

Reactions of Trivalent Iodine Reagents with Classic Iridium and Rhodium Complexes*
Mohammad Albayer A and Jason L. Dutton A BA Department of Chemistry and Physics, La Trobe Institute for Molecular Science, La Trobe University, Melbourne, Vic. 3086, Australia.
B Corresponding author. Email: j.dutton@latrobe.edu.au
Australian Journal of Chemistry 70(11) 1180-1187 https://doi.org/10.1071/CH17173
Submitted: 27 March 2017 Accepted: 7 July 2017 Published: 9 August 2017
Abstract
In this work, the reactions of iodine(iii) reagents (PhI(L)2: L = pyridine, acetate (OAc−), triflate (OTf−)) with iridium(i) and rhodium(i) complexes (Vaskas’s compound, Wilkinson’s catalyst, and bis[bis(diphenylphosphino)ethane]rhodium(i) triflate) are reported. In all cases, the reactions resulted in two-electron oxidation of the metal complexes. Mixtures of products were observed in the reactions of Iiii reagents with Vaska’s compound and Wilkinson’s catalyst via ligand exchange and anion scrambling. In the case of reacting Iiii reagents with chelating ligand-containing bis[bis(diphenylphosphino)ethane]rhodium(i) triflate, no scrambling was observed.
Introduction
Iodine(iii) reagents with a PhI(L)2 motif (L = –Cl, –OAc, pyridine) have attracted much attention as stoichiometric oxidants owing to their ease of handling, low toxicity, and ability to act as oxidizing reagents under mild conditions.[1] They have become a popular way to interrogate higher-oxidation-state late transition metal complexes.[2] For example, these reagents have been successfully used in isolating or accessing higher oxidation states of platinum group metals.[3–10] The Sanford group has demonstrated the use of these iodine reagents in investigating C–H bond activation at PdIV centres. The oxidation of compound 1 by either PhICl2 or PhI(OAc)2 resulted in the isolation and characterization of rare PdIV organometallic complexes 1Cl and 1OAc (Scheme 1). At elevated temperatures, these compounds undergo reductive elimination, with C–C, C–Cl, and C–O bond-forming reactions.[3,11]

|
Our group have been using dicationic Iiii reagents [PhI(L)2][OTf]2 (L = pyridine, 4-dimethylaminopyridine) to explore the chemistry of the II–IV redox couple for Pd and Pt and the I–III couple for Au (Schemes 2, 3).[6,12] These reagents were first synthesized by Weiss in 1994 and later investigated by Zhdankin.[13,14] These dicationic Iiii reagents typically simultaneously oxidize the transition metal and deliver the neutral ligands, allowing the isolation of charged metal complexes. The oxidation of compound 1 by [PhI(L)2][OTf]2 (2R) resulted in an unexpected outcome, as shown in Scheme 2. The reaction is proposed to proceed through oxidation of Pdii to give an unobserved PdIV intermediate, which undergoes reductive elimination, resulting in C–C bond formation by linking two 2-phenylpyridine ligands. Ligand redistribution then gives the observed products 3 and 4.[6]

|
Our group also reported a new class of tri-cationic gold(iii) complexes using the dicationic Iiii reagents, as shown in Scheme 3,[12] where the reaction of compound 1 with 2R resulted in simultaneously oxidizing Aui to Auiii and delivering pyridine ligands to form compound 6.

|
Ritter has reported the use of the dicationic Iiii reagent 2CN in the isolation of a PdIV complex (Scheme 4). The oxidation of compound 7 using 2CN resulted in the formation of PdIV with a single ligand transfer to give compound 8. This complex was then used in the formation of a late-stage fluorinating agent via displacement of pyridine with 18F− to generate complexes that have potential to be used in positron emission tomography (PET) imaging.[15]

|
Our group has been specifically avoiding phosphine-containing metal complexes in this chemistry because it was observed that phosphine ligands react with Iiii reagents, resulting in oxidation of the phosphine.[16] In the present study, we explored the reactions of selected phosphine-containing Iri and Rhi complexes with Iiii reagents containing anionic and neutral ligands (Fig. 1) and examined if the iodine(iii) reagents would be compatible with these phosphine-containing iridium and rhodium complexes. A very recent review on the area indicated very little work has been done examining the reactions of iridium and rhodium complexes with Iiii.[17]
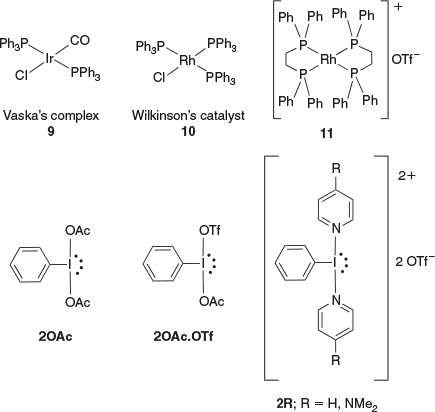
|
Experimental
Materials and Methods
NMR solvents were purchased from Cambridge Isotope Laboratories and dried by stirring for 3 days over CaH2, which was distilled and stored over 3-Å molecular sieves in a glovebox. CH2Cl2, CH3CN, n-hexane, toluene, and chloroform, which were purchased from Caledon Laboratories, were dried using an Innovative Technologies solvent purification system. All solvents were stored over 3-Å molecular sieves in a nitrogen-filled glovebox. All remaining reagents were ordered from Sigma–Aldrich and used as received. Ir(PPh3)2(CO)Cl,[18] Rh(PPh3)3Cl,[19] [Rh(dppe)2][Cl] (dppe = bis(diphenyl)phosphinoethane),[20] [PhI(Pyr)2][OTf]2 (pyr = pyridine), and [PhI(4-DMAP)2][OTf]2 (DMAP = 4-dimethylaminopyridine)[13] were prepared following published procedures. The metathesis to produce [Rh(dppe)2][OTf] was carried out by the addition of a 1 : 1 stochiometric ratio of TMS-OTf (OTf = triflate) to a CH2Cl2 solution of [Rh(dppe)2][Cl] followed by solvent removal at reduced pressure.
CCDC numbers 1539214–1539218 at the Cambridge Crystallographic Data Centre contain the crystallographic data for this manuscript in .cif format.
Reaction of 2OAc with 9
A solution of 2OAc (42 mg, 0.13 mmol) in 2 mL CDCl3 was added dropwise to a solution of 9 (100 mg, 0.13 mmol) in 5 mL CDCl3 and stirred for 3 h at room temperature. A colour change from bright yellow to light yellow was observed within 10 min. The solvent was reduced to half under reduced pressure, followed by addition of 10 mL n-hexane, which resulted in precipitation of a pale yellow solid. The solid was filtered, washed with n-hexane (2 × 10 mL), and dried under reduced pressure. δP (162 MHz, CDCl3) 2.0 (s), −1.6 (s), −9.6 (s), −10.9 (s), −12.6, −13.7 (s). See the Supplementary Material for the 1H NMR. m/z (electrospray ionization mass spectrometry (ESI-MS)) ([M]n+) 876.1 [Ir(PPh3)2(CO)(Cl)2(OAc)], 894.1 [Ir(PPh3)2(OAc)3].
Reaction of 2R with 9
A solution of 2R (0.13 mmol) in 5 mL CDCl3 was added dropwise to a solution of 9 (100 mg, 0.13 mmol) in 5 mL CDCl3 and stirred for 3 h at room temperature. A colour change from bright yellow to light yellow was observed within 30 min. The solvent was reduced to half under reduced pressure, followed by addition of 10 mL n-hexane, which resulted in precipitation of a pale yellow solid. The solid was filtered, washed with n-hexane (2 × 10 mL), and dried under reduced pressure. R = NMe2: δP (162 MHz, CDCl3) −2.4 (s), −8.4 (s), −15.4 (s). See the Supplementary Material for the 1H NMR. m/z (ESI-MS) ([M]n+)+ 797.1 [Ir(PPh3)(CO)(Cl)2(DMAP)2]+, 937.0 [Ir(PPh3)2(CO)(Cl)2(DMAP)]+. R = H: δP (162 MHz, CDCl3) 65.9 (s), 26.9 (s), −3.9 (s), −14.6 (s), −15.2 (s), −16.4 (s), −23.1 (s). m/z (ESI-MS) ([M]n+) 711.1 [Ir(PPh3)(CO)(Cl)2(Pyr)2]+, 894.1 [Ir(PPh3)2(CO)(Cl)2(Pyr)]+.
Reaction of 2OAc.OTf with 9
A mixture of 2OAc (20 mg, 0.064 mmol) and TMS-OTf (23 µL, 0.128 mmol) in 2 mL CDCl3 was added dropwise to a solution of 9 (50 mg, 0.064 mmol) in 2 mL CDCl3. A colour change from bright yellow to brown was observed in 10 min. An aliquot was removed for NMR and mass spectrometry analysis. δP (162 MHz, CDCl3) 14.4 (s), 8.0 (s), 5.3 (s), 2.6 (s), 0.4 (s), −3.6 (s), −8.1 (s), −15.6 (s), −21.4 (s). m/z (ESI-MS) ([M]n+) 745.2 [Ir(PPh3)2CO]+, 753.2 [Ir(PPh3)2Cl]+, 786.1 [Ir(PPh3)2Cl2]+, 803.1 [Ir(PPh3)2CO(OAc)]+, 839.1 [Ir(PPh3)2CO(OAc)Cl]+.
Reaction of 2OAc with 10
A solution of 2OAc (35 mg, 0.11 mmol) in 2 mL CDCl3 was added dropwise to a solution of 10 (100 mg, 0.11 mmol) in 5 mL CDCl3 and stirred for 3 h at room temperature. A colour change from burgundy to light red was observed. The solvent was reduced to half under reduced pressure, followed by addition of 10 mL n-hexane, which resulted in precipitation of a light orange solid. The solid was filtered, washed with n-hexane (2 × 10 mL), and dried under reduced pressure. δP (162 MHz, CDCl3) 24.5 (d, J 122). δH (400 MHz, CDCl3) 7.43–7.38 (m, 6H), 7.37–7.32 (m, 12H), 7.20–7.17 (m, 12H), 2.08 (s, 3H). δC (100 MHz, CDCl3) 191.24, 135.61, 130.94, 129.28, 127.71, 25.70. m/z (ESI-MS) ([M]n+) 721.1 [Rh(PPh3)2(Cl)(OAc)]+.
Reaction of 2R with 10
A solution of 2R (0.11 mmol) in 5 mL CDCl3 was added dropwise to a solution of 10 (100 mg, 0.11 mmol) in 5 mL CDCl3 and stirred for 3 h at room temperature. A colour change from burgundy to light red-brown was observed. The solvent was reduced to half under reduced pressure, followed by addition of 10 mL n-hexane, which resulted in precipitation of a light brown solid. The solid was filtered, washed with n-hexane (2 × 10 mL), and dried under reduced pressure. R = NMe2: δP (162 MHz, CH2Cl2) 28.2 (s), 14.1 (d, J 88), 11.9 (d, J 88). m/z (ESI-MS) ([M]n+) 803.0 [Rh(PPh3)(Cl)2(DMAP)3]+, 942.2 [Rh(PPh3)2(Cl)2(DMAP)2]+. R = H: δP (162 MHz, CH2Cl2) 65.0 (s), 14.0 (d, J 84), 9.7 (d, J 104). m/z (ESI-MS) ([M]n+) 673.6 [Rh(PPh3)(Cl)2(Pyr)3]+, 854.9 [Rh(PPh3)2(Cl)2(Pyr)2]+.
Reaction of 2OAc.OTf with 10
A mixture of 2OAc (17 mg, 0.054 mmol) and TMS-OTf (20 µL, 0.108 mmol) in 2 mL CDCl3 was added dropwise to a solution of 10 (50 mg, 0.054 mmol) in 2 mL CDCl3. A colour change from burgundy to brown was observed in 10 min. An aliquot was removed for NMR and mass spectrometry analysis. δP (162 MHz, CDCl3) 61.9 (s), 45.2 (dt, J 135), 23.4 (s), 19.8 (dt, J 100). m/z (ESI-MS) ([M]n+) 297.1 [PPh3Cl]+, 307.1 [RhIPh]+, 406.0 [RhPPh3NCCH3]+, 477.0 [RhPPh3Cl2NCCH3]+, 568.9 [PPh3RhIPh]+, 627.0 [Rh(PPh3)2]+, 697.0 [Rh(PPh3)2Cl2]+.
Reaction of 2OAc with 11
A mixture of 2OAc (31 mg, 0.095 mmol) and TMS-OTf (35 µL, 0.19 mmol) in 5 mL CH2Cl2 was added dropwise to a solution of 11 (100 mg, 0.095 mmol) in 5 mL CH2Cl2 and stirred for 1 h at room temperature. A colour change from bright yellow to yellow was observed within 5 min. The solvent was reduced to half under reduced pressure, followed by addition of 10 mL n-hexane, which resulted in precipitation of a yellow solid. The solid was filtered, washed with n-hexane (2 × 10 mL), and dried under reduced pressure (84 mg, 70 % yield). δP (162 MHz, CH2Cl2) 58.6 (dt, J 11, 84), 42.5 (dt, J 11, 115). δH (400 MHz, CD3CN) 7.81–7.76 (m, 8H), 7.68–7.60 (m, 8H), 7.58–7.56 (m, 12H), 7.39–7.34 (m, 4H), 7.24–7.20 (m, 4H), 7.18–7.15 (m, 4H), 2.68–2.56 (m, 8H), 1.96 (s, 3H). δC (100 MHz, CD3CN) 172.07, 134.55, 134.08, 133.55, 133.25, 130.45, 130.24, 126.05, 125.51, 24.37, 20.22, 16.68. m/z (ESI-MS) ([M]n+) 479.1 [Rh(dppe)2(OAc)]2+.
Reaction of 2NMe2 with 11
A solution of 2NMe2 (72 mg, 0.095 mmol) in 5 mL CDCl3 was added dropwise to a solution of 11 (100 mg, 0.095 mmol) in 5 mL CDCl3 and stirred for 3 h at room temperature. A colour change from bright yellow to yellow was observed. The solvent was reduced to half under reduced pressure, followed by addition of 10 mL n-hexane, which resulted in precipitation of a pale orange solid. The solid was filtered, washed with n-hexane (2 × 10 mL), and dried under reduced pressure (80 mg, 53 % crude yield). δP (162 MHz, CDCl3) 30.8 (s), 40.7 (dt, J 15, 113), 34.5 (dt, 15, 86). δH (400 MHz, CDCl3) 7.98–6.65 (m, 50H), 3.19 (s, 12H). m/z (ESI-MS) ([M]n+) 380.7 [Rh(dppe)2(DMAP)2]3+, 510.1 [Rh(dppe)2(DMAP)]2+.
Results and Discussion
Treatment of 9 with 2OAc in CDCl3 for 3 h resulted in a pale yellow solution. The 31P NMR spectrum of the reaction mixture gave six peaks between 2.0 and −13.7 ppm, indicating the presence of a mixture of products. Oxidation of the phosphine ligands as inferred from no strongly downfield-shifted signals was, however, not observed. The cationic ESI mass spectrum of the reaction mixture indicated the presence of compounds 12 and 13 at m/z 876.1 and 894.1 (Scheme 5). X-Ray diffraction studies were done on two single crystals of different morphologies obtained from vapour diffusion of Et2O into a concentrated CH2Cl2 solution of the mixture, which showed the crystals to be Iriii compounds 12 and 13 (Fig. 2). The bond distances and geometries of the complexes are all typical and therefore do not warrant any further discussion. The similar solubility of these products prevented their separation from each other. Compound 12 gained one chloride and one acetate whereas compound 13 lost a chloride and a CO, and gained three acetates. It is obvious that using Iiii oxidant with 9 resulted in a mixture of products, possibly arising from ligand exchange and anion scrambling. It has been reported that the use of Iiii oxidant in other systems such as Aui/Auiii resulted in significant ligand and anion scrambling.[12,21,22]
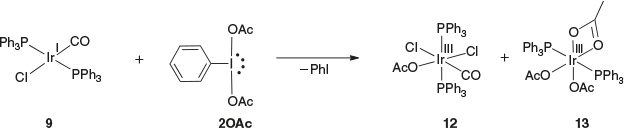
|

|
The reaction of 9 with 2OAc.OTf in a 1 : 1 ratio resulted in a colour change from bright yellow to brown within 10 min. The 31P NMR of the reaction mixture contained seven peaks between −22 and 15 ppm, indicating the presence of seven phosphorus-containing products. The positive ESI detection mass spectrum showed a similar scrambling pattern to that found from 2OAc with 9, with compound 12 being present.
Treatment of 9 with 2NMe2 in CDCl3 at room temperature for 3 h resulted in a colour change from bright yellow to light yellow. The 31P NMR spectrum of the isolated solid in CDCl3 gave three peaks at −2.4, −8.4, and −15.4 ppm indicating the presence of three phosphorus-containing products. Positive mode ESI-MS detection of a CH3CN solution of the isolated solid gave fragments that could be identified at [m/z]+ = 797.1 consistent with 14NMe2 and [m/z]+ = 937.1 consistent with 15NMe2 as shown in Scheme 6. The stereochemistry of 14NMe2 cannot be determined from the data. The presence of one unique P environment in 15NMe2 inferred by the lack of P–P coupling allows the assignment of PPh3 ligands as trans to one another, but the configuration of the other ligands relative to each other cannot be determined.

|
X-Ray diffraction studies were done on single crystals obtained from vapour diffusion of Et2O into concentrated CH2Cl2 solution of the isolated solid, which revealed compound 16. This compound has been synthesized previously by reacting IrCl(PPh3)3 with chlorine.[23] The use of the Iiii oxidant 2H resulted in a similar outcome, where the ESI-MS spectrum contained fragments consistent with 14R and 15R. The 31P NMR spectrum of the isolated solid contained signals between 65.9 and −23.1 ppm. The signal at 65.9 ppm is consistent with the formation of the chlorophosphonium cation ([Ph3PCl]+),[24] which may occur via reductive elimination from Iriii. Overall, the reaction of Vaska’s complex 9 with the chosen Iiii oxidants resulted in significant ligand and anion scrambling.
The reaction of 10 with 2OAc in CDCl3 at room temperature for 3 h resulted in a colour change to light reddish-brown. The 31P NMR spectrum of the reaction mixture contained two doublets at 24.5 and 28.3 ppm. The two doublets were taken to represent two chemically distinct phosphorus atoms coupling with 103Rh, which suggests the presence of two compounds, each with one unique 31P environment.
The most abundant signal in the mass spectrum ([m/z]+ 721.1) resulted from loss of one chloride from compound 17. A cationic fragment at [m/z]+ = 745.1 could result from the loss of one acetate from proposed compound 18. Compound 17 was isolated by dissolving the solid in a minimum amount of CH2Cl2 and placing at −35°C overnight. The solid then was filtered and washed with n-hexane. X-Ray diffraction studies performed on single crystals obtained from concentrated CH2Cl2 solution of the isolated solid at −35°C confirmed product 17 as shown in Fig. 3. The bond distances and geometry are all typical and therefore do not warrant further discussion. The reaction is proposed to proceed via oxidation of 10 to give an unobserved intermediate, which is followed by ligand redistribution and anion scrambling to give the observed products as shown in Scheme 7.
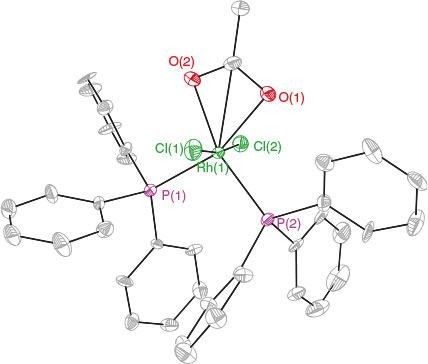
|

|
The dropwise addition of 2NMe2 to 10 at room temperature in CDCl3 resulted in a colour change from burgundy to light red over 3 h. The 31P NMR spectrum of the reaction mixture had two doublets at 14.1 and 11.9 ppm, indicating two phosphorus-containing products coupling to Rh in the mixture. A singlet at 28.2 ppm could not be identified. The positive ESI-MS detection of the mixture indicated the presence of 19 and 20 at [m/z]+ = 942.2 and 803.0, which also was consistent with the chlorine isotope patterns, as shown in Scheme 8. The similar solubilities of these products prevented their isolation. Using Iiii oxidant 2H resulted in similar Rh products as indicated by mass spectrometry and 31P NMR spectroscopy. As with the reaction of 9 with 2H, the 31P NMR also contained a singlet at 65.0 ppm, indicating the presence of the chlorophosphonium cation [Ph3PCl]+. The presence of doublets rather than doublets of doublets indicates the P atom environments are identical within each of the two compounds; therefore, the stereochemistry is proposed to be as depicted for 19R in Scheme 8, although for 20R, this cannot be ascertained.

|
The slow addition of 2OAc.OTf to 10 in 1 : 1 ratio resulted in a colour change from burgundy to brown within 10 min. The 31P NMR of the reaction mixture had four signals between 4 and 62 ppm. The major singlet at 62 ppm represents the chlorophosphonium cation [Ph3PCl]+, which was also observed in the mass spectrum of the reaction mixture at m/z 297.1. The two doublets at 45.2 (JP–Rh = 135 Hz) and 19.8 ppm (JP–Rh = 100 Hz) represent two P–Rh containing products. The cationic ESI mass spectrum of the reaction mixture contained fragments at m/z = 568.9 and 697.1, which were assigned to [Rh(PPh3)(Ph)i]+ and [Rh(PPh3)2(Cl)2]+ respectively. The former product indicates that it is possible for the I–Ph fragment of the iodine(iii) oxidant to also become involved in these reactions, which has also been observed in related chemistry with thiophenes, selenophenes, and tellurophenes using this family of oxidants.[25–27]
As with Vaska’s complex, the overall result using PhIL2 Iiii oxidants with Wilkinson’s catalyst resulted in significant scrambling and overall unproductive reactions.
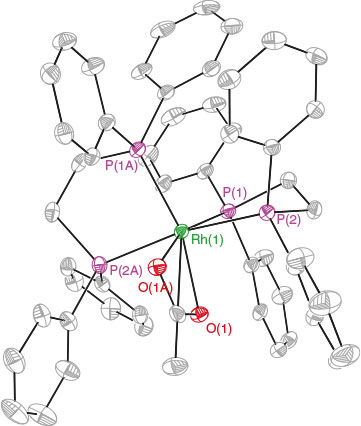
Treatment of homoleptic bis-dppe chelated Rhi compound 11 with 2OAc in CDCl3 at room temperature for 3 h resulted in no colour change. The 31P NMR spectrum of the reaction mixture also indicated no reaction had taken place, showing only starting material. The oxidation of this compound required the use of the stronger oxidizing reagent 2OAc.OTf reagent, prepared by the addition of 2 equivalents of TMS-OTf to 2OAc to give 2OAc.OTf (leaving a free equivalent of TMS-OTf).[16,28,29] The dropwise addition of a CH2Cl2 solution containing PhI(OAc)(OTf) and TMS-OTf to compound 11 resulted in a colour change from bright yellow to yellow and gave a yellow solid after a short workup. The positive ESI-MS spectrum of the isolated solid redissolved in CH3CN showed a major peak at [m/z]+ = 479.1 with a half-mass-unit peak separation that is consistent with 21 as shown in Scheme 9. Further characterization of compound 21 by X-ray crystallography confirmed that this complex contained one acetate group bound to the dicationic Rhiii in a bidentate fashion as shown in Fig. 4, with the two dppe ligands retained.
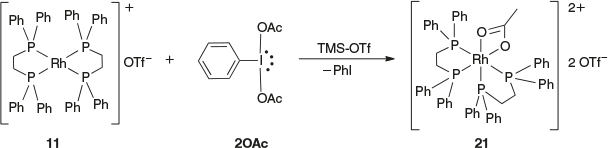
|
|
Treatment of compound 11 with 2NMe in CDCl3 at room temperature for 3 h followed by workup resulted in the isolation of a yellow solid. The 31P NMR spectrum of the isolated solid showed two doublets of triplets at 40.7 and 34.5 ppm, which is consistent with proposed product 22, as well as a singlet at 30.8 ppm. Searching the literature revealed that 1,2-bis(diphenylphosphinyl)ethane (23) has been reported to give an identical chemical shift to our observation (30.8 ppm).[30] The positive ESI-MS analysis of the isolated solid shows a major peak at [m/z]+ = 510.1 with a half-mass-unit peak separation and a minor peak at [m/z]+ = 380.7 with one-third mass-unit peak separation, which is in agreement with compound 22 as shown in Scheme 10.

|
Compound 23 and related phosphine oxide species have been observed to be generated from high-oxidation-state phosphorus triflate species.[16,31] Compounds 22 and 23 were shown to be in approximate ratio of 4 : 1 by 31P NMR, bearing in mind the semiquantitative nature of 31P NMR. A battery of efforts to selectively precipitate 22 to obtain an analytically pure sample unfortunately failed.
Conclusion
Reactions of trivalent iodine reagents containing a PhI(L)2 motif with classic iridium and rhodium complexes have been reported. The metal complexes under study were successfully oxidized from +1 to +3. Oxidations of Vaska’s compound generally resulted in a mixture of products via ligand exchange and anion scrambling. In the case of Wilkinson’s catalyst, the oxidation also resulted in a mixture of products due to ligand exchange and anion scrambling. Reactions of trivalent iodine reagents with homoleptic Rhi bound by chelating phosphines (compound 11) resulted in cleaner oxidation of Rhi to Rhiii and suggests that these systems have a better compatibility with Iiii reagents. In most cases, oxidized phosphine was not observed, likely resulting from reductive elimination from the high-oxidation-state metal complexes rather than direct oxidation of the phosphines, indicating Iiii is reasonably compatible with metal-bound phosphine-containing complexes. However, ligand scrambling in the oxidized species can clearly present a major problem in terms of accurately predicting reaction outcomes and isolating clean products in good yields.
Supplementary Material
Experimental details, 1H, 31P NMR spectra, and mass spectra are available on the Journal’s website.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgements
We thank La Trobe University, The Australian Research Council (JLD, FT16010007) and The Australian Commonwealth (MA, Australian Postgraduate Award PhD scholarship) for their generous funding of this work.
References
[1] A. Yoshimura, V. V. Zhdankin, Chem. Rev. 2016, 116, 3328.| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC28Xit1aksrg%3D&md5=4eae2f4431ae4478d804edda36ee2415CAS |
[2] A. J. Hickman, M. S. Sanford, Nature 2012, 484, 177.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XmtVers7o%3D&md5=0ee2fe36da54a5c896c133101f585eccCAS |
[3] J. M. Racowski, A. R. Dick, M. S. Sanford, J. Am. Chem. Soc. 2009, 131, 10974.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXmt1aru78%3D&md5=b3ed387e5816624006fff685f869c526CAS |
[4] N. R. Deprez, D. Kalyani, A. Krause, M. S. Sanford, J. Am. Chem. Soc. 2006, 128, 4972.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XislOnsr4%3D&md5=de2ab484bd56f7109841d008164f568aCAS |
[5] D. Kalyani, N. R. Deprez, L. V. Desai, M. S. Sanford, J. Am. Chem. Soc. 2005, 127, 7330.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXjvVeit78%3D&md5=8b01f3191d3dbdef50d3b9da42abc9c1CAS |
[6] R. Corbo, D. C. Georgiou, D. J. D. Wilson, J. L. Dutton, Inorg. Chem. 2014, 53, 1690.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXlslGhuw%3D%3D&md5=9302017c57f679779c953fcee382b7b3CAS |
[7] A. R. Dick, M. S. Remy, J. W. Kampf, M. S. Sanford, Organometallics 2007, 26, 1365.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhslajur8%3D&md5=d54c50b0014113191d157557b2c10e9aCAS |
[8] K. B. McMurtrey, J. M. Racowski, M. S. Sanford, Org. Lett. 2012, 14, 4094.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhtFWhsLjE&md5=89f0fee11d3f63cb2bf722475d322ea7CAS |
[9] J. M. Racowski, N. D. Ball, M. S. Sanford, J. Am. Chem. Soc. 2011, 133, 18022.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtlGjt7vL&md5=adaa5f5f6b8f12de81288b7909e09d80CAS |
[10] A. R. Dick, K. L. Hull, M. S. Sanford, J. Am. Chem. Soc. 2004, 126, 2300.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXosFKktQ%3D%3D&md5=fc76c6ec34fc2ac95c59aabad8b6db0fCAS |
[11] S. R. Whitfield, M. S. Sanford, J. Am. Chem. Soc. 2007, 129, 15142.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhtlWmu7vO&md5=d909d42926e78c30b2d9d2fed7a84370CAS |
[12] R. Corbo, T. P. Pell, B. D. Stringer, C. F. Hogan, D. J. D. Wilson, P. J. Barnard, J. L. Dutton, J. Am. Chem. Soc. 2014, 136, 12415.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXhtlalu7fL&md5=f1e692aee391631a44b7754ac44decc5CAS |
[13] R. Weiss, J. Seubert, J. Angew. Chem. Int. Ed. Engl. 1994, 33, 891.
| Crossref | GoogleScholarGoogle Scholar |
[14] N. S. Pirkuliyev, V. K. Brel, V. V. Zhdankin, N. S. Zefirov, Russ. J. Org. Chem. 2002, 38, 1224.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XosVKnurk%3D&md5=bad26f268b95efa83ed6d11a1a5614a2CAS |
[15] E. Lee, A. S. Kamlet, D. C. Powers, C. N. Neumann, G. B. Boursalian, T. Furuya, D. C. Choi, J. M. Hooker, T. Ritter, Science 2011, 334, 639.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtlyqu7vL&md5=b8d22079ce17c11f2312263fb2f8a518CAS |
[16] T. P. Pell, S. A. Couchman, S. Ibrahim, D. J. D. Wilson, B. J. Smith, P. J. Barnard, J. L. Dutton, Inorg. Chem. 2012, 51, 13034.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xhs1Kntb3L&md5=8bdd19b2d9d6cd61a1fa6f5a11474a7aCAS |
[17] F. C. Sousa e Silva, A. F. Tierno, S. E. Wengryniuk, Molecules 2017, 22, 780.
| Crossref | GoogleScholarGoogle Scholar |
[18] J. P. Collman, C. T. Sears, M. Kubota, A. Davison, E. T. Shawl, J. R. Sowa, R. J. Angelici, Inorg. Synth. 1990, 28, 92.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3MXhsFGqtrg%3D&md5=458b17eb53774915b407590d24130f92CAS |
[19] J. A. Osborn, G. Wilkinson, J. J. Mrowca, Inorg. Synth. 1990, 28, 77.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3MXit1Sjt7c%3D&md5=faabb6cf50c1b654ff020f2779eed77bCAS |
[20] A. J. Kunin, E. J. Nanni, R. Eisenberg, Inorg. Chem. 1985, 24, 1852.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2MXitlSrsL0%3D&md5=0f971e25857ed1c586d0866b7a630881CAS |
[21] M. Hofer, C. Nevado, Eur. J. Inorg. Chem. 2012, 1338.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsFaltLfO&md5=5f1eb879da8afcd278bb8af037941662CAS |
[22] M. Hofer, C. Nevado, Tetrahedron 2013, 69, 5751.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXmtlGnsLs%3D&md5=d2a5ca3269e16d852ddd29612ede5631CAS |
[23] M. A. Bennett, D. L. Milner, J. Am. Chem. Soc. 1969, 91, 6983.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE3cXhvVShtQ%3D%3D&md5=b213c291383ff553e1171bcba1f99509CAS |
[24] E. Krawczyk, A. Skowrońska, J. Michalski, J. Chem. Soc., Dalton Trans. 2002, 4471.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XovFKlsLw%3D&md5=855777b2f8886aed391f330aefa14acdCAS |
[25] M. Ito, C. Ogawa, N. Yamaoka, H. Fujioka, T. Dohi, Y. Kita, Molecules 2010, 15, 1918.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXjvFegtrc%3D&md5=c22551cc8446888f83a797b9e6303df9CAS |
[26] T. Dohi, M. Ito, K. Morimoto, Y. Minamitsuji, N. Takenage, Y. Kita, Chem. Commun. 2007, 4152.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhtFenur%2FP&md5=fde1c4c127d629d0c99789f8319cf272CAS |
[27] S. Egalahewa, M. Albayer, A. Aprile, J. L. Dutton, Inorg. Chem. 2017, 56, 1282.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2sXhtFOrsL4%3D&md5=9fa077a4bbb4300ce4c7a799cccd0177CAS |
[28] A. Aprile, K. J. Iversen, D. J. D. Wilson, J. L. Dutton, Inorg. Chem. 2015, 54, 4934.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXnsVeitb8%3D&md5=d35f8d44d3d453c2857ae6baca61431dCAS |
[29] U. Farid, T. Wirth, Angew. Chem. Int. Ed. 2012, 51, 3462.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xht1ymt74%3D&md5=dbd0d902ef5fc99f09266bc570df3f6fCAS |
[30] K. E. Fairfull-Smith, I. D. Jenkins, W. A. Loughlin, Org. Biomol. Chem. 2004, 2, 1979.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXls1ertLk%3D&md5=a8d8b7a4c094f737ec5c14bf7f45ca40CAS |
[31] K. E. Elson, I. D. Jenkins, W. A. Loughlin, Aust. J. Chem. 2004, 57, 371.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXivVKhs7g%3D&md5=3cef190c766b66807babe916711b6ef9CAS |
* Jason Dutton is the recipient of the 2016 Alan Sargeson Lectureship.