

Total colonic aganglionosis – long-segment Hirschsprung disease management successfully performed in Papua New Guinea
J. Mulu A * , O. H. Poki A B , I. Kevau A B and S. Fose CA
B
C
Abstract
Neonatal bowel obstruction is a common surgical pathology and is associated with neonatal sepsis. Medical management resolves most cases, however, a few cases cause challenges for the surgical division whereby resources are diverted by surgically treatable cases such as bowel atresia, Hirschsprung’s disease (HD), malrotation and even anorectal anomalies with imperforated anus. The diagnosis of HD is challenging but it can be diagnosed in the neonatal period. In low and middle-income countries (LMICs) the diagnosis is often missed, and the resulting late presentation is associated with significant co-morbidities. The following case report describes a neonate who was delivered in a village before being transferred to the hospital. A major surgical pathology was then corrected successfully using the limited resources that were available.
Keywords: aganglionosis, Hirschsprung’s disease (HD), neonatal bowel obstruction, Swenson pull through, total colonic, transitional zone.
Introduction
Hirschsprung’s disease is a congenital megacolon defect in which there is an absence of parasympathetic ganglionic cells in both the Meissner’s plexus and Auerbach’s plexus of the colon. It is a functional intestinal obstruction with an intact lumen. In 1886, Harald Hirschsprung1,2 described the condition of two infants who both died with swollen bellies. Autopsies showed pronounced dilatation and hypertrophy of the colon with an intact lumen. The definitive treatment of the disease came about 60 years later by Orvar Swenson in 1948.3
The incidence worldwide is approximately 1 in 5000 live births. Seventy percent (70%) of them are lower segment HD cases but total colonic aganglionosis (TCA) occurs in 8–10% of all HD patients. In rare cases it can involve the terminal ileum. Worldwide, the male-to-female ratio is approximately 4:1 for lower segment HD but as the distance of the aganglionosis from the anus lengthens, the ratio equalises between males and females. The clinical manifestations of short segment HD appear within the first 6 months of life but in long segment HD, it can arise at a much earlier age. Urla and colleagues reported 11 cases which were reviewed in a retrospective study and found that all had good qualities of life after total colectomy and ileoanal anastomosis.4 In the long segment case reported here, the aganglionic portion extended 10 cm proximal to the ileocecal junction and was managed successfully using the available resources.
Case presentation
Baby of R.A. (female, 3 days old) from Bereina in Central Province was delivered in the village by a para 2 gravida 2 (P2G2) un-booked mother on 24 February 2013. She was referred from the rural hospital and was admitted to Port Moresby General Hospital special care nursery on 27 February 2013. She presented with fever, abdominal distension and bilious vomiting. She was initially treated for neonatal sepsis with ampicillin and gentamicin 2 days prior to admission at the nearest district hospital. The 23-year-old para 2 mother denied any congenital anomalies within herself or in the family. She also denied any drug abuse. Upon examination this neonate was sick looking and septic but was not in any respiratory distress. The abdomen was shiny and distended with engorged abdominal veins (Fig. 1).
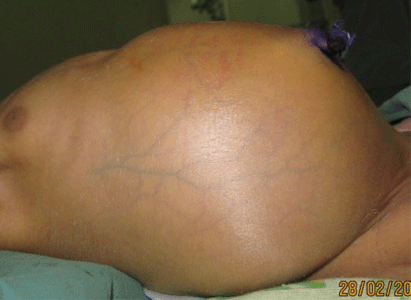
The perineum was normal and patent and there was no explosive sound upon inserting a lubricated thermometer into the anus. Air entry was equal bilaterally and no abnormal heart sound was detected. The other systems were basically normal.
She was admitted as a case of neonatal sepsis with a differential diagnosis of bowel obstruction due to meconium ileus/meconium plug syndrome, small gut atresia, volvulus or malrotation. Hirschsprung’s disease was not included in this differential diagnosis as it is a rare occurrence at this very age.
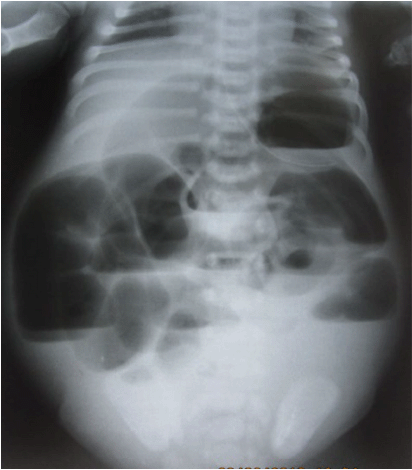
An erect abdominal X-ray revealed multiple air fluid levels, without pneumoperitoneum (Fig. 2). In a neonatal abdominal X-ray, it is difficult to demonstrate well the classical features of valvulae conniventes and haustrations in small and large bowel obstructions respectively, but the most important demonstration is to find the obvious air-fluid levels. This established, in this case, a mechanical obstruction for which surgical relief was the treatment of choice.
A plane erect abdominal X-ray showing multiple-air-fluid levels with a gasless pelvis. This revealed a surgical emergency.
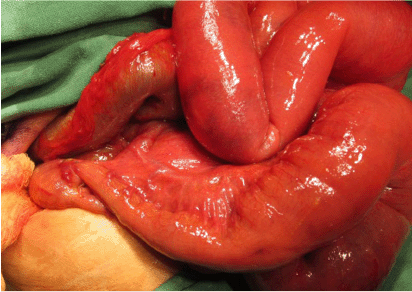
She was taken to the operating theatre on 1 March 2013, 2 days after hospitalisation. A sub-umbilical transverse incision was made under general anaesthesia. There was evidence of sepsis intra-abdominally with two perforations of the dilated small bowel. These perforations were sutured. There was a band at the ileocecal junction, collapsed segment at the terminal ileum with a transitional zone, 10 cm proximal to the ileocecal junction and a distended small bowel from the transitional zone (Fig. 3).
Intra-operative findings of the transitional zone with proximal dilatation and distal collapse at the proximal segment to the ileocecal junction.
Bowel resection (terminal 10 cm ileum, appendix and caecum including the ileocecal valve) and an ileostomy were done, and tissue specimen was sent to the histology department.
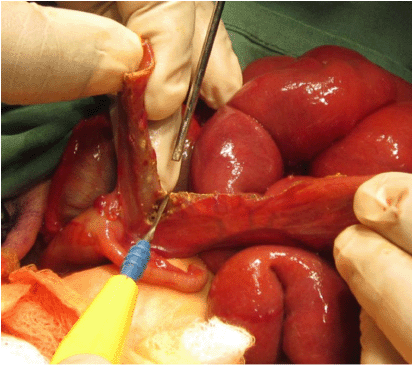
Post operatively the child was commenced on triple antibiotics (gentamicin, metronidazole and amoxicillin) and intravenous fluids accordingly. She was then kept in the ward as she had wound dehiscence on the medial aspect of the ileostomy site which needed to be dressed regularly. There were lots of excoriations around the ileostomy site which was to be expected. The excoriations were treated successfully with zinc oxide. The parents were informed of the high output, semi-fluid contents of the proximal stoma, which they understood well and have managed well by applying sufficient absorbents such as nappies. There wasn’t a sufficient supply of colostomy bags which makes it challenging for such parents (Fig. 4).
Resection of terminal ileum from the transitional zone extending to the ileocecal junction intra-operatively.
Four days later, histology results showed that there were no ganglion cells in the specimens (terminal ileum, appendix and the caecum). A diagnosis of total colonic aganglionosis extending into the terminal ileum was made. The patient recovered well in the ward. There were minimal signs of stoma excoriations, and she had an uneventful recovery. The child had no nutritional problem or electrolyte abnormalities during the period of stoma in situ. Three years later, a Swenson pull through procedure was performed in which the entire large colon was resected, and the terminal ileum was anastomosed to the anus, 2 cm above the dentate line. The perineum was well prepared by soiling the region with the stoma contents a month prior to the definitive procedure. The patient had an uneventful recovery and is a survivor of the rare condition of HD. She is passing well-formed stool with normal sphincter functioning. Currently she has no issues with voiding and defecation.
Discussion
Neural-crest-derived neuroblasts first appear in the fifth week of gestation and the normal migration of neuroblasts in a cephalo-caudal direction within the bowel reaches the rectum by 12 weeks of gestation5 In patients with HD, the caudal migration of cells from the neural crest cells is arrested at some point before reaching their destination. The two nerve roots that can be altered are the parasympathetic fibres of the sacral plexus and the terminal portion of the vagus nerve. The parasympathetic nerve fibres destined for the hindgut originate from the sacral plexus (S2–S4). In total aganglionosis, the vagus nerve can also be affected, as its innervations extends down to the proximal two-thirds of the transverse colon. At a molecular level, HD essentially appears to result from disruption of normal signalling during development. Such pathologies are termed neurocristopathies. Their rare occurrence is hardly surprising, as the signals governing cell migration and development in the embryo are extraordinarily complicated, and signalling molecules are notorious for crosstalk and redundancy. The reasons for the incomplete migration and development of ganglion cells is as yet not completely clear.
HD has a well-documented inherited familial predisposition (2.4–9%) and has also been reported in mono- and dizygotic twins. It has been described as a sex-linked heterogeneous disorder with various degrees of severity.6 It has a variable pattern of inheritance, which includes dominant, recessive, and multigenic patterns of transmission.6 Affected families are known to carry an approximately 200 times higher risk of delivering children with HD. The pattern of inheritance appears to vary in terms of the length of the affected segment. There are nine genes that are responsible for the development of HD and the two genes that have been best understood are the RET (rearranged during transfection) proto-oncogen and EDNRB (endothelin receptor B) gene. The long-segment Hirschsprung’s disease is considered to involve the RET oncogene with an autosomal dominant inheritance pattern that has complete penetrance. This means that when there is a mutation in the RET gene then there is 100% probability that the carrier will have HD. In contrast, short segment HD appears to be transmitted in an autosomal recessive manner and may be due to multiplicative effects of a number of other genes.5,7
The arrest of the migration of neural crest cells as it occurs in neurocristopathies5,7 leads to an absence of ganglion cells in the large colon and the terminal ileum as was seen in this case. Due to the absence of the post ganglionic parasympathetic neurons, there is no release of acetylcholine and there is an accumulation of the enzyme acetylcholine esterase.8 Peristalsis does not occur in the diseased section as there is absence of the neurons responsible for relaxation of the muscles of the gastro-intestinal tract. Hence it remains stenosed or contracted. The bowel proximal to the diseased section becomes dilated as there is peristalsis. In prolonged cases, the proximal segment becomes dilated and the musculature wall thickens due to excessive stimulation of the innervated muscles. Persistent dilatation of the proximal segment also reflects the faecal loading. In the present case, the transitional zone was in the terminal ileum. This resulted in the dilation of the small bowel mimicking small bowel obstruction. Due to the acute nature of the obstruction, the small bowel distended very quickly and there was evidence of thinning of the dilated small intestines resulting in perforation of a segment intra-operatively.
The classic histological picture of Hirschsprung’s disease is the absence of ganglion cells in intramuscular myenteric (Auerbach’s) plexus and the submucosal (Meissner’s) plexus.8 In addition, proliferation of peripheral nerve fibres may also be seen in the affected bowel. This mostly occurs in those in whom the disease presents late. In the present case, there was an absence of ganglion cells in the terminal ileum, the appendix and the caecum. All pre-ganglionic parasympathetic nerve fibres are cholinergic nerve fibres in which acetylcholine is the main neurotransmitter. The enzyme acetylcholinesterase breaks down the neurotransmitter into acetyl-CoA and choline. There is increase in the pre-ganglionic nerve stimulation and release of the neurotransmitter but due to the absence of the post parasympathetic ganglion, the enzyme persists. A test of acetylcholine esterase activity is not available in our setting so it was not done.
The clinical evaluation of a patient remains the most important diagnostic step in the diagnosis of HD. Normal babies pass meconium within 24 h, and a small percentage will also pass meconium by 48 h. A delay in passage of meconium is the most important neonatal observation and is present in 80% of cases of HD. A significant delay in the passage of meconium after birth indicates a congenital cause. This child did pass meconium after birth but then its passing ceased. Neonatal sepsis was the presumed diagnosis. In poorly resourced health situations, most cases of neonatal HD are overlooked and neonatal diagnoses may be made in as few as 10% of cases. Intestinal obstruction presents with bile-stained vomiting and abdominal distension on the second day of life.
Hirschsprung’s-associated enterocolitis (HAEC) presents with bloody diarrhoea and mucus associated with abdominal distension and vomiting. The majority of cases of enterocolitis present during the first 2–4 weeks after birth. This is an important diagnosis because it accounts for 53% of the mortality arising from Hirschsprung’s disease. Differential diagnosis on this child at this age were neonatal sepsis, neonatal intestinal dyspepsia, small left colon syndrome particularly in diabetic mothers, meconium plug syndrome in neonates or meconium ileus and other anglionosis of the gastrointestinal tract. Some literature talks about Ondine’s curse (congenital hypoventilation syndrome) and congenital neuroblastoma9 but was absent on this case.
The diagnostic accuracy of abdominal X-rays is 52%. Erect plates can demonstrate in air-fluid levels and a lateral view may demonstrate the narrow rectum. In this case, the child had signs of small bowel obstruction. An enema was not performed as she was septic and a further introduction of toxins into the child was risky. The absence of ganglions cells in the terminal ileum, appendix and the ascending colon confirmed the diagnosis of HD.
A principle of surgical management is determined by the level of transitional zone. A diverting colostomy is performed at 5 cm proximal to the transitional zone. For this child a terminal ileostomy was done. Complications from ileostomy including excoriations, wound sepsis, wound dehiscence, bleeding from the stoma and prolapse were carefully explained to the parents. She may develop electrolyte abnormalities and high output failure due to the exit of well-formed stool with water and electrolytes. In the growing infant, the remaining terminal ileum may not contain receptors for absorbing intrinsic factor combined with its associated vitamin B12 leading to megaloblastic anaemia.
Definitive surgical pull-through procedures that have received reasonably wide acceptance are the Swenson, Soave, Duhamel, Rehbein and the more recently developed transanal endorectal pull-through. The latter technique is a laparoscopic assisted single stage procedure without a diverting colostomy. Swenson’s pull-through was the original pull-through operation, described by Orvar Swenson in 1948.3 It involves resection of the aganglionic segment deep into the pelvis, and direct end-to-end anastomosis of the proximal ganglionic colon or terminal ileum for this case with the anorectal canal just 1.5–2 cm proximal to the dental line preserving the internal sphincters. In the present case, the child underwent this operation. Two weeks after the definitive procedure, examination under anaesthesia, dilatation and calibration was done to prevent scar and stenosis. The parents were involved in this procedure on the operating table and they were taught how to calibrate the candle and do the dilation. A specific treatment protocol was given to them so that they could do the dilatation at home.
Early postoperative complications appear to occur at a similar rate regardless of the age at surgery. Anastomotic insufficiency, stenosis, prolonged ileus, adhesive obstruction, intestinal obstruction, and retraction of the neorectum are common. Wound sepsis may be present to varying degrees, and other complications of sepsis or pelvic or presacral abscesses may be evident. Recto-peritoneal fistula formation during dilation protocol thus peritonitis is unavoidable. Acidosis may be associated with excessive fluid and electrolyte losses in long-segment cases.
Long term complications include stenosis of the anal canal is possible but if the dilatation protocol has been performed then there won’t be any stenotic complication. Weakness of the sphincter thus resulting in faecal incontinence can be present if during the procedure the nerves have been damaged or the sphincter has been damaged. This is prevented by sub-serousal dissection of the aganglionic colon and the site of transaction should be a 1.5–2 cm above the dentate line to prevent damaging nerves and the internal sphincter respectively. Damage to the vas deferens bilaterally can result in sterility for the males and damage to the nerves can result in incontinence as well as impotence. Reliable data and evaluation of late complications are largely unavailable regarding constipation incidence, continence, social integration and sexual function. Constipation and obstructive symptoms may be related to a number of possible causes, which include a residual aganglionic segment, sphincter achalasia, associated dysganglionosis of the enteric nervous system, strictures, restrictive cuff following ERP and retained spur following Duhamel procedures.4 Escoriations around the anus are inevitable but this can be prevented first by doing regular perineal washouts with the stoma output a week prior to the definitive procedure. After the actual procedure, the parents must be well informed of this sequalae and advised they have to clean the perineal area thoroughly after every passage of stool for a minimum of 4 weeks. Only then can this serious complication be avoided. Subsequently, patients are prone to develop pernicious anaemia, and other genetically predisposed pathologies such as MEN2B and neuroblastoma in individuals whose HD is RET oncogene-associated.10,11
Prognosis depends on the surgical technique of the procedure and experience in each respective case. HD is surgically correctable, and the majority of patients with the disease can live a productive and satisfying lives. The physical growth and development generally approximate normal. The intellectual function is mostly good. Most HD patients achieve acceptable anorectal function, up to 93%, given sufficient time to adjust.4
This all strongly suggests a multifactorial and multigenic HD aetiology, and the majority of HD cases can be classified as complex genetic disorders for which familial aggregation is observed with variable Mendelian inheritance.6 Genetic counselling via pedigree analysis alone is particularly difficult in such a multifactorial condition, as it may be affected by a number of other unrelated factors (e.g. small family size, poor history, and adoption, as well as the lack of an identifiable genetic mutation). As a result, practical therapeutic interventions still appear to be mostly unattainable. Nevertheless, apart from being an integral part of the ethics surrounding DNA testing, genetic counselling has become highly sought by families at risk but the fact of the matter is that DNA testing cannot be done in a country like Papua New Guinea. This will help to drive the process forward and DNA analysis of at least the major susceptibility genes in the family is an achievable goal. Should a major genetic defect be detected in this manner, the door is open for foetal genetic testing and evaluation of risk. For this child it may not be prudent to advice the parents of the possible genetic related risk factors that may be associated with HD but it is wise to get the details and follow them up for a considerable period of time to see whether they are developing these pathologies or not.
Data availability
The data that support this study will be shared upon reasonable request to the corresponding author.
Ethics
Ethical clearance was not required for this case write up, however verbal consent was obtained from the parents.
References
1 Ruhräh J. Harald Hirschsprung 1830-1916: a note on the history of hypertrophy of the colon. Am J Dis Child 1935; 50(2): 472-5.
| Crossref | Google Scholar |
2 Sergi C. Hirschsprung’s disease: Historical notes and pathological diagnosis on the occasion of the 100th anniversary of Dr. Harald Hirschsprung’s death. World J Clin Pediatr 2015; 4(4): 120-5 PMCID: PMC4637802.
| Crossref | Google Scholar | PubMed |
3 Swenson O, Bill AH, Jr. Resection of rectum and rectosigmoid with preservation of the sphincter for benign spastic lesions producing megacolon; an experimental study. Surgery 1948; 24(2): 212-20.
| Google Scholar | PubMed |
4 Urla C, Lieber J, Obermayr F, Busch A, Schweizer R, Warmann SW, Kirschner HJ, Fuchs J. Surgical treatment of children with total colonic aganglionosis: functional and metabolic long-term outcome. BMC Surg 2018; 18(1): 58 PMCID: PMC6094876.
| Crossref | Google Scholar | PubMed |
5 Burns AJ, Roberts RR, Bornstein JC, Young HM. Development of the enteric nervous system and its role in intestinal motility during fetal and early postnatal stages. Semin Pediatr Surg 2009; 18(4): 196-205.
| Crossref | Google Scholar | PubMed |
6 Moore SW. Chromosomal and related Mendelian syndromes associated with Hirschsprung’s disease. Pediatr Surg Int 2012; 28(11): 1045-58 Epub 23 September 2012.
| Crossref | Google Scholar | PubMed |
7 Butler Tjaden NE, Trainor PA. The developmental etiology and pathogenesis of Hirschsprung disease. Transl Res 2013; 162(1): 1-15 Epub 22 March 2013 PMCID: PMC3691347.
| Crossref | Google Scholar | PubMed |
8 Guinard-Samuel V, Bonnard A, De Lagausie P, Philippe-Chomette P, Alberti C, El Ghoneimi A, Peuchmaur M, Berrebi-Binczak D. Calretinin immunohistochemistry: a simple and efficient tool to diagnose Hirschsprung disease. Mod Pathol 2009; 22(10): 1379-84 Epub 31 July 2009.
| Crossref | Google Scholar | PubMed |
9 Roshkow JE, Haller JO, Berdon WE, Sane SM. Hirschsprung’s disease, Ondine’s curse, and neuroblastoma-manifestations of neurocristopathy. Pediatr Radiol 1988; 19(1): 45-9.
| Crossref | Google Scholar | PubMed |
10 Kapur RP. Multiple endocrine neoplasia type 2B and Hirschsprung’s disease. Clin Gastroenterol Hepatol 2005; 3(5): 423-31.
| Crossref | Google Scholar | PubMed |
11 Donis-Keller H, Dou S, Chi D, Carlson KM, Toshima K, Lairmore TC, Howe JR, Moley JF, Goodfellow P, Wells SA, Jr. Mutations in the RET proto-oncogene are associated with MEN 2A and FMTC. Hum Mol Genet 1993; 2(7): 851-6.
| Crossref | Google Scholar | PubMed |