

Recent evolutionary history of New Zealand’s North and South Island Kokako (Callaeas cinerea) inferred from mitochondrial DNA sequences
S. A. Murphy A C , I. A. Flux B and M. C. Double AA School of Botany and Zoology, Australian National University, ACT 0200, Australia.
B Research, Development and Improvement Division, Department of Conservation, Wellington, New Zealand.
C Corresponding author. Present address: Australian Wildlife Conservancy, PMB 925, Derby, WA 6728, Australia. Email: Steve@australianwildlife.org
Emu 106(1) 41-48 https://doi.org/10.1071/MU05007
Submitted: 2 February 2005 Accepted: 22 December 2005 Published: 10 March 2006
Abstract
The Kokako (Callaeas cinerea) is an endangered, forest-dependent bird belonging to the endemic New Zealand family Callaeidae, the New Zealand wattlebirds. Two subspecies of Kokako are recognised: the now extinct orange-wattled South Island Kokako (SI Kokako) and the blue-wattled North Island Kokako (NI Kokako). The latter is the subject of intense conservation management and several populations have now been established on offshore island reserves. This study aimed to investigate the recent evolutionary history of Kokako through an assessment of the sequence variation and geographical distribution of mitochondrial haplotypes. We sequenced ~400 bases of the Domain III of the mitochondrial control region for 28 NI Kokako and two SI Kokako. Among NI Kokako, nucleotide diversity was low (0.006) but haplotype diversity was high (0.93). The average nucleotide diversity between NI Kokako and SI Kokako was 0.049 and a phylogenetic analysis revealed well supported reciprocal monophyly between NI Kokako and SI Kokako but no robust structure within NI Kokako. A nested clade analysis detected significant geographical structure in the distribution of the 13 NI Kokako haplotypes but could not identify an evolutionary scenario to explain the distribution. We discuss these findings in the context of the recent climatic and geological history of New Zealand.
Introduction
The Kokako (Callaeas cinerea) is an endangered, endemic passerine that belongs to the Callaeidae family, a group also known as New Zealand wattlebirds. This is one of the few extant bird families thought to have been present in New Zealand when New Zealand separated from Australia some 65 million years ago (Fleming 1962; Williams 1976). Thus ancestral Kokako have persisted through a tumultuous period in New Zealand’s geological history that includes the marine incursions and tectonic activity of the Pliocene as well as the glacial phases and massive volcanic eruptions of the Pleistocene (McGlone 1985; Wardle 1991).
During the Pleistocene Epoch (1.8 million to 14 000 years ago) it is thought that throughout New Zealand the Kokako’s forested habitat repeatedly expanded and retracted and at times forested habitat was highly restricted (Fleming 1979; McGlone 1985; McGlone et al. 1993; Newnham 1999). Pollen analyses from the South Island suggest that at times during this period trees such as Nothofagus and podocarps (Podocarpaceae) were found only in moist, frost-free sites in a landscape otherwise dominated by shrub and grassland (McGlone 1985). In contrast, during warmer interglacial periods, forest covered almost all of New Zealand (McGlone et al. 1993, 2001). Massive volcanic explosions also occurred in New Zealand during the mid- and late Pleistocene and repeated pyroclastic eruptions in the Central Plateau region destroyed large areas of forest in the North Island (Stevens et al. 1995).
With such intense climatic and geological activity it is not surprising that recent phylogeographical studies of New Zealand fauna have identified Pleistocene glaciation as a significant factor influencing the distribution and structure of several species (Buckley et al. 2001; Trewick and Wallis 2001; Neiman and Lively 2004) although some authors argue that divergence of many animal lineages occurred much earlier and is most likely explained by the climatic and geological upheavals of the late Pliocene (~2.5 million years ago; Trewick et al. 2000; Trewick and Wallis 2001). There are few phylogeographical studies of New Zealand vertebrates but the fragmentation of habitat caused by the glacial periods of the Pleistocene has been proposed to account for the genetic divergence found among populations of brown kiwis (Apteryx spp.) and Short-tailed Bat (Mystacina tuberculata) (Baker et al. 1995; Lloyd 2003a).
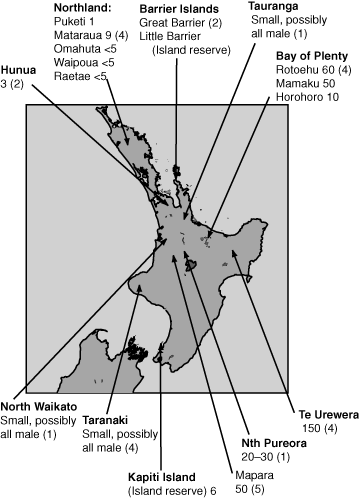
There are two recognised subspecies of Kokako, the North Island Kokako (NI Kokako, C. c. wilsoni) and South Island Kokako (SI Kokako, C. c. cinerea) but verified sightings of SI Kokako have not been made since the middle of the 1900s (Turbott 1967; Falla et al. 1981). At the time sampling for this study was completed (1998), NI Kokako only remained in ~15 declining or intensively managed, fragmented populations (Innes et al. 1999; Basse et al. 2003; Fig. 1). Small populations have also been established on predator-free islands and intensively managed mainland reserves (Innes et al. 1999; Hudson et al. 2000).
|
Through mitochondrial DNA sequencing this study aimed to: (1) assess the genetic diversity and structure within and among extant populations of NI Kokako; (2) determine if the relocation of NI Kokako between mainland populations or to offshore islands has merged previously distinct evolutionary lineages of NI Kokako; (3) determine if SI Kokako were genetically distinct from NI Kokako, and quantify the relative depth of any divergence; and (4) speculate on the recent evolutionary history of both NI Kokako and SI Kokako given the climatic and geological history of New Zealand.
Materials and methods
Study species
The family Callaeidae includes the Kokako, Saddleback (Philesturnus carunculatus) and the extinct Huia (Heteralocha acutirostris) (Heather and Robertson 1996). Previously, taxonomists have linked the Callaeidae with disparate avian groups such as the Ptilonorhynchidae (bowerbirds) and the Corvidae (Williams 1976; Sibley and Ahlquist 1990) but a recent study by Barker et al. (2004) found the Callaeidae to fall within a clade that also included the berrypeckers (Melanocharitidae) and the cnemophiline birds of paradise (Paradisaeidae: Cnemophilus, Loboparadisaea). The two subspecies of Kokako differed in size, plumage and the colouration of their wattles, although reports of orange-wattled Kokako exist from the North Island of New Zealand (Turbott 1967). The SI Kokako is considered to be ‘gravely endangered’ or extinct, with very few reported sightings since the middle of the1900s (Turbott 1967; Falla et al. 1981; McBride 1981; Buckingham 1987). The NI Kokako is endangered and its decline has been attributed primarily to habitat destruction and predation by introduced mammals (Lavers 1978; Leathwick et al. 1983; King 1984; Innes et al. 1999).
Sampling and DNA extraction
Blood and tissue samples from 28 NI Kokako from ten populations were made available for this study (Fig. 1). In addition, tissue samples from ten SI Kokako were taken from skins in collections at the National Museum of New Zealand in Wellington and the Naturhistorisches Museum in Vienna, Austria. D. Lambert and T. King of Massey University, New Zealand, supplied a DNA sample from a North Island Saddleback. NI Kokako DNA was extracted from either whole blood or tissue using a phenol : chloroform extraction protocol (Sambrook et al. 1990) whereas DNA was extracted from the tissue samples of SI Kokako using a QIAamp Blood and Tissue Kit (Qiagen, www.qiagen.com). All DNA extractions involving SI Kokako samples were performed in a laboratory not previously exposed to any Kokako or Saddleback samples.
PCR amplification and DNA sequencing
Initially we screened mitochondrial DNA sequences from cytochrome b and the control region which have both been shown to provide enough sequence variation to resolve population structure in other avian species (e.g. Wenink et al. 1994; Baker and Marshall 1997). A 323 base pair (bp) fragment of the cytochrome b gene was amplified from three NI Kokako from three geographically distant populations (Great Barrier Island, Mapara and Mataraua; Fig. 1) using the universal primers CB1 and CB3-H (Palumbi 1996). Each 25 µL PCR amplification contained: PCR buffer (final concentrations: 10 mm TRIS-HCl pH8.3; 50 mm KCl; 1.5 mm MgCl2; 0.001% gelatin (Applied Biosystems, www.appliedbiosystems.com.au)), 0.2 mm of each dNTP (Promega, www.promega.com), 0.2 µm of each primer, 0.75 U of Taq polymerase (Applied Biosystems), and 100 ng of template DNA. The PCR thermal profile started with 94°C for 3 min followed by 25 cycles of: 94°C for 45 s, 65°C for 30 s, and 72°C for 30 s.
Domains II and III of the mitochondrial control region were amplified using primers L437 and H1248 (Tarr 1995). Sequencing using the H1248 primer was problematic, so a full sequence was obtained using two forward primers. The second forward primer (L-DomIII: 3′-CTCACACTTTGCCCTGATGC-5′) was designed using the computer program Primer version 0.5 (Whitehead Institute of Biomedical Research, Cambridge, MA). The small size of the PCR fragment (406 bp) obtained using primers L-DomIII and H1248 also improved the PCR amplification of the heavily degraded DNA extracted from the tissue samples of SI Kokako. Each 25 µl PCR amplification contained: Opti-Prime PCR buffer (final concentrations: 25 mm KCl; 3.5 mm MgCl2; 10 mm Tris–HCl pH 9.2 (Stratagene, www.stratagene.com)), 0.2 mm of each dNTP (Promega), 0.2 µm of each primer, 0.75 U of Taq polymerase (Applied Biosystems), and up to100 ng of template DNA. Template concentration was determined by spectrophotometer GeneQuant (Biochrom, www.biochrom.co.uk). The PCR thermal profile started with 94°C for 3 min followed by 30 cycles of: 94°C for 30 s, 55°C for 30 s, and 72°C for 30 s.
All PCR products were run on 1% agarose gels which were then stained with ethidium bromide. The appropriate bands were excised and cleaned using a Bresaclean DNA Purification Kit (Geneworks, Adelaide, www.geneworks.com.au). Each product was sequenced using standard protocols for the ABI Prism BigDye Terminator Ready Reaction Kit (Applied Biosystems) and finally run on an ABI Prism 377 automated sequencer (Applied Biosystems).
Data analysis
All sequences were proof-read using Sequencher software (Gene Codes Corporation, Ann Arbor, MI, www.genecodes.com), and aligned using Clustal W (ver. 1.60; Thompson et al. 1994). Haplotype and nucleotide diversity was calculated using the software DNASP (Rozas et al. 2003) and MEGA3 (Kumar et al. 2004) with insertion/deletions (indels) recoded as single nucleotide polymorphisms. Estimates of standard error were based on 500 bootstrap replicates. Phylogenetic analyses were conducted in PAUP*4.0b10 (Swofford 1998) using both maximum parsimony and neighbour-joining criteria. A neighbour-joining tree was constructed using genetic distances based upon a model of molecular evolution (TRN + I, a variant of the GTR model) identified as the most appropriate for these sequences by Modelltest 3.7 (Posada and Crandall 1998). In all analyses, the Domain III sequence from the Saddleback was selected as the outgroup. Using maximum parsimony, a heuristic search was performed with ten random taxon addition replicates and Tree Bisection–Reconnection (TBR) branch swapping. Nodal support for both maximum parsimony and neighbour-joining trees was estimated based on 1000 bootstrap replicates with a value of 70% or greater considered to be strong support (Hillis and Bull 1993).
Geographical structure within the distribution of Kokako haplotypes was investigated using nested clade analysis (NCA; Templeton 1998). NCA is network-based phylogeographical analysis commonly applied to intraspecific data (e.g. Turner et al. 2000; Schultheis et al. 2002) and data from closely related species (e.g. Abbott and Double 2003). A statistical parsimony network with a 95% confidence limit was constructed using the software TCS v1.13 (Clement et al. 2000). Clades within this network were identified using the TCS software and following the guidelines described by Templeton and Sing (1993) and Crandall (1996). Statistical analysis of the geographical data overlaid upon the nested cladogram was performed using GeoDis software (Posada et al. 2000). GeoDis tests for non-random geographical distribution of haplotypes by: (a) performing clade-by-clade contingency analyses of haplotypes and sampled locations as categorical variables; and (b) a nested geographical distance analysis. This second analysis calculates the geographical spread within a clade (Dc), and the geographical spread of a clade in relation to other clades nested in the same higher-level category (Dn) (Posada et al. 2000). For this analysis both SI Kokako samples were given identical coordinates although the actual sampling location within the South Island was unknown for one sample (SI Kokako1; Table 1). Statistical significance was estimated by permutation that simulates a random geographical distribution of clades within a nesting category (Posada et al. 2000). Inference keys by Templeton (2004) were used to help interpret the population genetic or evolutionary implications of significant haplotype associations. The keys principally discriminate between three biological processes: restricted gene flow; range expansion and colonisation events; and fragmentation or splitting events. Recently, authors have argued that NCA may be misleading because it does not estimate the reliability of the inference (Creer et al. 2001; Knowles and Maddison 2002) and have highlighted the need to treat any predictions with caution and to look for independent lines of supporting evidence but Templeton (2004) discusses appropriate methods for evaluating such data.

|
Results
Mitochondrial sequences
The cytochrome b sequences of the three NI Kokako from Great Barrier Island, Mataraua and Mapara revealed only one variable site (0.3% variation in 323 bp; GenBank accession numbers AF433208–AF433209). Consequently we did not attempt to sequence cytochrome b from other samples.
We obtained 801 bp of Domain II and III control region sequences from 23 NI Kokako but only a 403 bp fragment of Domain III could be amplified from five other NI Kokako. This shorter region contained all but two of the polymorphic sites found in the larger fragment. We therefore decided to use the sequence data from the smaller fragment only for all subsequent analyses. Only two of the ten SI Kokako tissue samples provided DNA of reasonable quality for PCR and sequencing (SIKokako1 and 2; Table 1). SIKokako2 was collected from Milford Sound in 1888 but the collecting locality of SIKokako1 is not known.
The 403 bp fragment of Domain III amplified from NI Kokako was rich in adenine (t = 0.29; C = 0.28; A = 0.38; G = 0.06), a feature characteristic of the vertebrate mitochondrial control region (Brown et al. 1986). This region contained nine sites with single nucleotide polymorphism plus two 2-bp insertion/deletions (indels) and one 11-bp indel (Table 1; GenBank accession numbers AF433175–AF433206). These polymorphic sites defined 13 haplotypes among the 28 NI Kokako sample (Table 1). Haplotype diversity (Nei 1987) was high (0.93) with nine of the 13 haplotypes found in only one or two individuals. Only three of the 13 NI Kokako haplotypes were found in more than one population (Table 1). One haplotype (K) was widely distributed and found in the Mapara, Te Urewera, Hunua, and Taranaki populations (Fig. 1). Haplotypes containing the 11-bp indel (Table 1) were found in six of the nine populations sampled.
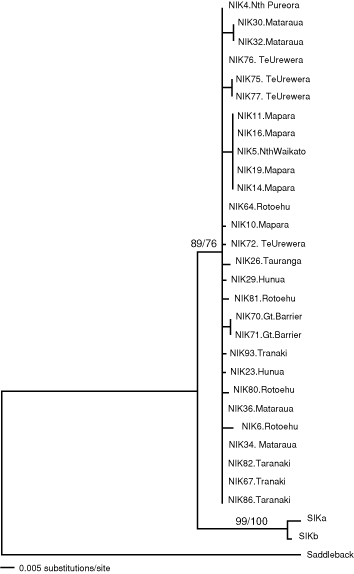
Within NI Kokako, nucleotide diversity (Nei 1987) was only 0.006 (s.e. = 0.002) reflecting the low average number of nucleotide differences between individuals (2.5; s.e. = 0.85). The two SI Kokako sequences defined two haplotypes that differed at three sites. The average genetic distance (uncorrected p) between NI Kokako and SI Kokako was 0.049 (s.e. = 0.01) whereas, for reference, the average genetic distance between the North Island Saddleback sequence and the NI Kokako and SI Kokako sequences was 0.14 (s.e. = 0.02) and 0.15 (s.e. = 0.02) respectively. Both neighbour-joining (Fig. 2) and maximum parsimony analyses recovered phylogenetic trees with strong bootstrap support for the reciprocal monophyly of NI and SI Kokako. No clades within NI Kokako were supported by bootstrap analyses (all < 70% of replicates) using either a neighbour-joining or maximum parsimony-based approach.
|
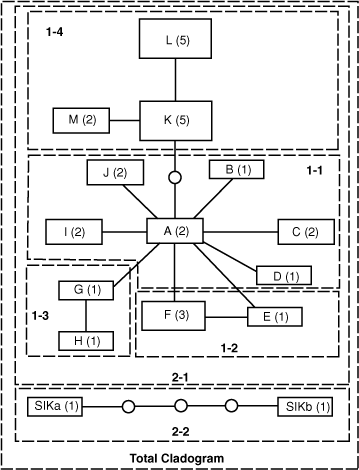
Separate haplotype networks were constructed for NI Kokako and SI Kokako because these groups could not be linked within the limits of 95% statistical parsimony (Fig. 3). The nesting procedure grouped the NI Kokako haplotypes into four one-step clades (Clades 1-1 to 1-4; Fig. 2). Of these, Clades 1-1 and 1-4 were found to have significantly non-random haplotype distributions (χ2 = 43.3, P < 0.001; and χ2 = 20.2, P = 0.005, respectively). Within Clade 1-4, haplotype L had a significantly small geographical distribution (Dc = 27.8, P = 0.01). Also the distance between sampling locations for haplotype L and the geographical centre of Clade 1-4 was significantly small (Dn = 66.7, P = 0.05) whereas Dn was significantly large for haplotype M (Dn = 278.7, P = 0.01). The inference key did not suggest an evolutionary scenario for Clade 1-1 but inferred restricted gene flow with isolation by distance to explain the haplotype distribution in Clade 1-4. The single two-step clade (2-1) contained all the NI Kokako haplotypes and also had significantly non-random distribution of haplotypes (χ2 = 46.7, P = 0.006), but all Dc and Dn values were non-significant. The inference key did not identify a likely evolutionary scenario to explain the geographical distribution of all NI Kokako haplotypes. Finally, the haplotypes within the total cladogram showed a significantly non-random haplotype distribution (χ2 = 30.0, P < 0.01). The Dc and Dn values were significantly small for Clade 2-1 (Dc = 142.7, P = 0.004; Dn = 161.3, P = 0.004) whereas Dn was significantly large for Clade 2-2 (Dn = 946.4, P = 0.004); the NCA identified allopatric fragmentation as the likely cause of this haplotype distribution.
|
Discussion
Sequencing of the mitochondrial control region revealed low nucleotide diversity among NI Kokako, with no two haplotypes separated by more than five mutational steps. This low nucleotide diversity was reflected in a haplotype network in which all samples of NI Kokako were contained within a single two-step clade and also in a phylogenetic analysis in which no clades within all NI Kokako sampled were well supported. The geographical distribution of haplotypes within the two major lower level clades (1-1 and 1-4) of the haplotype network showed significant structure (i.e. was non-random) and the NCA identified isolation by distance with restricted gene flow as a likely cause of the distribution within Clade 1-4. The significant non-random geographical distribution of haplotypes in Clade 2.1 (containing all NI Kokako) was largely generated by the absence of individuals from Clade 1-4 at the Mapara sampling site and the absence of individuals from Clade 1-1 in the Rotoehu sample. However, a geographical analysis based on distance rather than sampling location (Dc and Dn) of each clade nested within Clade 2.1 failed to detect a non-random distribution of haplotypes. This is because representatives from both Clades 1-1 and 1-4 were sampled at the same location in the northernmost (Mataraua), easternmost (Te Urewera) and southernmost (Taranaki) sampling locations, suggesting both clades have a widespread and largely overlapping distribution. The NCA did not identify the likely cause of the current geographical spread of haplotypes within either Clades 1-1 or Clade 2-1.
Such low nucleotide diversity and close evolutionary relationship between extant NI Kokako indicates that NI Kokako went through a relatively recent population bottleneck. However, characteristic of populations recovering from bottlenecks is not only low nucleotide diversity but also low haplotype diversity, with often only one or two common haplotypes within the population (e.g. Milá et al. 2000; Ruegg and Smith 2002). In contrast, our sample of NI Kokako showed low nucleotide diversity coupled with high haplotype diversity (0.93), a pattern more difficult to interpret. Unfortunately, owing to our low sample sizes at each sampling location it is not possible to accurately quantify haplotype frequencies. It is also possible that contemporary haplotype frequencies may now reflect rapid genetic drift induced by fragmentation and small population sizes rather than deeper evolutionary processes (Avise 2000).
The extensive deforestation during the glacial phases of the Pleistocene is thought to have forced forest-dependent species into isolated refugia. For example, studies by Lloyd (2003a, 2003b) found New Zealand’s endemic Short-tailed Bat contains six distinct mitochondrial lineages that probably diverged in the mid-Pleistocene owing to allopatric fragmentation. The North Island lineages show evidence of population expansion and the mixing in the central region of the Island but the distribution remains consistent with an isolation-by-distance dispersal model. Lloyd concluded that this pattern was most likely generated by secondary contact of populations expanding from multiple glacial refugia. NI Kokako by contrast show low nucleotide diversity implying a relatively recent population bottleneck and perhaps the existence of only one Pleistocene refuge. If NI Kokako existed in more than one Pleistocene refuge and genetically diverged (although divergence must have been limited) then the current distribution of haplotypes implies that representatives of each refuge have dispersed widely because individuals from the two major clades can be found throughout the current distribution of NI Kokako. However, the low samples sizes in each sampling location and the high haplotype diversity precludes analyses sensitive to subtle geographical structure in the haplotype distribution.
Hudson et al. (2000) also detected population genetic structure among contemporary NI Kokako populations. In a microsatellite-based study of the Rotoehu, Mapara and Te Urewera populations (separated by > 100 km) they reported low but significant differentiation between some but not all pairwise comparisons. This was surprising as Hudson et al. (2000) suggested that current gene flow was unlikely because Kokako are reluctant to fly over open habitat. It therefore seems likely that this lack of distinct structure may indicate that before deforestation NI Kokako showed relative genetic uniformity over large distances reflecting their ability to disperse widely through continuously forested habitat. This behaviour would also explain the widespread distribution of similar mitochondrial haplotypes.
Our data strongly suggest that North and South Island Kokako were phylogenetically distinct. Average nucleotide diversity between NI Kokako and SI Kokako was high (0.049) relative to only 0.006 within NI Kokako, and the phylogenetic analyses identified SI Kokako as a separate monophyletic clade. Studies of other forest-dwelling taxa from New Zealand, such as Short-tailed Bats (Lloyd 2003a), North Island and South Island kiwi (Baker et al. 1995), and a cicada (Buckley et al. 2001) have all revealed relatively large genetic distances between North and South Island clades and have estimated dates for divergence at between 0.89 and 0.9 million years ago. If we adopt the estimate divergence rate of the avian control region as 5% per million years, as suggested by Freeland and Boag (1999), then Kokako too would fit this pattern. However, recently Ho et al. (2005) showed that the rate of mitochondrial evolution is elevated and highly variable at these shorter divergence times and so cannot be expected to behave in clock-like manner. Nevertheless, these studies of forest-dependent species imply that little gene flow occurred between the two islands probably because the land bridge was likely to have been ‘limited and intermittent’ during the Pleistocene (Lloyd 2003a). Also these studies indicate that forested refugia existed in the South Island of New Zealand throughout the Pleistocene and thus allowed the survival of lineages distinct from those on the North Island.
Currently the mainland populations of NI Kokako are small and highly vulnerable to introduced predators. Therefore, conservation managers have relocated NI Kokako from mainland populations to create new populations on offshore islands. This action aims to bolster the number of viable populations and reduce the risk of extinction through stochastic processes. The results of this study and those from a microsatellite study by Hudson et al. (2000) suggest that before relocation the sampled populations of NI Kokako were closely related and that the movement of Kokako between these populations has not compromised the natural genetic architecture of this species.
Acknowledgments
We thank Te Papa, the National Museum of New Zealand and the Naturhistorisches Museum, Vienna, Austria, for foot-pad biopsies from Callaeidae species. We also thank Professor D. Lambert and Dr T. King of Massey University who kindly provided samples of Saddleback DNA. Quanah Hudson generously provided blood samples from Te Urewera Kokako. We also thank Cathryn Abbott, Nadeena Beck and Sarah Legge for their comments on the manuscript. New Zealand’s Department of Conservation and the Australian National University funded this study. South Island Kokako are a taonga of Poutini Ngai Tahu and North Island Kokako are taonga to northern Iwi. We thank them for their cooperation, and in some cases direct assistance, with the collection and processing of samples. We hope that the information contained in this paper strengthens their knowledge of these taonga.
Abbott, C. L. , and Double, M. C. (2003). Phylogeography of shy and white-capped albatrosses inferred from mitochondrial DNA sequences: implications for population history and taxonomy. Molecular Ecology 12, 2747–2758.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Baker, A. J. , Daugherty, C. H. , Colbourne, R. , and McLennan, J. L. (1995). Flightless brown kiwis of New Zealand possess extremely subdivided population structure and cryptic species like small mammals. Proceedings of the National Academy of Sciences of the United States of America 92, 8254–8258.
| PubMed |
Fleming, C. A. (1962). History of the New Zealand land bird fauna. Notornis 9, 270–274.
Freeland, J. R. , and Boag, P. T. (1999). Phylogenetics of Darwin’s finches: paraphyly in the tree-finches, and two divergent lineages in the Warbler Finch. Auk 116, 577–588.
Hillis, D. M. , and Bull, J. J. (1993). An empirical test of bootstrapping as a method for assessing confidence in phylogenetic analysis. Systematic Biology 42, 182–192.
Knowles, L. L. , and Maddison, W. P. (2002). Statistical phylogeography. Molecular Ecology 11, 2623–2635.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Milá, B. , Girman, D. J. , Kimura, M. , and Smith, T. B. (2000). Genetic evidence for the effect of a postglacial population expansion on the phylogeography of a North American songbird. Proceedings of the Royal Society of London Series B 267, 1033–1040.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Neiman, M. , and Lively, C. M. (2004). Pleistocene glaciation is implicated in the phylogeographical structure of Potamopyrgus antipodarum, a New Zealand snail. Molecular Ecology 13, 3085–3098.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Posada, D. , and Crandall, K. A. (1998). MODELTEST: testing the model of DNA substitution. Bioinformatics 14, 817–818.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Schultheis, A. S. , Weigt, L. A. , and Hendricks, A. C. (2002). Gene flow, dispersal, and nested clade analysis among populations of the stonefly Peltoperla tarteri in the southern Appalachians. Molecular Ecology 11, 317–327.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
Tarr, C. L. (1995). Primers for amplification and determination of mitochondrial control-region sequences in oscine passerines. Molecular Ecology 4, 527–529.
| PubMed |
Turner, T. F. , Trexler, J. C. , Harris, J. L. , and Haynes, J. L. (2000). Nested cladistic analysis indicates population fragmentation shapes genetic diversity in a freshwater mussel. Genetics 154, 777–785.
| PubMed |
Wenink, P. W. , Baker, A. J. , and Tilanus, M. G. J. (1994). Mitochondrial control-region sequences in two shorebird species, the turnstone and the dunlin, and their utility in population genetic studies. Molecular Biology and Evolution 11, 22–31.
| PubMed |