
The Genomics for Australian Plants (GAP) framework initiative – developing genomic resources for understanding the evolution and conservation of the Australian flora
Lalita Simpson A , David J. Cantrill B C , Margaret Byrne D , Theodore R. Allnutt B , Graham J. King E , Mabel Lum F , Ziad Al Bkhetan G , Rose L. Andrew H , William J. Baker I K , Matthew D. Barrett A , Jacqueline Batley L , Oliver Berry M , Rachel M. Binks D , Jason Bragg N , Linda Broadhurst O , Gillian Brown P , Jeremy J. Bruhl H , Richard J. Edwards Q R , Scott Ferguson S , Félix Forest I , Johan Gustafsson G , Timothy A. Hammer T U , Gareth D. Holmes B , Christopher J. Jackson B , Elizabeth A. James B , Ashley Jones S , Paul J. Kersey I , Ilia J. Leitch I , Olivier Maurin I J , Todd G. B. McLay B C V , Daniel J. Murphy B C W , Katharina Nargar A X , Lars Nauheimer A , Hervé Sauquet W Y , Alexander N. Schmidt-Lebuhn O , Kelly A. Shepherd Z AA , Anna E. Syme G , Michelle Waycott T U , Trevor C. Wilson Y and Darren M. Crayn A P X AB *
A , Jacqueline Batley L , Oliver Berry M , Rachel M. Binks D , Jason Bragg N , Linda Broadhurst O , Gillian Brown P , Jeremy J. Bruhl H , Richard J. Edwards Q R , Scott Ferguson S , Félix Forest I , Johan Gustafsson G , Timothy A. Hammer T U , Gareth D. Holmes B , Christopher J. Jackson B , Elizabeth A. James B , Ashley Jones S , Paul J. Kersey I , Ilia J. Leitch I , Olivier Maurin I J , Todd G. B. McLay B C V , Daniel J. Murphy B C W , Katharina Nargar A X , Lars Nauheimer A , Hervé Sauquet W Y , Alexander N. Schmidt-Lebuhn O , Kelly A. Shepherd Z AA , Anna E. Syme G , Michelle Waycott T U , Trevor C. Wilson Y and Darren M. Crayn A P X AB *
A
B
C
D
E
F
G
H
I
J
K
L
M
N
O
P
Q
R
S
T
U
V
W
X
Y
Z
AA
AB
Abstract
The generation and analysis of genome-scale data – genomics – is driving a rapid increase in plant biodiversity knowledge. However, the speed and complexity of technological advance in genomics presents challenges for the widescale use of genomics in evolutionary and conservation biology. We introduce and describe a national-scale collaboration conceived to build genomic resources and capability for understanding the Australian flora: the Genomics for Australian Plants (GAP) Framework Initiative. We outline (a) the history of the project including the collaborative framework, partners and funding; (b) GAP principles such as rigour in design, sample verification and documentation, data management and data accessibility; and (c) the structure of the consortium and the four associated activity streams (reference genomes, phylogenomics, conservation genomics and training), with the rationale and aims for each of these. We show, through discussion of successes and challenges, the value of this multi-institutional consortium approach and the enablers, such as well-curated collections and national collaborative research infrastructure, all of which have led to a substantial increase in capacity and delivery of biodiversity knowledge outcomes.
Keywords: Angiosperms353, Australian flora, conservation, genomics, phylogenomics, population genetics, reference genomes, systematics, taxonomy.
Introduction
Australia is a large, geologically reasonably stable landmass that is home to over 22,600 accepted vascular plant species (~6.3% of the world’s flora; Govaerts et al. 2021) in 2144 genera (Council of Heads of Australasian Herbaria, CHAH, Australian Plant Census, see https://biodiversity.org.au/nsl/services/search/taxonomy, accessed 28 June 2024). This flora is well known for diversity (>225 families) and endemism (>90% of species, Chapman 2009) spread across a wide range of biomes including the mesic south-east and south-west coasts to monsoon tropics across the north, and the large arid zone that dominates the inland part of the continent (Byrne et al. 2008, 2011; Bowman et al. 2010).
Documenting the diversity and relationships of the Australian flora has been a focus from the early days of plant collecting (1790s), whereas in recent decades, molecular systematics has been employed in an integrated approach to understanding phylogenetic relationships, particularly at genus and family levels. Indeed, molecular systematics has highlighted the role of isolation and nature of species radiations as important features in the evolutionary history of a great deal of the arid and mesic flora of Australia (Byrne et al. 2008, 2011; Crisp and Cook 2013), with immigration being more prominent in the monsoonal flora through the current proximity to south-east Asia (Crayn et al. 2015). At lower taxonomic levels, phylogeography has revealed a strong signal for persistence of species through Pleistocene climatic changes, providing insights into current species distributions (Byrne et al. 2008, 2011; Byrne and Murphy 2020), whereas population genetics has strengthened our understanding of the significance of diversity for conservation (Broadhurst et al. 2017).
A great deal of the taxonomic and systematic research on the Australian flora has been undertaken at herbaria and botanic gardens, as these institutions are centres for botanical collections and knowledge. Since the first local botanical institution (and Australia’s first scientific institution) was established in 1816 (now known as the Botanic Gardens of Sydney), Australia has developed 31 recognised national, state and university vascular plant herbaria (Atlas of Living Australia ‘collectory’ – collections.ala.org.au; Index Herbariorum, New York Botanical Garden’s Virtual Herbarium, see http://sweetgum.nybg.org/ih/, accessed October 2023), and 139 botanic gardens. Australia’s major herbaria were established by governments (State, Territory and Commonwealth), principally to research the flora and provide botanical information and advice. Many universities have developed herbaria to support research and education.
There has been a long history of collaboration among herbaria nationwide that became formalised in 1972 with the establishment of CHAH. Enabled by CHAH, Australian herbaria have been at the global forefront of plant biodiversity science and data delivery, exemplified by several major knowledge infrastructure projects and initiatives including a national specimen databasing initiative, Australia’s Virtual Herbarium (AVH, see www.avh.ala.org.au) that later spawned the Atlas of Living Australia (ALA, see www.ala.org.au), the Australian Plant Names Index (APNI, see https://biodiversity.org.au/nsl/services/) and the Australian Plant Census (APC, see https://biodiversity.org.au/nsl/services/) that is a national consensus checklist of accepted plant names.
In addition to leading in collections data and knowledge delivery, researchers at Australian herbaria and botanic gardens have been early adopters of molecular tools in systematics and evolution research (e.g. Gilmore et al. 1993). Nevertheless, the speed and complexity of technological advancement in molecular genomics and the concomitant challenges in data generation, analysis, management and use, have outpaced the capacity of many institutions and individual researchers to utilise these tools optimally (Attwood et al. 2019). The Genomics for Australian Plants (GAP) Framework Initiative is a consortium established to strategically address this gap and foster implementation of genomics technological applications to build plant knowledge across the research and wider community. In this introductory paper to the Genomics for Australian Plants Special Collection of Australian Systematic Botany, we outline the principles and approach taken in establishing this consortium, describe the aims and architecture of the research projects undertaken and describe progress to date against these aims. Several major milestones have been achieved to date: 35 reference genomes sequenced, a nuclear sequence data resource for 90% of Australian native angiosperm genera with an accompanying phylogenomic tree (Australian Angiosperm Tree of Life, AAToL) created and the resolution of 10 species complexes of conservation concern. These milestones clearly demonstrate that a consortium approach enables the plant biodiversity science community to achieve the scale required for effective national capacity building, utilisation of novel genomics technologies, and delivery of large-scale datasets and knowledge for understanding the evolution and conservation of the Australian flora.
Development of the Genomics for Australian Plants (GAP) Framework Initiative
Recognising the opportunity to enhance genomics research capability and infrastructure for the plant biodiversity sciences, Bioplatforms Australia (see https://bioplatforms.com/) initiated discussions in 2016 with the Australian herbarium and botanic gardens communities, and internationally with the Royal Botanic Gardens Kew, to explore the development of a consortium to undertake a collaborative, capacity-building genomics project.
Drawing on the findings from the Oz Mammals Genomics Framework Data Initiative (Eldridge et al. 2020), a draft proposal for the development of the consortium was circulated to Australian herbaria, interested individuals working in botanic gardens and universities, and subject-matter experts in other organisations. Participants at an initial workshop in Melbourne in August 2017 agreed to further develop, through subsequent meetings, the consortium model that led to the establishment of a Steering Committee and two Working Groups to advise the Steering Committee on strategy and methodological approaches: the Wet Lab Working Group and the Computational Working Group. The GAP Framework Initiative was formally launched at the Australasian Systematic Botany Society meeting in Brisbane in December 2018.
The development of the GAP Framework Initiative was undertaken in the context of, and enabled by, the resources, infrastructure, botanical knowledge and genomic expertise of the Australian national herbarium network. The GAP Framework Initiative is also aligned with commitments outlined in national strategic plans such as Australia’s Strategy for Nature (Interjurisdictional Biodiversity Working Group 2019) and the Decadal Plan for Taxonomy and Biosystematics (Taxonomy Decadal Plan Working Group 2018). The consortium model uses existing resources to enable and facilitate the development and adoption of innovative technological applications to generate data, and maximise the uptake and use of outputs (Fig. 1). Details of inputs, activities, outputs and outcomes are outlined below.
The Genomics for Australian Plants (GAP) Framework Initiative consortium approach. Inputs (data, people and institutional stakeholders) are the infrastructure that enables GAP data-generating activities that are undertaken in three streams: reference genomes, phylogenomics and conservation genomics. Training activities are embedded within and link across all three data streams. Together, these activities produce outputs and outcomes that enhance and enrich the human, knowledge and institutional resources that enabled the project.
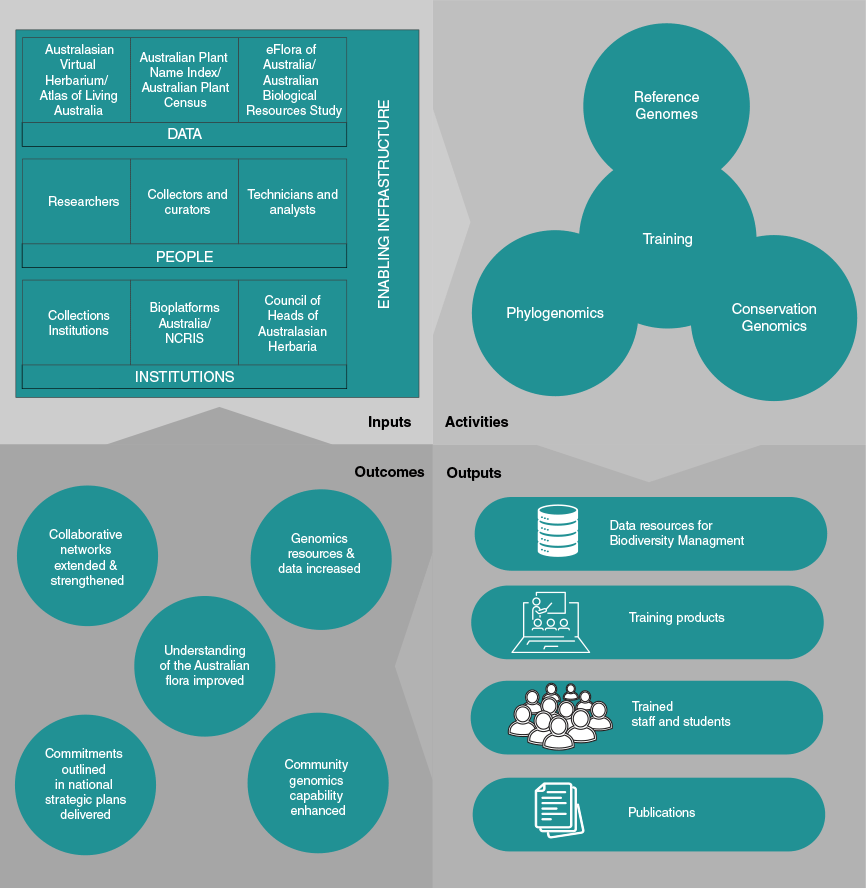
Inputs
The infrastructure that enabled the GAP Framework Initiative comprised people (a well-trained research workforce), knowledge (findable, accessible, interoperable and reusable data – FAIR; Wilkinson et al. 2016) and physical elements (world-class biological collections, laboratories, instruments and computational resources) (Fig. 1). Core funding was provided by Bioplatforms Australia (funded by the Australian Government’s National Collaborative Research Infrastructure Strategy fund – NCRIS), philanthropy (Ian Potter Foundation, Royal Botanic Gardens (Victoria) Foundation) and institutional partners (herbaria and botanical gardens). Further investment was provided through the contributions of the more than 210 systematists, taxonomists, herbarium and laboratory curators and technicians, project coordinators, bioinformaticians, ecologists and conservation scientists that comprised the GAP consortium. Collectively, these individuals represent 28 organisations, including government agencies, herbaria, botanical gardens, universities and genomics laboratories from all states and territories. Plant samples were sourced from curated botanical collections maintained in herbaria and botanical gardens, supplemented with wild collected material where required. Sample metadata curation and management were verified with existing biodiversity data infrastructure such as the AVH/ALA and National Species List (APNI/APC). Generation and management of genomic data is supported by Bioplatforms Australia’s genomics and bioinformatics infrastructure including the ACRF Biomolecular Resource Facility (BRF, see https://jcsmr.anu.edu.au/research/facilities/brf), the Australian Genomics Research Facility (AGRF, see https://www.agrf.org.au/), Genomics Western Australia (GWA, see https://www.genomicswa.com.au/), the Ramaciotti Centre for Genomics (see https://www.ramaciotti.unsw.edu.au/) and the Australian BioCommons (see https://www.biocommons.org.au/). Computational resources for data analysis are provided through the Australian BioCommons and partners such as the National Computational Infrastructure (NCI, see https://nci.org.au/). GAP partners and core funding providers are listed on the GAP website (see https://www.genomicsforaustralianplants.com/consortium/).
GAP aims and principles
Aims
The GAP Framework Initiative aimed to bring researchers and data specialists in plant science and conservation organisations together to increase and share biodiversity knowledge, accelerate species discovery, enhance biodiversity collections, and support strategic growth of taxonomic and biosystematic capacity and capabilities. These aims have been achieved by:
Generating plant genomic data across the plant tree of life to enable better conservation, utilisation and understanding of Australia’s unique plant diversity.
Building genomic capacity across Australian botanic gardens and herbaria to create networks that can collaborate on the collection, management, dissemination and application of genomic data for Australian plants.
Providing tools that enable the use of genomic data for identification and classification of biodiversity at a range of scales, and to use these tools to inform conservation management and enable better decision making.
Upskilling the plant science community in the use of new sequencing technological applications through the development of wet lab skills and bioinformatics training.
Principles
The GAP Framework Initiative has operated under a set of research and data standards as follows:
The taxonomic identity of all plant material used in the GAP Framework Initiative is determined by a taxonomic specialist.
Taxonomy conforms to the APC or recent peer reviewed taxonomic publications not yet considered by the APC.
Plant material was collected in Australia (including offshore territories).
Plant material was collected legally and is compliant with the Nagoya Protocol.1
Plant material was vouchered by specimens lodged in a recognised herbarium (Index Herbariorum, see http://sweetgum.nybg.org/ih/).
Samples comply with rigorous metadata standards that include herbarium voucher accession and genomic sample processing details.
All data are made available to consortium partners immediately through the Bioplatforms Australia Data Portal (see https://data.bioplatforms.com/) and following data publication are made freely accessible through public repositories (e.g. the European National Archive – ENA) in accordance with the FAIR principles (Findable, Accessible, Interoperable, Reusable; Wilkinson et al. 2016). Software, workflows and training materials are also made freely available.
Individual plants (or for outcrossing annuals, an individual of the same population or the closest heritable unit to the sampled ortet, or ramet) sampled for reference genome projects are conserved in living collections to enable future research projects to access the same individual or lineage.
For phylogenomics projects, type species of genera are sampled where possible.
GAP activities
The GAP consortium has undertaken data generation, workflow development and capacity building across four activity streams:
Although there has been a marked acceleration in the availability of plant nuclear genomic resources in recent years, most of these are for crop species and wild relatives, and forest trees. Thus, resources are very unevenly distributed across the plant tree of life, hampering use to understand patterns of genomic diversity and processes that drive genome evolution. The GAP Reference Genomes stream aimed to address this resource bias for Australian lineages of flowering plants (angiosperms). Initially, a Working Group was formed to consider: (1) which of the increasing array of short- and long-read genomic sequencing technologies are most appropriate for the task; and (2) how to prioritise taxa for sequencing.
To identify orders and families in the Australian angiosperm flora that lacked genomic resources, the GAP Steering Committee led the compilation of a list of all plant nuclear genomic resources available from public repositories (NCBI genomes, NCBI Sequence Read Archive, PlaBiPD), including completed genomes, draft genomes or assemblies and other sequence data (e.g. RNAseq and genome skimming). This list is available online (see links under GAP Data and Resources Availability section below). Using this list for priority setting, community participation in the GAP Reference Genomes stream was invited through Requests for Partnership in four phases. This approach enabled the consortium to adapt the program in response to new technological applications as these became available and progressively tackle larger genomes. Plants with diploid genomes up to 2 gigabases (Gb; 1C-value) were targeted as these were considered tractable for sequencing to sufficient depths for assembly. Additionally, the lower cost of sequencing for genomes in this size range compared with larger genomes meant that the GAP Framework Initiative could sequence more representatives to fill taxonomic gaps across the tree of life.
Extracting high molecular weight DNA suitable for reference genome assembly was a significant challenge for nearly all taxa and exceptionally difficult for some (Jones et al. 2021). To address this difficulty, members of the consortium developed dedicated protocols suitable for extracting high molecular weight DNA for long read sequencing of diverse non-model plants. This included virtual engagement with the wider scientific community for interactive protocol development, discussion and Open Access sharing through Protocols.io (Teytelman et al. 2016). Nevertheless, several projects could not be completed because obtaining DNA of sufficient quality and quantity proved intractable. All of the species for which nuclear genomes were sequenced through the GAP Framework Initiative are listed in Table 1, including those that are not yet completed at time of publication.
| Order | Family | Species | Estimated genome size (Gb, 1C-value) | Observed genome size (Gb) | Sequencing Method | Project status | Outputs | Living voucher location | |
|---|---|---|---|---|---|---|---|---|---|
| Arecales | DasypogonaceaeA | Kingia australis R.Br. | 0.5B | 1.5 | HiFi | Assembled | n/a | KPBG | |
| Asparagales | Asphodelaceae | Xanthorrhoea johnsonii A.T.Lee | 1.5C (T. McLay, unpubl. data) | HiFi, ONT, Hi-C, Illumina | Assembled | Ferguson et al. (2022) | ANBG | ||
| Asparagales | DoryanthaceaeA | Doryanthes excelsa Correa | 2.94 (Šmarda et al. 2014) | 2.6 | HiFi, ONT | Assembled | n/a | RBGV | |
| Asparagales | Orchidaceae | Thelymitra variegata (Lindl.) F.Muell. | – | – | HiFi | Assembled (low coverage) | n/a | KPBG | |
| Asterales | Asteraceae | Leucochrysum albicans (A.Cunn.) Paul G. Wilson | 0.9C (A. Schmidt-Lebuhn, unpubl. data) | – | HiFi | In assembly | n/a | ANBG | |
| Asterales | Campanulaceae | Wahlenbergia ceracea Lothian | 0.3D | 0.4 | HiFi, ONT, Illumina | Assembled | n/a | Unknown | |
| Asterales | Goodeniaceae | Dampiera purpurea R.Br. | 0.5D | 1.4 | HiFi | Assembled | n/a | RBGV-C | |
| AustrobaileyalesA | AustrobaileyaceaeA | Austrobaileya scandens C.T.White | 9.3 (Leitch and Hanson 2002) | 8.0 | HiFi, ONT | Assembled | n/a | CBotG | |
| BerberidopsidalesA | BerberidopsidaceaeA | Berberidopsis beckleri (F.Muell.) Veldkamp | 0.3D | 0.7 | HiFi | Assembled | n/a | NENP | |
| Brassicales | Cleomaceae | Areocleome oxalidea (F.Muell.) R.L.Barrett & Roalson | – | – | Project ended | Project ended – failure to culture plant material | n/a | n/a | |
| Canellales | Winteraceae | Tasmannia lanceolata (Poir.) A.C.Sm. | 1.2 Gb, tetraploid | HiFi | Assem bled | n/a | RBGV | ||
| Celastrales | Celastraceae | Denhamia bilocularis (F.Muell.) M.P.Simmons | 0.8B | 0.3 | HiFi | Assembled | n/a | RBGV-C | |
| DillenialesA | DilleniaceaeA | Hibbertia scandens (Willd.) Gilg | 2.64 (Garcia et al. 2010) | – | ONT, Illumina | In assembly | n/a | ABG | |
| Fabales | Fabaceae | Acacia pycnantha Benth. | 0.6C (McLay et al. 2022) | 0.6 | ONT, Illumina | Completed | Syme et al. (2021); McLay et al. (2022) | ANBG | |
| Fabales | Fabaceae | Gastrolobium racemosum (Turcz.) Crisp | 0.85C (Schmidt- Lebuhn and Cantrill 2023) | 0.8 | ONT | Assembled | n/a | RBGV-C | |
| Fabales | Fabaceae | Pultenaea daphnoides J.C.Wendl. | 0.7 (Morgan and Westoby 2005) | 0.6 | HiFi | Assembled | n/a | RBGV | |
| Fagales | Casuarinaceae | Gymnostoma australianum L.A.S.Johnson | 0.7B | 0.3 | HiFi | Assembled | n/a | RBGV-C | |
| Fagales | Nothofagaceae | Nothofagus cunninghamii (Hook.) Oerst. | 1E | 0.5 | HiFi | Assembled | n/a | RBGV-C | |
| Geraniales | Geraniaceae | Pelargonium australe Willd. | 0.32 (Weng et al. 2012) | 0.4 | HiFi | Assembled | n/a | ABG | |
| Lamiales | Scrophulariaceae | Eremophila drummondii F.Muell. | 0.8C (A. Schmidt-Lebuhn and R. Fowler unpubl. data) | 0.8 | HiFi, Illumina | Assembled | n/a | UMelb | |
| Laurales | MonimiaceaeA | Hedycarya angustifolia A.Cunn. | 0.7C (Schmidt-Lebuhn and Cantrill 2023) | 0.7 | HiFi | Assembled | n/a | RBGV-C | |
| Magnoliales | Eupomatiaceae | Eupomatia laurina R.Br. | 1.2 (Morawetz and Samuel, pers. comm., 1988, in Pellicer and Leitch 2020) | 0.9 | HiFi | Assembled | n/a | RBGV | |
| Malpighiales | Phyllanthaceae | Phyllanthus gunnii Hook.f. | 1.0C (Schmidt-Lebuhn and Cantrill 2023) | 1.0 | HiFi | Assembled | n/a | RBGV | |
| Myrtales | Myrtaceae | Archirhodomyrtus beckleri (F.Muell.) A.J.Scott | 0.8B | 0.4 | HiFi, ONT | Assembled | n/a | RBGV | |
| Oxalidales | CunoniaceaeA | Callicoma serratifolia Andrews | – | 0.3 | ONT | Assembled | n/a | RBGV | |
| Oxalidales | CunoniaceaeA | Eucryphia lucida (Labill.) Baill. | – | 0.3 | ONT | Assembled | n/a | RBGC | |
| Oxalidales | Elaeocarpaceae | Elaeocarpus reticulatus Sm. | 0.3B | – | HiFi | In assembly | n/a | RBGV | |
| ParacryphialesA | ParacryphiaceaeA | Quintinia fawkneri F.Muell. | 1.2C (Schmidt-Lebuhn and Cantrill 2023) | 1.0 | HiFi, ONT | Assembled | n/a | RBGV-C | |
| Poales | Cyperaceae | Lepidosperma gladiatum Labill. | 0.6B | 0.2 | HiFi | Assembled | n/a | RBGV | |
| Proteales | Proteaceae | Bellendena montana R.Br. | 4.86 (Jordan et al. 2015) | – | HiFi | In assembly | n/a | WP | |
| Proteales | Proteaceae | Isopogon anethifolius (Salisb.) Knight | 0.6D | 0.5 | ONT | Assembled | n/a | RBGV | |
| Proteales | Proteaceae | Telopea speciosissima (Sm.) R.Br. | – | 0.9 (Chen et al. 2022) | ONT, 10×, Hi-C | Completed | Chen et al. (2022) | n/a (plant died) | |
| Rosales | Urticaceae | Dendrocnide excelsa (Wedd.) Chew | 1.6B | 1.0 | HiFi | Assembled | n/a | RBGV | |
| Sapindales | Rutaceae | Flindersia xanthoxyla (A.Cunn. ex Hook.) Domin | 0.4C (Schmidt-Lebuhn and Cantrill 2023) | 0.4 | HiFi | Assembled | n/a | RBGV | |
| Sapindales | Rutaceae | Phebalium stellatum I.Telford & J.J.Bruhl | 1.5B | – | HiFi, ONT, Illumina | In assembly | Ferguson et al. (2022) | UNE | |
| Zygophyllales | Zygophyllaceae | Roepera similis (H.Eichler) Beier & Thulin | 1.2B | 0.9 | HiFi | Assembled | n/a | RBGV |
Sequencing methods are as follows: 10× indicates 10× Genomics (Pleasanton, CA, USA); ONT, Oxford Nanopore Technology, a long read single molecule sequencing approach (Oxford Nanopore, Oxford, UK); Hi-C, a genomic technique that captures chromatin conformation (Lieberman-Aiden et al. 2009); HiFi, high fidelity long read sequencing approach (PacBio, Menlo Park, CA, USA); and Illumina, short read sequencing approach (Illumina, San Diego, CA, USA). All species are diploid except Denhamia bilocularis (F.Muell.) M.P.Simmons (polyploid, ploidy level unknown), Flindersia xanthoxyla (A.Cunn. ex Hook.) Domin (tetraploid) and Tasmannia lanceolata (Poir.) A.C.Sm. (tetraploid).
Locations of living vouchers are as follows: ABG, Adelaide Botanic Gardens, Adelaide, SA; ANBG, Australian National Botanic Gardens, Canberra, ACT; KPBG, Kings Park Botanic Garden, Perth, WA; RBGV, Royal Botanic Gardens Victoria, Melbourne, Vic.; RBGV-C, Royal Botanic Gardens Victoria, Cranbourne, Vic.; CBotG, Cairns Botanic Gardens, Cairns, Qld; NENP, New England National Park, NSW; UMelb, Systems Garden, The University of Melbourne, Melbourne, Vic.; UNE, University of New England, Armidale, NSW; WP, Wellington Park, Tas. Observed genome size indicates that genome size was determined from genome assemblies.
To date, the GAP Reference Genomes stream has delivered: (1) new protocols for high-molecular weight DNA extraction, clean-up and size selection for long read sequencing (Jones et al. 2021; Jones and Schwessinger 2021); (2) an approach to base-caller selection to improve actual accuracy of nanopore sequencing (Ferguson et al. 2022); (3) case studies in the use of long read assemblies to reveal structural diversity in organellar genomes (Syme et al. 2021); (4) genome size estimates for 10 species (Chen et al. 2022; McLay et al. 2022; Schmidt-Lebuhn and Cantrill 2023; T. McLay, unpubl. data; A. Schmidt-Lebuhn and R. Fowler, unpubl. data); (5) an online concatenated database of published genomic resources compiled from public repositories; and (6) published genomic resources for Australia’s national floral emblem, the golden wattle, Acacia pycnantha Benth. (Syme et al. 2021; McLay et al. 2022) and the state floral emblem of New South Wales, the waratah, Telopea speciosissima (Sm.) R.Br. (Chen et al. 2022).
The status of all 35 reference genome projects is provided in Table 1. Together, these 35 projects provide genomic resources for 24 orders and 30 families, and no published genomic resources exist for 4 of these orders (Austrobaileyales Takht. ex Reveal, Berberidopsidales Doweld, Dilleniales DC. ex Bercht. & J.Presl, Paracryphiales Takht. ex Reveal) and 10 of these families (Austrobaileyaceae Croizat, Berberidopsidaceae Takht., Cunoniaceae R.Br., Dasypogonaceae Dumort., Doryanthaceae R.Dahlgren & Clifford, Dilleniaceae Salisb., Eupomatiaceae Orb., Monimiaceae Juss., Paracryphiaceae Airy Shaw, Winteraceae R.Br. ex Lindl.). Most of the genome projects that are nearing completion will be published in a single overarching and descriptive GAP genome paper.
An understanding of phylogenetic relationships underpins all evolutionary biology, including systematics. To date, plant molecular phylogenetic research in Australia has been largely directed by individual researcher or research institution needs and interests, rather than as a nationwide coordinated effort, with different approaches to marker choice and sampling across groups limiting the interoperability and reusability of data. The GAP Phylogenomics activity stream aimed to transform phylogenetic research on the Australian flora by producing a sequence data resource of common markers and a phylogenetic tree containing at least 95% of Australian native angiosperm genera (i.e. 2037 of 2144 total genera, APC, see https://biodiversity.org.au/nsl/services/, accessed 28 June 2024) – the AAToL. Progress has been achieved by supporting and coordinating collaborators in achieving research goals where contributions were made to building the AAToL.
Two working groups were formed to design and coordinate the GAP phylogenomics component. The Phylogenomics Working Group conceived the project aims and design, engaged the community and coordinated the sampling strategy. The Phylogenomics Bioinformatics Working Group provided guidance and recommendations for approaches to data analysis, development of bioinformatic tools for analysis of target sequence capture datasets and generation of the AAToL tree, and provided bioinformatics support.
The Phylogenomics Working Group advised the adoption of target capture (also known as hybrid enrichment, target enrichment and exon capture) as the sequencing approach that offers reliable retrieval of hundreds or thousands of loci from across the nuclear genome (Cronn et al. 2012). A decision was subsequently made to engage with the Plant and Fungal Trees of Life (PAFTOL; Baker et al. 2022) project led by the Royal Botanic Gardens Kew since 2015 that aims to build the tree of life for all angiosperm genera. Given the aligned approach, GAP and PAFTOL agreed to work in close partnership toward resolving the AAToL, with the GAP team reciprocating by making a subset of data available to the PAFTOL project (Zuntini et al. 2024). This has been achieved by coordinating sampling and methods for genomic data generation, and collaboration on scientific research outputs.
The aim was to construct the AAToL to genus level in two stages with community participation invited through Requests for Partnership:
Stage 1 – In collaboration with PAFTOL, resolve the AAToL to genus level using a single exemplar species to ensure representation for >95% of accepted Australian genera.
Stage 2 – Generate phylogenomic datasets densely sampled within selected genera, families or orders to address questions of monophyly, evolution and biogeography.
A pilot was undertaken to test the suitability of the Angiosperms353 (Johnson et al. 2019; Baker et al. 2021) universal target capture bait set for resolving the AAToL and refine sampling and data workflows at a national scale. Through a Request for Partnership, participants were invited to share existing DNA libraries for analysis. Six responses were received offering libraries representing 166 species from 134 genera and 67 families. Sequencing was carried out following the GAP phylogenomics protocol (see www.genomicsforaustralianplants.com/wp-content/uploads/2022/10/GAP-Phylogenomics-protocols_Oct2022.pdf). The Australian Angiosperm Tree of Life. In Stage 1, the Phylogenomics Working Group initially scoped the project by determining the genera in the Australian native angiosperm flora (based on the APC, CHAH, see https://biodiversity.org.au/nsl/services/search/taxonomy) for which suitable phylogenomics data from a sample collected in Australia were available, either from the GAP pilot study, the PAFTOL project (that had already sequenced more than 600 Australian genera) or other projects such as the One Thousand Plant Transcriptomes Initiative (1KP; One Thousand Plant Transcriptomes Initiative 2019). Six teams responded to the call for partnership, each nominating to sample a selection of the remaining unsampled genera based on the research interests of team members, institutional capability and availability of suitable samples. These six teams were led by members of the six largest Australian herbaria – AD, BRI, CANB, MEL, NSW and PERTH – and included members of five other herbaria – CNS, DNA, HO, MELU, UNE (abbreviations per Index Herbariorum, see http://sweetgum.nybg.org/ih/). Collectively, all states and territories were represented. Together, the six teams brought the total number of samples included in GAP AAToL stage 1 to 1861 (87% of genera). In Stage 2, 16 Requests for Partnership for the second stage of AATOL were accepted and projects initiated. These projects proposed to undertake dense taxonomic sampling within monophyletic groups (orders to genera) (Table 2). Based on results from AAToL Stage 1 (data not shown) and from other studies (e.g. Baker et al. 2021), Angiosperms353 baits were expected to underperform for certain groups and consequently, GAP supported the use of additional bait sets. Thus, the OZbaits set (Waycott et al. 2021) was used for Alismatales R.Br. ex Bercht. & J.Presl, Chamaelaucieae DC. (Myrtaceae Juss.), Drosera L. (Droseraceae), Hibbertia Andrews (Dilleniaceae), Lasiopetaleae Gay (Malvaceae Juss.), Santalaceae R.Br., and Lazarum A.Hay (Araceae Juss.), and the SaliBaits set (A. Žerdoner Čalasan, unpubl. data) was used for Tecticornia Hook.f.–Salicornia L. (Chenopodiaceae Vent.) (Table 2). In addition to the bespoke bait set data, Angiosperms353 data were generated for all samples in these projects to maximise combinability and reuse of datasets, and to enable head-to-head comparisons of bait set performance to provide evidence for bait choice and future bait design development.
| Monophyletic group | Higher taxon | Marker set | Status | Reference | |
|---|---|---|---|---|---|
| Acacia Mill. | Fabaceae | Angiosperms353 | Analysis underway | n/a | |
| Alismatales | Angiosperms353, OZbaits | Analysis underway | n/a | ||
| Boronieae | Rutaceae | Angiosperms353 | Analysis underway | n/a | |
| Chamaelaucieae | Myrtaceae | Angiosperms353, OZbaits | Complete | Nge et al. (2025) | |
| Drosera L. | Droseraceae | Angiosperms353, OZbaits | Complete | Williamson et al. (2025) | |
| Ericaceae | Ericales | Angiosperms353 | Analysis underway | n/a | |
| Hibbertia Andrews | Dilleniaceae | Angiosperms353, OZbaits | Complete | Hammer et al. (2025) | |
| Lasiopetaleae | Malvaceae | Angiosperms353, OZbaits | Analysis underway | n/a | |
| Mirbelieae | Fabaceae | Angiosperms353 | Analysis underway | n/a | |
| Persoonia Sm. | Proteaceae | Angiosperms353 | Analysis underway | n/a | |
| Santalaceae | Santalales | Angiosperms353, OZbaits | Complete | Anderson et al. (2025) | |
| Stylidiaceae | Asterales | Angiosperms353 | Analysis underway | n/a | |
| Tecticornia Hook.f.–Salicornia L. | Chenopodiaceae | Angiosperms353, SaliBaits | Analysis underway | n/a | |
| Teucrium L. | Lamiaceae | Angiosperms353 | Complete | Wilson and James (2025) | |
| Lazarum A.Hay (as syn. Typhonium Schott) | Araceae | Angiosperms353, OZbaits | Analysis underway | n/a | |
| Xanthorrhoea Sol. ex Sm. | Asphodelaceae | Angiosperms353 | Analysis underway | n/a |
The Phylogenomics Bioinformatics Working Group generated several key bioinformatics resources including: (1) a set of recommended approaches and software to use for preparation and analyses of target enrichment data for consortium-scale phylogenomics projects (link under the GAP Data and Resources Availability section below); (2) updated scripts for the recommended software (Yang and Smith 2014; Johnson et al. 2016) to provide novel options, remove bugs and seamlessly use the outputs of the former as inputs for the latter; (3) and a containerised analysis workflow for HybPiper (ver. 2.0, see https://github.com/mossmatters/HybPiper) and ParaGone (ver. 1.1, see https://github.com/chrisjackson-pellicle/ParaGone) (Jackson et al. 2023) to overcome challenges with software compatibility, unfamiliarity with relevant programming languages and the complexity involved in running numerous analysis steps required to use the recommended software (Jackson et al. 2023); (4) Python packages of HybPiper and ParaGone (links under the GAP Data and Resources Availability section below). Additionally, work on AAToL led to two methods and resources studies. The first study, undertaken in partnership with PAFTOL researchers, developed a new target file – ‘mega353’ – for recovery of sequences of Angiosperms353 loci from target capture data sets (McLay et al. 2021). The use of this new target file, rather than the original target file of Johnson et al. (2019), can substantially increase the percentage of on-target reads, locus recovery at 75% length and the total length of the concatenated loci compared to the default Angiosperms353 file. The second study explored conflict in a range of AAToL Stage 2 datasets and developed guidelines for researchers to detect, characterise, manage and interpret conflict in target capture datasets (Joyce et al. 2024, 2025).
Together, the pilot and Stage 1 of the AAToL generated Angiosperms353 sequence data for 2204 samples representing 2019 genera. Sequence data for 650 of these samples were provided by the PAFTOL project and data for 75 samples were sourced from genomic repositories such as the NCBI Sequence Read Archive (see https://www.ncbi.nlm.nih.gov/sra). A total of 2001 samples passed quality control, resulting in a draft phylogenomic tree including 1927 (90%) of 2144 native Australian angiosperm genera. Sequence data are available to the GAP consortium through the Bioplatforms Australia Data Portal. In a reciprocal data sharing arrangement with PAFTOL, data for 770 samples sequenced through the GAP Framework Initiative were made publicly accessible from the Kew Tree of Life Explorer (treeoflife.kew.org; Baker et al. 2022) that currently provides Angiosperms353 data for >10,000 species (data release 3.0, April 2023). AAToL contributed 763 samples (~8%) to a comprehensive analysis of the angiosperm tree of life including nearly 8000 (~60%) angiosperm genera that revealed new insights into angiosperm origins and diversification (Zuntini et al. 2024). The data have also contributed to several other publications produced by PAFTOL collaborators (e.g. Joyce et al. 2023; Elliott et al. 2024; Pérez-Escobar et al. 2024; Grass Phylogeny Working Group III 2025; Helmstetter et al. 2025). Samples released though the Kew Tree of Life Explorer are also publicly available from the Sequence Read Archive (European Nucleotide Archive – ENA – and National Centre for Biotechnology Information – NCBI; project PRJEB49212). The remaining data from Stage 1 of the AAToL will be made publicly available when the comprehensive phylogenetic analysis is completed and published.
The partnership with PAFTOL has greatly facilitated the achievement of GAP aims, most notably by (1) increasing the rate of progress toward completion of the AAToL; (2) expanding the professional networks of Australian scientists, particularly early to mid-career researchers through collaboration with a global team of leaders in plant phylogenomics; and (3) maximising the potential for future re-use of GAP data through adoption of a universal target capture bait set and contribution to global datasets (Baker et al. 2022) and analyses (e.g. Zuntini et al. 2024). Taken together these outcomes substantially advance the capacity for genomics data generation and use by the Australian herbarium community and enable a step change in our understanding of the relationships and evolution of Australia’s flora. In turn, GAP’s engagement has significantly helped expand the reach of PAFTOL and has functioned as a model for the collaborative, open data mindset that PAFTOL aims to foster.
AAToL Stage 2 generated Angiosperms353 sequence data for 10 species complexes (Table 2). In addition, sequence data were generated using OZbaits (Waycott et al. 2021) or SaliBaits (A. Žerdoner Čalasan, pers. comm.) kits for several groups in which those kits were known, or expected, to outperform Angiosperms353 (Table 2). Data analysis, including an assessment of the utility of each bait set and preparation of outputs will be reported in manuscripts focused on each taxonomic group. Papers on Chamelaucieae (Nge et al. 2025), Pultenaea Sm. (Barrett et al. 2024) and Minuria DC. (Schmidt-Lebuhn et al. 2024) have been published and manuscripts on Drosera L., Hibbertia Andrews, Santalaceae, Teucrium L. and on navigating conflict in target capture datasets have been submitted for this Special Collection (Table 2). Several other studies utilising AAToL Stage 1 and Stage 2 data have also been published (e.g. on Aglaia Lour., Cooper et al. 2023; Mirbelieae, Barrett et al. 2024; Pogonolepis Steetz, Schmidt-Lebuhn 2022; and Senecio L., Schmidt-Lebuhn et al. 2022).
The data and lessons from Stages 1 and 2 have enabled the community to commence planning for a continuation of the project beyond the GAP Framework Initiative: AAToL Stage 3. Stage 3 aims to resolve the AAToL to species level, complete for >95% of accepted Australian species. Avenues for resourcing this large undertaking (>18,000 species) and approaches for completion are currently being explored.
The Conservation Genomics stream aimed to provide genomic information to support conservation of the Australian flora. Conservation genomics covers a range of activities according to the management questions being addressed, including resolution of closely related taxa and species complexes to identify taxa of conservation concern; conservation units based on evolutionary lineages; genomic diversity to inform conservation interventions (e.g. augmentation, germplasm capture, translocation and restoration); hybridity and hybrid origin of species; mating system to understand level and pattern of outcrossing; and genotype-environment association analysis to identify signals of adaptation across climate gradients.
Studies among these aiming to resolve species complexes containing suspected taxa of conservation concern were deemed the most suitable for support through the GAP Framework Initiative. The Conservation Genomics Working Group was established to identify species complexes most in need of resolution due to conservation concerns and taxonomic complexity, and to engage the research community in identifying groups in which relevant taxonomic expertise could take advantage of the genomic input and data. The Conservation Working Group decided to take a specific molecular approach (double digest RADseq–ddRAD; Peterson et al. 2012) to create interoperable data sets and develop capability for population level data analyses. Projects to be supported were identified using the following criteria:
The species complex was expected to contain evolutionary units of conservation concern, and genomic data were required to inform taxonomic resolution.
Taxonomic expertise was available to utilise the genomic information.
Genomics expertise in data analysis and interpretation was available.
Samples were available as herbarium specimens or as fresh or dried material, and funding or other resourcing to obtain samples was available.
A Request for Partnership elicited 15 responses involving 25 institutions, including 7 state and institutional herbaria (Table 3). The Steering Committee was keen to stimulate collaboration among partners and the taxonomic and genomics communities, and assisted by linking up collaborators in cases in which proposals lacked key capability on the team (e.g. genomic data analysis expertise). Some projects did not proceed due to issues with DNA sample quality, particularly for projects that exclusively utilised herbarium samples. For four projects, the same samples were also sequenced using the Angiosperms353 bait kit to evaluate the comparative performance of a target capture approach for the resolution of species boundaries within species complexes.
| Order | Family | Species complex | Status | Data generated | Reference | |
|---|---|---|---|---|---|---|
| Asparagales | Orchidaceae | Paracaleana gracilicordata Hopper & A.P.Br. | Analysis underway | ddRAD | n/a | |
| Asparagales | Orchidaceae | Thelymitra variegata (Lindl.) F.Muell. | Project withdrawn | n/a | n/a | |
| Asterales | Asteraceae | Olearia ramulosa (Labill.) Benth. | Analysis underway | ddRAD | n/a | |
| Ericales | Ericaceae | Melichrus R.Br. | Ended due to insufficient DNA quality | n/a | n/a | |
| Fabales | Fabaceae | Cassia L. | Analysis underway | ddRAD, Angiosperms353 | n/a | |
| Fabales | Fabaceae | Gompholobium Sm. | ddRAD completed; Angiosperms353 analysis underway | ddRAD, Angiosperms353 | Simmons et al. (2025) | |
| Fagales | Casuarinaceae | Allocasuarina L.A.S.Johnson | Ended due to insufficient DNA quality | n/a | n/a | |
| Liliales | Colchicaceae | Wurmbea dioica subsp. alba T.D.Mcfarl. | Analysis underway | ddRAD, Angiosperms353 | n/a | |
| Poales | Cyperaceae | Lepidosperma fimbriatum Nees | Ended due to insufficient DNA quality | n/a | n/a | |
| Poales | Cyperaceae | Lepidosperma laterale R.Br. | Ended due to insufficient DNA quality | n/a | n/a | |
| Proteales | Proteaceae | Isopogon buxifolius R.Br. | ddRAD completed; Angiosperms353 analysis underway | ddRAD, Angiosperms353 | Anderson et al. (2024) | |
| Proteales | Proteaceae | Synaphea stenoloba A.S.George | Analysis underway | ddRAD | n/a | |
| Sapindales | Rutaceae | Geleznowia verrucosa Turcz. | Completed | ddRAD | Anderson et al. (2023) | |
| Sapindales | Rutaceae | Zieria Sm. | Completed | ddRAD | Orel et al. (2023a , 2023b) | |
| Sapindales | Simaroubaceae | Samadera bidwillii (Hook.f.) Oliv. | Analysis underway | ddRAD | n/a |
Ten of the fifteen projects proposed provided DNA of suitable quality for ddRAD sequencing whereas five projects, on Allocasuarina L.A.S.Johnson, Lepidosperma Labill. (x 2), Melichrus R.Br. and Thelymitra J.R.Forst. & G.Forst. did not proceed (Table 3). For most projects, herbarium specimens did not yield DNA of usable quality for ddRAD analysis and new collections were required to provide fresh material. The workflow for these projects included the assembly of sequence libraries and SNP calling using an optimised ipyrad (Eaton and Overcast 2020) approach, followed by custom filtering to provide high quality SNP data sets for downstream analyses. These analyses employed a combination of phylogenetic and population genetic approaches to obtain information on the genomic variation within and among the sampled populations of each species complex. This included concatenation and coalescent phylogenetic analyses, and ordination, admixture and distance-based population genetic analyses.
For each of the species complexes evaluated, the genetic results were interpreted in conjunction with morphological data to refine taxonomic hypotheses of relationship and identify undescribed, sometimes cryptic, taxa or hybrid populations – results that will directly underpin conservation assessments and management actions. Furthermore, the results provided insight into the morphological complexity within each group and the informative characters for species identification. Results for projects on Geleznowia Turcz., Isopogon R.Br. ex Knight and Zieria Sm. have been published (Anderson et al. 2023, 2024; Orel et al. 2023a, 2023b), and results for Gompholobium Sm. are included in this Special Collection (Simmons et al. 2025).
The GAP Framework Initiative aimed to build genomic capacity across Australian botanic gardens and herbaria, and provide tools to enable genomic data to be used to identify, classify and conserve biodiversity. Training and capacity building across the GAP consortium has been an inherent and integral part of the development and deployment of workflows undertaken at all stages of the reference genomes, phylogenomics and conservation genomics projects. This included sampling of curated collections for genomic analysis, sample metadata curation, wet lab sample preparation for genomic analysis, bioinformatic analysis of genomic data and preparation of manuscripts. The Wet Lab and Bioinformatics Working Groups reviewed current methods and available resources to develop best practice approaches and protocols, tools and training resources for GAP activities. Resources were also developed to train researchers in the use of the containerised bioinformatic pipelines (see below under the GAP Data and Resources Availability).
A webinar and workshop series for the analysis of target capture sequence data were delivered to 20 participants at the virtual Australasian Systematic Botany Society conference in July 2021, and the workshop materials and webinar recordings have been made publicly available (see below). Additionally, in response to key challenges identified in the early stages of the GAP Framework Initiative, resources were developed and made accessible to encourage and support best practice approaches for plant genomic initiatives and ongoing capacity building across the sector. These resources include: (a) protocols for the extraction of high molecular weight DNA for long read sequencing; (b) analysis guides for target sequence capture projects; (c) containerised bioinformatic software pipelines for the analysis of target sequence capture data (Jackson et al. 2023); and (d) scripts for preparing target sequence data for upload to ENA. Links for the first three of the above public resources are provided in the GAP Data and Resources Availability section, below. The last will be made available on a suitable Open Access platform in the near future.
In addition to the specific, measurable outputs detailed above, GAP has generated many intangible benefits for the community. A wide range of plant scientists, students and collections staff in herbaria, universities and botanic gardens – more than 210 people in total – was engaged in GAP activities. Mid-career and established researchers who have utilised legacy molecular approaches, such as Sanger sequencing or ISSR/SSR, for large parts of their careers have experienced a step change in their capacity to generate and interpret genome scale datasets. Staff of smaller institutions that lack capacity for genetic research of any kind have enjoyed unprecedented opportunities to participate in large genomics projects. Postgraduate students and early career researchers already well versed in genomic analysis have been able to undertake specific training in areas of need and access large volumes of data that would have been difficult to finance otherwise. All participants have benefited from opportunities to engage and collaborate with leading researchers globally, and through these collaborations have developed a greater understanding of the applications of genomic technological applications and data not only for evolution and systematics research, but also for collections development and management. These opportunities, critically enabled by the consortium model (Lowman et al. 2024) have undoubtedly led to higher-quality and more effective research outputs, and expanded professional networks, both of which are likely to have a positive influence on careers.
GAP outcomes
The GAP Framework Initiative has (a) extended and strengthened local, national and international collaborative networks; (b) increased genomics resources and data; (c) enhanced community genomics capability; (d) made progress on the national strategic plans, and e) improved understanding of the Australian flora (Fig. 1).
The GAP Framework Initiative consortium model has extended and strengthened collaborative networks across the sector nationally and internationally. Researchers and technical staff from the plant collections, systematics, genomics and bioinformatics communities were represented from the initial consultations undertaken to develop the consortium through to the assembly of the Steering Committee and advisory Working Groups. The wider consortium engagement across all GAP projects has strengthened cross-institutional collaboration and awareness on collections curation, wet lab and bioinformatics activities, and promoted coordination of approaches and activities to maximise efficiency and the potential for data sharing and re-use. Beyond Australia, GAP researchers have collaborated with hundreds of scientists across the global plant systematics and genomics community, principally through involvement in the PAFTOL consortium. These collaborations have produced a step change in global angiosperm phylogenomics and high-impact publications (e.g. Larridon et al. 2021a; McLay et al. 2021; Zuntini et al. 2024), and have sparked a large, diverse range of new projects with researchers worldwide (e.g. Maurin et al. 2023; Elliott et al. 2024; Saunders et al. 2024; Grass Phylogeny Working Group III 2025; Helmstetter et al. 2025). In turn, these relationships have influenced global thinking and practice, for example through the endorsement and uptake of universal target capture bait kits (e.g. McDonnell et al. 2021; Fonseca et al. 2024), and publication of new taxonomic frameworks (e.g. Larridon et al. 2021b; Pillon et al. 2024).
To date, the GAP Framework Initiative has generated reference genome sequence data for 35 species, including the first genomic resources for 10 families and 4 orders, target sequence capture datasets representing 87% of Australia’s native flowering plant genera and reduced representation genomic datasets for 10 species complexes of conservation concern. These outputs have considerably increased taxonomic coverage of genomic resources for Australian plants. All raw data are immediately released to GAP consortium partners on the Bioplatforms Australia Data Portal and made accessible on public genomic repositories upon publication of data papers. Through mandating that source material must be vouchered in an Australian herbarium, the GAP Framework Initiative has enhanced curated collections through the extended specimen concept (Webster 2017; Lendemer et al. 2020) by enriching physical collections with genomic data.
The GAP consortium includes curators, researchers, technicians and other staff from more than 30 institutions across Australia and internationally. Consortium members have benefited from this collaborative program of work through involvement in genomic data generation, analysis, interpretation and dissemination. These include sampling and curating metadata from herbarium collections, wet lab preparation, bioinformatics analysis, preparation of research outputs and participation in training workshops. These researchers constitute an upskilled workforce equipped with improved capacity to generate and leverage genomic resources to identify, classify and manage biodiversity, and to improve the management, development and use of collections. Some institutions have already implemented improvements to data and specimen management protocols based on GAP processes and workflows.
The outputs from the GAP Framework Initiative directly contributed to progress against various national science goals, in particular the Decadal Plan for Taxonomy and Biosystematics 2018–2027 (Taxonomy Decadal Plan Working Group 2018) through Strategic action 1.3 ‘…build a comprehensive framework to understand the evolution of the Australian and New Zealand biota’, and Australia’s Strategy for Nature through Objective 10: ‘Increase knowledge about nature to make better decisions’, specifically progress measures 10A (Explicit science and knowledge programs to support effective management of biodiversity) and 10C (Systems capturing data on the diversity of Australian nature and how ecosystems function).
Australia’s biological heritage includes a diverse flora with a complex evolutionary history, widespread and restricted species, both long term natural isolation and more recent fragmentation, and both old and young lineages. The GAP Framework Initiative has helped improve upon Australia’s world-class infrastructure for understanding and managing this heritage by enhancing and enriching knowledge, and human and institutional resources. The reference genomes are improving our understanding of genomic diversity, structure and evolution associated with major Australian plant radiations (e.g. Chen et al. 2022; McLay et al. 2022). The AAToL will provide a common phylogenomic dataset for the flora and ultimately, the phylogenetic backbone for the Flora of Australia project and data portals such as the Atlas of Living Australia. Finally, the conservation genomics projects are resolving the taxonomy and phylogeography of complex groups to enable species documentation and define units for conservation management (e.g. Anderson et al. 2023, 2024).
The GAP Framework Initiative has not only built capacity and delivered significant new data but also revealed where the approaches taken were fit for purpose and where not. For example, a key lesson was the recognition that large collaborative consortium approaches require dedicated human resourcing and robust governance structures. Critical to our success has been the provision of dedicated project management, community engagement and bioinformatics professionals, with Steering and Working groups overseeing and managing each of the major project domains. We learned the importance of converging on shared and scalable computational resources at the national level, while making use of common workflows tailored for GAP Framework Initiative aims and use cases. Another key lesson was understanding that a one-size fits all approach to source material and extraction protocols does not work across the broad range of molecular approaches employed in this project. For example, although herbarium specimens generally yielded DNA suitable for target capture sequencing, these were generally unsuitable for the ddRAD analyses undertaken through conservation genomics projects, such that new, fresh collections were required. For the reference genomes projects, most teams had access to adequate amounts of fresh tissue but still struggled to achieve the high quality, high molecular weight DNA extractions required for long read sequencing. Understanding and developing the most appropriate source material and extraction protocols for each approach was critical to success. Further investment in capacity building around DNA quality will be needed to further realise opportunities for genomics in collections science.
A final key lesson was the value of large-scale collaborative consortia in rapidly increasing capacity and uptake of genomic approaches across a broad scope of research in genome assembly, phylogeny, systematics and conservation genomics. The project developed and strengthened collaborations, and leveraged off the regional or local skills and specific taxonomic expertise across the group. The consortium approach used here provides a potential blueprint for similar initiatives in other countries, and a foundation for further development and application of genomics to plant research in Australia. Future directions for plant biodiversity genomics in Australia are discussed elsewhere in this Special Collection.
Data availability
Data generated by the GAP Framework Initiative adhere to the FAIR principles – Findability, Accessibility, Interoperability and Reusability (Wilkinson et al. 2016). These principles are achieved through: (a) immediate release of all data on the Bioplatforms Australia Data Portal to the GAP consortium, with an embargo period prior to the public release of the data; and (b) adopting genomic approaches in widespread global use that maximise the potential for reusability. GAP data and other outputs are available as follows: Phylogenomics Sampling and Data Protocol (see https://www.genomicsforaustralianplants.com/wp-content/uploads/2022/10/GAP-Phylogenomics-protocols_Oct2022.pdf); Phylogenomics Bioinformatics Recommendations (see https://www.genomicsforaustralianplants.com/wp-content/uploads/2022/02/2022-02-08-Bioinfo_WG_recommendations_v4_MLedit.pdf); phylogenomics bioinformatics workflow (see https://github.com/chrisjackson-pellicle/hybpiper-nf, https://github.com/chrisjackson-pellicle/paragone-nf and https://github.com/chrisjackson-pellicle/NewTargets); raw data (see https://data.bioplatforms.com/organization/about/bpa-plants); database of publicly available plant genome resources (see https://www.genomicsforaustralianplants.com/compilation-of-sequenced-plant-genomes/); reference genomes for Acacia pycnantha (see https://www.ebi.ac.uk/ena/browser/view/PRJNA752212) and Telopea speciosissima (see https://www.ebi.ac.uk/ena/browser/view/PRJNA712988); reference genomes, including the Australian Reference Genome Atlas (ARGA, see https://arga.org.au); target capture sequences (AAToL), e.g. samples released though the Kew Tree of Life Explorer (see treeoflife.kew.org) are available from ENA (project PRJEB49212) and NBCI’s Short Read Archive (project PRJEB49212), and additional GAP data are available at ENA under project PRJNA1075727; training resources, workshop materials and webinar recordings (see www.genomicsforaustralianplants.com/workshop-materials/, www.genomicsforaustralianplants.com/training-webinars/). A preprint version of this paper is available on EcoEvoRxiv (Simpson et al. 2024).
Conflicts of interest
J. Bruhl, D. Cantrill, D. Crayn, D. Murphy, K. Nargar and M. Waycott are editors for Australian Systematic Botany but were not involved in the peer review or decision-making process for this paper. Australian Systematic Botany encourages its editors to publish in the journal and has protocols that keep editors separate from the decision-making processes for their manuscripts. The authors have no conflicts of interests to declare that are relevant to the content of this article.
Declaration of funding
The Genomics for Australian Plants Framework Initiative consortium (see www.genomicsforaustralianplants.com/consortium/) is funded by Bioplatforms Australia (enabled by the National Collaborative Research Infrastructure Strategy – NCRIS), the Ian Potter Foundation, Royal Botanic Gardens Foundation (Victoria), Royal Botanic Gardens Victoria, the Royal Botanic Gardens and Domain Trust, the CHAH, CSIRO, the Centre for Australian National Biodiversity Research and the Department of Biodiversity, Conservation and Attractions, Western Australia. The Plant and Fungal Trees of Life (PAFTOL) project at the Royal Botanic Gardens Kew is funded by grants from the Calleva Foundation.
Acknowledgements
In addition to the funding partners listed above, we thank the leadership and staff of the many institutions that have made in-kind contributions including staff resources, access to collections, and use of laboratories and other infrastructure. We specifically thank the following herbaria, staff and collaborators who have collected, identified and curated the specimens used in this project: AD, BRI, CANB, CNS, DNA, MEL, MELU, NE, NSW and PERTH. For the PAFTOL data used in the AAToL project, detailed acknowledgements are provided in Zuntini et al. (2024). Kor-jent van Dijk, Rachael Fowler and Danielle Smith are thanked for technical assistance, and Anna Monro is thanked for advice on Australian Plant Census records. We acknowledge the provision of computing and data resources provided by the Australian BioCommons Leadership Share (ABLeS) program (Gustafsson et al. 2023), which is co-funded by Bioplatforms Australia (enabled by NCRIS), the National Computational Infrastructure and Pawsey Supercomputing Centre. Additional computing infrastructure and services were provided by various institutional facilities including The University of Melbourne and CSIRO.
Author contributions
M. Barrett, O. Berry, R. Binks, J. Bragg, L. Broadhurst, G. Brown, M. Byrne, D. Cantrill, D. Crayn, S. Ferguson, E. James, A. Jones, G. King, T. McLay, D. Murphy, K. Nargar, H. Sauquet and M. Waycott provided conceptualisation. T. Allnutt, M. Barrett, J. Bruhl, D. Crayn, R. Edwards, J. Gustafsson, T. Hammer, O. Maurin, T. McLay, L. Simpson, M. Waycott and T. Wilson contributed to the data curation. T. Allnutt, D. Crayn, T. McLay, D. Murphy and A. Schmidt-Lebuhn were responsible for the formal analysis. L. Broadhurst, M. Byrne, D. Cantrill and D. Crayn provided funding acquisition. T. Allnutt, R. Andrew, M. Barrett, J. Batley, D. Crayn, R. Edwards, S. Ferguson, G. Holmes, E. James, A. Jones, T. McLay, D. Murphy, K. Nargar, A. Schmidt-Lebuhn, H. Sauquet, K. Shepherd, L. Simpson, A. Syme and M. Waycott assisted with investigation. T. Allnutt, W. Baker, M. Barrett, R. Binks, J. Bragg, D. Cantrill, D. Crayn, R. Edwards, S. Ferguson, F. Forest, G. Holmes, A. Jones, P. Kersey, I. Leitch, T. McLay, L. Nauheimer, A. Schmidt-Lebuhn and A. Syme were responsible for the methodology. M. Barrett, W. Baker, J. Batley, R. Binks, J. Bragg, L. Broadhurst, M. Byrne, D. Cantrill, D. Crayn, F. Forest, J. Gustafsson, G. Holmes, A. Jones, G. King, M. Lum, D. Murphy, H. Sauquet, A. Schmidt-Lebuhn, K. Shepherd and L. Simpson supplied project administration. Z. Al Bkhetan, W. Baker, G. Brown, J. Bruhl, M. Byrne, D. Crayn, F. Forest, E. James, P. Kersey, I. Leitch, O. Maurin, K. Nargar, A. Schmidt-Lebuhn, K. Shepherd, M. Waycott and T. Wilson provided resources. Z. Al Bkhetan, T. Allnutt, R. Edwards, C. Jackson, T. McLay and A. Schmidt-Lebuhn contributed software. T. Allnutt, M. Barrett, T. McLay and A. Schmidt-Lebuhn provided data visualisations. D. Crayn was responsible for the writing the original draft, with contributions from M. Byrne, D. Cantrill and L. Simpson. T. Allnutt, R. Andrew, W. Baker, M. Barrett, O. Berry, R. Binks, J. Bragg, L. Broadhurst, G. Brown, J. Bruhl, M. Byrne, D. Cantrill, D. Crayn, R. Edwards, F. Forest, J. Gustafsson, T. Hammer, E. James, P. Kersey, G. King, I. Leitch, M. Lum, T. McLay, D. Murphy, K. Nargar, H. Sauquet, A. Schmidt-Lebuhn, K. Shepherd, L. Simpson, M. Waycott and T. Wilson reviewed and editied the written material.
References
Anderson BM, Binks RM, Byrne M, Crawford AD, Shepherd KA (2023) Using RADseq to resolve species boundaries in a morphologically complex group of yellow-flowered shrubs (Geleznowia, Rutaceae). Australian Systematic Botany 36, 277-311.
| Crossref | Google Scholar |
Anderson BM, Binks RM, Byrne M, Davis R, Hislop M, Rye BL (2024) Revised taxonomy for two species complexes of Western Australian Isopogon (Proteaceae) using RADseq. Taxon 73, 161-189.
| Crossref | Google Scholar |
Anderson BM, Edlund M, James SA, Lepschi BJ, Nickrent DL, Sultan A, Tate JA, Petersen G (2025) Evolutionary relationships in Santalales inferred using target capture with Angiosperms353, focusing on Australasian Santalaceae sensu lato. Australian Systematic Botany 38, SB24026.
| Crossref | Google Scholar |
Attwood TK, Blackford S, Brazas MD, Davies A, Schneider MV (2019) A global perspective on evolving bioinformatics and data science training needs. Briefings in Bioinformatics 20, 398-404.
| Crossref | Google Scholar | PubMed |
Baker WJ, Dodsworth S, Forest F, Graham SW, Johnson MG, McDonnell A, Pokorny L, Tate JA, Wicke S, Wickett NJ (2021) Exploring Angiosperms353: an open, community toolkit for collaborative phylogenomic research on flowering plants. American Journal of Botany 108, 1059-1065.
| Crossref | Google Scholar | PubMed |
Baker WJ, Bailey P, Barber V, Barker A, Bellot S, Bishop D, Botigué LR, Brewer G, Carruthers T, Clarkson JJ, Cook J, Cowan RS, Dodsworth S, Epitawalage N, Françoso E, Gallego B, Johnson MG, Kim JT, Leempoel K, Maurin O, McGinnie C, Pokorny L, Roy S, Stone M, Toledo E, Wickett NJ, Zuntini AR, Eiserhardt WL, Kersey PJ, Leitch IJ, Forest F (2022) A comprehensive phylogenomic platform for exploring the Angiosperm Tree of Life. Systematic Biology 71, 301-319.
| Crossref | Google Scholar | PubMed |
Barrett RL, Clugston JAR, Orthia LA, Cook LG, Crisp MD, Lepschi BJ, Macfarlane TD, Weston PH, Wilkins CF (2024) East rarely meets West: a revised delimitation for Pultenaea (Fabaceae: Mirbelieae) with reinstatement of Euchilus and three new genera from south-west Western Australia. Australian Systematic Botany 37, SB23029.
| Crossref | Google Scholar |
Bowman DMJS, Brown GK, Braby MF, Brown JR, Cook LG, Crisp MD, Ford F, Haberle S, Hughes J, Isagi Y, Joseph L, McBride J, Nelson G, Ladiges PY (2010) Biogeography of the Australian monsoon tropics. Journal of Biogeography 37, 201-216.
| Crossref | Google Scholar |
Broadhurst L, Breed M, Lowe A, Bragg J, Catullo R, Coates D, Encinas-Viso F, Gellie N, James E, Krauss S, Potts B, Rossetto M, Shepherd M, Byrne M (2017) Genetic diversity and structure of the Australian flora. Diversity and Distributions 23, 41-52.
| Crossref | Google Scholar |
Byrne M, Murphy DJ (2020) The origins and evolutionary history of xerophytic vegetation in Australia. Australian Journal of Botany 68, 195-207.
| Crossref | Google Scholar |
Byrne M, Yeates DK, Joseph L, Kearney M, Bowler J, Williams MA, Cooper S, Donnellan SC, Keogh JS, Leys R, Melville J, Murphy DJ, Porch N, Wyrwoll K-H (2008) Birth of a biome: insights into the assembly and maintenance of the Australian arid zone biota. Molecular Ecology 17, 4398-4417.
| Crossref | Google Scholar | PubMed |
Byrne M, Steane DA, Joseph L, Yeates DK, Jordan G, Crayn D, Aplin K, Cantrill DJ, Cook LG, Crisp MD, Keogh JS, Melville J, Moritz C, Porch N, Sniderman JMK, Sunnucks P, Weston P (2011) Decline of a biome: contraction, fragmentation, extinction and invasion of the Australian mesic zone biota. Journal of Biogeography 38, 1635-1656.
| Crossref | Google Scholar |
Chen SH, Rossetto M, van der Merwe M, Lu-Irving P, Yap JS, Sauquet H, Bourke G, Amos TG, Bragg JG, Edwards RJ (2022) Chromosome-level de novo genome assembly of Telopea speciosissima (New South Wales waratah) using long-reads, linked-reads and Hi-C. Molecular Ecology Resources 22, 1836-1854.
| Crossref | Google Scholar | PubMed |
Cooper WE, Crayn DM, Joyce EM (2023) Aglaia fellii W.E.Cooper & Joyce (Meliaceae), a new species for Cape York Peninsula. Australian Journal of Taxonomy 16, 1-9.
| Crossref | Google Scholar |
Crayn DM, Costion C, Harrington MG (2015) The Sahul–Sunda floristic exchange: dated molecular phylogenies document Cenozoic intercontinental dispersal dynamics. Journal of Biogeography 42, 11-24.
| Crossref | Google Scholar |
Crisp MD, Cook LG (2013) How was the Australian flora assembled over the last 65 million years? A molecular phylogenetic perspective. Annual Review of Ecology, Evolution, and Systematics 44, 303-324.
| Crossref | Google Scholar |
Cronn R, Knaus BJ, Liston A, Maughan PJ, Parks M, Syring JV, Udall J (2012) Targeted enrichment strategies for next-generation plant biology. American Journal of Botany 99, 291-311.
| Crossref | Google Scholar | PubMed |
Eaton DAR, Overcast I (2020) ipyrad: Interactive assembly and analysis of RADseq datasets. Bioinformatics 36, 2592-2594.
| Crossref | Google Scholar | PubMed |
Eldridge MDB, Deakin JE, MacDonald AJ, Byrne M, Fitzgerald A, Johnson RN, Moritz C, Palmer S, Young A (2020) The Oz Mammals Genomics (OMG) initiative: developing genomic resources for mammal conservation at a continental scale. Australian Zoologist 40, 505-509.
| Crossref | Google Scholar |
Elliott TL, Spalink D, Larridon I, Zuntini AR, Escudero M, Hackel J, Barrett RL, Martín-Bravo S, Márquez-Corro JI, Granados Mendoza C, Mashau AC, Romero-Soler KJ, Zhigila DA, Gehrke B, Andrino CO, Crayn DM, Vorontsova MS, Forest F, Baker WJ, Wilson KL, Simpson DA, Muasya AM (2024) Global analysis of Poales diversification – parallel evolution in space and time into open and closed habitats. The New Phytologist 242, 727-743.
| Crossref | Google Scholar | PubMed |
Ferguson S, McLay T, Andrew RL, Bruhl JJ, Schwessinger B, Borevitz J, Jones A (2022) Species-specific basecallers improve actual accuracy of nanopore sequencing in plants. Plant Methods 18, 137.
| Crossref | Google Scholar | PubMed |
Fonseca LHM, Asselman P, Goodrich KR, Nge FJ, Soulé V, Mercier K, Couvreur TLP, Chatrou LW (2024) Truly the best of both worlds: merging lineage-specific and universal probe kits to maximize phylogenomic inference. Applications in Plant Sciences 12, e11615.
| Crossref | Google Scholar | PubMed |
Garcia S, Garnatje T, Hidalgo O, Mas De Xaxars G, Pellicer J, Sánchez-Jiménez I, Vitales D, Vallès J (2010) First genome size estimations for some eudicot families and genera. Collectanea Botanica 29, 7-16.
| Crossref | Google Scholar |
Gilmore S, Weston P, Thomson J (1993) A simple, rapid, inexpensive and widely applicable technique for purifying plant DNA. Australian Systematic Botany 6, 139-148.
| Crossref | Google Scholar |
Govaerts R, Nic Lughadha E, Black N, Turner R, Paton A (2021) The World Checklist of Vascular Plants, a continuously updated resource for exploring global plant diversity. Scientific Data 8, 215.
| Crossref | Google Scholar | PubMed |
Grass Phylogeny Working Group III (2025) Nuclear phylogenomics of grasses (Poaceae) supports current classification and reveals repeated reticulation. New Phytologist 245, 818-834.
| Crossref | Google Scholar |
Gustafsson OJR, Al Bkhetan Z, Francis R, Manos S (2023) Enabling national step changes in bioinformatics through ABLeS, the Australian BioCommons Leadership Share. Zenodo 2023, version 3.0 [Preprint, published 15 November 2023].
| Crossref | Google Scholar |
Hammer TA, Biffin E, van Dijk K, Thiele KR, Waycott M (2025) A framework phylogeny of the diverse guinea-flowers (Hibbertia, Dilleniaceae) using high-throughput sequence data. Australian Systematic Botany 38(2), SB24009.
| Crossref | Google Scholar |
Helmstetter AJ, Ezedin Z, de Lírio EJ, de Oliveira SM, Chatrou LW, Erkens RHJ, Larridon I, Leempoel K, Maurin O, Roy S, Zuntini AR, Baker WJ, Couvreur TLP, Forest F, Sauquet H (2025) Towards a phylogenomic classification of Magnoliidae. American Journal of Botany 112, e16451.
| Crossref | Google Scholar | PubMed |
Interjurisdictional Biodiversity Working Group (2019) Australia’s Strategy for Nature 2019–2030. (Commonwealth of Australia) Available at https://www.australiasnaturehub.gov.au/sites/default/files/2020-11/australias-strategy-for-nature.pdf
Jackson C, McLay T, Schmidt-Lebuhn AN (2023) hybpiper-nf and paragone-nf: containerization and additional options for target capture assembly and paralog resolution. Applications in Plant Sciences 11, e11532.
| Crossref | Google Scholar | PubMed |
Johnson MG, Gardner EM, Liu Y, Medina R, Goffinet B, Shaw AJ, Zerega NJ, Wickett NJ (2016) HybPiper: extracting coding sequence and introns for phylogenetics from high-throughput sequencing reads using target enrichment. Applications in Plant Sciences 4, 1600016.
| Crossref | Google Scholar | PubMed |
Johnson MG, Pokorny L, Dodsworth S, Botigué LR, Cowan RS, Devault A, Eiserhardt WL, Epitawalage N, Forest F, Kim JT, Leebens-Mack JH, Leitch IJ, Maurin O, Soltis DE, Soltis PS, Wong GK, Baker WJ, Wickett NJ (2019) A universal probe set for targeted sequencing of 353 nuclear genes from any flowering plant designed using k-medoids clustering. Systematic Biology 68, 594-606.
| Crossref | Google Scholar | PubMed |
Jones A, Schwessinger B (2021) High-molecular weight DNA extraction from challenging fungi using CTAB for lysis and precipitation V.3. Protocols.io 2021, version 3 [Science methods, published 23 July 2021].
| Crossref | Google Scholar |
Jones A, Torkel C, Stanley D, Nasim J, Borevitz J, Schwessinger B (2021) High-molecular weight DNA extraction, clean-up and size selection for long-read sequencing. PLoS ONE 16, e0253830.
| Crossref | Google Scholar | PubMed |
Jordan GJ, Carpenter RJ, Koutoulis A, Price A, Brodribb TJ (2015) Environmental adaptation in stomatal size independent of the effects of genome size. The New Phytologist 205, 608-617.
| Crossref | Google Scholar | PubMed |
Joyce EM, Appelhans MS, Buerki S, Cheek M, de Vos JM, Pirani JR, Zuntini AR, Bachelier JB, Bayly MJ, Callmander MW, Devecchi MF, Pell SK, Groppo M, Lowry PP, Mitchell J, Siniscalchi CM, Munzinger J, Orel HK, Pannell CM, Nauheimer L, Sauquet H, Weeks A, Muellner-Riehl AN, Leitch IJ, Maurin O, Forest F, Nargar K, Thiele KR, Baker WJ, Crayn DM (2023) Phylogenomic analyses of Sapindales support new family relationships, rapid mid-Cretaceous hothouse diversification, and heterogeneous histories of gene duplication. Frontiers in Plant Science 14, 1063174.
| Crossref | Google Scholar | PubMed |
Joyce EM, Schmidt-Lebuhn AN, Orel HK, Nge FJ, Anderson BM, Hammer TA, McLay TGB (2024) Navigating phylogenetic conflict and evolutionary inference in plants with target capture data. EcoEvoRxiv
| Crossref | Google Scholar |
Joyce EM, Schmidt-Lebuhn AN, Orel HK, Nge FJ, Anderson BM, Hammer TA, McLAy TGB (2025) Navigating phylogenetic conflict and evolutionary inference in plants with target capture data. Australian Systematic Botany 38, SB24011.
| Crossref | Google Scholar |
Larridon I, Zuntini AR, Barrett RL, Wilson KL, Bruhl JJ, Goetghebeur P, Baker WJ, Brewer GE, Epitawalage N, Fairlie I, Forest F, Kikuchi I, Pokorny L, Spalink D, Simpson DA, Muasya AM, Roalson EH (2021a) Resolving the generic limits in Cyperaceae tribe Abildgaardieae using targeted sequencing. Botanical Journal of the Linnean Society 196, 163-187.
| Crossref | Google Scholar |
Larridon I, Zuntini AR, Léveillé-Bourret E, Barrett RL, Starr JR, Muasya AM, Villaverde T, Bauters K, Brewer GE, Bruhl JJ, Costa SM, Elliott TL, Epitawalage N, Escudero M, Fairlie I, Goetghebeur P, Hipp AL, Jiménez-Mejías P, Sabino Kikuchi IAB, Luceño M, Márquez-Corro JI, Martín-Bravo S, Maurin O, Pokorny L, Roalson EH, Semmouri I, Simpson DA, Spalink D, Thomas WW, Wilson KL, Xanthos M, Forest F, Baker WJ (2021b) A new classification of Cyperaceae (Poales) supported by phylogenomic data. Journal of Systematics and Evolution 59, 852-895.
| Crossref | Google Scholar |
Leitch IJ, Hanson L (2002) DNA C-values in seven families fill phylogenetic gaps in the basal angiosperms. Botanical Journal of the Linnean Society 140, 175-179.
| Crossref | Google Scholar |
Lendemer J, Thiers B, Monfils AK, Zaspel J, Ellwood ER, Bentley A, LeVan K, Bates J, Jennings D, Contreras D, Lagomarsino L, Mabee P, Ford LS, Guralnick R, Gropp RE, Revelez M, Cobb N, Seltmann K, Aime MC (2020) The Extended Specimen Network: a strategy to enhance US biodiversity collections, promote research and education. BioScience 70, 23-30.
| Crossref | Google Scholar | PubMed |
Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, Dekker J (2009) Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289-293.
| Crossref | Google Scholar | PubMed |
Lowman HE, DeSiervo M, Hall RO, Jr, Jahner JP, Jimoh SO, Laughlin DC, Patterson AC, Weiss-Lehman C, Barbosa CC, Bell KL, Blaszczak JR, Buerkle CA, Carter AM, Collins SM, DeLeo V, Dunkle M, Gannon D, Grames EM, Harrison JG, McFarlane SE, Oleksy I, Powers BF, Ray C, Stears A, Summers B, Torrens CL, Trentman M, Werner CM, Shoemaker LG (2024) Collaborative consortia can boost postdoctoral workforce development. Proceedings of the National Academy of Sciences 121, e2401812121.
| Crossref | Google Scholar | PubMed |
Maurin O, Anest A, Forest F, Turner I, Barrett RL, Cowan RC, Wang L-J, Tomlinson KW, Charles-Dominique T (2023) Drift in the tropics: phylogenetics and biogeographical patterns in Combretaceae. Global Ecology and Biogeography 32, 1790-1802.
| Crossref | Google Scholar |
McDonnell AJ, Baker WJ, Dodsworth S, Forest F, Graham SW, Johnson MG, Pokorny L, Tate J, Wicke S, Wickett NJ (2021) Exploring Angiosperms353: developing and applying a universal toolkit for flowering plant phylogenomics. Applications in Plant Sciences 9, e11443.
| Crossref | Google Scholar | PubMed |
McLay TGB, Birch JL, Gunn BF, Ning W, Tate JA, Nauheimer L, Joyce EM, Simpson L, Schmidt-Lebuhn AN, Baker WJ, Forest F, Jackson CJ (2021) New targets acquired: improving locus recovery from the Angiosperms353 probe set. Applications in Plant Sciences 9, e11420.
| Crossref | Google Scholar | PubMed |
McLay TGB, Murphy DJ, Holmes GD, Mathews S, Brown GK, Cantrill DJ, Udovicic F, Allnutt TR, Jackson CJ (2022) A genome resource for Acacia, Australia’s largest plant genus. PLoS One 17, e0274267.
| Crossref | Google Scholar | PubMed |
Morgan HD, Westoby M (2005) The relationship between nuclear DNA content and leaf strategy in seed plants. Annals of Botany 96, 1321-1330.
| Crossref | Google Scholar | PubMed |
Nge FJ, Biffin E, Rye BL, Wilson PG, van Dijk K, Thiele KR, Waycott M, Barrett MD (2025) Australian biogeography, climate-dependent diversification and phylogenomics of the spectacular Chamelaucieae tribe (Myrtaceae). Australian Systematic Botany 38, SB24014.
| Crossref | Google Scholar |
One Thousand Plant Transcriptomes Initiative (2019) One thousand plant transcriptomes and the phylogenomics of green plants. Nature 574, 679-685.
| Crossref | Google Scholar | PubMed |
Orel HK, McLay TGB, Guja LK, Duretto MF, Bayly MJ (2023a) Genomic data inform taxonomy and conservation of Critically Endangered shrubs: a case study of Zieria (Rutaceae) species from eastern Australia. Botanical Journal of the Linnean Society 205, 292-312.
| Crossref | Google Scholar |
Orel HK, Briggs JD, Duretto MF (2023b) Zieria nubicola (Rutaceae), a new and highly restricted species from New South Wales, Australia. Telopea 26, 169-174.
| Crossref | Google Scholar |
Pellicer J, Leitch IJ (2020) The Plant DNA C-values database (release 7.1): an updated online repository of plant genome size data for comparative studies. The New Phytologist 226, 301-305.
| Crossref | Google Scholar | PubMed |
Pérez-Escobar OA, Bogarín D, Przelomska NAS, Ackerman JD, Balbuena JA, Bellot S, Bühlmann RP, Cabrera B, Cano JA, Charitonidou M, Chomicki G, Clements MA, Cribb P, Fernández M, Flanagan NS, Gravendeel B, Hágsater E, Halley JM, Hu A-Q, Jaramillo C, Mauad AV, Maurin O, Müntz R, Leitch IJ, Li L, Negrão R, Oses L, Phillips C, Rincon M, Salazar GA, Simpson L, Smidt E, Solano-Gomez R, Parra-Sánchez E, Tremblay RL, van den Berg C, Tamayo BSV, Zuluaga A, Zuntini AR, Chase MW, Fay MF, Condamine FL, Forest F, Nargar K, Renner SS, Baker WJ, Antonelli A (2024) The origin and speciation of orchids. The New Phytologist 242, 700-716.
| Crossref | Google Scholar | PubMed |
Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE (2012) Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE 7, e37135.
| Crossref | Google Scholar | PubMed |
Pillon Y, Crayn DM, Streiff SJR, de Vos JM (2024) A suprageneric classification of Oxalidales. Swainsona 38, 153-160.
| Google Scholar |
Saunders TC, Larridon I, Baker WJ, Barrett RL, Forest F, Françoso E, Maurin O, Rokni S, Roalson EH (2024) Tangled webs and spiderflowers: phylogenomics, biogeography, and seed morphology inform the evolutionary history of Cleomaceae. American Journal of Botany 111, e16399.
| Crossref | Google Scholar | PubMed |
Schmidt-Lebuhn AN (2022) Sequence capture data support the taxonomy of Pogonolepis (Asteraceae: Gnaphalieae) and show unexpected genetic structure. Australian Systematic Botany 35, 317-325.
| Crossref | Google Scholar |
Schmidt-Lebuhn AN, Cantrill DJ (2023) Genome size estimates for Genomics for Australian Plants sequencing projects. Muelleria 41, 44-49.
| Crossref | Google Scholar |
Schmidt-Lebuhn AN, Egli D, Grealy A, Nicholls JA, Zwick A, Dymock JJ, Gooden B (2022) Genetic data confirm the presence of Senecio madagascariensis in New Zealand. New Zealand Journal of Botany 62, 1-13.
| Crossref | Google Scholar |
Schmidt-Lebuhn AN, Chen SH, Grealy A (2024) Elachanthus, Isoetopsis and Kippistia are nested in the genus Minuria (Asteraceae: Astereae). Australian Systematic Botany 37, SB23028.
| Crossref | Google Scholar |
Simmons CL, McLay TGB, Mathieson MT (2025) Wedged between two congeners: will the real Gompholobium nitidum (Fabaceae: Mirbelieae) please stand up? Australian Systematic Botany 38, SB24010.
| Crossref | Google Scholar |
Simpson L, Cantrill DJ, Byrne M, Allnutt TR, King GJ, Lum M, Al Bkhetan Z, Andrew RL, Baker WJ, Barrett MD, Batley J, Berry O, Binks RM, Bragg J, Broadhurst L, Brown G, Bruhl JJ, Edwards RJ, Ferguson S, Forest F, Gustafsson J, Hammer TA, Holmes GD, Jackson CJ, James EA, Jones A, Kersey PJ, Leitch IJ, Maurin O, McLay TGB, Murphy DJ, Nargar K, Nauheimer L, Sauquet H, Schmidt-Lebuhn AN, Shepherd KA, Syme AE, Waycott M, Wilson TC, Crayn DM (2024) The Genomics for Australian Plants (GAP) framework initiative – developing genomic resources for understanding the evolution and conservation of the Australian flora. EcoEvoRxiv 2024, 7349 [Preprint, published 9 July 2024].
| Crossref | Google Scholar |
Šmarda P, Bureš P, Horová L, Leitch IJ, Mucina L, Pacini E, Tichý L, Grulich V, Rotreklová O (2014) Ecological and evolutionary significance of genomic GC content diversity in monocots. Proceedings of the National Academy of Sciences 111, E4096-E4102.
| Crossref | Google Scholar | PubMed |
Syme AE, McLay TGB, Udovicic F, Cantrill DJ, Murphy DJ (2021) Long-read assemblies reveal structural diversity in genomes of organelles – an example with Acacia pycnantha. GigaByte 2021, gigabyte36.
| Crossref | Google Scholar | PubMed |
Taxonomy Decadal Plan Working Group (2018) Discovering Biodiversity: a decadal plan for taxonomy and biosystematics in Australia and New Zealand 2018–2028. (Australian Academy of Science: Canberra, ACT, Australia; and Royal Society Te Apārangi: Wellington, New Zealand) Available at https://www.science.org.au/support/analysis/decadal-plans-science/discovering-biodiversity-decadal-plan-taxonomy [Verified October 2023]
Teytelman L, Stoliartchouk A, Kindler L, Hurwitz BL (2016) Protocols.io: virtual communities for protocol development and discussion. PLoS Biology 14, e1002538.
| Crossref | Google Scholar | PubMed |
The Angiosperm Phylogeny Group (2016) An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Botanical Journal of the Linnean Society 181, 1-20.
| Crossref | Google Scholar |
Waycott M, van Dijk K, Biffin E (2021) A hybrid capture RNA bait set for resolving genetic and evolutionary relationships in angiosperms from deep phylogeny to intraspecific lineage hybridization. bioRxiv 2021, 2021.09.06.456727 [Preprint, published 7 September 2021].
| Crossref | Google Scholar |
Weng M-L, Ruhlman TA, Gibby M, Jansen RK (2012) Phylogeny, rate variation, and genome size evolution of Pelargonium (Geraniaceae). Molecular Phylogenetics and Evolution 64, 654-670.
| Crossref | Google Scholar | PubMed |
Wilkinson MD, Dumontier M, Aalbersberg IJ, Appleton G, Axton M, Baak A, Blomberg N, Boiten J-W, da Silva Santos LB, Bourne PE, Bouwman J, Brookes AJ, Clark T, Crosas M, Dillo I, Dumon O, Edmunds S, Evelo CT, Finkers R, Gonzalez-Beltran A, Gray AJG, Groth P, Goble C, Grethe JS, Heringa J, ’t Hoen PAC, Hooft R, Kuhn T, Kok R, Kok J, Lusher SJ, Martone ME, Mons A, Packer AL, Persson B, Rocca-Serra P, Roos M, van Schaik R, Sansone S-A, Schultes E, Sengstag T, Slater T, Strawn G, Swertz MA, Thompson M, van der Lei J, van Mulligen E, Velterop J, Waagmeester A, Wittenburg P, Wolstencroft K, Zhao J, Mons B (2016) The FAIR Guiding Principles for scientific data management and stewardship. Scientific Data 3, 160018.
| Crossref | Google Scholar | PubMed |
Williamson L, Biffin E, Hammer T, van Dijk K, Conran J, Waycott M (2025) Phylogenomics of Australian sundews (Drosera: Droseraceae). Australian Systematic Botany 38, SB24016.
| Crossref | Google Scholar |
Wilson T, James EA (2025) Phylogenetic and biogeographic insights into the reproductive evolution and taxonomy of Australasian Teucrium (Lamiaceae). Australian Systematic Botany 38, SB24027.
| Crossref | Google Scholar |
Yang Y, Smith SA (2014) Orthology inference in nonmodel organisms using transcriptomes and low-coverage genomes: improving accuracy and matrix occupancy for phylogenomics. Molecular Biology and Evolution 31, 3081-3092.
| Crossref | Google Scholar | PubMed |
Zuntini AR, Carruthers T, Maurin O, Bailey PC, Leempoel K, Brewer GE, Epitawalage N, Françoso E, Gallego-Paramo B, McGinnie C, Negrão R, Roy SR, Simpson L, Toledo Romero E, Barber VMA, Botigué L, Clarkson JJ, Cowan RS, Dodsworth S, Johnson MG, Kim JT, Pokorny L, Wickett NJ, Antar GM, DeBolt L, Gutierrez K, Hendriks KP, Hoewener A, Hu A-Q, Joyce EM, Kikuchi IABS, Larridon I, Larson DA, de Lírio EJ, Liu J-X, Malakasi P, Przelomska NAS, Shah T, Viruel J, Allnutt TR, Ameka GK, Andrew RL, Appelhans MS, Arista M, Ariza MJ, Arroyo J, Arthan W, Bachelier JB, Bailey CD, Barnes HF, Barrett MD, Barrett RL, Bayer RJ, Bayly MJ, Biffin E, Biggs N, Birch JL, Bogarín D, Borosova R, Bowles AMC, Boyce PC, Bramley GLC, Briggs M, Broadhurst L, Brown GK, Bruhl JJ, Bruneau A, Buerki S, Burns E, Byrne M, Cable S, Calladine A, Callmander MW, Cano Á, Cantrill DJ, Cardinal-McTeague WM, Carlsen MM, Carruthers AJA, de Castro Mateo A, Chase MW, Chatrou LW, Cheek M, Chen S, Christenhusz MJM, Christin P-A, Clements MA, Coffey SC, Conran JG, Cornejo X, Couvreur TLP, Cowie ID, Csiba L, Darbyshire I, Davidse G, Davies NMJ, Davis AP, van Dijk K, Downie SR, Duretto MF, Duvall MR, Edwards SL, Eggli , U, Erkens RHJ, Escudero M, de la Estrella M, Fabriani F, Fay MF, Ferreira PdL, Ficinski SZ, Fowler RM, Frisby S, Fu L, Fulcher T, Galbany-Casals M, Gardner EM, German DA, Giaretta A, Gibernau M, Gillespie LJ, González CC, Goyder DJ, Graham SW, Grall A, Green L, Gunn BF, Gutiérrez DG, Hackel J, Haevermans T, Haigh A, Hall JC, Hall T, Harrison MJ, Hatt SA, Hidalgo O, Hodkinson TR, Holmes GD, Hopkins HCF, Jackson CJ, James SA, Jobson RW, Kadereit G, Kahandawala IM, Kainulainen K, Kato M, Kellogg EA, King GJ, Klejevskaja B, Klitgaard BB, Klopper RR, Knapp S, Koch MA, Leebens-Mack JH, Lens F, Leon CJ, Léveillé-Bourret É, Lewis GP, Li D-Z, Li L, Liede-Schumann S, Livshultz T, Lorence D, Lu M, Lu-Irving P, Luber J, Lucas EJ, Luján M, Lum M, Macfarlane TD, Magdalena C, Mansano VF, Masters LE, Mayo SJ, McColl K, McDonnell AJ, McDougall AE, McLay TGB, McPherson H, Meneses RI, Merckx VSFT, Michelangeli FA, Mitchell JD, Monro AK, Moore MJ, Mueller TL, Mummenhoff K, Munzinger J, Muriel P, Murphy DJ, Nargar K, Nauheimer L, Nge FJ, Nyffeler R, Orejuela A, Ortiz EM, Palazzesi L, Peixoto AL, Pell SK, Pellicer J, Penneys DS, Perez-Escobar OA, Persson C, Pignal M, Pillon Y, Pirani JR, Plunkett GM, Powell RF, Prance GT, Puglisi C, Qin M, Rabeler RK, Rees PEJ, Renner M, Roalson EH, Rodda M, Rogers ZS, Rokni S, Rutishauser R, de Salas MF, Schaefer H, Schley RJ, Schmidt-Lebuhn A, Shapcott A, Al-Shehbaz I, Shepherd KA, Simmons MP, Simões AO, Simões ARG, Siros M, Smidt EC, Smith JF, Snow N, Soltis DE, Soltis PS, Soreng RJ, Sothers CA, Starr JR, Stevens PF, Straub SCK, Struwe L, Taylor JM, Telford IRH, Thornhill AH, Tooth I, Trias-Blasi A, Udovicic F, Utteridge TMA, Del Valle JC, Verboom GA, Vonow HP, Vorontsova MS, de Vos JM, Al-Wattar N, Waycott M, Welker CAD, White AJ, Wieringa JJ, Williamson LT, Wilson TC, Wong SY, Woods LA, Woods R, Worboys S, Xanthos M, Yang Y, Zhang Y-X, Zhou M-Y, Zmarzty S, Zuloaga FO, Antonelli A, Bellot S, Crayn DM, Grace OM, Kersey PJ, Leitch IJ, Sauquet H, Smith SA, Eiserhardt WL, Forest F, Baker WJ (2024) Phylogenomics and the rise of the angiosperms. Nature 629, 843-850.
| Crossref | Google Scholar | PubMed |
Footnotes
1 The Nagoya Protocol on Access to Genetic Resources and the Fair and Equitable Sharing of Benefits Arising from their Utilization to the Convention on Biological Diversity is an international agreement that aims to ensure that the benefits arising from the utilisation of genetic resources are shared in a fair and equitable way. It entered into force on 12 October 2014. Although not yet ratified in Australia, in practice, GAP participants demonstrate compliance with the spirit of the Nagoya Protocol by identifying the owner of the genetic material submitted for analysis. For samples obtained from public land, the owner is evidenced by collecting or scientific research permits from the relevant authority. In the case of samples collected on Indigenous or other land including land in private ownership, evidence may comprise a written agreement that demonstrates prior, informed consent was given by the landowner for genetic research to be conducted on the sample(s).