under reduced pressure and the crude product was purified using ‘dry-column’ flash chromatography with dichloromethane eluant, followed by trituration of the product with dichloromethane to yield the macrocycle 10a as a white solid (0.054 g, 62 %). Mp 296°C. Found: C 84.8, H 5.8, N 7.8; C50H40N4.0.5H2O requires: C 85.1, H 5.9 N 7.9 %. vmax (KBr)/cm−1 3060, 1460, 1350, 1250, 1205, 1170, 1020, 910, 740. λmax (CH2Cl2)/nm (ϵ/M−1 cm−1) 230 (124000), 291 (33500). Sample insoluble – unable to obtain 1H and 13C NMR measurement. m/z (MALDI) 696 (M+).
10,17,33,40-Tetraazaundecacyclo[38,6,1,212,15,235,38,13,10,117,24,126,33,04,9,018,23,027,32,041,46]tetrapentaconta-1(47),3(48),4,6,8,12,14,18,20,22,24(51),26(52),27,29,31,35,37,41,43,45,49,53-docosaene (10c)
To a stirred solution of the dialcohol 9c (0.20 g, 0.50 mmol) in anhydrous acetone (100 mL), a catalytic amount of p-toluenesulfonic acid monohydrate was added, and the mixture stirred for 1 h at room temperature. The solvent was removed under reduced pressure and the crude product was chromatographed with dichloromethane eluant to afford the macrocycle 10c as a white solid (0.12 g, 60 %). Mp 297–299°C. Found: C 84.5, H 5.7, N 7.7; C50H40N4.0.75H2O requires: C 84.5, H 5.9, N 7.9 %. vmax (KBr)/cm−1 1455, 1450, 1370, 1350, 1330, 1160, 1010, 915, 730. λmax (CH2Cl2)/nm (ϵ/M−1 cm−1) 231 (112000), 292 (25900). δH (300 MHz, CDCl3) 4.20 (4H, s, CH2), 5.16 (8H, s, CH2N), 6.71 (4H, s, H2 indolyl), 6.93 (8H, s, ArH xylyl), 7.08 (4H, t, J 7.4, ArH), 7.18 (4H, t, J 7.5, ArH), 7.27 (4H, d, J 8.7, ArH), 7.62 (4H, d, J 7.5, ArH). δC (75 MHz, CDCl3) 29.8 (CH2), 49.3 (CH2N), 109.3, 119.0, 119.4, 121.7, 126.3, 127.1 (ArCH), 115.1, 128.2, 136.7, 137.2 (ArC). m/z (MALDI) 696 (M+).
6,21,29,44-Tetrabenzyloxy-10,17,33,40-tetraazaundecacyclo[38,6,1,212,15,235,38,13,10,117,24,126,33,04,9,018,23,027,32,041,46]tetrapentaconta-1(47),3(48),4,6,8,12,14,18,20,22,24(51),26(52),27,29,31,35,37,41,43,45,49,53-docosaene (10e)
To a stirred solution of the dialcohol 9e (0.18 g, 0.30 mmol) in anhydrous acetone (200 mL), a catalytic amount of p-toluenesulfonic acid monohydrate was added, and the mixture was stirred for 1 h at room temperature. The solvent was removed under reduced pressure and the crude product was chromatographed with dichloromethane eluant to afford the macrocycle 10e as a white solid (0.10 g, 60 %). Mp 237–239°C. Found: C 82.6, H 6.0, N 4.9; C78H64N4O4.0.75H2O requires: C 82.6, H 6.0, N 4.9 %. vmax (KBr)/cm−1 2921, 2850, 1620, 1579, 1484, 1451, 1420, 1380, 1275, 1227, 1199, 1028. λmax (CH2Cl2)/nm (ϵ/M−1 cm−1) 232 (135900), 285 (43700), 308 (33000). δH (300 MHz, CDCl3) 4.10 (4H, s, CH2), 5.05 (8H, s, CH2N), 5.11 (8H, s, OCH2Ph), 6.69 (4H, s, ArH), 6.92 (8H, s, ArH), 7.15–7.18 (8H, m, ArH), 7.29–7.39 (16H, m, ArH), 7.43–7.45 (8H, m, ArH). δC (75 MHz, CDCl3) 22.6 (CH2), 49.4 (CH2N), 70.8 (OCH2Ph), 77.1, 102.6, 110.0, 112.4, 116.5, 126.8, 126.9, 127.5, 127.6, 128.3, 128.4 (ArCH), 114.3, 132.0, 137.2, 137.6, 152.9, 172.6 (ArC). m/z (MALDI) 1119 ([M – 2]+, 2 %).
6,21,29,44-Tetrabromo-10,17,33,40-tetraazaundecacyclo[38,6,1,212,15,235,38,13,10,117,24,126,33,04,9,018,23,027,32,041,46]tetrapentaconta-1(47),3(48),4,6,8,12,14,18,20,22,24(51),26(52),27,29,31,35,37,41,43,45,49,53-docosaene (10f)
To a stirred solution of the dialcohol 9f (0.28 g, 0.50 mmol) in anhydrous acetone (120 mL), a catalytic amount of p-toluenesulfonic acid monohydrate was added, and the mixture was stirred for 1 h at room temperature. The solvent was removed under reduced pressure and the crude product was chromatographed with dichloromethane eluant to afford the macrocycle 10f as a white solid (0.13 g, 50 %). Mp >300°C. Found: C 59.4, H 3.7, N 5.3; C50H36Br4N4 requires: C 59.3, H 3.6, N 5.5 %. vmax (KBr)/cm−1 1511, 1466, 1436, 1420, 1373, 1350, 1295, 1198, 1164, 1052, 865, 820, 790. λmax (CH2Cl2)/nm (ϵ/M−1 cm−1) 235 (126300), 297 (22300). δH (300 MHz, CDCl3) 4.08 (4H, s, CH2), 5.14 (8H, s, CH2N), 6.67 (4H, s, H2 indolyl), 6.89 and 6.91 (12H, 2s, H7 and ArH), 7.13 (4H, d, J 8.7, H6 indolyl), 7.71 (4H, d, J 1.5, H4 indolyl). δC (75 MHz, [D6]DMSO) 21.7 (CH2), 49.0 (CH2N), 112.4, 124.0, 127.3, 127.6, 128.9 (ArCH), 111.6, 113.5, 129.7, 135.3, 137.6 (ArC). m/z (MALDI) 1015 ([M + 1]+ 79,81,81,81Br, 2 %), 1013 [M + 1]+ 79,79,81,81Br, 5 %), 1011 ([M + 1]+ 79,79,79,81Br, 4 %), 1009 ([M + 1]+ 79,79,79,79Br, 3 %).
Ethyleneglycol-O,O′-Bis(2-(3-formylindol-1-ylmethylbenzene)) (12)
A mixture of indole-3-carbaldehyde 6a (0.15 g, 1.03 mmol) and freshly crushed potassium hydroxide (0.49 g, 8.73 mmol) in dry dimethyl sulfoxide (20 mL) was stirred for 1 h. 1,2-Bis(2-bromomethylphenoxy) ethane 11 (0.21 g, 0.52 mmol) was added and the mixture was stirred for an additional 2 h. Water was added and the yellowish product was filtered off, washed with water, and dried. The crude product was purified using gravity column chromatography with ethyl acetate/light petroleum (1 : 1) eluant to yield the dialdehyde 12 as a yellow solid (0.23 g, 85 %). Mp 177–178°C. Found: C 77.1, H 5.4, N 5.5; C34H28N2O4 requires: C 77.3, H 5.3, N 5.3 %. vmax (KBr)/cm−1 1640, 1450, 1370, 1240, 1150, 1115, 1030, 940, 920, 735. λmax (MeOH)/nm (ϵ/M−1 cm−1) 247 (30800), 303 (36100). δH (300 MHz, CDCl3) 4.11 (4H, s, CH2), 5.10 (4H, s, CH2O), 6.81 (2H, d, J 8.2, ArH), 6.97 (2H, t, ArH), 7.07 (2H, t, ArH), 7.13–7.22 (6H, m, ArH), 7.32 (2H, t, ArH), 7.53 (2H, s, H2), 8.24 (2H, d, J 7.7, ArH), 9.77 (2H, s, CHO). δC (75 MHz, CDCl3) 46.5 (CH2), 66.4 (CH2O), 110.3, 111.5, 121.4, 121.9, 122.8, 123.8, 130.0, 130.1, 138.8 (ArCH), 118.1, 123.7, 125.2, 137.6, 156.4 (ArC), 184.4 (CHO). m/z (EI) 529 ([M + 1]+, 2 %), 528 (M, 4), 500 (6), 499 (3), 234 (5), 158 (7), 144 (20), 133 (40), 107 (65), 91 (100), 78 (60), 77 (55).
Ethyleneglycol-O,O′-Bis(2-(3-hydroxymethylindol-1-ylmethylbenzene)) (13)
A mixture of dialdehyde 12 (0.10 g, 0.19 mmol) and sodium borohydride (0.10 g, 2.64 mmol) in absolute ethanol was stirred for 2h. The solvent was then evaporated almost to dryness, and the white residue was suspended in 5 % aqueous sodium hydroxide and the mixture cooled in ice. The product was filtered off, washed with water, and dried to give the dialcohol 13 as a white solid (0.09 g, 87 %). Mp 97–98°C. vmax (KBr)/cm−1 3320, 1610, 1450, 1390, 1240, 1180, 1130, 1040, 960, 750. λmax (CH2Cl2)/nm (ϵ/M−1 cm−1) 222 (63900), 281 (9100). δH (300 MHz, CDCl3) 4.29, 4.74, 5.16 (6H, 3s, CH2), 6.86–7.30 (16H, m, H2 and ArH), 7.68 (2H, d, J 6.7, ArH). δC (75 MHz, CDCl3) 45.1, 57.2, 66.7 (CH2), 109.9, 111.5, 119.1, 119.5, 121.3, 122.0, 127.1, 129.0, 129.1 (ArCH), 115.1, 126.0, 127.0, 137.0, 156.1 (ArC). m/z (MALDI) 555.75, [M + Na]+. m/z (HRMS ESI) [M + Na]+ 555.2253. C34H32N2NaO4 requires 555.2254.
10,29-Diazaheptacyclo[27,6,1,13,10,04,9,012,17,030,35]heptatriaconta-1(36),3(37),4,6,8,12,14,16,22,24,26,30,32,34-tetradecaene (14)
The dialcohol 13 (0.09 g, 0.16 mmol) in dichloromethane was treated with a catalytic amount of p-toluenesulfonic acid and stirred for 1 h. The pink solution was evaporated and the crude product was purified using gravity column chromatography with dichloromethane/light petroleum (1 : 1) eluant to afford the macrocycle 14 as a white solid (0.05 g, 68 %). Mp 228–229°C. vmax (KBr)/cm−1 1590, 1455, 1380, 1330, 1250, 1160, 1120, 1070, 1050, 960, 745. λmax (CH2Cl2)/nm (ϵ/M−1 cm−1) 229 (103200), 281 (27000), 293 (23500). δH (300 MHz, CDCl3) 4.14 (2H, s, CH2), 4.26 (4H, s, CH2), 5.21 (4H, s, CH2), 6.87–7.59 (18H, m, ArH). δC (75 MHz, CDCl3) 20.1, 45.7, 67.9 (CH2), 109.4, 112.6, 118.6, 118.9, 121.2, 121.3, 126.9, 129.7, 131.8 (ArCH), 113.7, 125.9, 128.4, 136.4, 157.4 (ArC). m/z (MALDI) 484.60, M+. m/z (HRMS ESI) [M + H]+ 485.2224. C33H29N2O2 requires 485.2229.
4-(3-(Indol-1-ylpropoxy)-3-methoxybenzene)methanol (16a)
Indole 5a (0.21 g, 1.79 mmol) and freshly crushed potassium hydroxide (0.42 g, 7.49 mmol) were stirred for 1 h in dry dimethyl sulfoxide. 4-(3-Iodopropoxy)-3-methoxybenzenemethanol[26] 15 (1.20 g, 3.72 mmol) was added and the mixture was stirred for 1 h at room temperature. The mixture was diluted with water and extracted with ethyl acetate. The combined extracts were washed with brine, dried over magnesium sulfate, and concentrated. The crude product was purified using gravity column chromatography with ethyl acetate/light petroleum (1 : 1) eluant to afford the alcohol 16a as a colourless oil (0.47 g, 84 %). vmax (KBr)/cm−1 3400, 1600, 1450, 1410, 1370, 1260, 1180, 1150, 1130, 1030, 850, 800, 740. λmax (CH2Cl2)/nm (ϵ/M−1 cm−1) 231 (41600), 283 (14800), 326. δH (300 MHz, CDCl3) 2.33 (2H, quin, CH2), 3.89–3.93 (5H, m, CH2 and OCH3), 4.42 (2H, t, CH2), 4.61 (2H, s, CH2OH), 6.52 (1H, d, J 2.6, ArH), 6.76 (d, J 8.2, ArH), 6.84 (1H, d, J 8.2, ArH), 6.98 (1H, s, ArH), 7.13–7.26 (3H, m, ArH), 7.43 (1H, d, J 8.2, ArH), 7.68 (1H, d, J 7.7, ArH). δC (75 MHz, CDCl3) 29.7, 42.5, 64.9, 65.6 (CH2), 55.8 (OCH3), 101.0, 109.3, 110.9, 113.5, 119.2, 119.3, 120.8, 121.3, 128.1 (ArCH), 128.5, 134.2, 135.8, 147.5, 149.6 (ArC). m/z (EI) 312 ([M + 1]+, 25 %), 311 (M, 90), 174 (5), 158 (90), 130 (100), 117 (25), 77 (30).
4-(3-(5′-Benzyloxy-1′-indolylpropoxy)-3-methoxybenzene)methanol (16e)
5-Benzyloxyindole 5e (0.65 g, 2.91 mmol) and freshly crushed potassium hydroxide (0.66 g, 11.8 mmol) were stirred in dry dimethyl sulfoxide (40 mL) for 1 h at room temperature. 4-(3-Iodopropoxy)-3-methoxybenzenemethanol[26] 15 (1.88 g, 5.84 mmol) was added and the mixture was stirred overnight. The mixture was diluted with water and extracted with dichloromethane and again with ethyl acetate. The combined extracts were washed several times with brine, dried over magnesium sulfate, and concentrated under reduced pressure. The crude product was purified using gravity column chromatography with ethyl acetate/light petroleum (1 : 1) eluant to afford the alcohol 16e as a colourless oil, which solidified after storing in the refrigerator to become a white solid (1.02 g, 84 %). Mp 92–93°C. Found: C 74.3, H 6.6, N 3.2; C26H27NO4 requires: C 74.8, 6.5, 3.4 %. vmax (KBr)/cm−1 3450, 1590, 1480, 1450, 1370, 1260, 1230, 1200, 1180, 1150, 1040, 1000, 840, 820, 790, 750, 700. λmax (MeOH)/nm (ϵ/M−1 cm−1) 225 (40000), 277 (10100). δH (300 MHz, CDCl3) 2.30 (2H, quin, CH2), 3.89–3.93 (5H, m, CH2 and OCH3), 4.37 (2H, t, CH2), 4.63 (2H, d, CH2OH), 5.10 (2H, s, CH2Ph), 6.38 (1H, d, J 2.6, ArH), 6.74–6.97 (4H, m, ArH), 7.08 (1H, d, J 3.1, ArH), 7.17 (1H, d, J 2.0, ArH), 7.26–7.49 (6H, m, ArH). δC (75 MHz, CDCl3) 29.9, 42.9, 65.3, 65.7, 71.0 (CH2), 55.9 (OCH3), 100.8, 104.3, 110.1, 111.0, 112.6, 113.7, 119.5, 127.5, 127.7, 128.5, 128.7 (ArCH), 128.9, 131.5, 134.3, 137.8, 147.7, 149.8, 153.2 (ArC). m/z (EI) 418 ([M + 1]+, 10 %), 417 (30), 400 (17), 326 (30), 236 (10), 174 (50), 146 (100), 117 (35), 91 (95), 77 (15).
Supplementary Material
1H NMR and 13C NMR spectra verifying the identity of new compounds are available on the Journal’s website.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgements
We thank the University of New South Wales for use of their NMR and bioanalytical mass spectrometry facilities and support in this project. We would also like to thank the Australian Research Council (ARC) for their financial support and the Australian Government for the Australian Postgraduate Award (APA).
References
[1] D. StC. Black, M. C. Bowyer, N. Kumar, P. S. R. Mitchell, J. Chem. Soc. Chem. Commun. 1993, 819.| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3sXmslWisLc%3D&md5=4cd4eed4f79cffb747e001d15c7b3b09CAS |
[2] D. StC. Black, D. C. Craig, N. Kumar, Tetrahedron Lett. 1995, 36, 8075.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXovFyit7k%3D&md5=513a7dfd73609865162563857d4740caCAS |
[3] D. E. Ames, R. E. Bowman, D. D. Evans, W. A. Jones, J. Chem. Soc. 1956, 1984.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaG2sXisFCltA%3D%3D&md5=c9ba687cd2f18bcb3f284b74e45b7449CAS |
[4] E. Leete, J. Am. Chem. Soc. 1959, 81, 6023.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaF3cXjsVajtQ%3D%3D&md5=54fb7fa1daa4c285cce52838370e4062CAS |
[5] A. R. Mattocks, J. Chem. Soc., Perkin Trans. 1 1978, 896.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE1MXmtFKq&md5=9165dd9c50ac54a52472be29f53ce3caCAS |
[6] J. Bergman, S. Högberg, O. J. Lindström, Tetrahedron 1970, 26, 3347.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE3cXkslKqtbc%3D&md5=b2fca5c4b13e391601d96b6c353b9895CAS |
[7] M. Santoso, K. Somphol, N. Kumar, D. StC. Black, Tetrahedron 2009, 65, 5977.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXotFCksL4%3D&md5=d6426f94f3922ffdca63116ad8647802CAS |
[8] J. Bloxham, C. J. Moody, A. M. Z. Slawin, Tetrahedron 2002, 58, 3709.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XjtVeksL4%3D&md5=a36997c56f1c881cdc82b01c969faf0cCAS |
[9] S. Muthusamy, C. Gunanathan, E. Suresh, Tetrahedron 2004, 60, 7885.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXmsVajtrk%3D&md5=7d39f8664b059b4f624ffcaad4f71051CAS |
[10] C. Gonzalez, R. Greenhouse, Heterocycles 1985, 23, 1127.
| Crossref | GoogleScholarGoogle Scholar |
[11] P. Rajakumar, M. Gayatri Swaroop, S. Jayavelu, K. Murugesan, Tetrahedron 2006, 62, 12041.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XhtFyqurbM&md5=2302ddb6c0d354e4c73ff882d1e9924fCAS |
[12] P. Rajakumar, M. Gayatri Swaroop, Tetrahedron Lett. 2004, 45, 6165.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXlvVSrtb4%3D&md5=05506ce04ca35536bfd9efb3d10151d9CAS |
[13] M. R. Buemi, L. De Luca, A. Chimirri, S. Ferro, R. Gitto, J. Alvarez-Builla, R. Alajarin, Bioorg. Med. Chem. 2013, 21, 4575.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXps12jtb8%3D&md5=2f2de8fd2cfedf5cc012d100e467e8c8CAS |
[14] I. W. J. Still, J. R. Strautmanis, Tetrahedron Lett. 1989, 30, 1041.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL1MXmtFCku78%3D&md5=7979fac93d2e6531a17ccab3066fc9f7CAS |
[15] D. StC. Black, M. A. Horsham, M. Rose, Tetrahedron 1995, 51, 4819.
| Crossref | GoogleScholarGoogle Scholar |
[16] E. Leete, L. Marion, Can. J. Chem. 1953, 31, 775.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaG2cXls1KgtA%3D%3D&md5=31ab0a0386c2445a565b3c844ba50667CAS |
[17] J. Thesing, Chem. Ber. 1954, 87, 692.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaG2MXkvFCksA%3D%3D&md5=3736ae3f67c538aa8bd7bccb2ea58d86CAS |
[18] E. Leete, J. Am. Chem. Soc. 1959, 81, 6023.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaF3cXjsVajtQ%3D%3D&md5=54fb7fa1daa4c285cce52838370e4062CAS |
[19] W.-R. Chao, D. Yean, K. Amin, C. Green, L. Jong, J. Med. Chem. 2007, 50, 3412.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXns1Ohtr4%3D&md5=d84ddd9827c3d4a0cae2f7a6b7992e1fCAS |
[20] M. S. C. Pedras, V. K. Sarma-Mamillapalle, Bioorg. Med. Chem. 2012, 20, 3991.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XotVOrtL0%3D&md5=875bb2f63570e8d68cf51c142377b94dCAS |
[21] M. Santoso, K. Somphol, N. Kumar, D. StC. Black, Tetrahedron 2009, 65, 5977.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXotFCksL4%3D&md5=d6426f94f3922ffdca63116ad8647802CAS |
[22] M. Bingul, B. B. Cheung, N. Kumar, D. StC. Black, Tetrahedron 2014, 70, 7363.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXhtFKqu7%2FI&md5=c60d6743d6f2f1f69d998f2fe9b20aaeCAS |
[23] D. StC. Black, D. C. Craig, R. Rezaie, Chem. Commun. 2002, 810.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XislSgsLk%3D&md5=f1975d08827cba0a08c09fb1fc70195fCAS |
[24] K. Somphol, R. Chen, M. Bhadbhade, N. Kumar, D. StC. Black, Tetrahedron Lett. 2013, 54, 24.
[25] R. Chen, K. Somphol, M. Bhadbhade, N. Kumar, D. StC. Black, Synlett 2013, 1497.
| Crossref | GoogleScholarGoogle Scholar |
[26] J. Canceill, A. Collet, G. Gottarelli, P. Palmieri, J. Am. Chem. Soc. 1987, 109, 6454.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2sXlvFOiu7s%3D&md5=c4151e7aaa7b8768babc74a93de19057CAS |
* David StClair Black was awarded the Australian Academy of Science David Craig Medal for 2017.