Australian Journal of Chemistry
Volume 63 Number 12 2010
RESEARCH FRONT: Heron 5
CH103895th Heron Island Conference on Reactive Intermediates and Unusual Molecules
Curt Wentrup and Craig Williams
pp. 1597-1597

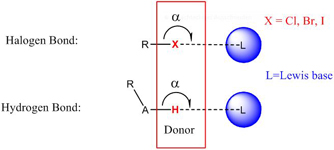
CH10259Directional Weak Intermolecular Interactions: σ-Hole Bonding
Jane S. Murray, Kevin E. Riley, Peter Politzer and Timothy Clark
pp. 1598-1607
CH10325UV-photoelectron Spectroscopy of Unhindered Germylenes and Carbon-arsenic Multiple-bonded Species
Anna Chrostowska, Alain Dargelos and Alain Graciaa
pp. 1608-1614

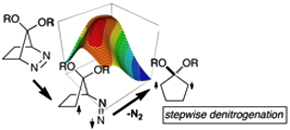
CH10281Notable Substituent Effects on the Rate Constant of Thermal Denitrogenation of Cyclic Azoalkanes: Strong Evidence for a Stepwise Denitrogenation Mechanism
Chizuko Ishihara and Manabu Abe
pp. 1615-1618
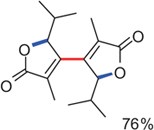
CH10342Gold-Catalysis: Reactions of Organogold Compounds with Electrophiles
A. Stephen K. Hashmi, Tanuja Dondeti Ramamurthi, Matthew H. Todd, Althea S.-K. Tsang and Katharina Graf
pp. 1619-1626
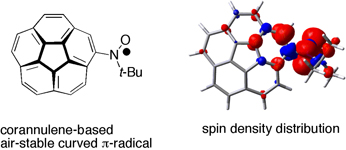
CH10280Air-stable Curved π-Radical Based on Corannulene: Dynamic Electronic-spin Structure Induced by Temperature-dependent Conformational Changes
Akira Ueda, Kanako Ogasawara, Shinsuke Nishida, Kozo Fukui, Kazunobu Sato, Takeji Takui, Kazuhiro Nakasuji and Yasushi Morita
pp. 1627-1633
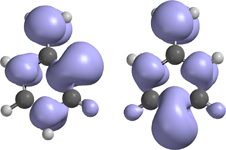
CH10348Electron Paramagnetic Resonance Spectroscopic Characterization of α,2- and α,4-Didehydrotoluene
Patrik Neuhaus, Stefan Henkel and Wolfram Sander
pp. 1634-1637
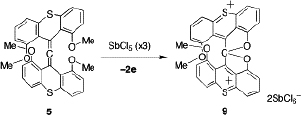
CH10297Demethylation of an Allene Bearing Two Dimethoxythioxanthene Groups by Oxidation via a Vinyl Cation Intermediate
Torahiko Yamaguchi, Shin-ichi Fuku-en, Shun Sugawara, Satoshi Kojima and Yohsuke Yamamoto
pp. 1638-1644
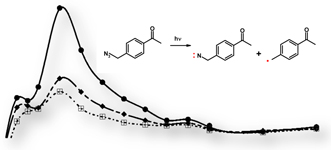
CH10331Competition Between Azido Cleavage and Triplet Nitrene Formation in Azidomethylacetophenones
Ranaweera A. A. Upul Ranaweera, Yu Zhao, Sivaramakrishnan Muthukrishnan, Christopher Keller and Anna D. Gudmundsdottir
pp. 1645-1655
CH10318New Syntheses and Ring Expansion Reactions of Cyclobutenimines
Ernst Schaumann, Gerrit Oppermann, Michael Stranberg and Harold W. Moore
pp. 1656-1664
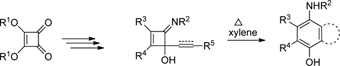
CH10359The Pd-Catalyzed Alder–Ene Reactions of N-Protected and Propargylated 1-Amino-2-aryl-2-cyclohexenes as a New Route to C3a-Arylhexahydroindoles: Towards the Total Synthesis of Tazettine
Anna L. Lehmann, Anthony C. Willis and Martin G. Banwell
pp. 1665-1678
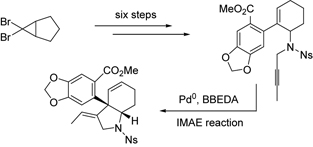
CH10345Usual Molecules in Unusual Environments Displaying Unusual Properties
Ernesto Brunet
pp. 1679-1685
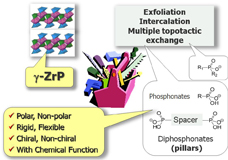
CH10303Structure, Stability, and Generation of CH3CNS
Melinda Krebsz, Balázs Hajgató, Gábor Bazsó, György Tarczay and Tibor Pasinszki
pp. 1686-1693
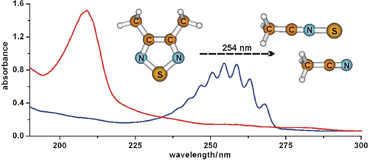
CH10340Thioketenes and Iminopropadienethiones RN=C=C=C=S from Isoxazolones
David Kvaskoff and Curt Wentrup
pp. 1694-1702
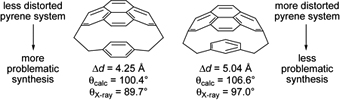
CH10356Mixed [2.2]Cyclophanes of Pyrene and Benzene
Rudolf J. Vermeij, David O. Miller, Louise N. Dawe, Ivan Aprahamian, Tuvia Sheradsky, Mordecai Rabinovitz and Graham J. Bodwell
pp. 1703-1716
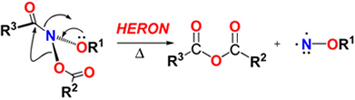
CH10350Thermal Decomposition of N-Acyloxy-N-alkoxyamides – a New HERON Reaction
Jennifer P. Johns, Arjan van Losenoord, Clément Mary, Pierre Garcia, Damian S. Pankhurst, Adam A. Rosser and Stephen A. Glover
pp. 1717-1729