

A method to determine silver partitioning and lability in soils
Lara Settimio A C , Mike J. McLaughlin A B , Jason K. Kirby B and Kate A. Langdon BA Waite Research Institute, School of Agriculture, Food and Wine, University of Adelaide, Adelaide, Waite Road, SA 5064, Australia.
B CSIRO Land and Water, Contaminant Chemistry and Ecotoxicology program, Minerals Down Under Flagship, Waite Campus, Waite Road, Adelaide, SA 5064, Australia.
C Corresponding author. Email: lara.settimo@adelaide.edu.au
Environmental Chemistry 11(1) 63-71 https://doi.org/10.1071/EN13163
Submitted: 28 August 2013 Accepted: 3 December 2013 Published: 21 February 2014
Environmental context. Soils contaminated with silver can have detrimental environmental effects because of silver’s toxicity to a range of soil-dwelling organisms. The total concentration of silver in soil, however, is often not a good indicator of potential toxicity as it does not account for variations in bioavailability. We report a method for soil analysis that measures the amount of silver available for uptake by soil-dwelling organisms, and hence could provide data that better reflect potential toxicity.
Abstract. There is increasing potential for pollution of soils by silver because of an increased use of this metal in consumer and industrial products. Silver may undergo reactions with soil components that mitigate its availability and potential toxicity, so that the total concentration of this metal in soil is not a useful indicator of potential risk. We developed an isotopic dilution method to simultaneously measure the partitioning (Kd-value) and lability (E-value) of Ag in soils, using the 110mAg isotope. An equilibration solution containing 10 mM Ca(NO3)2 was used along with a cation exchange resin to correct for possible interferences from non-isotopically exchangeable Ag associated with soil colloids in suspension (Er-value). The quantification limits for Kd and Er will depend on the amounts of radioisotope spiked and daily detection limits of inductively coupled plasma-mass spectrometry instrumentation but are typically >4000 L kg–1 and <0.92 mg kg–1. Measurement of Kd values for Ag in a range of soils indicated strong partitioning to the solid phase is positively associated with soil cation-exchange capacity or total organic carbon and pH. The concentrations of labile Ag in soils geogenically enriched in Ag were not detectable indicating occlusion of the Ag within poorly soluble solid phases. Measurement of labile Ag in soils spiked with a soluble Ag salt and aged for 2 weeks indicated rapid conversion of soluble Ag into non-isotopically exchangeable forms, either irreversibly adsorbed or precipitated in the soil. These results indicate that measurement of labile Ag will be important to estimate toxicity risks to soil organisms or to predict bioaccumulation through the food chain.
Additional keywords: E-value, isotope dilution, Kd, partition coefficient.
Introduction
Silver is a precious metal that has long been used in the manufacture of coins, mirrors, jewellery, utensils, photographic film and electronics. Anthropogenic sources of Ag to the environment include emissions from mining and smelting operations, manufacture and disposal of photographic materials and electronics and coal combustion.[1] Historically the photographic industry has accounted for a high proportion of all Ag discharged into the environment from anthropogenic sources.[2] Inputs of Ag from the photographic industry into the environment have rapidly declined in recent years because of the lower demand for colour film with advancements in digital technology. Emerging applications for Ag are in solar energy, water purification, medicine and nanotechnology.[3] Nanotechnology is a rapidly emerging area, with Ag nanoparticles (AgNPs) one of the most commonly used nanomaterials in consumer products (e.g. washing machines and clothing) and medical applications.[4,5] The anti-bacterial properties that make Ag and AgNPs desirable in products may also lead to human and environmental risks following their release into the environment.[6]
The major sink for Ag entering into the environment is terrestrial ecosystems. During the mining process, Ag losses to the environment can occur through tailings, slag, smelting, dust and leachate.[7–10] The major contamination pathway for Ag from urban areas (e.g. consumer products and industries) will occur through wastewater treatment plants (WWTPs), predominately by land application of biosolids.[8,11] If biosolid materials produced in WWTPs have elevated concentrations of Ag, they may pose a significant risk to terrestrial environments. The presence of Ag in the environment is of concern because of its potential toxicity to a range of organisms such as plants, invertebrates, microbes and bacteria living in the soil.[12–16] In comparison to aquatic environments, there has been limited research undertaken on the fate and behaviour of Ag in soil. The predicted increase in Ag concentrations (e.g. because of increased use of Ag and AgNP containing products) requires the development of sensitive techniques that can be used to examine the fate and potential bioavailability (e.g. partitioning and lability) of Ag in soils.
Solid–solution partitioning, which can be quantified using a distribution coefficient (Kd-value), can be used to provide a simple measure of the partitioning of a metal in soil between the solid phase and the soil solution phase.[17] The Kd-value can provide information on the total metal that is mobile and possibly in a bioavailable form, which can assist in evaluating the potential risks of metals in soil. The partitioning of Ag in soils has been reported to range from 10 to 32 000 L kg–1 [18,19] and is strongly influenced by soil properties such as pH, clay content, organic matter content and chloride (Cl–) concentration (possibly because of Ag+ being precipitated as cerargyrite (AgCl)).[18] In addition, the composition of soil organic matter (SOM) is important because ionic Ag (Ag+), being a soft metal cation, can form strong complexes with reduced sulfur and thiol groups in SOM.[20]
The total concentration of metals in soils is known to be a poor indicator of the potential bioavailable fraction.[21] Methods that measure the total labile metal fraction (in solution and on solid phases in rapid exchange) such as chemical extractants, resin techniques and isotope dilution (ID) methods have been shown to be better indicators of the bioavailable fraction of metals in soils.[22,23] ID methods have been shown to be a powerful tool to determine the labile fraction of metals in soils.[24–26] These ID methods provide a measure of the total labile fraction of an element in soil, also known as an E-value. The E-value is calculated using the distribution of an added stable or radioactive isotope between the soil solution and exchangeable surfaces on solid phases in rapid equilibrium over an operationally defined time period e.g. 24–72 h.[27,28] The E-value technique assumes that the labile element in solution and associated with the soil solid phase remains in equilibrium throughout the measurement period.[29] The accuracy of the ID method in determining the labile pool of metals in soils can be influenced by isotope fixation,[25] changes in speciation, differences in solution–solid phase partitioning[30] and the presence of non-exchangeable metal associated with colloidal materials in soil solutions.[31,32] These potential errors in E-value determinations can be corrected through an understanding of the solution and solid phase speciation of the metal in soils.[30–32]
The aim of this study was to develop an accurate and sensitive ID method to simultaneously measure the partitioning (Kd-value) and lability (E-value) of Ag in soils. This method will provide a powerful tool to examine the long-term fate of Ag-containing materials added to soils, either from the perspective of examining the transformation of soluble Ag salts into non-labile forms, or for examining the dissolution of sparingly soluble Ag compounds into labile forms.
Materials and methods
Soil sampling and characterisation
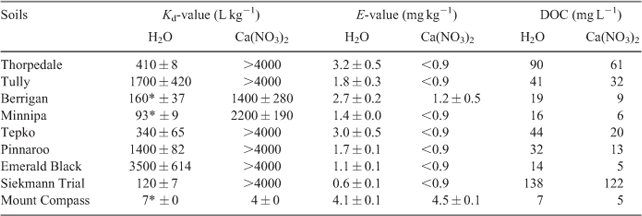
This study used soils that were sourced from ten locations in Australia and one location in France. The soils were collected from 0–10-cm depth, air-dried, homogenised and sieved to <2 mm. The soils were then characterised (Table 1) before use in all experiments.

|
Soil pH was measured using a soil-to-solution ratio of 1 : 5 (m/v), with ultrapure deionised water (Milli-Q, Millipore) and shaking for 1 h. Total organic carbon (TOC) contents, cation exchange capacity (CEC), particle size and oxalate-extractable aluminium (Alox) and iron (Feox) contents were determined according to standard methods.[33,34] Concentrations of dissolved organic carbon (DOC) were measured (Shimadzu TOC-VCSH/CSN + TNM-1, Shimadzu, Kyoto, Japan) using a 1 : 5 (m/v) soil-to-solution ratio with ultrapure deionised water (Milli-Q) and shaking for 1 h, with the suspension filtered through a 0.2-µm cellulose acetate filter (Sartorius, Goettingen, Germany). Concentrations of soluble chloride were measured (Dionex ICS-2500, Dionex, Sunnyvale, CA, USA) using a 1 : 5 (m/v) soil-to-solution ratio with ultrapure deionised water (Milli-Q) and shaking for 1 h, with the suspension filtered through a 0.2-µm cellulose acetate filter (Sartorius, Germany). Total Ag concentrations in soils were determined using a microwave-assisted aqua regia (US-EPA 3051A) procedure. This was conducted by weighing soil (0.25 g) into Teflon vessels and adding HNO3 and HCl at a ratio of 1 : 3 (v/v). The soils were allowed to digest at room temperature overnight and then heated for 45 min at 175 °C (after a 10-min ramp period) in a microwave system (Mars Express, 1600 W, CEM, Matthews, NC, USA). Each sample was filtered through a 0.2-µm cellulose acetate filter (Sartorius, Germany) and Ag+ was measured by inductively coupled plasma–optical emission spectroscopy (ICP-OES) (Spectro ARCOS, Spectro, Kleve, Germany) or inductively coupled plasma–mass spectrometry (ICP-MS) (Agilent 7500ce, Agilent, Santa Clara, CA, USA). The limit of detection of the soil digestion method was 0.04 mg kg–1. A digest blank and a certified reference material (NRC-CNRC PACS-2) with a Ag concentration of 1.22 ± 0.14 mg kg–1 were included for quality control in all batches of digests. Blank solutions were consistently lower than the limit of detection for Ag by ICP-MS of 0.15 µg L–1 and the certified reference material was determined to have a Ag concentration of 1.15 ± 0.16 mg kg–1.
Soil spiking with soluble silver
Soils (50–200 g) were spiked to the desired Ag concentrations using silver nitrate (Sigma–Aldrich, ≥99 %) in ultrapure deionised water (Milli-Q). The Ag spiking solutions were aspirated onto dry soils using a glass nebuliser, with spiking solutions made in the required volume of water to hydrate the soils to 70 % of field capacity (FC, determined at pF 2.0 using a tension plate technique[35]). Soils were incubated at 22 °C in the dark, aerated daily and maintained at 70 % FC by weight for 2 weeks before the commencement of experiments. At the completion of all experimental work, the total Ag concentrations in the spiked soils were confirmed using the microwave assisted aqua regia digest method outlined above (confirmed Ag concentrations are reported in tables and figures).
ID method to determine the partitioning coefficient (Kd-value) and isotopically exchangeable silver (E-value)
Soils (2.0 ± 0.02 g) (n ≥ 3) were weighed into 50-mL polypropylene centrifuge tubes (LabServ) and mixed end-over-end for 48 h with 20 mL of equilibration solution (selection of equilibration solutions is outlined below). Following this initial equilibration period, each sample was spiked with 12 kBq of 110mAg in 100 µL of carrier-free solution (in the form of 110mAgNO3 (t1/2 = 250 days)). The 110mAg spiked soil suspensions were mixed on an end-over-end shaker for a further 72 h, followed by centrifugation at 1200g for 20 min at room temperature (22 °C) and the supernatants filtered through 0.2-µm cellulose acetate filters (Sartorius, Germany). Preliminary experiments indicated that isotope exchange was essentially complete after 3 days of equilibration of 110mAg with the soil suspensions (data not shown). The pH of soil suspensions was determined before 110mAg addition and after the spike equilibrium period and no changes in pH were observed. The 110mAg activity in the samples was then determined by gamma spectroscopy (WIZARD2, Perkin Elmer) and total Ag concentrations by ICP-OES or ICP-MS. As 110mAg decays to 110Cd emitting both gamma and beta radiation, it can also be determined using scintillation counting[36] – we preferred gamma spectroscopy because of the lower cost, absence of stability issues with scintillants and limited generation of chemical waste.
The 110mAg activity in the samples was used to determine the partitioning of Ag in the soils (Kd-value) using the following equation:
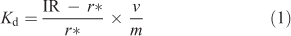
where Kd is the solid–solution partitioning coefficient of Ag in soils (L kg–1), IR is the initial spiked 110mAg radioactivity (Bq), r* is the 110mAg solution radioactivity after equilibration (Bq), v is the solution volume (L) and m is the sample mass (kg).
The isotopically exchangeable (labile) fraction of Ag in soils (E-value) was determined using the following equation[29]:
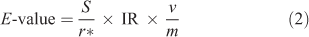
where the E-value is the isotopically labile Ag concentration in soils (mg kg–1) and S is the solution Ag concentration (mg L–1).
A potential error in E-value determinations can occur as a result of the presence of non-labile (fixed) elements associated with colloidal material in soil suspensions. This potential error on E-value determinations can be corrected using resin techniques.[26,31] An 8-mL aliquot of the <0.2-μm soil suspensions was pipetted into 15-mL centrifuge tubes with ~0.1 g of Chelex 100 resin (Bio-Rad Laboratories). The resin had previously been converted from sodium into calcium form using calcium nitrate (Sigma–Aldrich). The 0.2-µm filtered samples with resin were mixed end-over-end for 24 h, centrifuged at 1200g for 20 min at room temperature (22 °C) and the supernatants discarded. The resins were rinsed twice with ultrapure deionised water (Milli-Q) and eluted with 6–12 mL of 1 M nitric acid (this was found to elute 100 ± 9 % of the Ag sorbed to the resin). The eluted solutions were analysed for 110mAg by gamma spectroscopy and total Ag concentrations by ICP-OES or ICP-MS. The E-value accounting for the possible presence of non-exchangeable Ag in soil solutions (Er-value) was determined using Eqn 3.
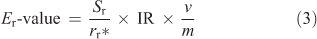
where the Er-value is the resin-corrected isotopically labile Ag concentration in soils (mg kg–1), Sr is the solution Ag concentration in the resin eluate (mg L–1) and rr* is the 110mAg solution radioactivity in the resin eluate (Bq).
Optimisation of ID method for determination of Kd and E-values
A series of experiments were undertaken to optimise the ID method for the determination of Kd and E-values (E and Er) for Ag in soils. The selected soils that were used for each experimental component are indicated in Table 1.
Experiment 1. Effect of equilibration solution on partitioning and lability of silver
The initial experiment was conducted to identify the most suitable equilibration solution to determine Kd and E-values for Ag. Three solutions were initially compared (Milli-Q H2O, 10 mM Ca(NO3)2 and 10 mM CaCl2, Merck Millipore, Billerica, MA, USA), which were chosen because they are commonly used for measuring partitioning and lability of metals in soils.[37,38]
To compare the three equilibration solutions, soils with varying physical and chemical properties (Mount Compass, Tepko and Minnipa) were spiked to a concentration of 100 mg kg–1, before the determination of Kd and E-values using the method outlined above. Following this, the equilibration solutions of H2O and 10 mM Ca(NO3)2 were selected to determine Kd and E-values on all soils (excluding Bordeaux and Charleston), following spiking with a lower concentration of Ag (5 mg kg–1) and equilibration for 2 weeks at 70 % FC.
Experiment 2. Effect of Ca concentration on partitioning and lability of silver
The extent to which varying concentrations of Ca(NO3)2 affected the resulting Kd and E-values was determined by following the ID method using equilibration solutions with increasing concentrations of Ca(NO3)2 (0, 5, 10, 20 and 50 mM). This was performed using two soils with contrasting soil properties (Bordeaux and Charleston) which had been spiked to a Ag concentration of 100 mg kg–1 and equilibrated for 2 weeks at 70 % FC.
Experiment 3. Potential for colloidal interferences in determination of silver lability
The final optimisation experiment was conducted to examine the effect of non-exchangeable Ag colloids in soil suspensions on the accurate determinations of E-values for Ag. For this experiment, two strongly adsorbing soils (high Kd-values) with high concentrations of TOC (Charleston and Millicent) were spiked with Ag to concentrations of 10, 50 and 100 mg kg–1. After 2 weeks of equilibration, E and Er-values were determined in the soils using 10 mM Ca(NO3)2 as an equilibration solution. Differences in the E and Er-values were then used to determine if a resin clean up technique is required in the method to correct for colloidal interferences in the soil suspensions.
Statistical analysis
Analyses of variance (ANOVA) were conducted using Genstat 11 to determine if significant differences were present between various experimental treatments. All statistical analyses were conducted at a significance level of 0.05. A multiple linear regression analysis was used to determine if soil properties significantly affected soil Kd-values using Genstat 11.
Results and discussion
Effect of equilibration solution on partitioning and lability of silver
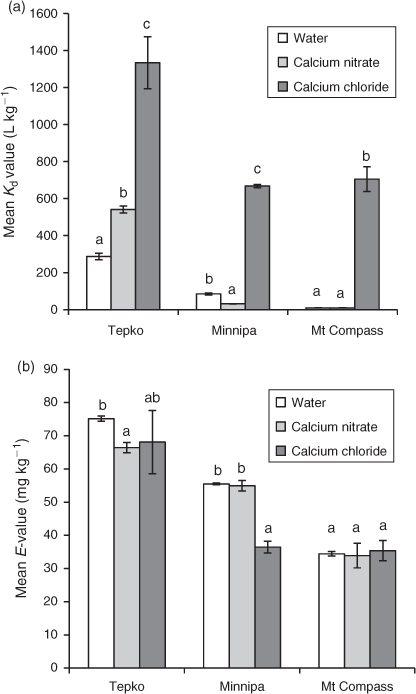
The variation in Kd-values determined using different equilibration solutions highlights their importance when measuring Ag partitioning (Fig. 1). The Kd-values for Ag were significantly higher (up to 70 fold) when CaCl2 was used as the equilibrating solution compared to H2O and Ca(NO3)2 (Fig. 1a). There were no consistent trends in Kd-values when H2O and Ca(NO3)2 were used as the equilibration solutions, with Ca(NO3)2 producing significantly higher Kd-values in the Tepko soil, lower values in the Minnipa soil and similar values in the Mount Compass soil (Fig. 1a). Across the three soils (Fig. 1a) there was a large variation in Kd-values, suggesting soil properties may be important in controlling partitioning of Ag in soil.
|
When the E-values were compared from the three equilibration solutions, they were found to be similar across all three soils when H2O and Ca(NO3)2 were used, and were either similar or lower when CaCl2 was used (Fig. 1b). The inconsistencies in the overall Kd and E-values when CaCl2 was used as an equilibration solution compared to the other solutions (H2O and Ca(NO3)2) is likely a result of precipitation of Ag+ as cerargyrite (AgCl) in the soil suspensions. Measured solution concentrations of Ag in the suspensions varied from 0.05 to 1.7 mg L–1, and modelling of solution speciation using Visual MINTEQ[39] confirmed that cerargyrite was indeed supersaturated in these solutions. Using 10 mM CaCl2 as the background electrolyte effectively limits solution Ag concentrations to 0.11 mg L–1 because of precipitation of cerargyrite. This solution is therefore not suitable as a background electrolyte solution for measuring partitioning and lability of Ag in soils. The presence of Cl– highlights the limit on Ag lability in saline soils where it is likely to precipitate as cerargyrite. Based on this outcome, further experimentation focussed on H2O and Ca(NO3)2 as equilibration solutions.
Partitioning and lability of Ag was measured in H2O and Ca(NO3)2 solutions in a wider range of soils at a lower Ag spiking concentration of 5 mg kg–1 (Table 2). When H2O was used as the equilibration solution Kd-values were determined in all soils. In contrast, when Ca(NO3)2 was used as the equilibration solution Kd-values could only be determined in the Berrigan, Minnipa and Mount Compass soils (Table 2). This was because of the 110mAg activity (r*) in solution being below the quantification limits of the gamma counter for the remaining soils. Similarly it was difficult to determine E-values in the suspensions using Ca(NO3)2 because of the extremely low concentrations of total Ag (<0.1 μg L–1) and activities (<1.6 Bq mL–1) of 110mAg in these soil solutions (Table 2).
|
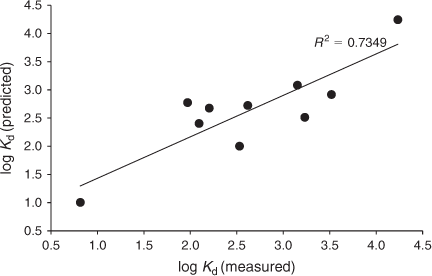
To demonstrate the effect of soil type on the partitioning of Ag, the Kd-values measured in the H2O solutions were analysed using multiple linear regression, with the various soil properties as predictive variables. Based on this analysis, the Kd-values were found to be positively correlated with CEC or TOC content and pH of the soils (Fig. 2). The positive correlation of the Kd-values with soil TOC and pH is also consistent with other studies.[40] In addition to Ag binding being positively correlated with TOC, it is likely there will be complexation of Ag with DOC, resulting in increased Ag being found in the solution phase of the soil suspensions in soils with higher DOC. This process will be dependent on the partitioning of soil organic carbon (OC) to the solution phase. Indeed, we found a positive relationship between the Ag Kd-values and Kd-values for the TOC (Figs S1, S2 in Supplementary material), indicating that when TOC was more soluble, Ag Kd-values were low
|
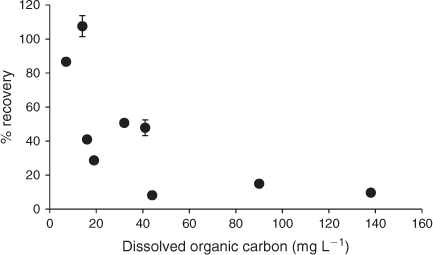
To further investigate this hypothesis, ionic Ag (Ag+) from the H2O solutions was extracted using a cation exchange resin. Following elution of the Ag+ from the resin with 1 M HNO3 the concentrations of Ag were determined. Comparison of the initial and final concentration of Ag from the H2O solutions indicated that the percentage of total Ag present as Ag+ decreased with increasing DOC concentration (Fig. 3). This supports the hypothesis of strong binding of Ag+ by the DOC in the H2O soil suspensions, which is similar to that observed for DOC in natural waters in aquatic ecosystems.[41–43]
|
As water suspensions of soils are well known to have higher concentrations of DOC than dilute Ca2+ suspensions (because of Ca2+ ions suppressing the mobilisation of DOC), which we also observed (Table 2), the effect of DOC on the outcome of partitioning studies with Ag are likely to be more pronounced when H2O is used as an equilibration solution. In addition, it is generally accepted that using a dilute electrolyte like Ca(NO3)2 compared with deionised H2O provides a more realistic representation of a soil solution. Based on this, Ca(NO3)2 is considered to be the most suitable equilibration solution for experimental work determining partitioning and lability of Ag in soils. To overcome low concentrations of 110mAg activity in strongly sorbing soils when using Ca(NO3)2, the amount of initial spiked 110mAg can be increased from 12 to 24 kBq per sample.
Effect of Ca concentration in the equilibration solution on silver partitioning in soils
To examine the effect of Ca2+ concentration on the partitioning of Ag, two soils were chosen; Bordeaux having acidic pH, low clay content and low concentrations of DOC in water suspensions and Charleston, having a similar pH but higher concentrations of clay and DOC (Table 1). When Kd-values were determined in the soils using equilibration solutions with Ca(NO3)2 concentrations of 0, 5, 10, 20 and 50 mM, there was contrasting behaviour between the two soils (Fig. 4). In the Bordeaux soil, Ag adsorption was reduced with increasing Ca concentrations (Fig. 4a) concurrent with a decrease in pH of the suspensions from 5.3 to 4.4 because of increasing Ca concentration. In contrast, in the Charleston soil, partitioning to the solid phase increased dramatically with a small increase in Ca(NO3)2 concentration (5 mM), and then slowly declined following a similar trend to that observed in the Bordeaux soil (Fig. 4b). We interpret these trends as an interaction between three factors. First, increasing Ca2+ concentrations may compete with Ag+ for weaker sorption sites on the soil. Second, increasing ionic strength of the equilibration solution decreases suspension pH which may decrease Ag+ sorption. These two processes explain the observed reductions in Kd-values for Ag noted in the Bordeaux soil with increasing concentrations of Ca(NO3)2 and in the Charleston soil at concentrations of Ca(NO3)2 greater than 5 mM. Third, increasing Ca2+ may flocculate organic and mineral colloids in H2O suspensions and hence affect Ag sorption as outlined previously. This last effect was strongly apparent in the Charleston soil where a small (5 mM) increase in concentration of Ca(NO3)2 significantly increased Ag+ sorption compared to H2O suspensions (0 mM), concomitant with reductions in concentrations of DOC from 65 to 20 mg L–1. The effect was not observed in the Bordeaux soil because of its low DOC concentration.

|
These results further confirm that use of H2O suspensions to determine Ag+ partitioning may lead to erroneously low Kd-values in soils with higher DOC concentrations. We therefore recommend that a Ca(NO3)2 solution be used as a background electrolyte for determinations of Ag+ partitioning and lability in soils. A 10 mM solution is recommended as this provides a Ca2+ concentration and ionic strength similar to that in the soil solution of most aerobic temperate region soils[44,45] but lower concentrations of Ca(NO3)2 (e.g. 5 mM) could be used in highly weathered soils.[46]
Effect of colloids in the equilibration solution on silver lability in soils
As outlined above, the use of Ca(NO3)2 as the equilibration solution will reduce the error in E-values introduced by non-labile Ag in colloidal forms in soil suspensions, but may not remove completely the need for a resin clean-up. Possible errors in determination of E-values of elements in soils can occur when soil suspensions have significant concentrations of mineral or organic colloids (defined as size <0.22- or <0.45-μm diameter, depending on filtration used) containing non-labile elements. A fundamental assumption of the ID method requires that the determined solution concentration (S) is fully isotopically exchangeable. These errors are likely to be more prevalent at lower concentrations of Ag in soils, where low solution concentrations of Ag occur because of strong solid phase adsorption and solution measurements of Ag can be easily affected by small amounts of non-labile Ag in suspension.
We examined this possibility by spiking two soils (Charleston and Millicent) with a range of Ag concentrations (10, 50 and 100 mg kg–1) and then determined labile Ag using 10 mM Ca(NO3)2 as an equilibration solution. To determine if Ag in non-labile colloids in soil solutions could lead to erroneous estimates of the labile Ag fraction in soils, we compared E-values determined with and without a cation exchange resin clean-up step (Er and E-value respectively). The results showed that at the two lowest Ag concentrations (10 and 50 mg kg–1) in both soils, the Er-values were significantly lower when compared to the E-values (Fig. 5). At the highest rate of 100 mg kg–1 the Er-value was only significantly lower in the Charleston soil (Fig. 5a), whereas in the Millicent soil there was no difference (Fig. 5b). These results indicate the potential for errors in determination of labile Ag in soils as a result of the presence of non-labile colloidal Ag in suspensions. The lack of difference between the E and Er-value in the Millicent soil is likely attributable to the higher concentrations of Ag in suspension being less affected by small concentrations of non-labile Ag. This effect was confirmed by spiking soils with higher rates of soluble Ag and determine E and Er-values – no differences were observed at high Ag concentrations in soil (data not shown). Based on these results, we recommend the use of the resin clean-up step for the determination of labile Ag in soils of varying Ag concentration, even when 10 mM Ca(NO3)2 is used as the background electrolyte. Using 10 mM Ca(NO3)2 and the resin clean-up step we calculate a detection limit (dependent on the concentration of Ag+ in solution) for labile Ag (Er) of 0.92 mg kg–1.

|
Partitioning and lability of silver in soils
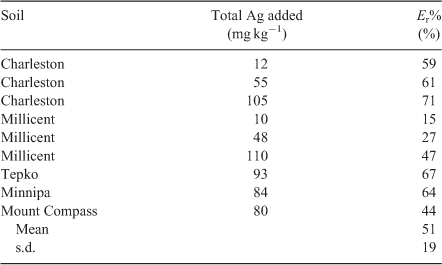
Using the optimised ID method Er-values were determined in several soils used throughout this study (Charleston, Millicent, Tepko, Minnipa and Mount Compass). The change in lability of added soluble metals usually requires consideration of the lability of background metals in the soil, but in all the soils used in this study the background concentrations of Ag were negligible. Hence the lability of added Ag+ was calculated as a percentage of total Ag in soil as follows:
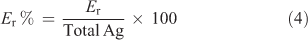
Values of Er% varied from 15 to 71 % depending on soil type and spiking concentration (Table 3) but it is evident that soluble Ag+ quickly partitioned to pools in soil not rapidly in equilibrium with Ag+ in soil solution. This has important implications for studies of Ag+ ecotoxicity when soils are spiked with soluble Ag salts, as significant amounts of the added Ag will be transformed into non-labile forms over a short time period. It is likely that additional reactions may take place in these soils over time, further reducing the labile concentrations of Ag from those reported in this study. These long-term aging reactions of Ag in soil are the subject of a further study in our laboratory.
|
Finally, we used the optimised ID method to examine the partitioning and lability of Ag in several European soils geochemically enriched in Ag, sampled as part of the GEMAS program.[47] These soils had total Ag concentrations varying from 1 to 5 mg kg–1. The Kd-values were high and varied from 250 to over 4000 L kg–1. This was consistent with the high Kd-values that were observed for Ag throughout the optimisation experiments conducted as part of this study. These results are in agreement with earlier studies on Ag retention in soils which also found strong retention by the solid phase.[40,48–51] Compared to other metals, Ag+ which has a median Kd of 1400 L kg–1 (calculated from this study) appears to be retained with a similar strength as Zn or As in soils.[17]
We could detect no labile Ag in the European GEMAS soils (because total Ag+ in solution was below the limit of detection of ICP-MS) indicating that most of the geogenic Ag was occluded in crystal lattices or precipitated/reduced (e.g. as AgCl or Ag0) and not in forms likely to be readily available to terrestrial organisms. This outcome is expected as in the soils spiked with soluble Ag+ (Table 1) in this study, the Ag+ was rapidly converted into non-labile pools in these soils following a short incubation period (2 weeks) (Table 3). These results indicate that when Ag is present in soils a high proportion is likely to be non-bioavailable; however, this will be influenced by soil and organism properties. This outcome will be useful in gaining an overall understanding of the potential risks associated with Ag in the terrestrial environment.
Conclusions
We present a method for determining the partitioning and lability (Er-value) of Ag in soils using an ID method with a 110mAg tracer equilibrated in soil suspensions for 72 h. The use of a 10 mM Ca(NO3)2 electrolyte solution is recommended as it provides a Ca2+ concentration and ionic strength similar to that in the soil solution of most aerobic temperate region soils and reduces errors in E-value determinations by suppressing colloid mobilisation (potentially containing non-labile Ag). We recommend a resin clean-up step for the determination of labile Ag in soils of varying Ag concentration to correct for possible interference in determinations from non-exchangeable Ag in soil solutions.
This ID method should prove useful to examine the long-term fate of Ag-containing materials added to soils, either from the perspective of examining the transformation of soluble Ag salts into non-labile forms, or for examining the dissolution of sparingly soluble Ag compounds into labile forms (e.g. AgNPs). We found partitioning of Ag+ to the solid phase was strongest for soils with high CEC or TOC and pH. The presence of Cl– in the equilibration solution or in the soil increased partitioning to the solid phase, likely because of precipitation of AgCl(s) and highlights the implications for Ag lability in saline soils. The Er-values expressed as a percentage of the total Ag concentration in soils also demonstrated that not all Ag added to the soils remained in the labile pool – significant aging of Ag occurred in a relatively short period of time (2 weeks). Further studies will focus on exploring aging mechanisms because Ag+ is known to form strong complexes with reduced sulfur functional groups on organic matter or in solution which may cause precipitation of Ag2S and reduction to Ag0, and the role of environmental conditions (e.g. salinity and wet–drying cycles) on the partitioning and lability of Ag in soils.
Acknowledgements
The authors thank the Precious Metals and Rhenium Consortium for financial support. They also thank Cathy Fiebiger, Gill Cozens and Claire Wright for expert technical assistance.
References
[1] R. Eisler, Silver hazards to fish, wildlife, and invertebrates: a synoptic review, in Contaminant Hazard Reviews 1997, pp. 2–5 (US Department of the Interior: Washington, DC).[2] I. C. Smith, B. L. Carson, Volume 2: Silver, in Trace Metals in the Environment 1977, p. 204 (Ann Arbor Science Publishers: Ann Arbor, MI).
[3] Silver Uses 2011 (The Silver Institute). Available at http://www.silverinstitute.org/silver_uses.php [Verified 29 May 2011].
[4] J. R. Morones, J. L. Elechiguerra, A. Camacho, K. Holt, J. B. Kouri, J. T. Ramírez, M. J. Yacaman, The bactericidal effect of silver nanoparticles. Nanotechnology 2005, 16, 2346.
| The bactericidal effect of silver nanoparticles.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXht1CiurjJ&md5=4c2b48daa4c25552131adb6c4d84b36eCAS | 20818017PubMed |
[5] T. Benn, B. Cavanagh, K. Hristovski, J. D. Posner, P. Westerhoff, The release of nanosilver from consumer products used in the home. J. Environ. Qual. 2010, 39, 1875.
| The release of nanosilver from consumer products used in the home.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsVKlu7zO&md5=665579b3e77d38985564355f31a44340CAS | 21284285PubMed |
[6] C. L. Doolette, M. J. McLaughlin, J. K. Kirby, D. J. Batstone, H. H. Harris, H. Q. Ge, G. Cornelis, Transformation of PVP coated silver nanoparticles in a simulated wastewater treatment process and the effect on microbial communities. Chem. Cent. J. 2013, 7, 46.
| Transformation of PVP coated silver nanoparticles in a simulated wastewater treatment process and the effect on microbial communities.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXpvVKlsrs%3D&md5=f3a87cce8f06c4d2557451407b16abaeCAS | 23497481PubMed |
[7] M. J. Eckelman, T. E. Graedel, Silver emissions and their environmental impacts: a multilevel assessment. Environ. Sci. Technol. 2007, 41, 6283.
| Silver emissions and their environmental impacts: a multilevel assessment.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXot1ahsr0%3D&md5=9f7da99683e892561f9ba63c9181397cCAS | 17937316PubMed |
[8] J. Johnson, J. Jirikowic, M. Bertram, D. Van Beers, R. B. Gordon, K. Henderson, R. J. Klee, T. Lanzano, R. Lifset, L. Oetjen, T. E. Graedel, Contemporary anthropogenic silver cycle: a multilevel analysis. Environ. Sci. Technol. 2005, 39, 4655.
| Contemporary anthropogenic silver cycle: a multilevel analysis.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXjvFKjsrc%3D&md5=1646ddb9d6bd62d7795d6d7fcbef8c78CAS | 16047806PubMed |
[9] H. G. Petering, C. J. McClain, Silver, in Metals and Their Compounds in the Environment: Occurrence, Analysis and Biological Relevance (Ed. E. Merian) 1991, pp. 1191–1202 (VCH: Weinham, Germany).
[10] B. Elvers, S. Hawkins, W. Russey, G. Schulz, Silver, silver compounds and silver alloys, in Ullmann’s Encyclopedia of Industrial Chemistry (Ed. H. Renner) 1993, pp. 1–77 (VCH: Weinham, Germany).
[11] I. W. Oliver, M. J. McLaughlin, G. Merrington, Temporal trends of total and potentially available element concentrations in sewage biosolids: a comparison of biosolid surveys conducted 18 years apart. Sci. Total Environ. 2005, 337, 139.
| Temporal trends of total and potentially available element concentrations in sewage biosolids: a comparison of biosolid surveys conducted 18 years apart.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXktFOr&md5=1839f0d8dd79ce49796c30259b50d165CAS | 15626385PubMed |
[12] H. T. Ratte, Bioaccumulation and toxicity of silver compounds: a review. Environ. Toxicol. Chem. 1999, 18, 89.
| Bioaccumulation and toxicity of silver compounds: a review.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXkt1Or&md5=3d5a7fbb056a3de8b9b7c5de7810058cCAS |
[13] J. Y. Roh, S. J. Sim, J. Yi, K. Park, K. H. Chung, D. Y. Ryu, J. Choi, Ecotoxicity of silver nanoparticles on the soil nematode Caenorhabditis elegans using functional ecotoxicogenomics. Environ. Sci. Technol. 2009, 43, 3933.
| Ecotoxicity of silver nanoparticles on the soil nematode Caenorhabditis elegans using functional ecotoxicogenomics.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXkvVSktLs%3D&md5=e233e9535dcd28e8b0395413a44ea842CAS | 19544910PubMed |
[14] F. P. C. Blamey, P. M. Kopittke, J. B. Wehr, T. B. Kinraide, N. W. Menzies, Rhizotoxic effects of silver in cowpea seedlings. Environ. Toxicol. Chem. 2010, 29, 2072.
| Rhizotoxic effects of silver in cowpea seedlings.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXht12jsL3L&md5=457626e3547e42a05ba115f8104115ffCAS |
[15] I. N. Throbäck, M. Johansson, M. Rosenquist, M. Pell, M. Hansson, S. Hallin, Silver (Ag+) reduces denitrification and induces enrichment of novel Nir-k genotypes in soil. FEMS Microbiol. Lett. 2007, 270, 189.
| Silver (Ag+) reduces denitrification and induces enrichment of novel Nir-k genotypes in soil.Crossref | GoogleScholarGoogle Scholar | 17250758PubMed |
[16] T. Murata, M. Kanao-Koshikawa, T. Takamatsu, Effects of Pb, Cu, Sb, In and Ag contamination on the proliferation of soil bacterial colonies, soil dehydrogenase activity, and phospholipid fatty acid profiles of soil microbial communities. Water Air Soil Pollut. 2005, 164, 103.
| Effects of Pb, Cu, Sb, In and Ag contamination on the proliferation of soil bacterial colonies, soil dehydrogenase activity, and phospholipid fatty acid profiles of soil microbial communities.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXmvFOksrw%3D&md5=d7d646269d79a35ac23110624dfb16b7CAS |
[17] S. Sauvé, W. Hendershot, H. E. Allen, Solid-solution partitioning of metals in contaminated soils: dependence on pH, total metal burden, and organic matter. Environ. Sci. Technol. 2000, 34, 1125.
| Solid-solution partitioning of metals in contaminated soils: dependence on pH, total metal burden, and organic matter.Crossref | GoogleScholarGoogle Scholar |
[18] G. Cornelis, J. K. Kirby, D. Beak, D. Chittleborough, M. J. McLaughlin, A method for determination of retention of silver and cerium oxide manufactured nanoparticles in soils. Environ. Chem. 2010, 7, 298.
| A method for determination of retention of silver and cerium oxide manufactured nanoparticles in soils.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtFOnsrw%3D&md5=d2dca43d470834db8ab85d37c7384b3bCAS |
[19] J. D. Allison, T. L. Allison, Partition coefficients for metals in surface water, soil and waste. EPA/600/R-05/074 2005, (US Environmental Protection Agency: Washington, DC).
[20] L. J. Evans, S. J. Barabash, Molybdenum, silver, thallium and vanadium, in Trace Elements in Soils (Ed. P. S. Hooda) 2010, pp. 516–49 (Blackwell Publishing Ltd: Wiltshire).
[21] K. Lock, C. R. Janssen, Influence of aging on metal availability in soils, in Reviews of Environmental Contamination and Toxicology, Vol 178 (Ed. G. W. Ware) 2003, pp. 1–21 (Springer: New York).
[22] S. D. Young, H. Zhang, A. M. Tye, A. Maxted, C. Thums, I. Thornton, Characterizing the availability of metals in contaminated soils. I. The solid phase: sequential extraction and isotopic dilution. Soil Use Manage. 2005, 21, 450.
| Characterizing the availability of metals in contaminated soils. I. The solid phase: sequential extraction and isotopic dilution.Crossref | GoogleScholarGoogle Scholar |
[23] H. Zhang, S. D. Young, Characterizing the availability of metals in contaminated soils. II. The soil solution. Soil Use Manage. 2005, 21, 459.
| Characterizing the availability of metals in contaminated soils. II. The soil solution.Crossref | GoogleScholarGoogle Scholar |
[24] A. L. Nolan, Y. B. Ma, E. Lombi, M. J. McLaughlin, Measurement of labile Cu in soil using stable isotope dilution and isotope ratio analysis by ICP-MS. Anal. Bioanal. Chem. 2004, 380, 789.
| Measurement of labile Cu in soil using stable isotope dilution and isotope ratio analysis by ICP-MS.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXhtVarsr3I&md5=dbdc43c60a159352ff975f5ed9bbab50CAS | 15517206PubMed |
[25] L. A. Wendling, Y. B. Ma, J. K. Kirby, M. J. McLaughlin, A predictive model of the effects of aging on cobalt fate and behavior in soil. Environ. Sci. Technol. 2009, 43, 135.
| A predictive model of the effects of aging on cobalt fate and behavior in soil.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtl2ltb7E&md5=3d8593a6392aa3a570676c7564413f80CAS | 19209596PubMed |
[26] J. K. Kirby, M. J. McLaughlin, Y. B. Ma, B. Ajiboye, Aging effects on molybdate lability in soils. Chemosphere 2012, 89, 876.
| Aging effects on molybdate lability in soils.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XoslGqsrk%3D&md5=d4b1e8f94fae5f95915cb44a2c561e4cCAS | 22704209PubMed |
[27] S. D. Young, N. M. J. Crout, J. Hutchinson, A. Tye, S. Tandy, L. Nakhone, Techniques for measuring attenuation: isotopic dilution methods, in In Natural Attenuation of Trace Element Availability in Soils 2005, pp. 19–37 (Society of Environmental Toxicology and Chemistry (SETAC): Pensacola, FL).
[28] R. E. Hamon, D. R. Parker, E. Lombi, Advances in isotopic dilution techniques in trace element research: a review of methodologies, benefits and limitations. Adv. Agron. 2008, 99, 289.
| Advances in isotopic dilution techniques in trace element research: a review of methodologies, benefits and limitations.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhslWjsr0%3D&md5=aeb25b675e7d479d5105ea0135ef7a99CAS |
[29] R. E. Hamon, I. Bertrand, M. J. McLaughlin, Use and abuse of isotopic exchange data in soil chemistry. Aust. J. Soil Res. 2002, 40, 1371.
| Use and abuse of isotopic exchange data in soil chemistry.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXlvFeisA%3D%3D&md5=a2335fd5c2d0289139864b888b130ceaCAS |
[30] R. E. Hamon, E. Lombi, P. Fortunati, A. L. Nolan, M. J. McLaughlin, Coupling speciation and isotope dilution techniques to study arsenic mobilization in the environment. Environ. Sci. Technol. 2004, 38, 1794.
| Coupling speciation and isotope dilution techniques to study arsenic mobilization in the environment.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXht1Wis7s%3D&md5=b9c61ce59f7859c73ee88c02dccdd7c0CAS | 15074691PubMed |
[31] L. A. Wendling, J. K. Kirby, M. J. McLaughlin, A novel technique to determine cobalt exchangeability in soils using isotope dilution. Environ. Sci. Technol. 2008, 42, 140.
| A novel technique to determine cobalt exchangeability in soils using isotope dilution.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhtlaktbrI&md5=7f617c14f6d3e7fdb7edd64bf4e06647CAS | 18350888PubMed |
[32] E. Lombi, R. E. Hamon, S. P. McGrath, M. J. McLaughlin, Lability of Cd, Cu, and Zn in polluted soils treated with lime, beringite, and red mud and identification of a non-labile colloidal fraction of metals using isotopic techniques. Environ. Sci. Technol. 2003, 37, 979.
| Lability of Cd, Cu, and Zn in polluted soils treated with lime, beringite, and red mud and identification of a non-labile colloidal fraction of metals using isotopic techniques.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXosVWgtg%3D%3D&md5=28c89196f94c22ce8d328a07efb8c1a5CAS | 12666929PubMed |
[33] G. W. Gee, J. W. Bauder, Particle size analysis, in Methods of soil analysis. Part 1. Physical and mineralogical methods 1986, pp. 383–411 (American Society of Agronomy Inc. and Soil Science Society of America Inc.: Madison, WI).
[34] G. E. Rayment, F. R. Higginson, Australian Laboratory Handbook of Soil and Water Chemical Methods 1992 (Inkata Press: Melbourne).
[35] T. J. Marshall, J. W. Holmes, C. W. Rose, Soil Physics 1979 (Cambridge University Press: Cambridge, UK).
[36] L. R. Rodríguez Barquero, J. M. Los Arcos, Study of the stability problems of 110mAg samples in several commercial liquid scintillators. Appl. Radiat. Isot. 2000, 52, 679.
| Study of the stability problems of 110mAg samples in several commercial liquid scintillators.Crossref | GoogleScholarGoogle Scholar |
[37] E. Smolders, K. Brans, A. Foldi, R. Merckx, Cadmium fixation in soils measured by isotopic dilution. Soil Sci. Soc. Am. J. 1999, 63, 78.
| Cadmium fixation in soils measured by isotopic dilution.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXitFGgsb8%3D&md5=28a1fbf366eb37c5b3dab4e2fe8aa7e3CAS |
[38] S. D. Young, A. Tye, A. Carstensen, L. Resende, N. Crout, Methods for determining labile cadmium and zinc in soil. Eur. J. Soil Sci. 2000, 51, 129.
| Methods for determining labile cadmium and zinc in soil.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXisVKmsb0%3D&md5=0bbe8f1e0e696f858eee8491f18bb3bfCAS |
[39] J. Gustafsson, Visual Minteq, Version: 3.0 2010 (KTH Royal Institute of Technology: Stockholm, Sweden). Available at http://www2.lwr.kth.se/English/OurSoftware/vminteq/ [Verified 7 February 2014].
[40] G. Cornelis, C. Doolette, M. Thomas, M. J. McLaughlin, J. K. Kirby, D. G. Beak, D. Chittleboroug, Retention and dissolution of engineered silver nanoparticles in natural soils. Soil Sci. Soc. Am. J. 2012, 76, 891.
| Retention and dissolution of engineered silver nanoparticles in natural soils.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XosFejur0%3D&md5=fc4482ad314a7eac6744f5f27411d0c6CAS |
[41] D. S. Smith, R. A. Bell, J. R. Kramer, Metal speciation in natural waters with emphasis on reduced sulfur groups as strong metal binding sites. Comp. Biochem. Physiol. C: Pharmacol. Toxicol 2002, 133, 65.
| Metal speciation in natural waters with emphasis on reduced sulfur groups as strong metal binding sites.Crossref | GoogleScholarGoogle Scholar |
[42] J. R. Kramer, R. A. Bell, D. S. Smith, Determination of sulfide ligands and association with natural organic matter. Appl. Geochem. 2007, 22, 1606.
| Determination of sulfide ligands and association with natural organic matter.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXpt1yls74%3D&md5=2147babe9913b78475b919dc9e1bbddaCAS |
[43] N. W. H. Adams, J. R. Kramer, Silver speciation in wastewater effluent, surface waters, and pore waters. Environ. Toxicol. Chem. 1999, 18, 2667.
| Silver speciation in wastewater effluent, surface waters, and pore waters.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXns1GlsL8%3D&md5=6499af5799b870255728a065b248c84aCAS |
[44] F. N. Ponnamperuma, E. M. Tianco, T. A. Loy, Ionic strengths of the solutions of flooded soils and other natural aqueous solutions from specific conductance. Soil Sci. 1966, 102, 408.
| Ionic strengths of the solutions of flooded soils and other natural aqueous solutions from specific conductance.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaF2sXhtVCgtLk%3D&md5=5eeb3fc33795a7715203c55a0576d2c7CAS |
[45] S. Larsen, A. E. Widdowson, Chemical composition of soil solution. J. Sci. Food Agric. 1968, 19, 693.
| Chemical composition of soil solution.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaF1MXls1Onsw%3D%3D&md5=c8b61050fbcc5785a76b901d6c437a05CAS |
[46] G. P. Gillman, L. C. Bell, Soil solution studies on weathered soils from tropical north Queensland. Aust. J. Soil Res. 1978, 16, 67.
| Soil solution studies on weathered soils from tropical north Queensland.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE1cXksFyqsb0%3D&md5=c79eca00a855eddb90ee2b143aaf206aCAS |
[47] P. de Caritat, C. Reimann, Comparing results from two continental geochemical surveys to world soil composition and deriving Predicted Empirical Global Soil (PEGS2) reference values. Earth Planet. Sci. Lett. 2012, 319–320, 269.
| Comparing results from two continental geochemical surveys to world soil composition and deriving Predicted Empirical Global Soil (PEGS2) reference values.Crossref | GoogleScholarGoogle Scholar |
[48] K. C. Jones, B. E. Davies, P. J. Peterson, Silver in Welsh soils – physical and chemical distribution studies. Geoderma 1986, 37, 157.
| Silver in Welsh soils – physical and chemical distribution studies.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL28XhslWqs78%3D&md5=c3225c4f13806802dabf95a5ddc6e3d9CAS |
[49] G. Szabó, J. Guczi, J. Valyon, R. A. Bulman, Investigations of the sorption characteristics of radiosilver on some natural and artificial soil particles. Sci. Total Environ. 1995, 172, 65.
| Investigations of the sorption characteristics of radiosilver on some natural and artificial soil particles.Crossref | GoogleScholarGoogle Scholar |
[50] A. R. Jacobson, M. B. McBride, P. Baveye, T. S. Steenhuis, Environmental factors determining the trace-level sorption of silver and thallium to soils. Sci. Total Environ. 2005, 345, 191.
| Environmental factors determining the trace-level sorption of silver and thallium to soils.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXks1Klurk%3D&md5=5ed5dd6d3dad249c5036fb2788b1699dCAS | 15919539PubMed |
[51] P. Ciffroy, J. M. Garnier, L. Benyahya, Kinetic partitioning of Co, Mn, Cs, Fe, Ag, Zn and Cd in fresh waters (Loire) mixed with brackish waters (Loire estuary): experimental and modelling approaches. Mar. Pollut. Bull. 2003, 46, 626.
| Kinetic partitioning of Co, Mn, Cs, Fe, Ag, Zn and Cd in fresh waters (Loire) mixed with brackish waters (Loire estuary): experimental and modelling approaches.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXjsVaqtLc%3D&md5=0daa90c5e46a73d20d297628edcd940cCAS | 12735960PubMed |