

Long-read Pore-C shows the 3D structure of the cattle genome
Loan T. Nguyen A * , Hyungtaek Jung A , Jun Ma B , Stacey Andersen B and Elizabeth Ross A
A * , Hyungtaek Jung A , Jun Ma B , Stacey Andersen B and Elizabeth Ross A
A Queensland Alliance for Agriculture and Food Innovation, The University of Queensland, Brisbane, Qld, Australia.
B Genome Innovation Hub, The University of Queensland, Brisbane, Qld, Australia.
Animal Production Science 63(11) 972-982 https://doi.org/10.1071/AN22479
Submitted: 4 January 2023 Accepted: 9 March 2023 Published: 24 April 2023
© 2023 The Author(s) (or their employer(s)). Published by CSIRO Publishing. This is an open access article distributed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License (CC BY-NC-ND)
Abstract
Context: Recent advances in molecular technology have allowed us to examine the cattle genome with an accuracy never before possible. Genetic variations, both small and large, as well as the transcriptional landscape of the bovine genome, have both been explored in many studies. However, the topological configuration of the genome has not been extensively investigated, largely due to the cost of the assays required. Such assays can both identify topologically associated domains and be used for genome scaffolding.
Aims: This study aimed to implement a chromatin conformation capture together with long-read nanopore sequencing (Pore-C) pipeline for scaffolding a draft assembly and identifying topologically associating domains (TADs) of a Bos indicus Brahman cow.
Methods: Genomic DNA from a liver sample was first cross-linked to proteins, preserving the spatial proximity of loci. Restriction digestion and proximity ligation were then used to join cross-linked fragments, followed by nucleic isolation. The Pore-C DNA extracts were then prepped and sequenced on a PromethION device. Two genome assemblies were used to analyse the data, namely, one generated from sequencing of the same Brahman cow, and the other is the ARS-UCD1.2 Bos taurus assembly. The Pore-C snakemake pipeline was used to map, assign bins and scaffold the draft and current annotated bovine assemblies. The contact matrices were then used to identify TADs.
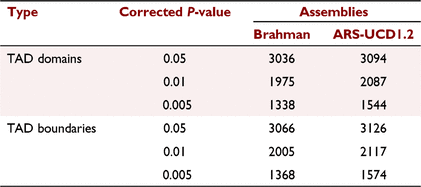
Key results: The study scaffolded a chromosome-level Bos indicus assembly representing 30 chromosomes. The scaffolded assembly showed a total of 215 contigs (2.6 Gbp) with N50 of 44.8 Mb. The maximum contig length was 156.8 Mb. The GC content of the scaffold assembly is 41 ± 0.02%. Over 50% of mapped chimeric reads identified for both assemblies had three or more contacts. This is the first experimental study to identify TADs in bovine species. In total, 3036 and 3094 TADs across 30 chromosomes were identified for input Brahman and ARS-UCD1.2 assemblies respectively.
Conclusions: The Pore-C pipeline presented herein will be a valuable approach to scaffold draft assemblies for agricultural species and understand the chromatin structure at different scales.
Implications: The Pore-C approach will open a new era of 3D genome-organisation studies across agriculture species.
Keywords: 3D chromatin conformation capture, Bos indicus, Bos taurus, Brahman cattle, multi-way contacts, nanopore sequencing technologies, Pore-C, topologically associating domains.
Introduction
Cattle and their products are one of the top global agricultural commodities. Over the past 20 years, total global consumption of beef meat has been steadily growing at an average of 1.1% annually (OECD/FAO 2022). In 2021, Australia was listed as the fourth-largest beef exporter (IHS Markit 2023). Importantly, the beef industry is one of Australia’s major agricultural industries, accounting for 22% of total farm export income in 2018–2019 (Meat & Livestock Australia 2022). Bos indicus cattle are extensively farmed in northern Australia due to their ability to withstand the harsh environmental conditions. However, their reproductive performance is lower than that of Bos taurus cattle (Johnston et al. 2009), which limits profitability and the rate of genetic gain in the industry. Therefore efforts are underway to characterise genetic and transcriptomic variation in beef cattle that could result in economic gains.
As a result of the rapid development of next-generation sequencing, especially long-read sequencing technologies such as PacBio and Oxford Nanopore Technologies (ONT), numerous genome assemblies for agricultural species have been recently published (Jain et al. 2018; Choi et al. 2020; Moss et al. 2020; Upadhyay et al. 2020). For example, an assembly of a Simmental animal, a Bos taurus breed, has been generated using ONT and Hi-C (Heaton et al. 2021). Long-read technology, which can sequence the entire length of DNA fragments, offers an opportunity for assembling a genome that exceeds the continuity of the current annotated Bos taurus reference assembly. Additionally, assembly contiguity can be further increased by the additional information on the folded state of a genome by using chromatin conformation-capture assays (Jiao et al. 2017).
Generally, the Hi-C, a chromatin conformation-capture (3C) assay, uses short-read Illumina sequencing technology to investigate interactions between genomic loci that are not adjacent in the primary sequence (Belton et al. 2012). By using cross-linking and proximity ligation, the Hi-C protocol has been applied to understand the 3D structure of the genome of important agricultural species such as rice, cattle, goat (Bickhart et al. 2017; Dong et al. 2018; Low et al. 2020). The utilisation of Hi-C for genome scaffolding could assist the exploration of compartments, topologically associating domains (TADs) and loops (Luo et al. 2021). However, how DNA folds and coordinates functional activities is extremely complex and may be insufficiently explained by pair-wise interactions generated from short-read sequencing technologies. Transcriptional machinery may require the interaction of three or more DNA loci. Additionally, the true linear genomic distance between the interacted DNA loci varies greatly. The maximum distance could be a few million bases, which could prevent Hi-C paired-reads from accurately scaffolding a draft genome assembly (Luo et al. 2021).
Recently, ONT has developed a 3C method combined with long-read sequencing of concatemers, called Pore-C, to capture the long-range information of DNA–DNA interactions (Oxford Nanopore Technologies). This method has proved its efficiency in the scaffolding of a rice genome assembly (Choi et al. 2020). The complete sample-to-answer ONT protocol for rice assembly and scaffolding took about 1 week, yielding a draft assembly with 527 contigs and an N50 length of 3.7 Mb. Additionally, TADs are known to be associated with gene expression (Dixon et al. 2012), but the putative experimental TADs in bovine are not well defined. Here, we aimed to employ the Pore-C pipeline for scaffolding and TAD identifications of a Bos indicus sample.
Materials and methods
Sample information and ethics
The liver sample was collected from a Brahman cow under relevant guidelines and regulations approved by the Queensland Department of Agriculture and Fisheries Animal Ethics Committee. This sample was collected at the abattoir post-commercial slaughter, snap-frozen in liquid nitrogen, transported to the laboratory on dry ice and stored at −80°C until use.
Pore-C DNA extraction
Cross-linking
The frozen liver sample was ground into a fine powder by using a pre-chill mortar and pestle with liquid nitrogen. Approximately 100 mg of cryo-ground liver tissue was transferred to a 50 mL falcon tube and resuspended in 1 mL of chilled 1× phosphate-buffered saline. The prepared sample was centrifuged at 300g at 4°C for 5 min. Pellet was resuspended with 1% formaldehyde solution and incubated for 10 min at room temperature for cross-linking.
Cell lysis and chromatin digestion
For cell lysis, 550 μL protease inhibitor cocktail-permeabilisation solution (10 mM Tris-HCl pH 8.0, 10 mM NaCl, 0.2% IGEPAL CAS630 and 50 μL of protease inhibitors) was added to one freshly cross-linked sample pellet, followed by 15 min incubation on ice. The suspension was spun down for 10 min at 500g at 4°C. The supernatant was discarded, and the pellet was resuspended in a pre-chilled 1.5× digestion reaction buffer by gently pipetting up and down. To denature the chromatin, 1% sodium dodecyl sulfate (SDS) was added directly to the sample suspension, and the mixture was incubated at 65°C for 10 min at 300 rpm. The tube was then put on ice and 10% ECOSURF EH-9 was added and mixed to quench SDS. Chromatin was subsequently digested overnight at 37°C by adding 45 units NlaIII (New England BioLabs, NEB).
Proximity ligation
The restriction enzyme was inactivated by incubating the sample suspension at 65°C with 300 rpm for 20 min. The proximity ligation reaction was then added to the mixture and incubated at 16°C with 300 rpm for 6 h.
Reserve cross-link and DNA purification
To reserve cross-links and degrade protein, the protein degradation reaction (5% Tween-20, 0.5% SDS and proteinase K) was added to the sample suspension. The mixture was incubated at 56°C with 300 rpm for 18 h. The reaction mixture was then cooled on ice to room temperature. The DNA was extracted by adding pre-chilled phenol:chloroform:isoamyl alcohol 25:24:1. After mixing by inverting and centrifugation for 15 min at 16 000g at 15°C, the supernatants were transferred to new Eppendorf tubes. To precipitate the DNA, 0.2 volumes of 5 M NaCl and 0.1 volumes of 3 M sodium acetate were added and mixed well before adding three volumes of ice-cold 100% ethanol. Tubes were inverted several times to mix properly and were incubated at ×80°C for at least 1 h. Next, the Eppendorf tubes were spun at 4°C for 30 min at 16 000g. The supernatant was discarded, and each DNA pellet was washed with 70% ethanol before resuspending in the Tris–EDTA buffer.
Quality control of extract DNA
The quantity of Pore-C DNA extracts was measured using an Invitrogen Qubit 4.0 device (Thermo Fisher Scientific). The size of extracted DNA samples was visualised by running on Tapestation (Sage Science).
Nanopore sequencing
Pore-C DNA extracts and genomic DNA extracted using Gentra Puregene Tissue Kit (Qiagen, Hilden, Germany) were prepared for sequencing, following the protocol in the genomic sequencing kit SQK-LSK110 (Oxford Nanopore Technologies, Oxford, UK), with some modifications. Briefly, approximately 3 μg of Pore-C DNA extracts and 8 μg of genomic liver DNA were end-repaired and deoxyadenosine (dA)-tailed by using the Ultra II end-repair module (New England BioLabs, NEB, Australia). Sequencing adapters (Oxford Nanopore Technologies) were ligated using blund/T4 ligase (New England BioLabs, NEB). Libraries from end-repair reaction and ligation steps were clean-up with AMPureXP beads (Beckman Coulter, Australia). All incubation times at the end-repair and ligation steps were extended. After cleaning, each prepped library was resuspended in 24 μL of elution buffer (Oxford Nanopore Technologies, ONT) before being combined with 75 μL of sequencing buffer (SQB, Oxford Nanopore Technologies) and 51 μL of loading bead and loaded on a promethION flow cell (FLO-PRO102, R9.4.1 version). Sequencing was performed for 96 h with a promethION sequencer using high accuracy base-calling model (Guppy ver. 6.0.7) for each library.
Pore-C pipeline for scaffolding
Input assemblies
A draft Brahman genome assembled by Ross et al. (2022a) was used for scaffolding in this study. Briefly, this draft assembly was created by integrating short-read Illumina sequencing, PacBio, ONT, Chicago and Dovetail HiC. The study also tested the effect of Pore-C data for scaffolding current annotated Bos taurus genome reference: ARS-UCD1.2 (Rosen et al. 2020).
Pore-C snakemake pipeline
Pore-C data were analysed using the Pore-C analysis pipeline (https://github.com/nanoporetech/Pore-C-Snakemake) previously developed by Oxford Nanopore Technologies (ONT), available with snakemake workflow (Köster and Rahmann 2012). Briefly, the reference genome or draft assembly was pre-processed to create virtual in silico restriction maps of the reference genome. In this study, the virtual digests of a draft Brahman assembly (Ross et al. 2022a) and annotated Bos taurus reference genome (ARS-UCD1.2) were generated using the pore_c refgenome virtual-digest tool. The Pore-C reads were first aligned to either the draft Bos indicus reference genome (Ross et al. 2022a) or Bos taurus reference genome (ARS-UCD1.2) by using bwa ver. 0.7.17 (Li and Durbin 2009). The mapped files were filtered to remove spurious alignments. On the basis of an in silico restriction digest map, the filtered alignments were then annotated with the set of reference-genome restriction fragments. From those fragment sets, each alignment was assigned to a single fragment on the basis of the position of the alignment mid-point, eliminating direct non-chimeric pairs from either cognate re-ligation or incomplete digestion. The assignment of each alignment along a Pore-C read resulted in a multi-way contact, which was then decomposed into pairwise contacts. Conventional Hi-C workflow was then used to assign the pairwise contacts to genome bins (Ramírez et al. 2018; Abdennur and Mirny 2019). The resulted bed format contact data were used to scaffold the draft assembly with a 3D de novo assembly pipeline (3D-DNA; Dudchenko et al. 2017) and curated using the juicebox software (Robinson et al. 2018).
Identification of topologically associating domains (TADs)
The Pore-C data at 50 kbp resolution was used to calculate the TAD separation score and define the TAD domains and boundaries. Briefly, TAD domains and boundaries were identified using the hicFindTADs function from the hicexplorer tool (Ramírez et al. 2018). Generally, the TADs were called as those domains having a local TAD separation score minimum, reflecting reduced contact frequencies between upstream and downstream loci. The TAD separation score was calculated using the z-score of the Pore-C contact matrix and was defined as the mean z-score of all the matrix contacts between the left and right regions. To find TADs, the Pore-C contact matrix was first transformed into a z-score matrix. Each contact frequency in the matrix was transformed into a z-score based on the distribution of all contacts at the same genomic distance. For each bin, the contacts between an upstream and downstream region were in z-score submatrix. The TAD separation score was computed for each matrix bin. Bins having a local minimum of the TAD separation score were evaluated with respect to the surrounding bins to assign a P-value. The Wilcoxon rank-sum test was used to compute the P-value, and the Bonferroni method was used to correct the P-value. Only TAD boundaries and domain with a minimum local minima depth of 0.01 with a corrected P-value threshold of 0.05 were used for comparison with published TADs.
Compare bovine experimental TADs with published TADs
Putative bovine TAD genomic coordinates based on dog TADs provided by Wang et al. (2018) were converted to the bovine genome Bos taurus UMD3.1.1 and then Bos taurus ARS.UCD1.2 by using liftover (Kuhn et al. 2013). The overlap in TAD regions between published bovine TADs (Wang et al. 2018) and TADs identified in this study by using ARS-UCD1.2 assembly was identified using the bedtools software (Quinlan and Hall 2010). The output bed file containing TAD domains at a corrected P-value of 0.05 was used in this task.
Visualisation of TADs with other genome tracts
The identified TADs from both assemblies were visualised with two sets of data from the same animal and tissue, namely, PacBio full-length Isoseq transcript sequencing (methods have been published in Ross et al. (2022b) and Nguyen et al. (2022)) and reduced representation bisulfite sequencing (unpubl. data) using hicPlotTADs option from hicexplorer tool (Ramírez et al. 2018).
Results
Nanopore sequencing of chromatin concatemers
The Pore-C library generated 99.5 passed Gbp of sequence data, equivalent to 36× of genome-wide read depth from one flow cell. The read length N50 of Pore-C data was 2.8 kbp (Fig. 1a). The length N50 was in the expected range reported by nanopore communities, where Pore-C DNA extract length was within 7–10 kb. Compared with normal nanopore sequencing, the same liver sample generated long-read sequenced data with an N50 length of 12.7 kb (Fig. 1b).

|
When the draft Brahman assembly was used for mapping and constructing in silico digestion maps, a total of 552.9 million concatemers with a density of 6.5 million contacts per giga base was detected (Table 1). The equivalent metrics were slightly lower when using the Bos taurus reference genome, resulting in 517.8 million concatemers with a density of 6.2 (Table 1). An expectable ratio between long-range cis- and trans-interactions was also detected from the Pore-C data, regardless of the input assembly used. In addition to the pair-wise contact, the Pore-C data were able to detect >50% of contacts with order of three or greater in both assemblies (Table 1), that is, chromatin from more than three genome regions was connected and sequenced.
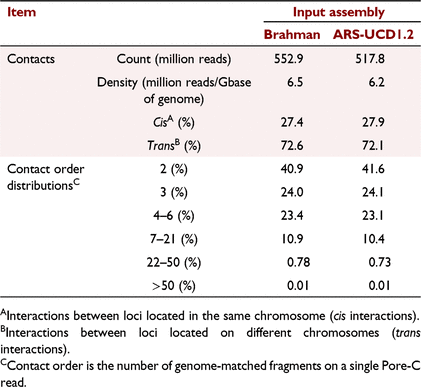
|
Nanopore scaffolding of draft genome assemblies
The Pore-C data were used to assess the ability of the Pore-C pipeline to faithfully reflect linear genome structure by scaffolding draft Brahman genome assembly. To assess the effectiveness of the Brahman Pore-C data to different bovine subspecies, the study also tested the Pore-C data against the annotated Bos taurus reference genome (ARS-UCD1.2). A slight improvement of the Brahman assembly was observed after scaffolding with Pore-C data (Table 2), whereas there was no improvement obtained when using ARS-UCD1.2 as input assembly. The percentage of the genome contained in gaps in the Brahman reference genome was reduced from 0.012% to 0.005%, while it increased from 0.001% to 0.167% in scaffolded ARS-UCD1.2 assembly. The length of the Brahman assemblies was 2.64 Gb, while the length of scaffolded ARS-UCD1.2 was 2.76 Gb, slightly longer than the input ARS-UCD1.2 assembly (2.72 Gb). Interestingly, using the Pore-C pipeline, the scaffolded Brahman assembly (UQ_Brahman_2) resulted in a contig N50 of 44.8 Mbp, a significant improvement. The number of contigs was reduced by 145 when scaffolding the draft Brahman assembly.

|
Visualisation of pair-wise contact maps
Despite having fewer pairwise contacts and no improvement in the final scaffolded assembly for the Bos taurus reference genome, the heatmap generated by mapping the Pore-C scaffolded and ARS-UCD1.2 reference using juicebox (Robinson et al. 2018) showed significant concordance (Fig. 2, Supplementary material Fig. S1). Interestingly, disagreements were observed when visualising chromatin interaction between the studied Pore-C data with ARS-UCD1.2 assembly in the X chromosome (Fig. 2c). These disagreements could be explained by the different structural variants (SVs) among subspecies as well as the specialisation of X chromosome after divergence of Bos species (Fortes et al. 2020).

|
TAD identifications
This study is the first report of the experimental putative TADs in cattle. On the basis of Pore-C matrices, there were more than 3000 TADs identified in both assemblies with a corrected P-value of 0.05 (Table 3). As expected, this study observed highly significant correlation between the number of TADs per chromosome and the chromosome size for both assemblies (Fig. 3a, Supplementary Table S1).
|

|
Comparison of bovine experimental TADs with published TADs
We compared the Pore-C Brahman TADs with a number of published TADs detected in liver samples from different species (Rudan et al. 2015). At a corrected P-value of 0.05, the Brahman data showed a number of TADs similar to that in other investigations (3643 and 3315 of TADs identified in mice and dogs respectively). Wang et al. (2018) utilised genomic synteny to map TADs from dogs and mice to bovine genome (UMD3.1), resulting in 2979 and 3507 TADs in cattle (Wang et al. 2018). Of 3094 TADs identified in this study for the ARS-UCD1.2 assembly, 958 TADs were not overlapping with the bovine TADs set identified by using dog TADs provided Wang et al. (2018). TADs with a minimum overlap fraction of 50% and 100% between our TADs and putative bovine TADs identified by Wang et al. (2018) resulted in a total of 1198 and 318 TAD regions respectively.
Visualisation of TADs with other genome tracts
We investigated the overlap between methylation and expression data and the TADs we have discovered on Chromosome 1 of Brahman assembly. We observed agreement of isoforms and methylation sites along Chromosome 1 (Fig. 3b). Interestingly, TADs having high separation scores seem to be transcriptionally inactive with high methylation profiles and low gene expression.
Discussion
The availability of highly complete and contiguous genome assemblies in agricultural species will enable functional and evolutionary genomic analyses, providing precise identification of genetic variations, from single nucleotide polymorphisms to large SVs that drive the difference in traits of interest among species. A highly contiguous assembly of several agricultural species has been established using the Pore-C pipeline, including rice, Arabidopsis and Ohia plant (Choi et al. 2020; Li et al. 2022). In rice species, the assembly contiguity increased at eight-fold when using Pore-C workflow (Choi et al. 2020). More importantly, this complete sample-to-answer pipeline took about 1 week, making it readily adaptable to any 3D genome-organisation studies across agriculture species. In cattle, having a high-quality genome assembly for Bos indicus species is optimal, which can eliminate the reference bias when using a taurine assembly. The Brahman reference genome could serve as a valuable resource for gene editing or genome selection of causal variants, especially for SVs for beef cattle.
Compared with Hi-C, Pore-C library preparation is much simpler by removing the biotin labelling, fragmentation, and purification steps. In situ ligation also facilitates retaining the interaction information in Pore-C reads in different cells. More importantly, Pore-C can directly capture the multi-way contacts compared with the pair-wise contacts obtained by Hi-C, paving the way to improve the genome assemblies. In this study, the Pore-C pipeline generated more than 50% of reads containing multi-way interactions (contact order of equal or greater than three), consistent with Pore-C libraries generated for Arabidopsis (44%) and human cells (47.56%) (Ulahannan et al. 2019; Li et al. 2022).
The Brahman genome assembly presented here is a continuous chromosome-scale reference genome. The final scaffolded Brahman assembly (UQ_Brahman_2) consists of 215 contigs assembled into 31 scaffolds. The quality of contigs N50 of 44.8 Mbp and scaffold N50 of 104.8 Mb (max scaffold 157.8 Mb) exceeds the current annotated Brahman and Bos taurus reference genomes. The scaffold N50 of the final Brahman assembly (UQ_Brahman_2) was 104 Mb, exceeding the continuity of the current annotated ARS-UCD1.2 assembly (103 Mb) and the ARS_Simm1.0 assembly (102 Mb). The UQ_Brahman_2 assembly had only 31 scaffolds compared with 2211 for Hereford, 1250 for UOA_Brahman_1 (GCA_003369695.2) and 1584 for the Brahman assembly that used the haplotype-resolved genome approach. To validate the effectiveness and robustness of the Pore-C pipeline for producing exemplary quality reference assemblies, scaffolding using draft assembly generated by whole genome long-read nanopore sequencing could be also performed.
TADs have been known as regulatory units of the genome and tend to be conserved between cell types, across species and along evolution. More than 50% of TAD boundaries identified in human cells were reported to be found at homologous locations in mouse genomes (Dixon et al. 2012, 2016). However, despite this conservation of TADs across species, the underlying regulatory systems for traits of interest seem substantially to be divergent, implying that understanding complex traits need species-specific information (Schmidt et al. 2010; Cheng et al. 2014; Stergachis et al. 2014). Additionally, study by Foissac et al. (2019) also indicated that TADs boundaries under stronger selective pressure play more important role in genome architecture and regulatory function.
The only available TAD information for cattle is from the genomic synteny map of human, mouse and dog TADs that were mapped to the Bos taurus UMD3.1 assembly (Wang et al. 2018). The computation method applied by Wang et al. (2018) relied on quality of genome assemblies as well as the conserved nature of TADs. Therefore, to the best of our knowledge, this was the first study providing experimental TAD structures in the bovine sample, especially using a platinum assembly of Brahman cattle (UQ_Brahman_2). When comparing our findings with published TADs, a low level of concordance between the studies was observed. The dissimilarity of TAD regions could be caused by differences among species, breeds and samples, as well as computational methods. As such, the divergence/genetic diversity among subspecies as well as a long evolutionary distance between Bos taurus taurus and Bos taurus indicus (over 210 000 years ago) will make our dataset valuable for understanding the underlying regulatory systems and genome variation across species and evolutionary distances (Krefting et al. 2018; Foissac et al. 2019).
Chromatin topological associating domains and loop maps generated by the Pore-C pipeline offer numerous genomic applications. As such, the study by Wang et al. (2018) noted that having TAD information could decrease the search space for identifying functional causative variants that affect the complex traits in cattle. In humans, genetic variants in TADs have been shown to contribute to complex human diseases (McArthur and Capra 2021). The simple and scalable Pore-C pipeline presented herein could also be used to show active and in-active compartments when case and control samples are utilised in the study. The low cost of the consumables also means that multiple samples could be assayed, allowing multiple biological replicates to be included in future studies.
Although this study did not investigate the analysis of base modifications on Pore-C data, the methylation signals could be retrieved from this Pore-C dataset and used to discover the relationship between methylation states and chromatin structures. A study by Li et al. (2022) noted the high correlation between methylation profiles called by Pore-C and whole genome bisulfite sequencing for Arabidopsis samples. In humans, the concatemers of higher-order contacts in highly expressed genes were preferentially demethylated, suggesting the modulation of methylation by gene expression levels. The PCR-free and long-read sequencing approach of Pore-C also enables the detection of SVs, as stated by (Deshpande et al. 2022). Such applications would make Pore-C a robust and scalable tool for studying high-order chromatin structure, genome assembly, methylation profiling and SVs in one assay.
Conclusions
This study implemented the Pore-C pipeline for improving the scaffolding of draft assembly in mammalian species and revealing higher-order 3D chromatin structure at the chromatin domains scale. With this method, the final chromosome-scale Brahman assembly (UQ_Brahman_2) resulted in 31 scaffolds and 215 contigs, with contig N50 of 44.8 Mbp. Additionally, a number of TADs were identified, and their overlap with methylation islands and gene expression was observed. This study demonstrated that Pore-C is a simple, accurate, effective, and scalable approach for exploring multi-way chromatin interactions and genome scaffolding.
Supplementary material
Supplementary material is available online.
Data availability
Bos indicus raw Pore-C sequencing data were deposited in Sequence Read Archive (SRA) with the project accession PRJNA941083.
Conflicts of interest
The authors declare no conflicts of interest.
Declaration of funding
The authors thank the UQ Genomic Innovation Hub grant for the funding for this research project ‘Multi-contact Pore-C’; and the Research Strategic Package 3 funding from the University of Queensland.
Author contributions
LTN, HJ and ER conceptualised the project and acquired funding. LTN and JM performed all molecular works. LTN, HJ, JM, SA and ER administered the project. HJ performed the Pore-c snakemake pipeline. LTN performed downstream analysis and visualised the data. LN wrote the manuscript. All authors reviewed and edited and approved the final manuscript.
Acknowledgements
We thank Dr Mike Yarski and ONT support team for advising on molecular tasks and providing support with Pore-c snakemake workflow scripts. We also acknowledge Dr Subash Rai for his support with running Tapestation of Liver Pore-C DNA extract. We thank our colleagues in the Centre for Animal Science, QAAFI, UQ, for their support and enthusiasm.
References
Abdennur N, Mirny LA (2019) Cooler: scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics 36, 311–316.| Cooler: scalable storage for Hi-C data and other genomically labeled arrays.Crossref | GoogleScholarGoogle Scholar |
Belton J-M, McCord RP, Gibcus JH, et al. (2012) Hi-C: a comprehensive technique to capture the conformation of genomes. Methods 58, 268–276.
| Hi-C: a comprehensive technique to capture the conformation of genomes.Crossref | GoogleScholarGoogle Scholar |
Bickhart DM, Rosen BD, Koren S, et al. (2017) Single-molecule sequencing and chromatin conformation capture enable de novo reference assembly of the domestic goat genome. Nature Genetics 49, 643–650.
| Single-molecule sequencing and chromatin conformation capture enable de novo reference assembly of the domestic goat genome.Crossref | GoogleScholarGoogle Scholar |
Cheng Y, Ma Z, Kim B-H, et al. (2014) Principles of regulatory information conservation between mouse and human. Nature 515, 371–375.
| Principles of regulatory information conservation between mouse and human.Crossref | GoogleScholarGoogle Scholar |
Choi JY, Lye ZN, Groen SC, et al. (2020) Nanopore sequencing-based genome assembly and evolutionary genomics of circum-basmati rice. Genome Biology 21, 21
| Nanopore sequencing-based genome assembly and evolutionary genomics of circum-basmati rice.Crossref | GoogleScholarGoogle Scholar |
Deshpande AS, Ulahannan N, Pendleton M, et al. (2022) Identifying synergistic high-order 3D chromatin conformations from genome-scale nanopore concatemer sequencing. Nature Biotechnology 40, 1488–1499.
| Identifying synergistic high-order 3D chromatin conformations from genome-scale nanopore concatemer sequencing.Crossref | GoogleScholarGoogle Scholar |
Dixon JR, Selvaraj S, Yue F, et al. (2012) Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380.
| Topological domains in mammalian genomes identified by analysis of chromatin interactions.Crossref | GoogleScholarGoogle Scholar |
Dixon JR, Gorkin DU, Ren B (2016) Chromatin domains: the unit of chromosome organization. Molecular Cell 62, 668–680.
| Chromatin domains: the unit of chromosome organization.Crossref | GoogleScholarGoogle Scholar |
Dong Q, Li N, Li X, et al. (2018) Genome-wide Hi-C analysis reveals extensive hierarchical chromatin interactions in rice. The Plant Journal 94, 1141–1156.
| Genome-wide Hi-C analysis reveals extensive hierarchical chromatin interactions in rice.Crossref | GoogleScholarGoogle Scholar |
Dudchenko O, Batra SS, Omer AD, et al. (2017) De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95.
| De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds.Crossref | GoogleScholarGoogle Scholar |
Foissac S, Djebali S, Munyard K, et al. (2019) Multi-species annotation of transcriptome and chromatin structure in domesticated animals. BMC Biology 17, 108
| Multi-species annotation of transcriptome and chromatin structure in domesticated animals.Crossref | GoogleScholarGoogle Scholar |
Fortes MRS, Porto-Neto LR, Satake N (2020) X chromosome variants are associated with male fertility traits in two bovine populations. Genetics Selection Evolution 52, 46
| X chromosome variants are associated with male fertility traits in two bovine populations.Crossref | GoogleScholarGoogle Scholar |
Heaton MP, Smith TPL, Bickhart DM, et al. (2021) A reference genome assembly of Simmental Cattle, Bos taurus taurus. Journal of Heredity 112, 184–191.
| A reference genome assembly of Simmental Cattle, Bos taurus taurus.Crossref | GoogleScholarGoogle Scholar |
Jain M, Koren S, Miga KH, et al. (2018) Nanopore sequencing and assembly of a human genome with ultra-long reads. Nature Biotechnology 36, 338–345.
| Nanopore sequencing and assembly of a human genome with ultra-long reads.Crossref | GoogleScholarGoogle Scholar |
Jiao W-B, Accinelli GG, Hartwig B, et al. (2017) Improving and correcting the contiguity of long-read genome assemblies of three plant species using optical mapping and chromosome conformation capture data. Genome Research 27, 778–786.
| Improving and correcting the contiguity of long-read genome assemblies of three plant species using optical mapping and chromosome conformation capture data.Crossref | GoogleScholarGoogle Scholar |
Johnston DJ, Barwick SA, Corbet NJ, Fordyce G, Holroyd RG, Williams PJ, Burrow HM (2009) Genetics of heifer puberty in two tropical beef genotypes in northern Australia and associations with heifer- and steer-production traits. Animal Production Science 49, 399–412.
| Genetics of heifer puberty in two tropical beef genotypes in northern Australia and associations with heifer- and steer-production traits.Crossref | GoogleScholarGoogle Scholar |
Krefting J, Andrade-Navarro MA, Ibn-Salem J (2018) Evolutionary stability of topologically associating domains is associated with conserved gene regulation. BMC Biology 16, 87
| Evolutionary stability of topologically associating domains is associated with conserved gene regulation.Crossref | GoogleScholarGoogle Scholar |
Kuhn RM, Haussler D, Kent WJ (2013) The UCSC genome browser and associated tools. Briefings in Bioinformatics 14, 144–161.
| The UCSC genome browser and associated tools.Crossref | GoogleScholarGoogle Scholar |
Köster J, Rahmann S (2012) Snakemake – a scalable bioinformatics workflow engine. Bioinformatics 28, 2520–2522.
| Snakemake – a scalable bioinformatics workflow engine.Crossref | GoogleScholarGoogle Scholar |
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760.
| Fast and accurate short read alignment with Burrows–Wheeler transform.Crossref | GoogleScholarGoogle Scholar |
Li Z, Long Y, Yu Y (2022) Pore-C simultaneously captures genome-wide multi-way chromatin interaction and associated DNA methylation status in Arabidopsis. Plant Biotechnology Journal 20, 1009–1011.
| Pore-C simultaneously captures genome-wide multi-way chromatin interaction and associated DNA methylation status in Arabidopsis.Crossref | GoogleScholarGoogle Scholar |
Low WY, Tearle R, Liu R, et al. (2020) Haplotype-resolved genomes provide insights into structural variation and gene content in Angus and Brahman cattle. Nature Communnication 11, 2071
| Haplotype-resolved genomes provide insights into structural variation and gene content in Angus and Brahman cattle.Crossref | GoogleScholarGoogle Scholar |
Luo J, Wei Y, Lyu M, et al. (2021) A comprehensive review of scaffolding methods in genome assembly. Briefings in Bioinformatics 22, bbab033
| A comprehensive review of scaffolding methods in genome assembly.Crossref | GoogleScholarGoogle Scholar |
IHS Markit (2023) Beef Market Analysis. Available at https://www.spglobal.com/commodityinsights/en/ci/products/food-commodities-food-manufacturing-proteins-beef.html
McArthur E, Capra JA (2021) Topologically associating domain boundaries that are stable across diverse cell types are evolutionarily constrained and enriched for heritability. The American Journal of Human Genetics 108, 269–283.
| Topologically associating domain boundaries that are stable across diverse cell types are evolutionarily constrained and enriched for heritability.Crossref | GoogleScholarGoogle Scholar |
Meat & Livestock Australia (2022) Australia’s red meat and livestock industry well placed to capitalise on exceptional conditions. State of the Industry Report. Meat & Livestock Australia.
Moss EL, Maghini DG, Bhatt AS (2020) Complete, closed bacterial genomes from microbiomes using nanopore sequencing. Nature Biotechnology 38, 701–707.
| Complete, closed bacterial genomes from microbiomes using nanopore sequencing.Crossref | GoogleScholarGoogle Scholar |
Nguyen LT, Cheng Y, Kuo R, et al. (2022) Predicted isoforms of a Brahman cow revealed by full-length transcript sequencing. In ‘Proceedings of 12th World Congress on Genetics Applied to Livestock Production’. (Eds RF Veerkamp, Y de Haas) pp. 2130–2133. https://doi.org/10.3920/978-90-8686-940-4
OECD/FAO (2022) OECD-FAO Agricultural Outlook, OECD Agriculture statistics (database). Available at http://dx.doi.org/10.1787/agr-outl-data-en
Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842.
| BEDTools: a flexible suite of utilities for comparing genomic features.Crossref | GoogleScholarGoogle Scholar |
Ramírez F, Bhardwaj V, Arrigoni L, et al. (2018) High-resolution TADs reveal DNA sequences underlying genome organization in flies. Nature Communications 9, 189
| High-resolution TADs reveal DNA sequences underlying genome organization in flies.Crossref | GoogleScholarGoogle Scholar |
Robinson JT, Turner D, Durand NC, et al. (2018) Juicebox.js provides a cloud-based visualization system for Hi-C data. Cell Systems 6, 256–258.e1.
| Juicebox.js provides a cloud-based visualization system for Hi-C data.Crossref | GoogleScholarGoogle Scholar |
Rosen BD, Bickhart DM, Schnabel RD, et al. (2020) De novo assembly of the cattle reference genome with single-molecule sequencing. GigaScience 9, giaa021
| De novo assembly of the cattle reference genome with single-molecule sequencing.Crossref | GoogleScholarGoogle Scholar |
Ross EM, Nguyen LT, Lamb HJ, et al. (2022a) The genome of tropically adapted Brahman cattle (Bos taurus indicus) reveals novel genome variation in production animals. bioRxiv 2022.02.09.479458
| The genome of tropically adapted Brahman cattle (Bos taurus indicus) reveals novel genome variation in production animals.Crossref | GoogleScholarGoogle Scholar |
Ross EM, Sanjana H, Nguyen LT, et al. (2022b) Extensive variation in gene expression is revealed in 13 fertility-related genes using RNA-Seq, ISO-Seq, and CAGE-Seq from Brahman Cattle. Frontiers in Genetics 13, 784663
| Extensive variation in gene expression is revealed in 13 fertility-related genes using RNA-Seq, ISO-Seq, and CAGE-Seq from Brahman Cattle.Crossref | GoogleScholarGoogle Scholar |
Rudan MV, Barrington C, Henderson S, et al. (2015) Comparative Hi-C reveals that CTCF underlies evolution of chromosomal domain architecture. Cell Reports 10, 1297–1309.
| Comparative Hi-C reveals that CTCF underlies evolution of chromosomal domain architecture.Crossref | GoogleScholarGoogle Scholar |
Schmidt D, Wilson MD, Ballester B, et al. (2010) Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science 328, 1036–1040.
| Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding.Crossref | GoogleScholarGoogle Scholar |
Stergachis AB, Neph S, Sandstrom R, et al. (2014) Conservation of trans-acting circuitry during mammalian regulatory evolution. Nature 515, 365–370.
| Conservation of trans-acting circuitry during mammalian regulatory evolution.Crossref | GoogleScholarGoogle Scholar |
Ulahannan N, Pendleton M, Deshpande A, Schwenk S, Behr JM, Dai X (2019) Nanopore sequencing of DNA concatemers reveals higher-order features of chromatin structure. bioRxiv 833590
Upadhyay M, Hauser A, Kunz E, et al. (2020) The first draft genome assembly of snow sheep (Ovis nivicola). Genome Biology and Evolution 12, 1330–1336.
| The first draft genome assembly of snow sheep (Ovis nivicola).Crossref | GoogleScholarGoogle Scholar |
Wang M, Hancock TP, Chamberlain AJ, et al. (2018) Putative bovine topological association domains and CTCF binding motifs can reduce the search space for causative regulatory variants of complex traits. BMC Genomics 19, 395
| Putative bovine topological association domains and CTCF binding motifs can reduce the search space for causative regulatory variants of complex traits.Crossref | GoogleScholarGoogle Scholar |