

Assessing the colloidal properties of engineered nanoparticles in water: case studies from fullerene C60 nanoparticles and carbon nanotubes
Kai Loon Chen A D , Billy A. Smith B , William P. Ball A and D. Howard Fairbrother B CA Department of Geography and Environmental Engineering, Johns Hopkins University, Baltimore, MD 21218-2686, USA.
B Department of Chemistry, Johns Hopkins University, Baltimore, MD 21218-2686, USA.
C Department of Materials Science and Engineering, Johns Hopkins University, Baltimore, MD 21218-2686, USA.
D Corresponding author. Email: kailoon.chen@jhu.edu

Kai Loon Chen is an Assistant Professor in the Department of Geography and Environmental Engineering at Johns Hopkins University in Baltimore, Maryland. He completed his B.Eng. and M.Eng. degrees in civil engineering at the National University of Singapore in 2001 and 2003 respectively. He joined the Environmental Engineering Program at Yale University in 2003 and received his Ph.D. in 2008. His current research focusses on understanding the fate and transport of engineered nanoparticles in natural and engineered aquatic systems. He is also interested in utilising nanotechnology for water purification and environmental remediation. |

Billy A. Smith is a graduate research assistant pursuing his doctorate in Chemistry in the research group of Professor Howard Fairbrother, Johns Hopkins University (JHU), Baltimore, Maryland. He earned his B.S. in chemistry from Stevenson University (previously known as Villa Julie College) in 2005, and currently holds a masters degree in Chemistry from JHU. In the Fairbrother group, he has used surface analytical techniques in conjunction with time-resolved dynamic light scattering to study the role that oxygen containing functional groups play in determining the colloidal stability and transport properties of oxidised carbon nanotubes. |

William P. Ball (P.E., Ph.D., BCEE) is a Professor of environmental engineering in the Department of Geography and Environmental Engineering at Johns Hopkins University. He received his B.S. from the University of Virginia in 1976 and his M.S. and Ph.D. in environmental engineering from Stanford University in 1977 and 1989. Between his M.S. and Ph.D., Professor Ball worked for six years for James M. Montgomery Consulting Engineers. He was previously on the faculty at Duke University and in 1992 joined the faculty at Johns Hopkins University. Professor Ball’s research program is focussed on physical and chemical processes affecting pollutant fate and treatment in natural environments and engineered systems, with focus on complex aquatic systems. |

D. Howard Fairbrother is a Professor of Chemistry at Johns Hopkins University in Baltimore, Maryland. He received his B.S. degree from Oxford University, England, in 1989, and his Ph.D. in chemistry from Northwestern University in 1994. After completing a postdoctoral position with Professor Gabor Somorjai at the University of California, Berkeley, he joined the faculty in the Chemistry Department at Johns Hopkins University (JHU) in 1997. His research program at JHU is focussed on surface chemistry, with particular emphasis on characterising the functional groups on environmentally relevant materials and understanding the role of surface chemistry on the behaviour of engineered nanomaterials in aquatic environments. |
Environmental Chemistry 7(1) 10-27 https://doi.org/10.1071/EN09112
Submitted: 1 September 2009 Accepted: 11 January 2010 Published: 22 February 2010
Environmental context. The fate and bioavailability of engineered nanoparticles in natural aquatic systems are strongly influenced by their ability to remain dispersed in water. Consequently, understanding the colloidal properties of engineered nanoparticles through rigorous characterisation of physicochemical properties and measurements of particle stability will allow for a more accurate prediction of their environmental, health, and safety effects in aquatic systems. This review highlights some important techniques suitable for the assessment of the colloidal properties of engineered nanoparticles and discusses some recent findings obtained by using these techniques on two popular carbon-based nanoparticles, fullerene C60 and multi-walled carbon nanotubes.
Abstract. The colloidal properties of engineered nanoparticles directly affect their use in a wide variety of applications and also control their environmental fate and mobility. The colloidal stability of engineered nanoparticles depends on their physicochemical properties within the given aqueous medium and is ultimately reflected in the particles’ aggregation and deposition behaviour. This review presents some of the key experimental methods that are currently used to probe colloidal properties and quantify engineered nanoparticle stability in water. Case studies from fullerene C60 nanoparticles and multi-walled carbon nanotubes illustrate how the characterisation and measurement methods are used to understand and predict nanoparticle fate in aquatic systems. Consideration of the comparisons between these two classes of carbon-based nanoparticles provides useful insights into some major current knowledge gaps while also revealing clues about needed future developments. Key issues to be resolved relate to the nature of near-range surface forces and the origins of surface charge, particularly for the reportedly unmodified or ‘pure’ carbon-based nanoparticles.
Additional keywords: aggregation, deposition, DLVO, dynamic light scattering, X-ray photoelectron spectroscopy.
Introduction
Over the last decade, nanotechnology has matured into a prominent, interdisciplinary field that influences almost every major branch of science.[1] A fundamental building block of nanotechnology is the engineered nanoparticle (ENP). These particles are by definition of a size between 1 and 100 nm in a least one dimension,[2] and come in a variety of geometries such as spheres,[3] cylinders,[4] and planes.[5] As a subset of traditional colloids (particles between 1 nm and a few micrometres in diameter), nanomaterials are unique because they are on a length scale in which surface and quantum effects become increasingly important.[2] Indeed, these effects often provide ENPs with unusual reactivity as well as novel optical, mechanical, and electrical properties. Compositionally, ENPs may be broadly categorised as being either carbonaceous (e.g. fullerene C60 nanoparticles and carbon nanotubes (CNTs)) or non-carbonaceous (e.g. zero-valent iron nanoparticles, silver nanoparticles, and quantum dots).
The combination of chemical and physical attributes exhibited by ENPs has led to their increased use in commercial applications.[6] This is reflected by the fact that their global market has grown steadily from US$7.5 billion in 2003 to US$12.7 billion in 2008. Over the next 5 years, their market value is expected to exceed US$27 billion.[7,8] Current estimates suggest that more than 800 consumable products, including cell phone batteries, sporting equipment, and cosmetics, already contain ENPs.[9] In addition to these everyday items, work is being conducted to investigate the potential of ENPs in environmental remediation strategies[10–15] and advanced medical procedures.[16,17] For example, the US Environmental Protection Agency (US EPA) recently (2008) released data on 26 sites across North America in which nanomaterials were being used to remove contaminants such as chlorinated solvents from groundwater.[18] In nanomedicine, ENPs are being studied for use in tissue scaffolds, as targeted drug delivery agents, and as components of gene therapy.[16]
In many applications, the interfacial properties of ENPs (e.g. cohesion and adhesion, wettability and adsorption) are tailored using either covalent or non-covalent surface-modification strategies. For instance, colloidal dispersions of ENPs are commonly desired for industrial applications, but such dispersions can be impossible to prepare with unmodified ENPs owing to the fact that the attractive forces between particles induce aggregation. One approach to overcome this obstacle is to covalently graft functional groups into the ENP surface. Alternatively, surfactants may be physisorbed onto the surface of the ENP in a non-covalent modification process. In either case, interparticle repulsion is increased and dispersion properties are improved.[14,19] Surface modification techniques are also used to deliberately modify other properties (e.g. sorption and wettability) and to create points of attachment for more complex hierarchical nanostructures.[20,21]
As the global market for nanomaterials and nanomaterial-containing products continues to expand, increasing quantities of ENPs are expected to enter the environment from manufacturing effluents, discarded nanoproducts, or incidental spills.[22,23] The environmental fate, transport, and bioavailability of these ENPs are dependent on their aggregation and deposition behaviour. Comparative toxicity studies have also been conducted to better understand the environmental risks associated with carbonaceous and inorganic nanoparticles in different fluid phase environments.[24–28] In these studies, particular attention has been paid to ENP composition, dispersion state, size distributions, chemical treatments, and surface chemistry, as well as the chemistry of the aquatic environment (e.g. pH, ionic strength, and the concentration of natural organic matter (NOM)). The upshot from these studies is that while some ENPs are toxic to some organisms, the degree of toxicity depends on both the intrinsic properties of the manufactured ENPs and their physicochemical state in the environment, as also influenced by solution chemistry and the presence of co-contaminants. It is interesting to note that the same variables play a determinant role in the colloidal stability of ENPs.
Thus, it is clear that to develop models that allow us to anticipate and adequately address the environmental risks posed by different ENPs, more detailed information on their behaviour in aquatic environments is required. Structure–property relationships that relate the ENPs’ environmental behaviour to their physicochemical properties (e.g. size, shape, and surface chemistry) are also needed. In this review, we will highlight some of the experimental methods currently being used to characterise ENPs and probe their colloidal properties. We then show how several of these techniques have been applied to evaluate the colloidal properties of two important carbonaceous nanoparticles – fullerene C60 nanoparticles and multiwalled carbon nanotubes (MWNTs). To conclude the review, we describe the implications of these findings on general issues of ENP fate and transport and suggest directions for further research.
Colloidal stability
Aggregation
A suspension with superior colloidal stability is one in which the suspended colloidal particles are able to resist aggregation for an indefinite period of time. In this context, aggregation refers to the association of colloidal particles to form larger clusters.[29] The fate and transport of ENPs when released into natural and engineered aquatic systems is largely determined by their aggregation behaviour. Specifically, the rate of ENP aggregation will influence their rate of sedimentation and thus their removal from the aqueous phase.
Depending on the colloidal stability of the ENPs and the chemistry of the surrounding environment (e.g. pH and electrolyte composition), ENPs can undergo aggregation in solution among themselves before encountering any natural constituents. This process of association among colloidal particles of the same kind is referred to as homoaggregation. In systems containing more than one type of suspended particles, heteroaggregation (or association between particles of different types) can take place either exclusively or simultaneously with homoaggregation. The frequency of homoaggregation and heteroaggregation will depend on the particle concentrations, physical and chemical properties of the particles, and solution chemistry.[30–33] In natural aquatic systems, the fate of ENPs is more likely to be controlled by heteroaggregation between ENPs and naturally occurring colloidal particles because the concentration of naturally occurring colloids will greatly exceed that of ENPs. Nevertheless, homoaggregation is a suitable starting point for investigating the colloidal stability of ENPs because a single-component suspension is a much simpler system to study systematically in the laboratory and to understand conceptually.
Aggregation may be conceptualised as a two-stage process: transport and attachment.[29] First, ENPs and other colloidal particles must be transported towards each other before aggregation can occur. The mechanisms of achieving particle–particle contact can be broadly classified as Brownian diffusion (perikinetic aggregation), fluid motion (orthokinetic aggregation), and differential sedimentation. Further details regarding these transport mechanisms can be found elsewhere.[29] The focus of the present review paper, however, is on the attachment stage of aggregation. The likelihood of a permanent attachment resulting from the approach of two particles is controlled by short-range interparticle forces of interaction, which are in turn dependent on the surface chemistry and composition of the particles as well as the solution chemistry.
Deposition
Another key process that controls the fate and transport of ENPs in natural and engineered aquatic systems is deposition. Deposition refers to the process in which ENPs collide and stick over time to an immobile solid (collector) surface, such as sand, sediments, and rocks. As the solid surface is composed of a different material from the ENPs and can be treated as an infinitely large stationary particle, deposition of ENPs can be considered as an extreme form of heteroaggregation.[29] Deposition can take place in different natural environments, such as surface water and groundwater, and is especially important in systems where solid surfaces for attachment are readily available. Thus, ENP deposition is expected to play a crucial role in influencing the transport of ENPs in groundwater flow through porous media, owing to the vast opportunities for ENP attachment to collector surfaces.
As with aggregation, deposition begins when ENPs are transported to the collector surface. Transport mechanisms for deposition are analogous to those for aggregation and include Brownian diffusion, interception, and sedimentation, with details described elsewhere.[29,34] In the attachment stage, both hydrodynamic effects and colloidal interactions are important factors that will influence the adherence of ENPs to solid surfaces.[29]
Derjaguin–Landau–Verwey–Overbeek (DLVO) theory
The colloidal stability of charged particles dispersed in aqueous solutions is often controlled by their van der Waals and electric double layer interactions.[29,35] The van der Waals interactions between colloidal particles are the result of the interactions between the permanent or induced dipoles within the ENPs and are dependent on the particles’ composition, size and geometry. For most particles in aqueous environments, van der Waals interactions are attractive and thus promote aggregation.
Conversely, electric double layer interactions occur owing to charges that reside on the particle surface. The origins of surface charge vary and depend on the surface composition of ENPs. Surface charge is often a consequence of dissociable surface functional groups.[29,35] Some examples of ENPs with surface functional groups include oxidised CNTs,[36] which we will later discuss extensively in this paper, citrate-coated silver nanoparticles,[37,38] and metal oxide nanoparticles such as titanium,[39,40] cerium,[41] and iron oxides.[42] The sign and magnitude of the charge imparted by surface functional groups will depend on their surface site density and solution pH. From a coulombic standpoint, ENPs exhibiting charge of the same sign will repel one another, whereas oppositely charged particles will attract. As counterions in the solution tend to accumulate at the charged interface, they screen the ENP surface charge and effectively reduce the electrostatic repulsion between particles. The degree of charge screening is dependent on electrolyte concentration and valence of counterions. Charge neutralisation can also occur as a result of specific adsorption, when divalent and trivalent counterions or polyelectrolytes adsorb on the ENP surface within the Stern layer and reduce the effective surface charge.[29,35,43] Excess sorption of these counterions or polyelectrolytes in the Stern layer can result in charge reversal and particle restabilisation.[29,35]
The classic Derjaguin–Landau–Verwey–Overbeek (DLVO) theory of colloidal stability is commonly invoked to describe the interfacial forces experienced between charged colloidal particles.[44,45] In essence, the theory states that the interaction energy between colloidal particles is the sum of van der Waals and electric double layer interactions. While van der Waals interactions are insensitive to changes in solution chemistry, the electric double layer interaction is strongly dependent on solution pH and ionic strength.[35] As van der Waals and electric double layer interactions follow power-law and exponential-law relationships as a function of separation distance respectively, the magnitude of van der Waals interactions always exceeds electrostatic interactions at very small separation distances.[29,35] For similarly charged particles and sufficiently low ionic strength conditions, the sum of these interaction energies results in the presence of an energy barrier that must be overcome by the kinetic energy of the particles before aggregation in the primary minimum can occur, as shown in Fig. 1. As the ionic strength increases, the energy barrier decreases until it is eliminated. At this point, the salt concentration has exceeded the critical coagulation concentration (CCC), resulting in favourable aggregation. The same logic applies in the case of deposition of colloids onto surfaces of larger solids. In this context, the pivotal salt concentration is known as the critical deposition concentration (CDC). At intermediate salt concentrations, an additional secondary minimum may be formed (Fig. 1).[46] Aggregation or deposition can take place through particle accumulation within the interfacial region of the secondary minimum (i.e. at this distance from the particle surface), but adhesion under these circumstances is much weaker than that which occurs when the two surfaces are sufficiently close to experience the energies of the primary minimum.[35]
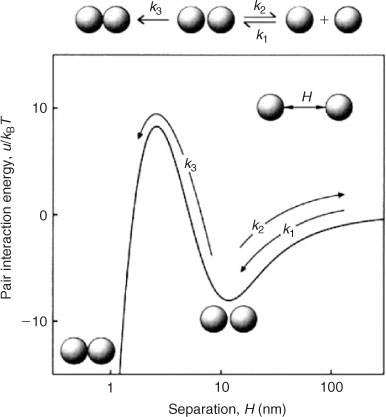
|
Discrepancies between experimental results and predictions based on DLVO theory have been observed over the past few decades, particularly under conditions where repulsive electric double layer interactions predominate.[29,47–49] Under such conditions, the predicted aggregation and deposition kinetics (i.e. inverse stability ratios or attachment efficiencies) based on DLVO theory are frequently orders of magnitude lower than those obtained experimentally. Another discrepancy is that experiments have shown that colloidal stability is independent of particle size even when DLVO theory predicts that larger particles should be more stable than smaller ones.[47–49] These inconsistencies between experiments and theory have been attributed to surface roughness, surface charge heterogeneity, and non-DLVO forces such as hydrophobic interactions and structural forces.[29,47–49]
Steric stabilisation
Steric stabilisation refers to the enhancement in colloidal stability that occurs as a result of polymers adsorbed or grafted on the particle surface. As the particles approach each other, the interpenetration of the hydrophilic portion of the polymers that extends into the solution phase causes the displacement of water molecules into the bulk phase, resulting in an increase in free energy.[43] This repulsive interaction, which results from the polymer interpenetration, can be large enough to keep colloidal particles stable.
Steric stabilisation is the only mechanism at play for non-ionic polymers,[50] and the degree of stabilisation will be dependent on the surface density and the molecular weight of the polymers as well as the solubility of that portion of the polymer that extends into solution. As the extension of non-ionic polymers is largely insensitive to the presence of electrolytes,[43] the colloidal stability of particles coated with these polymers is not impacted by even large variations in salt concentrations. In cases where particles are only partially coated with polymers, bridging between particles (or flocculation) may occur instead.[51]
Conversely, when polyelectrolytes are adsorbed or grafted on the surface of particles, both steric and electrostatic repulsion could result to enhance the stability of particles.[52] The effect of both contributions is known as electrosteric stabilisation. Unlike steric stabilisation, the electrosteric effect is dependent on solution chemistry. The variation of solution pH, ionic strength, and divalent cation concentration can result in the conformational change of the surface-immobilised polyelectrolytes, as well as the overall surface charge of the coated particles.[53–55] Indeed, the challenge in modelling electrosteric repulsion is that both contributions are interdependent.[52,55] For instance, the degree of polyelectrolyte adsorption and polyelectrolyte conformation are dependent on the electric double layer, while the adsorption of polyelectrolytes itself can influence the double layer.[52]
In many cases, ENPs are provided (during synthesis or in subsequent modification) with surface coatings that enhance stability. For example, quantum dots can be coated with biocompatible polymers,[56,57] while zero-valent iron (ZVI) nanoparticles used for the treatment of groundwater contamination can be coated with synthetic or natural polymers to increase their mobility.[58–60] Silver nanoparticles are often synthesised in the presence of polymeric capping agents in order to control their sizes.[61,62] In the natural environment, ENPs will also encounter NOM, and the effects of NOM on the stability of colloidal particles have been extensively investigated over the past few decades.[63–67] The nature of NOM varies significantly, however, as a result of the wide diversity of molecular weights and conformations, as well as from the negative polyelectrolytic nature of the humic and fulvic substances and polysaccharides that comprise a substantial fraction of NOM. Studies with these substances indicate that ENPs released into natural aquatic systems can adsorb NOM and that this can impart steric or electrosteric stabilisation. The situation becomes more complex, however, when NOM and ENPs also interact with other aqueous constituents, including multivalent cations and naturally occurring colloids.
Analytical and experimental methods
A comprehensive approach to understanding the environmental behaviour of ENPs is to correlate their physicochemical and electrokinetic properties with their colloidal stability. In the discussion that follows, we examine some prominent ENP characterisation techniques and highlight a few of the experimental methods used to measure colloidal stability. In this section, the discussion of particle imaging is treated in a cursory fashion, while techniques used to measure chemical composition, electrokinetic property, and aggregate structure are more thoroughly examined. Many but not all of the techniques presented here are used in the case studies of C60 nanoparticles and MWNTs discussed later in the review.
Characterisation techniques
Particle imaging
ENP morphology, size distribution, and dispersion state (i.e. primary particles, doublets, and higher-order aggregates) in solution may play important roles in controlling colloidal behaviour. To characterise these attributes, techniques for direct visualisation, such as transmission electron microscopy (TEM),[68,69] scanning electron microscopy (SEM),[70,71] and atomic force microscopy (AFM)[72,73] can be used. TEM and SEM require ultra-high vacuum conditions and use electrons to render images (micrographs) of the ENPs being studied. AFM is performed under ambient or solution-phase conditions and renders images based on interactions between particles and a sharp tip. In all three techniques, ENP morphologies and size distributions may be assessed. In addition, qualitative appraisals of ENP dispersion states are also possible. Of the three techniques, TEM provides the highest magnification power and can be used to attain more detailed information on ENP structure. However, a drawback with all of these techniques is that sampling preparation methods and drying effects may change the aggregation state of the ENPs.
Elemental (bulk) composition
Energy dispersive X-ray spectroscopy (EDS)[70] can be used to measure the elemental (bulk) composition of an entire ENP sample or a localised region, depending on the focus and magnification of the system.[74] In the EDS process, an incident electron beam initiates the ejection of core-level electrons and the emission of element-dependent X-rays whose energy can be analysed to determine the ENPs’ chemical composition. The analysis depth of EDS is ~1.5 μm (although the exact depth will depend on the electron beam energy and the elements present in the particle) and the spot size is generally larger than 100 nm. It is important, however, to recognise the limitations of EDS. Given the analysis depth and spot size, data acquired on individual ENPs could also include signal from the substrate.[75] Additional techniques that can be used to determine the bulk composition of ENPs are inductively coupled plasma mass spectrometry (ICP-MS)[76,77] and combustion analysis (CA).[78] These techniques, which suffer similar problems of differentiation among particles, are also not especially useful for determining individual particle properties in heterogeneous materials.
Surface composition
X-ray photoelectron spectroscopy (XPS)[79] is commonly used to characterise an ENP’s surface composition. In this technique, a sample is irradiated with X-rays and the measured binding energies of the ejected photoelectrons provide information on the elements present at the surface. The surface specificity (3–10 nm) of this technique is a consequence of the extremely short inelastic mean free path of the ejected photoelectrons. For nanomaterials smaller than 10 nm, the surface specificity of XPS diminishes, and the compositional information obtained is comparable with the bulk measurements acquired with EDS. XPS for nanomaterials requires relatively large amounts of sample to sufficiently cover the entire analysis region (1 cm2) and reduce unwanted contributions from the substrate. As an internal check, XPS results should be consistent with reasonable expectations. For example, in the case of oxidised CNTs, the only elements detected should be oxygen and carbon. The detection of other elements is a strong indication that drying effects have led to the precipitation or adsorption of unwanted impurities on the sample surface.
In addition to surface composition, XPS can also be used to identify the nature and concentration of surface functional groups using either peak deconvolution methods or chemical derivatisation (CD) techniques. These two methods are described below.
-
Peak deconvolution. Although the binding energy of photoelectrons ejected during X-ray irradiation is largely determined by the element and the core level from which they are ejected, their binding energies are also influenced (albeit to a lesser degree) by the element’s local chemical environment. For example, photoelectrons ejected from carbon atoms bound to fluorine exhibit higher binding energies than photoelectrons ejected from hydrocarbons. In principle, this information can be used to deconvolute an XPS spectral envelope, such as the C(1s) region into its various components. Such interpretations can often be ambiguous, however, because of the close binding energy of different species present (e.g. C–O and C=O) on the surface, coupled with the limited resolution of X-ray photoelectron spectrometers. To overcome this problem, CD methods have been developed.
-
Chemical derivatisation. As an alternative to XPS peak-fitting, CD techniques[80,81] can be used to more accurately and precisely quantify specific functional groups present on surfaces. In CD, a targeted surface functional group reacts with a specific chemical (derivatising) reagent that contains a unique chemical tag that is not present on the ENP surface. The chemical tag is chosen so that its concentration can be quantified either by XPS or another analytical technique, such as fluorescence. When CD is used in conjunction with XPS, fluorine is often used as the chemical tag because it is not normally found in most ENPs and has a high X-ray photoelectron (XP) cross-section. By measuring the concentration of fluorine using XPS after a derivatising reaction, the concentration of the targeted functional groups can be determined. Fig. 2 shows the different derivatising reactions that have been used to titrate the concentration of hydroxyl, carbonyl, and carboxyl groups on oxidised CNTs. An analogous protocol has also been developed using 3,3,3-(trifluoropropyl)dimethylchlorosilane to assay the concentration of free hydroxyl groups on silica surfaces.[82] Compared with XPS peak-fitting methods, CD offers a far more quantitative method to assay those functional groups for which selective derivatising reactions exist.
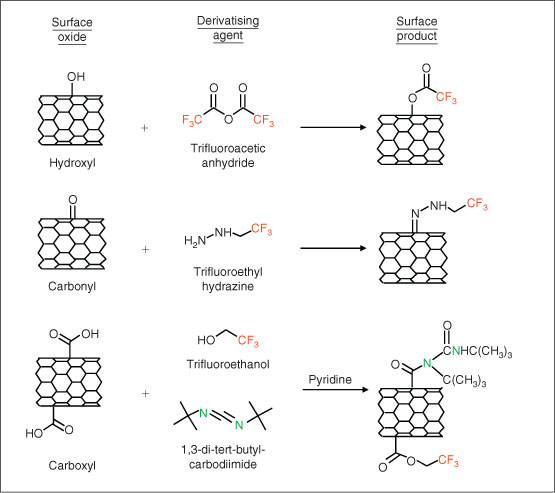
|
Surface charge
ENP surface charge (C m–2) can be calculated by dividing an ENP’s specific charge (C g–1) by its specific surface area (m2 g–1). Specific charge may be determined by titrating a known ENP mass with acid or base and solving a charge-balance equation.[83] An ENP’s specific surface area can be estimated using the Brunauer–Emmett–Teller (BET) method,[84] although care should to be taken when interpreting BET measurements because the method is not believed to have absolute accuracy when pore sizes below 20 nm are being probed.[85] Also, the drying of ENP samples under vacuum conditions during BET measurements may cause the ENPs to aggregate in ways that prevent N2 from entering occluded surfaces. This occlusion will result in an underestimation of the actual surface area available when ENPs are suspended in aqueous solutions. For these reasons, the interpretation of N2 adsorption data with the BET approach is only suitable for providing relative comparison between ENP samples with full appreciation of its limitations. Care must also be taken in conducting and interpreting titration experiments with inorganic ENPs to account for possible particle dissolution, especially at low pH conditions. The advantage of potentiometric titration is that it provides a direct measurement of surface charge using readily available, cost-effective equipment. The technique does, however, require comparatively large quantities of ENPs (0.1–2 g L–1).[86]
Electrokinetic properties
In electrokinetic techniques, an ENP’s surface potential is approximated by its zeta (ζ) potential, as determined from some comparatively straightforward measures of electrophoretic mobility (EPM).[29,43] Particle electrophoresis is a popular method used to determine a particle’s EPM. This method involves the measurement of a particle’s migration rate in an electric field of known strength, and for ENPs, this is most often measured using electrophoretic light-scattering systems. For spherical particles, EPM values may be converted to ζ potentials using well-known equations[29] or tabulated values.[87] In contrast to surface-charge titration, EPM measurements are conducted on dilute suspensions, so only small quantities of ENPs are needed. Other than electrophoresis, other options, such as electroacoustic[88] and agar-gel,[89] are also available.
Surface forces
Over the past two decades, particle–particle and particle–flat surface interactions have been investigated by performing force measurements using the AFM.[90] After attaching a colloidal particle on a tipless AFM cantilever,[91] the colloidal probe can be brought towards the substrate of interest and retracted from the substrate after contact occurs. By monitoring the deflection of the cantilever, the forces between the colloidal probe and substrate can be derived as a function of separation distance. As these measurements can be performed in aqueous media, they allow the investigation of the interfacial interactions (including DLVO-type interactions and steric or electrosteric repulsion for polymer-coated surfaces) that determine the particle’s aggregation and deposition behaviour. In principle, this method can also be used to probe ENP–ENP and ENP–solid-surface interactions. As the conventional method of attaching a colloidal particle to the AFM cantilever requires the manipulation of the particle under an optical microscope, a practical difficulty lies in the preparation of a nanometre-sized colloidal probe suitable for force measurements.[92] However, some progress has been made in this regard, as demonstrated by studies that have used inorganic nanoparticles and CNTs attached to AFM cantilevers for force measurements as well as for imaging.[92–94] Alternatively, AFM force measurements between a colloidal probe whose composition is of interest and a ‘lawn’ of ENPs deposited on a flat substrate can be extrapolated to elucidate ENP–ENP and ENP–solid-surface interactions.[95]
Aggregate structure
When ENPs undergo homoaggregation in natural aquatic systems, the aggregate structure will influence their rate of sedimentation and transport behaviour. It is well known that the compactness of the aggregate structure is dependent on homoaggregation kinetics – diffusion-limited (favourable) aggregation results in open fractal structures, whereas reaction-limited (unfavourable) aggregation produces compact fractal structures.[96–98] Static light scattering (SLS) is a common technique used to study these aggregate structures.[99–102] It is a non-invasive technique that involves the irradiation of a suspension containing the aggregates with a laser beam and the measurement of the intensities of the scattered light at various angles. The scattering intensities are plotted against the magnitude of the scattering vectors (which is a function of the scattering angle), and the slope obtained in the power-law region yields the fractal dimension, which can then be used as a metric to quantify the compactness of the aggregate structures. Typically, compact and open structures will yield fractal dimensions of ~2.1 and 1.8 respectively.[97] In a recent study, Chen et al.[103] determined the fractal dimensions of aggregates comprised of as-received and acid-treated single-walled CNTs to be 2.27 and 2.50 respectively by using SLS. Their results show that the shortened, acid-treated CNTs can form more compact aggregate structures compared with the longer, as-received CNTs. SLS can also be used to investigate the restructuring of aggregates that has been found to occur, as evident from the change in fractal dimensions with time.[99,102] For binary systems (i.e. systems comprising two types of colloidal particles), it is possible to obtain the fractal dimensions of heteroaggregates using SLS.[104,105] Care must be taken, however, to perform these measurements under solution conditions in which heteroaggregation occurs exclusively.
In addition to SLS, fractal dimensions of aggregates can be obtained by analysing the TEM or SEM images of aggregates.[101] However, this method involves the drying of the aggregates on a substrate, which is likely to result in the collapse of the aggregate structures. Also, a direct analysis of the TEM or SEM images would only allow the derivation of a two-dimensional fractal dimension, which may not be truly representative of the three-dimensional aggregate structure.
Measurements of aggregation and deposition kinetics
Sedimentation properties
Sedimentation refers to the settling of particles and aggregates due to gravitation or forces of centrifugation. For a given settling force, the rate of sedimentation will depend on aggregate size, shape, and particle densities. Sedimentation experiments are frequently used to make qualitative, visual assessments of colloidal stability under different aquatic conditions. In conjunction with UV-visible spectroscopy or turbidity measurements, kinetic experiments may be conducted and compared among samples.[106–108] Through sedimentation experiments, for example, CCCs can be approximated by determining the minimum concentrations of electrolyte needed to remove a substantial amount of particles from the suspensions.[109,110] One drawback of the technique, however, is that quantitative comparisons with other studies cannot be made unless the particle concentrations and sedimentation conditions (e.g. time allowed for sedimentation to occur) are well controlled and carefully replicated.[111]
Homoaggregation kinetics
Time-resolved dynamic light scattering (TR-DLS) can measure temporal changes in the hydrodynamic diameter (Dh) of aggregates during perikinetic homoaggregation. In these experiments, homoaggregation is typically induced by adding an electrolyte to the colloidal suspension being studied. As shown in Fig. 3, TR-DLS data can be used to construct aggregation profiles. In this profile, a linear increase in Dh with time is observed during the initial period of aggregation during which doublet formation is predominant.[54,112–114] However, during later stages of aggregation, the observed growth rate slows down owing to particle depletion.
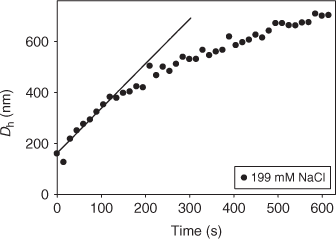
|
By fitting the initial linear region of Fig. 3, the initial particle growth rate, (dD/dt)t→0, can be determined and used to obtain understanding about the aggregation rate constant, k, with Eqn 1:
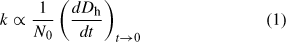
where N0 is the initial primary particle concentration.
For TR-DLS experiments that are conducted at the same initial particle concentration, Eqn 2 shows that (dDh/dt)t→0 values collected under different salt concentrations can be normalised to the diffusion-limited (dDh/dt)t→0 value ((dDh/dt)t→0,fast) to yield an empirical inverse stability ratio, 1/W (or attachment efficiency, α):
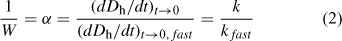
where the subscript ‘fast’ refers to diffusion-limited (favourable) aggregation conditions.
When the inverse stability ratio is plotted as a function of electrolyte concentration, the result is an inverse stability profile, which may be fitted empirically to yield the CCC.[36,113] As the CCC represents the minimum amount of electrolyte needed to destabilise the suspension such that diffusion-limited aggregation occurs, it provides a useful metric of colloidal stability for ENPs.
Deposition kinetics
As ENPs can undergo heteroaggregation with other types of colloidal particles, it is of interest to measure and model the kinetics of heteroaggregation. Currently, a multiangle static and dynamic laser light-scattering technique exists for the determination of heteroaggregation rate constants from laboratory measurements.[30,31,33] A principal limitation, however, is that this technique requires a sophisticated light-scattering setup with multiple detectors that may not be commonly accessible in many laboratories.
In recent years, the quartz crystal microbalance (QCM) has emerged as an alternative method to measure the interactions of ENPs with other solid surfaces. The QCM comprises a flow cell that houses a quartz crystal. Because quartz is a piezoelectric material, the crystal can be excited to oscillate at its resonance and overtone frequencies under an applied electric potential difference. To investigate the interactions between ENPs and solid surfaces, an ENP suspension of interest is directed across the crystal surface under a constant flow rate. Depending on the solution chemistry, some ENPs will deposit on the crystal surface and cause a shift in the resonance and overtone frequencies. Within certain limits, the increase in total mass of the crystal due to ENP deposition is proportional to the shift in the resonance and overtone frequencies.[115] Hence, the deposition kinetics of the ENPs on the crystal surface can be derived from the rate of frequency shift.
Traditionally, the interactions between colloidal particles and other solid surfaces have been investigated through column filtration experiments. However, the interpretation of results from such systems is more complex because of the multiple processes affecting the retention of colloidal particles within the filter media.[116,117] These processes will depend not only on the surface-scale factors that influence DLVO interactions between particles and collectors, but also on physical properties such as the geometry and size of the particles and collectors and the hydrodynamics within the column. Usually, the flow chambers of the QCM use much simpler geometries, such as in radial stagnation point and parallel-plate flow systems. To date, the QCM has been used to study the deposition of fullerene C60 nanoparticles,[113,118] titanium dioxide nanoparticles,[119] quantum dots,[120] viruses,[121] and ZVI nanoparticles[122,123] on silica surfaces.
Case studies
Fullerene C60 nanoparticles
Buckminsterfullerene C60 exhibits unique physical, chemical, and electronic properties due to its spherical molecular structure. Since its discovery,[124] there has been growing interest in utilising this novel molecule for applications in diverse fields, including biomedical and environmental engineering.[19,125] The major constraint to the use of fullerene C60, however, is its extreme hydrophobicity, which limits its aqueous solubility.[126,127] Despite the relative insolubility of C60 molecules, the molecules are able to form colloidally stable C60 nanoparticles in aqueous solutions using well-established preparation methods. Because C60 is likely to take the form of nanoparticles when released into aqueous systems, the colloidal properties of these nanoparticles are expected to control their environmental fate and transport. As evidence from recent studies has shown that C60 nanoparticles can be toxic,[128–131] a better understanding of their colloidal properties will also provide insights into nanoparticle–cell interactions, which may in turn help to elucidate the mechanisms of toxicity.
Preparation of C60 colloids
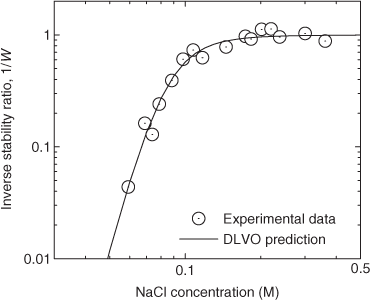
In one of the earliest reports on the preparation of a C60 colloidal suspension, Scrivens et al.[132] devised a synthesis method that involved first the dissolution of C60 in benzene, followed by sequential dilution with tetrahydrofuran (THF) and acetone. The solvent–C60 mixture was then introduced into water, which led to the formation of C60 colloidal particles. The final suspension was distilled to remove most of the organic solvents. This method of ‘solvent exchange’ has since been used and modified by other researchers to synthesise C60 nanoparticles. Other organic solvents that have been used include toluene[54] and ethanol,[133] and these solvents have been used either on their own or in various combinations. The type of solvents used and the parameters employed for the synthesis influenced the geometries and size distributions of the suspended C60 nanoparticles. For instance, solvent-exchange using THF produces angular nanoparticles, whereas exchange using toluene produces nanoparticles that are more rounded (Fig. 4). The reasons for these differences in C60 nanoparticle structure are, however, not fully understood.

|
In more recent years, it has been shown that colloidally stable C60 nanoparticles can be produced by a ‘top–down’ approach that agitates bulk C60 crystals, breaking them into smaller C60 particles, some of which are expected to be in the nanometre-scale range. Some possible methods include the stirring of C60 crystals in water for a prolonged period of time (from 2 weeks to 5 months)[134–138] and ultrasonication of C60 crystals in water.[139,140]
Colloidal stability
The unusual colloidal stability of the supposedly hydrophobic C60 nanoparticles has been highlighted in numerous studies.[132,141–143] Specifically, C60 nanoparticles are found to be extremely stable to aggregation under low ionic-strength conditions regardless of the synthesis method used, and even in the absence of surfactants or stabilisers, they have been reported to remain colloidally stable for months. The electrokinetic properties of C60 nanoparticles produced using various methods have been investigated through EPM measurements. Interestingly, these nanoparticles are found to exhibit negative EPMs under a wide range of pH conditions (from 2 to 12),[136,142–144] with the synthesis method playing an important role in determining the magnitude of the mobility. For instance, C60 nanoparticles produced through extended stirring in water were more negatively charged than those produced through solvent-exchange methods using THF[144] and toluene.[136] One possibility is that the difference in EPMs could be due to dissimilar surface chemistries resulting from the various preparation methods.
Influence of pH
More systematic electrokinetic studies have varied solution pH with the aim of understanding the source of the nanoparticle surface charge. C60 nanoparticles produced via either extended stirring in water or solvent exchange (with either THF or toluene) have exhibited increasingly negative EPM as pH is raised from 2 to 12 in the presence of monovalent electrolytes.[136,144] These results seem to imply that surface functional groups are a possible origin of C60 nanoparticle surface charge. It has also been suggested that the preferential adsorption of OH– ions on C60 nanoparticle surfaces could potentially cause the nanoparticles to be more negatively charged under higher pH conditions owing to the greater availability of OH– ions.[145] However, Brant et al.[144] measured the pH of the solution before and after the prolonged stirring required to disperse C60 in water and did not find any significant difference in the pH. At least for this system, it is therefore unlikely that the adsorption of OH– ions is the origin of surface charge.
Influence of electrolytes
The electrokinetic properties of C60 nanoparticles in different electrolytes have also been extensively investigated. In several studies, an increase in monovalent electrolyte concentration caused the EPM to become less negative,[136,144] consistent with charge screening effects. In most cases, divalent electrolytes were found to reduce the EPMs of C60 nanoparticles more effectively compared with monovalent electrolytes, either owing to improved charge screening or possible specific adsorption of divalent cations.[136,144,146] However, in the presence of low concentrations of CaCl2, Brant et al.[144] reported that charge reversal took place for C60 nanoparticles produced through solvent exchange with THF. These C60 nanoparticles, originally negatively charged at 0.01 mM CaCl2, became positively charged at 0.1 mM CaCl2, and subsequently became negatively charged again at ~0.3 mM CaCl2. The authors attributed the charge reversal and re-reversal to the preferential adsorption of Ca2+ and Cl– ions respectively. In a separate study, Wang et al.[146] presented contradictory results, showing that nanoparticles produced by solvent exchange remained negatively charged over the same range of CaCl2 concentrations studied by Brant et al. Further investigation is clearly required to verify the phenomenon of charge reversal (and re-reversal) in the presence of Ca2+.
Aggregation kinetics
The first reported study on the aggregation behaviour of C60 nanoparticles was conducted by Mchedlov-Petrossyan et al.[142] The authors determined the aggregation kinetics of C60 nanoparticles synthesised through solvent exchange with toluene by monitoring the absorbance of aggregating C60 suspensions at 500 nm. At pH 5–6, the CCCs were found to be 85 mM NaCl and 4.1 mM CaCl2. In a more recent study, TR-DLS was employed to investigate the aggregation kinetics of C60 nanoparticles produced through a similar method over a range of NaCl and CaCl2 concentrations at pH 5.2.[113] Fig. 5 presents the inverse stability ratios (1/W) obtained as a function of NaCl concentration. The results from the latter study revealed reaction-limited (i.e. 1/W < 1) and diffusion-limited (i.e. 1/W = 1) aggregation in the presence of NaCl and CaCl2, demonstrating that C60 nanoparticles undergo classical aggregation behaviour typical of charged colloidal systems in aqueous solutions. The CCCs obtained (120 mM NaCl and 4.8 mM CaCl2) were similar to, albeit slightly higher than the ones obtained by Mchedlov-Petrossyan et al. The dissimilarity in CCCs is likely due to slight variations in the preparation methods as well as the different experiments used to derive the aggregation kinetics. Through TR-DLS, C60 nanoparticles produced by extended stirring in water were also shown to undergo classical aggregation behaviour.[136] However, C60 nanoparticles produced by extended stirring were found to exhibit a much higher CCC (166 mM KCl) than C60 nanoparticles produced through solvent exchange with toluene (40 mM KCl), both at pH 5.5. These results corroborate findings from EPM measurements that the colloidal stability of C60 nanoparticles is influenced by the preparation method.
|
In their investigation of the aggregation kinetics of C60 nanoparticles produced from solvent exchange with toluene as well as extended stirring in water, Chen and Elimelech[113,136] compared the experimentally obtained inverse stability ratios with theoretical DLVO predictions. The authors found that particle stabilities for C60 nanoparticles produced by either solvent exchange or water stirring were in good agreement with the DLVO predictions (Fig. 5), which is very uncommon in studies of aggregation and deposition kinetics of colloidal particles under conditions where repulsive electrostatic interactions predominate.[29,47–49] Because DLVO theory is based on the simplifying assumption that the surface charge of the colloidal particle is uniformly distributed over the particle surface,[29] discrepancies between experimental results and theoretical predictions have often been attributed to heterogeneities in surface charge. Hence, one possible reason for the good agreement between DLVO theory and experimental data in Chen and Elimelech’s studies[113,136] could be that the charges are evenly distributed on the C60 nanoparticle surface.
Interactions with solid surfaces
The QCM has been used to investigate the deposition behaviour of C60 nanoparticles prepared through solvent exchange with toluene.[118] The deposition kinetics of the nanoparticles on silica surfaces were found to increase with increasing NaCl and CaCl2 concentrations until transport-limited deposition occurred at the CDCs. At salt concentrations equal to or greater than the CDC value, the surface charges of the nanoparticles and solid surfaces are sufficiently screened such that favourable deposition occurs. The CDC was found to be 32.1 mM in NaCl (at pH 5.5), much higher than NaCl concentrations in freshwater systems, while the CDC in CaCl2 was found to be 0.7 mM (at pH 5.5), which falls within the Ca2+ concentrations of hard freshwaters. Hence, the attachment of C60 nanoparticles to silica surfaces is more likely to be controlled by the concentration of divalent cations in freshwater systems. These results from QCM experiments are in agreement with the ones from column filtration experiments that have also shown C60 nanoparticle retention increasing with increasing salt concentrations.[146,147]
Influence of natural organic matter
In one of the earliest studies on the influence of NOM, Terashima and Nagao[148] stirred C60 material in solutions containing humic and fulvic acids. They reported that C60 colloidal particles were produced in much higher concentrations in the presence of the organic matter and that the particles were smaller than the ones produced in pure water.[148] Similar findings on the reduction in C60 particle sizes in the presence of organic matter have also been observed in other studies.[137,149] The influence of humic acid was also found to be much greater than fulvic acid.[148] This observation was attributed to humic acid having a higher affinity with C60 because it has a lower charge density and a larger aromatic backbone compared with fulvic acid.
The influence of NOM on the colloidal stability of C60 nanoparticles was systematically investigated by Chen and Elimelech through TR-DLS.[95] A low concentration of humic acid (1 mg L–1 total organic carbon) was sufficient to significantly reduce the aggregation kinetics of C60 nanoparticles (at a concentration of 0.3 mg L–1) in the presence of NaCl or MgCl2 electrolytes. Even in a high ionic strength solution (650 mM NaCl), the inverse stability ratio obtained in the presence of humic acid was only ~0.1. Under such conditions, the charges on the humic acid-coated nanoparticles are likely to be effectively screened, indicating that the key stabilising mechanism is steric repulsion. However, in the presence of high concentrations of CaCl2 electrolyte (>10 mM), bridging between the humic acid-coated C60 nanoparticles occurred, resulting in enhanced aggregation. These results clearly demonstrate that the influence of NOM on colloidal stability greatly depends on the solution chemistry.
Carbon nanotubes
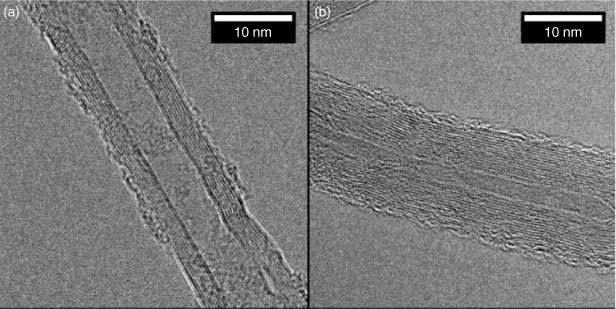
CNTs are long, thin, hollow cylinders, composed of one (single-walled CNTs, SWNTs) or many (MWNTs) concentric layers of graphenic carbon.[4] Representative TEM images of MWNTs are presented in Fig. 6. Because of their unique electronic and mechanical properties, CNTs are being used in a growing number of commercial applications such as composite materials,[150] electronic devices,[151] and fuel cells.[152] For unmodified CNTs, surface hydrophobicity prohibits their dispersion in water, making them difficult to use in many applications. This behaviour of pristine CNTs is consistent with the relative insolubility of C60 molecules. In contrast to C60 nanoparticles, however, stable dispersions of CNTs are not created by stirring CNTs in water or conducting solvent exchange. Instead, their surfaces must be modified either by covalently attaching dissociable, hydrophilic functional groups (e.g. carboxylic acids, hydroxyls, amines, or amides) or by adsorbing stabilising macromolecules or surfactants. In the present discussion, we focus on the colloidal properties of MWNTs.
|
Surface oxidation
To create stable CNTs, the pristine nanomaterial is often treated with aggressive oxidants (e.g. HNO3, HNO3/H2SO4, KMnO4, or H2O2),[153–156] cleaned, and dispersed using sonication. The principal effect of these oxidising treatments is to graft charged, hydrophilic functional groups (e.g. carboxyls and hydroxyls) into the exposed graphene sidewalls and at the tube ends. In aqueous suspensions, these functional groups facilitate electrostatic stabilisation and favourable interactions with water molecules, thus promoting stability.
Influence of electrolytes
Both EPM measurements and surface-charge titrations have shown that acid treatments impart a negative charge to the CNT surface.[86,157] Consistent with charge screening and charge neutralisation effects, increasing concentrations of monovalent and divalent electrolytes result in more rapid sedimentation of acid-washed MWNTs.[109] To shed additional light on the aggregation behaviour of MWNTs leading to sedimentation, Saleh et al.[158] and Smith et al.[36,86] have used TR-DLS to investigate MWNTs dispersed by high-power ultrasonication and acid treatment respectively. It is important to note that DLS measures particle size on the basis of a spherical particle approximation, far from the rod-like structure of CNTs. Although semi-flexible rod models have been developed for DLS that can provide information on MWNT length in non-aggregating systems,[159] a spherical approximation has been invoked to monitor the change in the particle size over time in the studies conducted on CNTs described here.
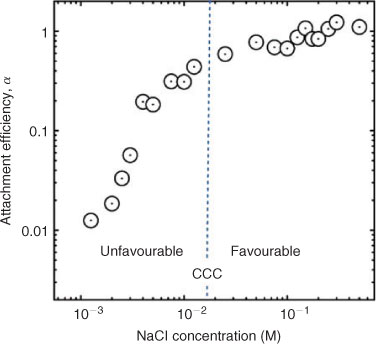
The results obtained using TR-DLS show that MWNTs exhibit reaction- and diffusion-limited aggregation regimes, comparable with trends observed in sedimentation studies (Fig. 7).[36,86,158] As stated, although DLVO theory can be applied in a quantitative way to spherical particles such as C60 nanoparticles (Fig. 5), its direct applicability to elongated particles and complex aggregates is less apparent and needs further investigation. However, results from studies on MWNT aggregation[36,86,158] show that qualitative tenets from DLVO theory do apply to electrolyte effects on the aggregation of MWNTs.
|
In the presence of NaCl, the CCC of acid-washed MWNTs (93 mM) is much higher than that of MWNTs prepared by high-power ultrasonication (25 mM) (Table 1). In similar solution chemistries, the EPMs for the acid-washed and ultrasonicated MWNTs are –3.5 × 10–8 and –1.9 × 10–8 m2 V–1 s–1 respectively. Thus, the larger absolute EPM values exhibited by the acid-washed MWNTs are consistent with their superior colloidal stability, which is believed to be a reflection of their higher surface concentration of dissociable functional groups.

|
Furthermore, analysis of Table 1 reveals that the acid-washed MWNTs appear to follow the Schulze–Hardy rule (SHR), which states that the CCC of highly charged particles is proportional to the counterion valence (Z) to the power of negative six (i.e. CCC ∝ Z–6).[29,43] Adherence to the SHR has also been observed in sedimentation studies with acid-washed CNTs.[109,110,160] In contrast, MWNTs prepared through high-power ultrasonication deviated from the SHR, yielding a CCC ∝ Z–3.3 relationship. This difference in the exponent on Z may be a reflection of the relatively low EPM exhibited by MWNTs prepared by ultrasonication, because the CCC is predicted to be proportional to Z–2 for particles with low ζ potentials.[29,43]
Influence of pH
The colloidal stability of acid-treated CNTs is sensitive to pH, and EPM studies have been conducted by several research groups[36,157,161–164] to determine the underlying cause. Typical trends for acid-treated CNTs are shown in Fig. 8. In this study, Schierz et al.[161] used MWNTs that were oxidised using a 3 : 1 H2SO4/HNO3 mixture for 16 h (solid triangles) and 42 h (hollow triangles). As shown in the figure, ζ potentials increase in magnitude as pH is increased from 1 to 4. This is likely due to an increase in the number of charged carboxylate groups (–COO–) with increasing pH. Above pH 4, the ζ potential asymptotically levels out, although Saleh et al.[158] reported a steady increase in the magnitude of EPM of ultrasonicated MWNTs as the pH increased from 3 to 12.
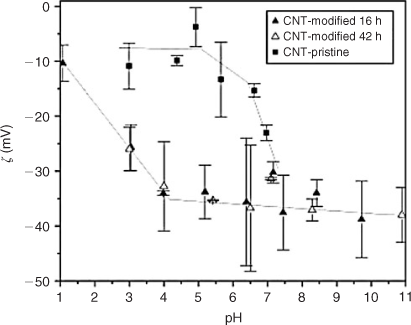
|
Based on the many studies that have shown that the EPM of MWNTs remains constant above pH ~6,[36,157,163,164] the colloidal stability of MWNTs should be independent of pH in basic solutions. This hypothesis, however, contradicts visual observations from sedimentation studies that show decreases in the rates of MWNT aggregation and sedimentation with increasing pH.[36,165] Furthermore, Smith et al.[36] showed with TR-DLS that the CCCs of the acid-washed MWNTs increased linearly from 25 mM NaCl at pH 3 to 239 mM NaCl at pH 10. Saleh et al.[158] reported similar pH-dependent increases in MWNT stability using TR-DLS. Although variations in EPM with pH cannot explain the observed increases in MWNT colloidal stability under basic conditions, Smith et al.[36] have noted that the titrated surface charge increases systematically with increasing pH and correlates well with stability. The fundamental reasons why titrations provide a metric for colloidal stability while EPM measurements fail are unknown. Interestingly, a similar discrepancy between the variations in EPM and charge densities (calculated from potentiometric titration) with pH has been observed for colloidal silica.[166]
Influence of surface oxygen concentration
It is well known that oxygen-containing functional groups help stabilise CNTs in suspension. Indeed, qualitative assessments of colloidal stability due to oxidation have been made by conducting sedimentation studies.[163] Increased concentrations of protic oxygen functional groups (as measured by titration methods such as Boehm titrations[126,165]) have generally resulted in suspensions that were more resistant to electrolyte-induced aggregation.[165]
More quantitative structure–property relationships linking surface chemistry to colloidal behaviour have recently been developed by Smith et al.[86] using a suite of oxidised MWNTs. In this study, XPS was used to quantify the surface oxygen concentration and determine the distribution of carboxyl, hydroxyl, and carbonyl groups on the MWNT surface. As shown in Fig. 9, the CCC increased linearly with MWNTs’ surface oxygen concentration at three different pH values. Furthermore, Smith et al.[86] showed that negatively charged surface carboxyl groups played the dominant role in stabilising CNT suspensions, although the other oxygen-containing functionalities were also found to contribute to stability.
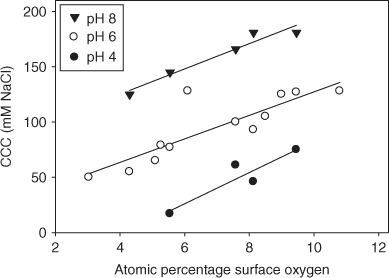
|
Influence of natural organic matter
Recent investigations have shown that the colloidal stability of pristine CNTs is significantly enhanced by the adsorption of NOM. Indeed, Hyung et al.[167] reported that adsorbed NOM stabilised MWNTs more effectively than the surfactant sodium dodecyl sulfate (SDS). Similar to previously discussed results from NOM-C60 studies,[148] results from studies in NOM-MWNT systems showed that colloidal suspensions of individually dispersed MWNTs could be produced by mere agitation (stirring and shaking). Similar results have been noted using a tannic acid surrogate for NOM.[168] These observations are in marked contrast to the case of oxidised MWNTs where aggressive sonication methods were required for dispersion in the absence of NOM.
To develop a mechanistic understanding of the effect that NOM has on MWNT stability, the sorption of humic and fulvic acids with MWNTs has been studied.[169–171] Results from these studies reveal that MWNT sorption capacity for humics is greater than that for fulvics,[169] similarly to the findings of Terashima and Nagao[148] in their study of NOM–C60 suspensions. The stronger affinity observed for humics was attributed to their greater aromatic character, which resulted in enhanced π–π interactions with the MWNTs’ surface.[169,170] The adsorption of fulvics was also shown to increase with increasing ionic strength and decrease with increasing pH.[171] Using TR-DLS, Saleh et al.[158] were able to compare the inverse stability ratios of MWNTs in the presence and absence of adsorbed humic acid. With the humic coating, the inverse stability ratios were one to two orders of magnitude lower than those observed for uncoated MWNTs, indicating a significant increase in colloidal stability. Despite the differences in colloidal behaviour, EPM measurements yielded similar results for both samples. This observation suggested that the stabilisation effect of NOM is purely a result of steric stabilisation.
In general, owing to the dynamic nature of sorption under various solution-phase conditions and the fact that fulvics and humics adsorb to different extents, predicting the stability of NOM-coated CNTs remains a challenge. However, Hyung et al.[169] have suggested that the stability conferred to the CNTs by NOM depends primarily on the concentration of NOM and not its exact composition. Clearly, additional studies are needed to uncover the detailed interactions of different NOM fractions with CNTs.
Comparing origins of surface charge of C60 nanoparticles and CNTs
Much more is known about the origin of surface charge for CNTs than for C60 nanoparticles. Studies that have systematically varied the degree of CNT surface oxidation have verified surface functional groups to be an important cause of surface charge for these materials. Various surface-charging mechanisms for C60 nanoparticles have been hypothesised and discussed,[136,143–145] and one clue is found in the apparently uniform charge distribution, as evidenced by the consistency of aggregation behaviour with DLVO theory.[113,136] Some of the suggested mechanisms include the presence of dissociable surface functional groups, the preferential adsorption of OH– or other anions, and charge transfer from solvent molecules. It is important to realise that there could be more than one origin of surface charge and that the contribution from each origin could be different for C60 nanoparticles synthesised through different methods. Experimental attempts to identify the origins of the surface charge on C60 nanoparticles have recently been made. For example, the EPMs of C60 nanoparticles synthesised through solvent exchange and extended stirring in water were measured and compared in solutions containing different anions (including Cl–, I– and SO42–).[136,144] The EPMs were not significantly different in the various electrolytes,[136] and no characteristic maximum (an indication of preferential adsorption[172]) was observed when EPMs were measured as a function of electrolyte concentration.[144] Thus, it seems unlikely that anion adsorption is the origin of the surface charge. Based on the results obtained by Brant et al.,[144] who observed no significant change in pH after the mixing process, the adsorption of OH– ions has also been ruled out as a possible charging mechanism for C60 nanoparticles formed from extended stirring in water.
Prior studies have shown that oxidative treatments can result in the formation of oxygen-containing functional groups on CNTs. Because the degree of curvature for CNTs is confined to only a single dimension, C60 is expected to be more susceptible to chemical modification than CNTs.[19] In a recent study, Chen and Elimelech[136] used XPS analysis to reveal an increase in oxygen content on the surface of C60 material that had been stirred in water for a prolonged period of time. In another study, Labille et al.[140] reported the presence of hydroxyl functional groups on C60 nanoparticles produced through sonication in water based on their results from Fourier transform infrared (FTIR) and solid-state 1H nuclear magnetic resonance (NMR) analyses. In addition, numerous electrokinetic studies have shown that C60 nanoparticles become more negatively charged at higher pH conditions.[136,144,145] Thus, all evidence seems to point to the fact that ionic surface functional groups are at least in part responsible for the surface charge on C60 nanoparticles. However, it is still unclear why the nanoparticles exhibit negative charge even at pH 2 when most functional groups should remain protonated.[135,136,144]
Clearly, detailed and quantitative knowledge of the surface chemistry of C60 nanoparticless is a necessary step to reveal the origin of the surface charge on these ENPs. However, to date, no studies have been conducted to provide functional group-specific information on the surface chemistry of C60 nanoparticles. In this regard, XPS performed in conjunction with CD would be a potential method suitable to test for and identify the nature and concentration of surface functionalities on C60 nanoparticles. The key challenge is to produce sufficient amounts of dried C60 nanoparticles (at least a few milligrammes for XPS analysis) especially when the current synthesis techniques only yield C60 nanoparticle suspensions with concentrations ranging from a few to tens of milligrams per litre.
Research needs and future directions
It is clear from the discussion in this review article that one necessary requirement to rationalise the colloidal stability of ENPs is that the physicochemical properties of the ENPs themselves must be rigorously characterised and understood. In particular, there is a need for a better understanding of the surface chemistry of ENPs, which is known to depend on the synthesis and preparation techniques employed as well as subsequent modifications. In addition to deliberate surface modification, ENPs released into natural and engineered systems may undergo light-based transformations, oxidations or interactions with other aquatic species that can result in changes to their surface composition.[173–175] In this review, we have shown that XPS used in conjunction with CD can be a powerful technique for the determination of the surface functionalities of ENPs. The ability to accurately identify and quantify the surface functional groups appears to open the door to develop structure–property relationships that can be used to predict the colloidal stability as well as other properties (e.g. toxicity and reactivity) of ENPs based on their surface chemistry.
Currently, most studies have focussed on the homoaggregation kinetics of ENPs. However, heteroaggregation between ENPs and naturally occurring colloids is almost certain to be a major process in natural aquatic systems. To date, quantitative studies on the heteroaggregation kinetics of ENPs are limited owing to the complexity of the systems involved. QCM has recently emerged as an option that can allow the direct measurements of ENP deposition kinetics on solid surfaces, but this approach still lacks the desired ability to capture the heteroaggregation kinetics between ENP and colloidal particles of varying types and sizes. Thus, there is a need for the development of novel and more accessible experimental techniques for the study of heteroaggregation between ENPs and other colloidal particles.
The homoaggregation and heteroaggregation (deposition) behaviour of ENPs are controlled by ENP–ENP and ENP–solid-surface interactions respectively. Even though current experimental results have shown that the aggregation and deposition behaviour of ENPs is qualitatively in agreement with DLVO theory, more study on the effects of non-DLVO forces is required. The most direct approach is to probe the interfacial forces between ENPs (or between ENPs and solid surfaces) under varying solution conditions. Recent developments have allowed the use of AFM force measurements between ENPs and solid surfaces. However, the preparation of a nanometre-sized colloidal probe for such measurements still remains a complicated task and an as yet unsolved challenge for many materials.
The mobility, bioavailability, and toxicity of ENPs are controlled by the aggregate structure in which they reside. Even though the influence of aggregation kinetics on the compactness of aggregate structures formed from monodisperse spherical particles is well known, few studies have been conducted on the aggregate structures formed from suspensions of polydisperse and non-spherical particles, which are arguably the more typical cases for ENP use and release. For instance, it is expected that the bundling of CNTs will result in a more compact structure than the random cross-linking between them. Similar questions can be asked about the heteroaggregate structures formed from particles of different shapes and sizes, such as the possible combination of CNTs and naturally occurring silica nanoparticles. Cryogenic TEM (Cryo-TEM) offers the potential to acquire detailed molecular-scale images of ENP aggregation processes as well as the aggregates formed in situ under aqueous conditions.[176–178]
Acknowledgements
K. L. Chen acknowledges financial support from Oak Ridge Associated Universities. D. H. Fairbrother and W. P. Ball acknowledge financial support from the National Science Foundation (grant no. BES0731147), the Environmental Protection Agency (grant no. RD-8338570-0), and the Institute for Nanobiotechnology (INBT) at Johns Hopkins University (JHU). The authors would also like to acknowledge the scientific discussions and insights provided by Professor Charlies O’Melia (Department of Geography and Environmental Engineering, JHU).
[1]
L. C. Qin ,
X. L. Zhao ,
K. Hirahara ,
Y. Miyamoto ,
Y. Ando ,
S. Iijima ,
Materials science – the smallest carbon nanotube.
Nature 2000
, 408, 50.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[Verified 22 January 2010]
[8]
[9]
[10]
J. Theron ,
J. A. Walker ,
T. E. Cloete ,
Nanotechnology and water treatment: applications and emerging opportunities.
Crit. Rev. Microbiol. 2008
, 34, 43.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[Verified 22 January 2010]
[19]
M. S. Mauter ,
M. Elimelech ,
Environmental applications of carbon-based nanomaterials.
Environ. Sci. Technol. 2008
, 42, 5843.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |

[20]
L. Li ,
Y. Xing ,
Pt-Ru nanoparticles supported on carbon nanotubes as methanol fuel cell catalysts.
J. Phys. Chem. C 2007
, 111, 2803.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[21]
Y. Xing ,
Synthesis and electrochemical characterization of uniformly dispersed high-loading Pt nanoparticles on sonochemically treated carbon nanotubes.
J. Phys. Chem. B 2004
, 108, 19255.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[22]
A. R. Köhler ,
C. Som ,
A. Helland ,
F. Gottschalk ,
Studying the potential release of carbon nanotubes throughout the application life cycle.
J. Clean. Prod. 2008
, 16, 927.
| Crossref | GoogleScholarGoogle Scholar |
[23]
N. C. Mueller ,
B. Nowack ,
Exposure modeling of engineered nanoparticles in the environment.
Environ. Sci. Technol. 2008
, 42, 4447.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[24]
X. Zhu ,
L. Zhu ,
Z. Duan ,
R. Qi ,
Y. Li ,
Y. Lang ,
Comparative toxicity of several metal oxide nanoparticle aqueous suspensions to zebrafish (Danio rerio) early developmental stage.
J. Environ. Sci. Health Part A Tox. Hazard. Subst. Environ. Eng. 2008
, 43, 278.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[25]
S. Kang ,
M. S. Mauter ,
M. Elimelech ,
Microbial cytotoxicity of carbon-based nanomaterials: implications for river water and wastewater effluent.
Environ. Sci. Technol. 2009
, 43, 2648.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[26]
S. Kang ,
M. S. Mauter ,
M. Elimelech ,
Physicochemical determinants of multiwalled carbon nanotube bacterial cytotoxicity.
Environ. Sci. Technol. 2008
, 42, 7528.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[27]
B. J. Panessa-Warren ,
M. M. Maye ,
J. B. Warren ,
K. M. Crosson ,
Single-walled carbon nanotube reactivity and cytotoxicity following extended aqueous exposure.
Environ. Pollut. 2009
, 157, 1140.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[28]
T. M. Sager ,
D. W. Porter ,
V. A. Robinson ,
W. G. Lindsley ,
D. E. Schwegler-Berry ,
V. Castranova ,
Improved method to disperse nanoparticles for in vitro and in vivo investigation of toxicity.
Nanotoxicology 2007
, 1, 118.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[29]
[30]
W. L. Yu ,
M. Borkovec ,
Distinguishing heteroaggregation from homoaggregation in mixed binary particle suspensions by multiangle static and dynamic light scattering.
J. Phys. Chem. B 2002
, 106, 13106.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[31]
W. Lin ,
M. Kobayashi ,
M. Skarba ,
C. D. Nu ,
P. Galletto ,
M. Borkovec ,
Heteroaggregation in binary mixtures of oppositely charged colloidal particles.
Langmuir 2006
, 22, 1038.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[32]
P. Galletto ,
W. Lin ,
M. Borkovec ,
Measurement of heteroaggregation rate constants by simultaneous static and dynamic light scattering.
Phys. Chem. Chem. Phys. 2005
, 7, 1464.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[33]
W. L. Yu ,
E. Matijević ,
M. Borkovec ,
Absolute heteroaggregation rate constants by multiangle static and dynamic light scattering.
Langmuir 2002
, 18, 7853.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[34]
K. M. Yao ,
M. M. Habibian ,
C. R. O’Melia ,
Water and waste water filtration – concepts and applications.
Environ. Sci. Technol. 1971
, 5, 1105.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[35]
[36]
B. Smith ,
K. Wepasnick ,
K. E. Schrote ,
A. H. Bertele ,
W. P. Ball ,
C. O’Melia ,
D. H. Fairbrother ,
Colloidal properties of aqueous suspensions of acid-treated, multi-walled carbon nanotubes.
Environ. Sci. Technol. 2009
, 43, 819.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[37]
D. Fornasiero ,
F. Grieser ,
The kinetics of electrolyte-induced aggregation of Carey Lea silver colloids.
J. Colloid Interface Sci. 1991
, 141, 168.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[38]
Z. S. Pillai ,
P. V. Kamat ,
What factors control the size and shape of silver nanoparticles in the citrate ion reduction method?
J. Phys. Chem. B 2004
, 108, 945.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[39]
Y. Rong ,
H. Z. Chen ,
G. Wu ,
M. Wang ,
Preparation and characterization of titanium dioxide nanoparticle/polystyrene composites via radical polymerization.
Mater. Chem. Phys. 2005
, 91, 370.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[40]
K. Yang ,
B. S. Xing ,
Sorption of phenanthrene by humic acid-coated nanosized TiO2 and ZnO.
Environ. Sci. Technol. 2009
, 43, 1845.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[41]
L. K. Limbach ,
Y. C. Li ,
R. N. Grass ,
T. J. Brunner ,
M. A. Hintermann ,
M. Muller ,
D. Gunther ,
W. J. Stark ,
Oxide nanoparticle uptake in human lung fibroblasts: effects of particle size, agglomeration, and diffusion at low concentrations.
Environ. Sci. Technol. 2005
, 39, 9370.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[42]
A. K. Gupta ,
M. Gupta ,
Synthesis and surface engineering of iron oxide nanoparticles for biomedical applications.
Biomaterials 2005
, 26, 3995.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[43]
[44]
B. V. Derjaguin ,
L. D. Landau ,
Theory of the stability of strongly charged lyophobic sols and of the adhesion of strongly charged particles in solutions of electrolytes.
Acta Physicochim. URSS 1941
, 14, 733.
[45]
[46]
S. H. Behrens ,
M. Borkovec ,
Influence of the secondary interaction energy minimum on the early stages of colloidal aggregation.
J. Colloid Interface Sci. 2000
, 225, 460.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[47]
M. Elimelech ,
C. R. O’Melia ,
Effect of particle-size on collision efficiency in the deposition of Brownian particles with electrostatic energy barriers.
Langmuir 1990
, 6, 1153.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[48]
S. H. Behrens ,
M. Borkovec ,
P. Schurtenberger ,
Aggregation in charge-stabilized colloidal suspensions revisited.
Langmuir 1998
, 14, 1951.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[49]
S. H. Behrens ,
D. I. Christl ,
R. Emmerzael ,
P. Schurtenberger ,
M. Borkovec ,
Charging and aggregation properties of carboxyl latex particles: experiments versus DLVO theory.
Langmuir 2000
, 16, 2566.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[50]
D. H. Napper ,
Steric stabilization.
J. Colloid Interface Sci. 1977
, 58, 390.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[51]
E. Dickinson ,
L. Eriksson ,
Particle flocculation by adsorbing polymers.
Adv. Colloid Interface Sci. 1991
, 34, 1.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[52]
M. B. Einarson ,
J. C. Berg ,
Electrosteric stabilization of colloidal latex dispersions.
J. Colloid Interface Sci. 1993
, 155, 165.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[53]
A. Pettersson ,
G. Marino ,
A. Pursiheimo ,
J. B. Rosenholm ,
Electrosteric stabilization of Al2O3, ZrO2, and 3Y-ZrO2 suspensions: effect of dissociation and type of polyelectrolyte.
J. Colloid Interface Sci. 2000
, 228, 73.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[54]
K. L. Chen ,
S. E. Mylon ,
M. Elimelech ,
Aggregation kinetics of alginate-coated hematite nanoparticles in monovalent and divalent electrolytes.
Environ. Sci. Technol. 2006
, 40, 1516.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[55]
G. Fritz ,
V. Schadler ,
N. Willenbacher ,
N. J. Wagner ,
Electrosteric stabilization of colloidal dispersions.
Langmuir 2002
, 18, 6381.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[56]
I. Yildiz ,
B. McCaughan ,
S. F. Cruickshank ,
J. F. Callan ,
F. M. Raymo ,
Biocompatible CdSe-ZnS core-shell quantum dots coated with hydrophilic polythiols.
Langmuir 2009
, 25, 7090.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[57]
W. W. Yu ,
E. Chang ,
J. C. Falkner ,
J. Y. Zhang ,
A. M. Al-Somali ,
C. M. Sayes ,
J. Johns ,
R. Drezek ,
V. L. Colvin ,
Forming biocompatible and non-aggregated nanocrystals in water using amphiphilic polymers.
J. Am. Chem. Soc. 2007
, 129, 2871.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[58]
T. Phenrat ,
N. Saleh ,
K. Sirk ,
H. J. Kim ,
R. D. Tilton ,
G. V. Lowry ,
Stabilization of aqueous nanoscale zero-valent iron dispersions by anionic polyelectrolytes: adsorbed anionic polyelectrolyte layer properties and their effect on aggregation and sedimentation.
J. Nanopart. Res. 2008
, 10, 795.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[59]
A. Tiraferri ,
K. L. Chen ,
R. Sethi ,
M. Elimelech ,
Reduced aggregation and sedimentation of zero-valent iron nanoparticles in the presence of guar gum.
J. Colloid Interface Sci. 2008
, 324, 71.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[60]
S. R. Kanel ,
R. R. Goswami ,
T. P. Clement ,
M. O. Barnett ,
D. Zhao ,
Two-dimensional transport characteristics of surface stabilized zero-valent iron nanoparticles in porous media.
Environ. Sci. Technol. 2008
, 42, 896.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[61]
H. H. Huang ,
X. P. Ni ,
G. L. Loy ,
C. H. Chew ,
K. L. Tan ,
F. C. Loh ,
J. F. Deng ,
G. Q. Xu ,
Photochemical formation of silver nanoparticles in poly(N-vinylpyrrolidone).
Langmuir 1996
, 12, 909.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[62]
L. Quaroni ,
G. Chumanov ,
Preparation of polymer-coated functionalized silver nanoparticles.
J. Am. Chem. Soc. 1999
, 121, 10642.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[63]
C. L. Tiller ,
C. R. O’Melia ,
Natural organic-matter and colloidal stability – models and measurements.
Colloids Surf. A Physicochem. Eng. Asp. 1993
, 73, 89.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[64]
E. Tipping ,
D. C. Higgins ,
The effect of adsorbed humic substances on the colloid stability of hematite particles.
Colloids Surf. 1982
, 5, 85.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[65]
R. Amal ,
J. A. Raper ,
T. D. Waite ,
Effect of fulvic acid adsorption on the aggregation kinetics and structure of hematite particles.
J. Colloid Interface Sci. 1992
, 151, 244.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[66]
I. Heidmann ,
I. Christl ,
R. Kretzschmar ,
Aggregation kinetics of kaolinite–fulvic acid colloids as affected by the sorption of Cu and Pb.
Environ. Sci. Technol. 2005
, 39, 807.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[67]
J. Buffle ,
K. J. Wilkinson ,
S. Stoll ,
M. Filella ,
J. W. Zhang ,
A generalized description of aquatic colloidal interactions: the three-colloidal component approach.
Environ. Sci. Technol. 1998
, 32, 2887.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[68]
[69]
G. G. Leppard ,
Nanoparticles in the environment as revealed by transmission electron microscopy: detection, characterisation and activities.
Current Nanoscience 2008
, 4, 278.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[70]
[71]
Y. Sun ,
Y. Xia ,
Shape-controlled synthesis of gold and silver nanoparticles.
Science 2002
, 298, 2176.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[72]
J. R. Lead ,
K. J. Wilkinson ,
Aquatic colloids and nanoparticles: current knowledge and future trends.
Environ. Chem. 2006
, 3, 159.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[73]
C. N. R. Rao ,
K. Biswas ,
Characterization of nanomaterials by physical methods.
Annu. Rev. Anal. Chem. 2009
, 2, 435.
|
CAS |
[74]
H. Yang ,
P. H. Holloway ,
Enhanced photoluminescence from CdS : Mn/ZnS core/shell quantum dots.
Appl. Phys. Lett. 2003
, 82, 1965.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[75]
H. W. Kim ,
S. H. Shim ,
Branched structures of tin oxide one-dimensional nanomaterials.
Vacuum 2008
, 82, 1395.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[76]
H. J. Lee ,
S. Y. Yeo ,
S. H. Jeong ,
Antibacterial effect of nanosized silver colloidal solution on textile fabrics.
J. Mater. Sci. 2003
, 38, 2199.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[77]
R. H. Yang ,
L. W. Chang ,
J. P. Wu ,
M. H. Tsai ,
H. J. Wang ,
Y. C. Kuo ,
T. K. Yeh ,
C. S. Yang ,
P. Lin ,
Persistent tissue kinetics and redistribution of nanoparticles, Quantum Dot 705, in mice: ICP-MS quantitative assessment.
Environ. Health Perspect. 2007
, 115, 1339.
|
CAS |
PubMed |
[78]
C. Baleizão ,
B. Gigante ,
H. Garcia ,
A. Corma ,
Vanadyl salen complexes covalently anchored to single-wall carbon nanotubes as heterogeneous catalysts for the cyanosilylation of aldehydes.
J. Catal. 2004
, 221, 77.
| Crossref | GoogleScholarGoogle Scholar |
[79]
[80]
L. A. Langley ,
D. H. Fairbrother ,
Effect of wet chemical treatments on the distribution of surface oxides on carbonaceous materials.
Carbon 2007
, 45, 47.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[81]
L. A. Langley ,
D. E. Villanueva ,
D. H. Fairbrother ,
Quantification of surface oxides on carbonaceous materials.
Chem. Mater. 2006
, 18, 169.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[82]
N. M. Washton ,
S. L. Brantley ,
K. T. Mueller ,
Probing the molecular-level control of aluminosilicate dissolution: a sensitive solid-state NOM proxy for reactive surface area.
Geochim. Cosmochim. Acta 2008
, 72, 5949.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[83]
T. Preoanin ,
N. Kallay ,
Application of ‘mass titration’ to determination of surface charge of metal oxides.
Croat. Chem. Acta 1998
, 71, 1117.
[84]
S. Brunauer ,
P. H. Emmett ,
E. Teller ,
Adsorption of gases in multimolecular layers.
J. Am. Chem. Soc. 1938
, 60, 309.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[85]
F. Li ,
Y. Wang ,
D. Wang ,
F. Wei ,
Characterization of single-wall carbon nanotubes by N2 adsorption.
Carbon 2004
, 42, 2375.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[86]
B. Smith ,
K. Wepasnick ,
K. E. Schrote ,
H. H. Cho ,
W. P. Ball ,
D. H. Fairbrother ,
Influence of surface oxides on the colloidal stability of multiwalled carbon nanotubes: a structure–property relationship.
Langmuir 2009
, 25, 9767.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[87]
R. H. Ottewill ,
J. N. Shaw ,
Electrophoretic studies on polystyrene latices.
J. Electroanal. Chem. 1972
, 37, 133.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[88]
Y. P. Sun ,
X. Q. Li ,
W. X. Zhang ,
H. P. Wang ,
A method for the preparation of stable dispersion of zero-valent iron nanoparticles.
Colloids Surf. 2007
, 308, 60.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[89]
M. L. Usrey ,
N. Nair ,
D. E. Agnew ,
C. F. Pina ,
M. S. Strano ,
Controlling the electrophoretic mobility of single-walled carbon nanotubes: a comparison of theory and experiment.
Langmuir 2007
, 23, 7768.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[90]
H. J. Butt ,
B. Cappella ,
M. Kappl ,
Force measurements with the atomic force microscope: technique, interpretation and applications.
Surf. Sci. Rep. 2005
, 59, 1.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[91]
W. A. Ducker ,
T. J. Senden ,
R. M. Pashley ,
Direct measurement of colloidal forces using an atomic force microscope.
Nature 1991
, 353, 239.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[92]
Q. K. Ong ,
I. Sokojov ,
Attachment of nanoparticles to the AFM tips for direct measurements of interaction between a single nanoparticle and surfaces.
J. Colloid Interface Sci. 2007
, 310, 385.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[93]
S. Akita ,
Y. Nakayama ,
S. Mizooka ,
Y. Takano ,
T. Okawa ,
Y. Miyatake ,
S. Yamanaka ,
M. Tsuji ,
T. Nosaka ,
Nanotweezers consisting of carbon nanotubes operating in an atomic force microscope.
Appl. Phys. Lett. 2001
, 79, 1691.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[94]
H. J. Dai ,
J. H. Hafner ,
A. G. Rinzler ,
D. T. Colbert ,
R. E. Smalley ,
Nanotubes as nanoprobes in scanning probe microscopy.
Nature 1996
, 384, 147.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[95]
K. L. Chen ,
S. E. Mylon ,
M. Elimelech ,
Enhanced aggregation of alginate-coated iron oxide (hematite) nanoparticles in the presence of calcium, strontium, and barium cations.
Langmuir 2007
, 23, 5920.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[96]
J. Rarity ,
Flocculation – colloids stick to fractal rules.
Nature 1989
, 339, 340.
| Crossref | GoogleScholarGoogle Scholar |
[97]
M. Y. Lin ,
H. M. Lindsay ,
D. A. Weitz ,
R. C. Ball ,
R. Klein ,
P. Meakin ,
Universality in colloid aggregation.
Nature 1989
, 339, 360.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[98]
D. A. Weitz ,
J. S. Huang ,
M. Y. Lin ,
J. Sung ,
Limits of the fractal dimension for irreversible kinetic aggregation of gold colloids.
Phys. Rev. Lett. 1985
, 54, 1416.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[99]
Z. K. Zhou ,
P. Q. Wu ,
B. J. Chu ,
Cationic surfactant-induced fractal silica aggregates – a light-scattering study.
J. Colloid Interface Sci. 1991
, 146, 541.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[100]
R. Amal ,
J. A. Raper ,
T. D. Waite ,
Fractal structure of hematite aggregates.
J. Colloid Interface Sci. 1990
, 140, 158.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[101]
J. W. Zhang ,
J. Buffle ,
Multi-method determination of the fractal dimension of hematite aggregates.
Colloid. Surface A 1996
, 107, 175.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[102]
Z. K. Zhou ,
B. J. Chu ,
Light-scattering study on the fractal aggregates of polystyrene spheres – kinetic and structural approaches.
J. Colloid Interface Sci. 1991
, 143, 356.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[103]
Q. Chen ,
C. Saltiel ,
S. Manickavasagam ,
L. S. Schadler ,
R. W. Siegel ,
H. C. Yang ,
Aggregation behavior of single-walled carbon nanotubes in dilute aqueous suspension.
J. Colloid Interface Sci. 2004
, 280, 91.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[104]
A. Y. Kim ,
J. C. Berg ,
Fractal heteroaggregation of oppositely charged colloids.
J. Colloid Interface Sci. 2000
, 229, 607.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[105]
J. M. López-López ,
A. Schmitt ,
A. Moncho-Jordá ,
R. Hidalgo-Álvarez ,
Stability of binary colloids: kinetic and structural aspects of heteroaggregation processes.
Soft Matter 2006
, 2, 1025.
| Crossref | GoogleScholarGoogle Scholar |
[106]
M. A. Chappell ,
A. J. George ,
K. M. Dontsova ,
B. E. Porter ,
C. L. Price ,
P. Zhou ,
E. Morikawa ,
A. J. Kennedy ,
J. A. Steevens ,
Surfactive stabilization of multiwalled carbon nanotube dispersions with dissolved humic substances.
Environ. Pollut. 2009
, 157, 1081.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[107]
T. Phenrat ,
N. Saleh ,
K. Sirk ,
R. D. Tilton ,
G. V. Lowry ,
Aggregation and sedimentation of aqueous nanoscale zero-valent iron dispersions.
Environ. Sci. Technol. 2007
, 41, 284.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[108]
A. S. Teot ,
S. L. Daniels ,
Flocculation of negatively charged colloids by inorganic cations and anionic polyelectrolytes.
Environ. Sci. Technol. 1969
, 3, 825.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[109]
A. N. Giordano ,
H. Chaturvedi ,
J. C. Poler ,
Critical coagulation concentrations for carbon nanotubes in non-aqueous solvent.
J. Phys. Chem. C 2007
, 111, 11583.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[110]
M. Sano ,
J. Okamura ,
S. Shinkai ,
Colloidal nature of single-walled carbon nanotubes in electrolyte solution: the Schulze–Hardy Rule.
Langmuir 2001
, 17, 7172.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[111]
B. Tezak ,
E. Matijević ,
K. Schulz ,
Coagulation of hydrophobic sols in statu nascendi. I. Determination of coagulation values.
J. Phys. Chem. 1951
, 55, 1557.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[112]
H. Holthoff ,
S. U. Egelhaaf ,
M. Borkovec ,
P. Schurtenberger ,
H. Sticher ,
Coagulation rate measurements of colloidal particles by simultaneous static and dynamic light scattering.
Langmuir 1996
, 12, 5541.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[113]
K. L. Chen ,
M. Elimelech ,
Aggregation and deposition kinetics of fullerene (C60) nanoparticles.
Langmuir 2006
, 22, 10994.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[114]
Y. T. He ,
J. Wan ,
T. Tokunaga ,
Kinetic stability of hematite nanoparticles: the effect of particle sizes.
J. Nanopart. Res. 2008
, 10, 321.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[115]
G. Sauerbrey ,
Verwendung von Schwingquarzen zur Wagung Dunner Schichten und zur Mikrowagung.
Z. Phys. 1959
, 155, 206.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[116]
S. A. Bradford ,
S. R. Yates ,
M. Bettahar ,
J. Simunek ,
Physical factors affecting the transport and fate of colloids in saturated porous media.
Water Resour. Res. 2002
, 38, 1327.
| Crossref | GoogleScholarGoogle Scholar |
[117]
S. A. Bradford ,
J. Simunek ,
M. Bettahar ,
M. T. Van Genuchten ,
S. R. Yates ,
Modeling colloid attachment, straining, and exclusion in saturated porous media.
Environ. Sci. Technol. 2003
, 37, 2242.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[118]
K. L. Chen ,
M. Elimelech ,
Interaction of fullerene (C60) nanoparticles with humic acid and alginate-coated silica surfaces: measurements, mechanisms, and environmental implications.
Environ. Sci. Technol. 2008
, 42, 7607.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[119]
J. Fatisson ,
R. F. Domingos ,
K. J. Wilkinson ,
N. Tufenkji ,
Deposition of TiO2 nanoparticles onto silica measured using a quartz crystal microbalance with dissipation monitoring.
Langmuir 2009
, 25, 6062.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[120]
I. R. Quevedo ,
N. Tufenkji ,
Influence of solution chemistry on the deposition and detachment kinetics of a CdTe quantum dot examined using a quartz crystal microbalance.
Environ. Sci. Technol. 2009
, 43, 3176.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[121]
B. L. Yuan ,
M. Pham ,
T. H. Nguyen ,
Deposition kinetics of bacteriophage MS2 on a silica surface coated with natural organic matter in a radial stagnation point flow cell.
Environ. Sci. Technol. 2008
, 42, 7628.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[122]
K. M. Sirk ,
N. B. Saleh ,
T. Phenrat ,
H. J. Kim ,
B. Dufour ,
J. Ok ,
P. L. Golas ,
K. Matyjaszewski ,
G. V. Lowry ,
R. D. Tilton ,
Effect of adsorbed polyelectrolytes on nanoscale zero-valent iron particle attachment to soil surface models.
Environ. Sci. Technol. 2009
, 43, 3803.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[123]
N. Saleh ,
K. Sirk ,
Y. Q. Liu ,
T. Phenrat ,
B. Dufour ,
K. Matyjaszewski ,
R. D. Tilton ,
G. V. Lowry ,
Surface modifications enhance nanoiron transport and NAPL targeting in saturated porous media.
Environ. Eng. Sci. 2007
, 24, 45.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[124]
H. W. Kroto ,
J. R. Heath ,
S. C. Obrien ,
R. F. Curl ,
R. E. Smalley ,
C60: buckminsterfullerene.
Nature 1985
, 318, 162.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[125]
A. W. Jensen ,
S. R. Wilson ,
D. I. Schuster ,
Biological applications of fullerenes.
Bioorg. Med. Chem. 1996
, 4, 767.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[126]
Q. C. Ying ,
J. Zhang ,
D. H. Liang ,
W. Nakanishi ,
H. Isobe ,
E. Nakamura ,
B. Chu ,
Fractal behavior of functionalized fullerene aggregates. I. Aggregation of two-handed tetraaminofullerene with DNA.
Langmuir 2005
, 21, 9824.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[127]
R. G. Alargova ,
S. Deguchi ,
K. Tsujii ,
Stable colloidal dispersions of fullerenes in polar organic solvents.
J. Am. Chem. Soc. 2001
, 123, 10460.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[128]
C. M. Sayes ,
J. D. Fortner ,
W. Guo ,
D. Lyon ,
A. M. Boyd ,
K. D. Ausman ,
Y. J. Tao ,
B. Sitharaman ,
et al. The differential cytotoxicity of water-soluble fullerenes.
Nano Lett. 2004
, 4, 1881.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[129]
J. D. Fortner ,
D. Y. Lyon ,
C. M. Sayes ,
A. M. Boyd ,
J. C. Falkner ,
E. M. Hotze ,
L. B. Alemany ,
Y. J. Tao ,
et al. C60 in water: nanocrystal formation and microbial response.
Environ. Sci. Technol. 2005
, 39, 4307.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[130]
S. J. Klaine ,
P. J. J. Alvarez ,
G. E. Batley ,
T. F. Fernandes ,
R. D. Handy ,
D. Y. Lyon ,
S. Mahendra ,
M. J. McLaughlin ,
J. R. Lead ,
Nanomaterials in the environment: behavior, fate, bioavailability, and effects.
Environ. Toxicol. Chem. 2008
, 27, 1825.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[131]
R. D. Handy ,
F. von der Kammer ,
J. R. Lead ,
M. Hassellov ,
R. Owen ,
M. Crane ,
The ecotoxicology and chemistry of manufactured nanoparticles.
Ecotoxicology 2008
, 17, 287.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[132]
W. A. Scrivens ,
J. M. Tour ,
K. E. Creek ,
L. Pirisi ,
Synthesis of 14C-labeled C60, its suspension in water, and its uptake by human keratinocytes.
J. Am. Chem. Soc. 1994
, 116, 4517.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[133]
A. Dhawan ,
J. S. Taurozzi ,
A. K. Pandey ,
W. Q. Shan ,
S. M. Miller ,
S. A. Hashsham ,
V. V. Tarabara ,
Stable colloidal dispersions of C60 fullerenes in water: evidence for genotoxicity.
Environ. Sci. Technol. 2006
, 40, 7394.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[134]
X. K. Cheng ,
A. T. Kan ,
M. B. Tomson ,
Naphthalene adsorption and desorption from aqueous C60 fullerene.
J. Chem. Eng. Data 2004
, 49, 675.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[135]
D. Bouchard ,
X. Ma ,
C. Isaacson ,
Colloidal properties of aqueous fullerenes: isoelectric points and aggregation kinetics of C60 and C60 derivatives.
Environ. Sci. Technol. 2009
, 43, 6597.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[136]
K. L. Chen ,
M. Elimelech ,
Relating colloidal stability of fullerene (C60) nanoparticles to nanoparticle charge and electrokinetic properties.
Environ. Sci. Technol. 2009
, 43, 7270.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[137]
L. K. Duncan ,
J. R. Jinschek ,
P. J. Vikesland ,
C60 colloid formation in aqueous systems: effects of preparation method on size, structure, and surface charge.
Environ. Sci. Technol. 2008
, 42, 173.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[138]
J. A. Brant ,
J. Labille ,
J. Y. Bottero ,
M. R. Wiesner ,
Characterizing the impact of preparation method on fullerene cluster structure and chemistry.
Langmuir 2006
, 22, 3878.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[139]
D. Jakubczyk ,
G. Derkachov ,
W. Bazhan ,
E. Lusakowska ,
K. Kolwas ,
M. Kolwas ,
Study of microscopic properties of water fullerene suspensions by means of resonant light scattering analysis.
J. Phys. D Appl. Phys. 2004
, 37, 2918.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[140]
J. Labille ,
A. Masion ,
F. Ziarelli ,
J. Rose ,
J. Brant ,
F. Villieras ,
M. Pelletier ,
D. Borschneck ,
M. R. Wiesner ,
J. Y. Bottero ,
Hydration and dispersion of C60 in aqueous systems: the nature of water–fullerene interactions.
Langmuir 2009
, 25, 11232.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[141]
G. V. Andrievsky ,
M. V. Kosevich ,
O. M. Vovk ,
V. S. Shelkovsky ,
L. A. Vashchenko ,
On the production of an aqueous colloidal solution of fullerenes.
J. Chem. Soc. Chem. Commun. 1995
, 12, 1281.
| Crossref | GoogleScholarGoogle Scholar |
[142]
N. O. Mchedlov-Petrossyan ,
V. K. Klochkov ,
G. V. Andrievsky ,
Colloidal dispersions of fullerene C60 in water: some properties and regularities of coagulation by electrolytes.
J. Chem. Soc., Faraday Trans. 1997
, 93, 4343.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[143]
S. Deguchi ,
R. G. Alargova ,
K. Tsujii ,
Stable dispersions of fullerenes, C60 and C70, in water. Preparation and characterization.
Langmuir 2001
, 17, 6013.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[144]
J. Brant ,
H. Lecoanet ,
M. Hotze ,
M. Wiesner ,
Comparison of electrokinetic properties of colloidal fullerenes (n-C60) formed using two procedures.
Environ. Sci. Technol. 2005
, 39, 6343.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[145]
X. Ma ,
D. Bouchard ,
Formation of aqueous suspensions of fullerenes.
Environ. Sci. Technol. 2009
, 43, 330.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[146]
Y. G. Wang ,
Y. S. Li ,
K. D. Pennell ,
Influence of electrolyte species and concentration on the aggregation and transport of fullerene nanoparticles in quartz sands.
Environ. Toxicol. Chem. 2008
, 27, 1860.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[147]
B. Espinasse ,
E. M. Hotze ,
M. R. Wiesner ,
Transport and retention of colloidal aggregates of C60 in porous media: effects of organic macromolecules, ionic composition, and preparation method.
Environ. Sci. Technol. 2007
, 41, 7396.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[148]
M. Terashima ,
S. Nagao ,
Solubilization of [60]fullerene in water by aquatic humic substances.
Chem. Lett. 2007
, 36, 302.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[149]
B. Xie ,
Z. H. Xu ,
W. H. Guo ,
Q. L. Li ,
Impact of natural organic matter on the physicochemical properties of aqueous C60 nanoparticles.
Environ. Sci. Technol. 2008
, 42, 2853.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[150]
T. E. Chang ,
L. R. Jensen ,
A. Kisliuk ,
R. B. Pipes ,
R. Pyrz ,
A. P. Sokolov ,
Microscopic mechanism of reinforcement in single-wall carbon nanotube/polypropylene nanocomposite.
Polymer 2005
, 46, 439.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[151]
R. H. Baughman ,
A. A. Zakhidov ,
W. A. de Heer ,
Carbon nanotubes – the route toward applications.
Science 2002
, 297, 787.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[152]
R. P. Raffaelle ,
B. J. Landi ,
J. D. Harris ,
S. G. Bailey ,
A. F. Hepp ,
Carbon nanotubes for power applications.
Mater. Sci. Eng. B 2005
, 116, 233.
| Crossref | GoogleScholarGoogle Scholar |
[153]
S. Banerjee ,
S. S. Wong ,
Rational sidewall functionalization and purification of single-walled carbon nanotubes by solution-phase ozonolysis.
J. Phys. Chem. B 2002
, 106, 12144.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[154]
Y. Peng ,
H. Liu ,
Effects of oxidation by hydrogen peroxide on the structures of multiwalled carbon nanotubes.
Ind. Eng. Chem. Res. 2006
, 45, 6483.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[155]
I. D. Rosca ,
F. Watari ,
M. Uo ,
T. Akasaka ,
Oxidation of multiwalled carbon nanotubes by nitric acid.
Carbon 2005
, 43, 3124.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[156]
H. Hiura ,
T. W. Ebbesen ,
K. Tanigaki ,
Opening and purification of carbon nanotubes in high yields.
Adv. Mater. 1995
, 7, 275.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[157]
H. Hu ,
A. Yu ,
E. Kim ,
B. Zhao ,
M. E. Itkis ,
E. Bekyarova ,
R. C. Haddon ,
Influence of the zeta potential on the dispersability and purification of single-walled carbon nanotubes.
J. Phys. Chem. B 2005
, 109, 11520.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[158]
N. B. Saleh ,
L. D. Pfefferle ,
M. Elimelech ,
Aggregation kinetics of multiwalled carbon nanotubes in aquatic systems: measurements and environmental implications.
Environ. Sci. Technol. 2008
, 42, 7963.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[159]
H. S. Lee ,
C. H. Yun ,
Translational and rotational diffusions of multiwalled carbon nanotubes with static bending.
J. Phys. Chem. C 2008
, 112, 10653.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[160]
[161]
A. Schierz ,
H. Zänker ,
Aqueous suspensions of carbon nanotubes: surface oxidation, colloidal stability and uranium sorption.
Environ. Pollut. 2009
, 157, 1088.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[162]
M. Cox ,
A. A. Pichugin ,
E. I. El-Shafey ,
Q. Appleton ,
Sorption of precious metals onto chemically prepared carbon from flax shive.
Hydrometallurgy 2005
, 78, 137.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[163]
M. Li ,
M. Boggs ,
T. P. Beebe ,
C. P. Huang ,
Oxidation of single-walled carbon nanotubes in dilute aqueous solutions by ozone as affected by ultrasound.
Carbon 2008
, 46, 466.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[164]
K. Esumi ,
M. Ishigami ,
A. Nakajima ,
K. Sawada ,
H. Honda ,
Chemical treatment of carbon nanotubes.
Carbon 1996
, 34, 279.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[165]
Y.-T. Shieh ,
G.-L. Liu ,
H.-H. Wu ,
C.-C. Lee ,
Effects of polarity and pH on the solubility of acid-treated carbon nanotubes in different media.
Carbon 2007
, 45, 1880.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[166]
[167]
H. Hyung ,
J. D. Fortner ,
J. B. Hughes ,
J.-H. Kim ,
Natural organic matter stabilizes carbon nanotubes in the aqueous phase.
Environ. Sci. Technol. 2007
, 41, 179.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[168]
D. Lin ,
B. Xing ,
Tannic acid adsorption and its role for stabilizing carbon nanotube suspensions.
Environ. Sci. Technol. 2008
, 42, 5917.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[169]
H. Hyung ,
J.-H. Kim ,
Natural organic matter (NOM) adsorption to multiwalled carbon nanotubes: effect of NOM characteristics and water quality parameters.
Environ. Sci. Technol. 2008
, 42, 4416.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[170]
X. Wang ,
S. Tao ,
B. Xing ,
Sorption and competition of aromatic compounds and humic acid on multiwalled carbon nanotubes.
Environ. Sci. Technol. 2009
, 43, 6214.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[171]
K. Yang ,
B. S. Xing ,
Adsorption of fulvic acid by carbon nanotubes from water.
Environ. Pollut. 2009
, 157, 1095.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[172]
M. Elimelech ,
C. R. O’Melia ,
Effect of electrolyte type on the electrophoretic mobility of polystyrene latex colloids.
Colloids Surf. 1990
, 44, 165.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[173]
W. C. Hou ,
C. T. Jafvert ,
Photochemistry of aqueous C60 clusters: evidence of 1O2 formation and its role in mediating C60 phototransformation.
Environ. Sci. Technol. 2009
, 43, 5257.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[174]
Q. L. Li ,
B. Xie ,
Y. S. Hwang ,
Y. J. Xu ,
Kinetics of C60 fullerene dispersion in water enhanced by natural organic matter and sunlight.
Environ. Sci. Technol. 2009
, 43, 3574.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[175]
H. Hyung ,
J. H. Kim ,
Dispersion of C60 in natural water and removal by conventional drinking water treatment processes.
Water Res. 2009
, 43, 2463.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[176]
C. Y. Wang ,
C. Bottcher ,
D. W. Bahnemann ,
J. K. Dohrmann ,
A comparative study of nanometer-sized Fe(III)-doped TiO2 photocatalysts: synthesis, characterization and activity.
J. Mater. Chem. 2003
, 13, 2322.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[177]
J. H. Choi ,
F. T. Nguyen ,
P. W. Barone ,
D. A. Heller ,
A. E. Moll ,
D. Patel ,
S. A. Boppart ,
M. S. Strano ,
Multimodal biomedical imaging with asymmetric single-walled carbon nanotube/iron oxide nanoparticle complexes.
Nano Lett. 2007
, 7, 861.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[178]
L. F. Shen ,
A. Stachowiak ,
S. E. K. Fateen ,
P. E. Laibinis ,
T. A. Hatton ,
Structure of alkanoic acid stabilized magnetic fluids. A small-angle neutron and light scattering analysis.
Langmuir 2001
, 17, 288.
| Crossref | GoogleScholarGoogle Scholar |
CAS |