

The influence of season, habitat and diet on the faecal microbiome of the yellow-footed rock-wallaby (Petrogale xanthopus xanthopus)
Lauren Elizabeth Werner A * , Shannon Leilani Kleemann A , Stacey Kathleen Dix B , Korjent van Dijk C D , Edward Biffin D , Michelle Waycott C D , Raphael Eisenhofer A E and David Augustine Taggart B F
A * , Shannon Leilani Kleemann A , Stacey Kathleen Dix B , Korjent van Dijk C D , Edward Biffin D , Michelle Waycott C D , Raphael Eisenhofer A E and David Augustine Taggart B F
A
B
C
D
E
F
Abstract
Knowledge of an animal’s microbiome is becoming increasingly recognised as an important consideration in the conservation of threatened species, particularly in the face of wide-spread changes to climate and rainfall patterns. The yellow-footed rock-wallaby (YFRW; Petrogale xanthopus xanthopus) is endemic to the semiarid regions of South Australia and New South Wales. This study aimed to characterise the faecal microbial diversity and its relationship to diet and season/rainfall in two geographically separated South Australian YFRW populations with differing habitat characteristics. Sequencing targeting the 16S rRNA gene revealed that location was the greatest driver of faecal microbial differences (P < 0.01), with season (P < 0.01) and the interaction of location × season also statistically significant (P < 0.01). The main phyla identified throughout were Firmicutes and Bacteroidota. Diet varied between individuals with Acacia species commonly detected at each study site, which appeared to be linked with an increase in the proportion of Firmicutes present, although further sampling is required to confirm this. Further research and continued long term monitoring is required to understand microbial functions, how the role of these microbes may affect an individual’s health, and how faecal microbiome can be manipulated to increase a species’ resilience to dry times and drought.
Keywords: ASV diversity, composition, diet, Faecal microbiome, microbial diversity, rainfall, richness, rock-wallaby.
Introduction
The gut microbiome, that is the community of microorganisms residing in the digestive tract, has been linked with the general health and wellbeing of humans and other animals (Haller 2018; Amato et al. 2021; Huang et al. 2022; Pannoni et al. 2022; Ribas et al. 2023). Deviations from a normal gut microbial composition can result in health issues such as nutritional deficiencies (Kau et al. 2011), decreased immunological function (Round and Mazmanian 2009), and abnormal physical development (Sommer and Bäckhed 2013). The importance of gut or faecal microbes in wildlife conservation has recently been recognised (Banerjee et al. 2020), with studies suggesting that the microbiome could act as an early indicator of ecosystem declines, aid in increasing species fecundity, and help improve reintroduction success (Redford et al. 2012; West et al. 2019; Ribas et al. 2023). As a result, wildlife microbiome studies are increasing in popularity and are often associated with conservation priority species such as the critically endangered indri (Indri indri) (Correa et al. 2021), black-and-white ruffed lemur (Varecia variegata) (Donohue et al. 2019), and the endangered Malayan tapir (Tapirus indicus) (Arumugam et al. 2023).
In Australia, however, the number of gut or faecal microbiome studies are still relatively few, with investigations conducted on only a small subset of native species such as the Tasmanian devil (Sarcophilus harrisii) (Cheng et al. 2015), koala (Phascolarctos cinereus) (Brice et al. 2019; Blyton et al. 2023; Eisenhofer et al. 2023), tammar wallaby (Notamacropus eugenii) (Chhour et al. 2010) and the southern hairy-nosed wombat (SHNW) (Lasiorhinus latifrons) (Eisenhofer et al. 2021), with some of these also characterising the microbiome of other body sites (i.e. skin, pouch). Building the understanding of faecal microbial characteristics of Australian native species is integral for assessing how microbiome manipulation can be used to assist conservation programs, such as how microbiome is naturally altered upon wild release in reintroduction programs in conjunction with dietary changes. This is particularly pertinent as climate change is predicted to result in higher average temperatures, decreased annual rainfall, longer and more severe droughts, and more extreme climatic events (Department for Environment and Water 2022), forcing wildlife to adapt to new environmental conditions, move their current distribution, or risk significant population decline (Davis et al. 2005; Mills 2012; Höglund et al. 2019).
Habitat specialists are predicted to be at highest risk under progressive climate change (Klamt et al. 2011; Adams-Hosking et al. 2012; Gibson et al. 2018; Marshall et al. 2018). The yellow-footed rock-wallaby (YFRW) (Petrogale xanthopus xanthopus), a semiarid-dwelling, medium-sized macropodid marsupial is one such species. The YFRW utilises foregut fermentation to extract short-chain fatty acids (SCFAs) from their herbivorous diet (Hume 1999; Armati et al. 2006). Currently, there are no direct studies on energy metabolism for YFRWs, but macropodids in general are thought to produce approximately 42% of their total digestible energy intake from fermentation in the foregut region (Armati et al. 2006). However, no microbiome examinations have been completed on the YFRW, leaving the microbial communities that acquire their energy requirements relatively unknown.
In South Australia, the YFRW is found in the Flinders, Gammon, Gawler and Olary Ranges in rocky habitats, which they rely upon for shelter from environmental extremes and predation (Copley 1981; Short 1982; Copley 1983; Lim et al. 1987; Ruykys 2017; Michael and Lindenmayer 2018). The current conservation status of the YFRW in South Australia is Vulnerable (EPBC Classification; Baker and Gynther 2023). This study aimed to examine the faecal microbiome of YFRWs from two geographically separated regions with differing habitat characteristics, within their current distribution in South Australia. As faecal microbiome is directly associated with and dependent on dietary intake (Chen et al. 2014; Maria Moise 2017), we also assessed how dietary composition interacts with microbial diversity and structure in YFRWs, a factor that has not been assessed in-depth in other faecal microbiome studies.
Understanding the interactions between habitat connectivity, diet, rainfall, and faecal microbiome may prove to be important information for managing the YFRW in the future, particularly as the impacts of climate change intensify.
Methods
Ethical statement
This project has been approved by the University of Adelaide’s Animal Ethics Committee (S-2020-097), and the Department of Environment and Water (Scientific Research Permit #Y26992-1).
Study sites
This study was undertaken at two sites in South Australia (Italowie Gap and Bimba Hill), where trapping was conducted three times across a 15-month period (March 2021 to May 2022) to collect faecal microbiome and dietary samples from captured YFRWs.
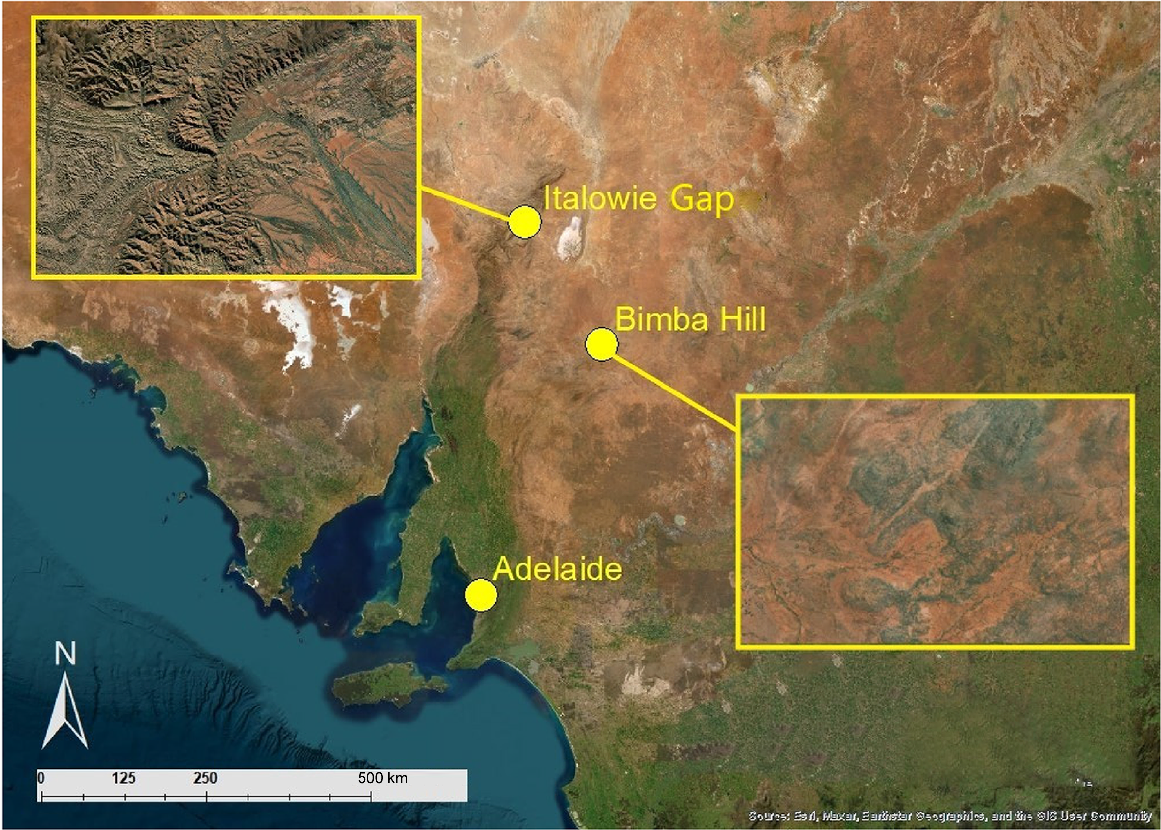
Italowie Gap in the Vulkathunha-Gammon Ranges National Park (Fig. 1; −30.558691, 139.164285) is approximately 7.5 h from Adelaide. Long-term data indicate that this area receives approximately 257 mm of rainfall annually; however, during this study it received 470 mm of rainfall. Rainfall in the 3 months prior to each trapping period was recorded as this was considered most likely to influence the vegetation and gut microbiome of the animal caught (Table 1; Fig. 2; Bureau of Meteorology, data from the closest weather station, Arkaroola). The habitat at this site is characterised by steep escarpments with simple stratified sandstone, siltstone, and quartzite rock formations with few areas of high rock complexity, interspersed with loose earth and scree slopes. The vegetation is mostly confined to the adjacent creek bed, dominated by river red gum trees (Eucalyptus camaldulensis), and the slopes, where there is a scattering of rock fuchsia bush (Eremophila freelingii) and white cypress pine (Callitris glaucophylla). During times of high rainfall, the hills are dominated by rock sida (Sida petrophila) and desert lantern-bush (Abutilon leucopetalum) with small herbaceous plants and grasses growing within the rocks.
The location of the yellow-footed rock-wallaby (Petrogale xanthopus xanthopus) study sites, Italowie Gap in the Gammon Ranges, and Bimba Hill in the Olary Ranges, in South Australia, Australia. Inset images show a closer aerial view of each study site.
| Rainfall (mm) in 3-month period prior to sampling | ||||
|---|---|---|---|---|
| Autumn 2021 | Spring 2021 | Autumn 2022 | ||
| Italowie Gap | 64.4 | 23.6 | 84.5 | |
| Bimba Hill | 45.1 | 32.7 | 62.8 | |
The amount of rainfall (mm) that was received in the three-month period prior to trapping and the collection of yellow-footed rock-wallaby (Petrogale xanthopus xanthopus) scats from Italowie Gap and Bimba Hill, South Australia. Rainfall data are presented for the three different trapping periods per site: autumn 2021, spring, and autumn 2022.
The average monthly rainfall (mm) received preceding and during the study period at Italowie Gap (top) and Bimba Hill (bottom), South Australia. Months of higher-than-average rainfall (  ), conditions at each site (drought, dry, or wet), and yellow-footed rock-wallaby (Petrogale xanthopus xanthopus) trapping periods indicated (downward arrow). Rainfall data were obtained from the Bureau of Meteorology.
), conditions at each site (drought, dry, or wet), and yellow-footed rock-wallaby (Petrogale xanthopus xanthopus) trapping periods indicated (downward arrow). Rainfall data were obtained from the Bureau of Meteorology.
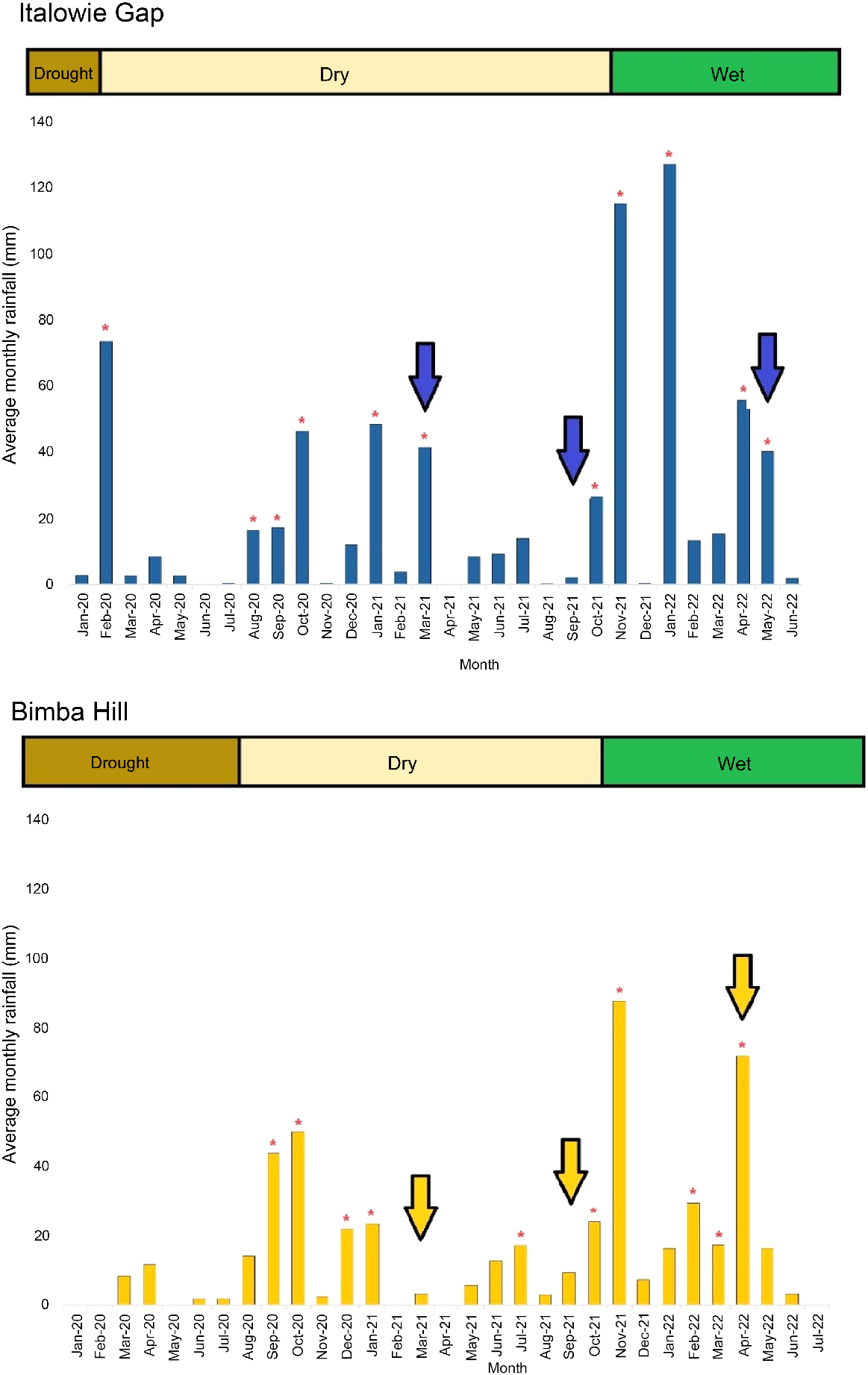
The Bimba Hill complex on Bimbowrie Conservation Park in the Olary Ranges of South Australia (Fig. 1; −31.983341, 140.211817) is approximately 5 h from Adelaide and receives, on average, 203 mm of rainfall annually. During this study, however, it received 311 mm of rainfall, and the rainfall received in the preceding three-month period before trapping was recorded (Table 1; Fig. 2; Bureau of Meteorology, data from Bimbowrie weather station). The habitat at this site consists of isolated monzogranite outcrops, comprising flat rock sheets and large, complex boulder jumbles interspersed with earthen slopes. The vegetation is dominated by rock fuchsia bush (Eremophila freelingii), with the occasional mulga (Acacia aneura) and dead finish (Acacia tetragonophylla). After rainfall, the slopes are dominated by rock sida (Sida petrophila) and Solanum species, with herbaceous plants and a variety of grasses growing in between the rocks.
Sample acquisition
Faecal samples (scats) were collected as part of a larger YFRW trapping program during autumn 2021, spring 2021, and autumn 2022 (Fig. 2). During each time point, YFRWs were trapped for four nights using soft-sided, steel framed traps, which were baited with a mixture of kangaroo pellets (Lauke Mills), carrot, apple, and sweet potato. Traps were cleared by 0800 hours each trapping morning and animals processed as per the objectives of the trapping program.
Scats were collected from 31 trapped YFRWs (Italowie Gap n = 15, Bimba Hill n = 16) across the trapping period by extracting fresh scats from the bottom of the trap. A summary of sample acquisition during each trapping period is provided in Table 2. Individual scats were used for microbiome and dietary assessment due to different storage requirements. Scat samples for microbiome analysis (n = 42) were opened, and an internal portion of the scat collected using a sterile stainless-steel scoop, and stored in 90–100% ethanol in a 1.5 mL Eppendorf tube, to fix microbial community composition. For dietary analysis, scats (n = 24) were stored in individual plastic snap-lock bags. Both types of samples were kept frozen (−4°C to −20°C) until laboratory processing. Only fresh scats (less than 3 h after deposition) were collected to minimise microbial die-off after deposition, and were collected by inverting a snap-lock bag over the collector’s hand to avoid human contamination. As presented, matched faecal microbiome and diet samples were collected from trapped individuals, but this was not possible for all animals captured.
| Italowie Gap | Bimba Hill | ||||
|---|---|---|---|---|---|
| Microbiome | Diet | Microbiome | Diet | ||
| Autumn 2021 | 6 | 5 | 0 | 0 | |
| Spring 2021 | 7 | 6 | 9 | 4 | |
| Autumn 2022 | 6 | 6 | 8 | 4 | |
The number of scats that were acquired for faecal microbiome and dietary analyses from each study location (Italowie Gap or Bimba Hill) during three trapping events (autumn 2021, spring 2021, and autumn 2022).
Laboratory processing
Faecal microbiome samples (n = 42) were processed in a pre-PCR microbiology laboratory in 5% sodium hypochlorite cleaned and UV irradiated hoods to avoid contamination of samples. Faecal samples were processed using the QIAGEN DNEasy PowerSoil kit following the manufacturer’s protocol. Extracted samples were then kept frozen at −20°C until further processing.
Extracted microbial samples were amplified by PCR targeting the 16S ribosomal RNA gene V4 region (Caporaso et al. 2011). The primers used were the forward primer 515F (AATGATACGG CGACCACCGAGATCTACACTATGGTAATTGTG TGCCAGCMGCCGCGGTAA) and barcoded reverse primer 806R (CAAGCAGAAGACGGCAT-ACGA GATnnnnnnnnnnnnAGTCAGTCAGCCGGAC TACHVGGGTW TCTAAT), where the n’s are unique to each barcode sequence. PCRs were set up using a master mix of 19.2 μL of dH2O, 2.5 μL 10× buffer, 1 μL MgSO4, 0.2 μL dNTP mix (100 mM), 0.5 μL of forward primer (10 μM), and 0.1 μL of Platinum™ Taq high fidelity DNA polymerase (Invitrogen™) for each sample. Finally, 1 μL of sample and 0.5 μL of a uniquely barcoded reverse primer (10 μM) were added to make up a final PCR volume of 25 μL. PCR conditions were an initial denaturing step of 94°C for 3 min, followed by 35 cycles of 94°C for 45 s, 50°C for 60 s and 68°C for 90 s, with a final adenylation step of 68°C for 10 min. PCR products were then assessed on a 3.5% agarose gel and visualised on a ChemiDoc imager (Bio-Rad Laboratories, Australia) for successful PCR amplification.
PCR products were then quantified using a Qubit 2.0 fluorometer (Thermo Fisher Scientific, Australia) at a 1:199 ratio of sample to Qubit working solution. Qubit results were then used to bulk samples in equimolar pools. DNA Extraction negative control samples were pooled separately because of their lower DNA concentration than positive samples. Each pool was cleaned using AxyPrep (Axygen) following the manufacturer’s protocol and visualised using the Agilent TapeStation 4150 to assess the quality and quantity of products. Pools were then bulked together in equimolar concentrations and reassessed on the TapeStation before being sent to the South Australian Genomics Centre (SAHMRI, South Australia) for sequencing on the Illumina MiSeq (v2) using a 2× 150 bp run.
Individual scat samples (n = 24) were prepared for DNA extraction using a Labconco FreeZone 4.5L −50°C freeze-drier (Labconco Corporation, USA) for 48–72 h. Samples were then pulverised using a mortar and pestle to form a powder. Subsets of the pulverised samples were then processed using the Isolate II Plant DNA kit (Meridian Bioscience, USA) following the standard protocol and lysis buffer PA1. The eluted DNA was then quantified using a Quantus Fluorometer (Promega, Australia) before being stored frozen at −20°C.
Genomic libraries compatible with Illumina sequencing were generated using the extracted DNA (n = 24) and the NEBNext® Ultra™ II FS DNA Library Prep Kit (New England Biolabs, Australia). In brief, DNA was fragmented enzymatically, end-repaired, a-tailed, and custom stubby Y-adaptors ligated to the size selected inserts. All preparation was performed using an Eppendorf epMotion 5075 (Eppendorf, USA) liquid handler. A detailed description of the methodology can be found in Waycott et al. (2021). Libraries were PCR amplified over 21 cycles and the resulting intermediate-stage library was then visualised on a 1.5% Agarose gel.
The library gel images were assessed visually for intensity to determine the relative fractions of each sample added into a pool. Pools were then purified with MagNA beads (Rohland and Reich 2012) at a 1:1 ratio and eluted in 35 μL of 1× TE.
Target capture was performed using the OzBaits_Cp bait set (Waycott et al. 2021) on the library pools following the manufacturer’s instructions (myBaits® targeted NGS capture; Manual version 4.01, Arbor Biosciences, USA), and hybridization conducted at 65°C for 24 h. The bait design targets approximately 50 regions of the chloroplast genome and can capture these regions for any vascular plant species (Waycott et al. 2021). After hybridisation and clean-up, the resulting enriched libraries were finalised (made compatible with Illumina sequencing flow cell and sequencing primers) by performing a PCR for 18 cycles with indexed P5 and P7 fusion primers (Waycott et al. 2021). Following this PCR stage, the products were purified with MagNA beads at a 1:1 ratio.
The finalised libraries were then analysed on an Agilent TapeStation 4150 (Agilent Technologies, USA) with high-sensitivity tapes to assess molarity and fragment size distribution (region setting set to 300–700 bp). All libraries were then pooled in equimolar concentration based on TapeStation results. The pooled library was then size selected on a Pippin Prep (Sage Science) using a 2% Agarose gel cassette with marker ‘L’ and range set to 300–600 bp. The final library was again visualised on the Agilent TapeStation 4150 to ensure the quality and molarity of the sequencing products.
The final pooled library was then sent to the Australian Genome Research Facility, Melbourne, for sequencing as 2× 150 bp reads across one lane of an Illumina NovaSeq X 10B flow cell.
Sequence preparation and informative analysis
Raw 16S rRNA gene sequencing data were processed and analysed using QIIME2 (Bolyen et al. 2019). Reads were imported into QIIME2 using the deblur plug-in (Amir et al. 2017) with a 150 bp trim length, and amplicon sequence variants (ASVs) were identified. The QIIME2 feature-classifier plug-in (naïve Bayesian approach) on the pretrained SILVA (Quast et al. 2013) 132 V4 region classifier was then used to assign ASVs to taxonomic units (Bokulich et al. 2018). Classified sequences were then imported into R (R Core Team 2023) and filtered to remove chloroplast DNA sequences (9.5% present in total sequences), low abundance ASVs (threshold = 0.05%), and ASVs present in only a single sample (37.2% ASVs removed). The filtered sequences were then analysed using the packages microbiome, phyloseq (McMurdie and Holmes 2013) and ggplot2 (Wickham 2016) to calculate and visualise Alpha diversity metrics, ASV richness and evenness (Pielou), and Beta diversity metrics (weighted and unweighted UniFrac distances: Lozupone and Knight 2005; Lozupone et al. 2007).
For statistical analysis, data were first examined to assess normality using a Shapiro–Wilk test (shapiro.test function). Once normality was established, Alpha diversity statistical differences were calculated using an ANOVA with the lme4 package (Bates et al. 2015). Statistical differences in Beta diversity metrics (microbial composition) were conducted using an Adonis test with the vegan (Oksanen et al. 2022) package.
Sequencing of samples returned paired-end sequence reads that were demultiplexed using Axe (Murray and Borevitz 2018) by recognition of the internal barcodes imbedded in the ligated stubby Y-adaptors, and was followed by quality and adaptor trimming using FASTP (Chen 2023). Trimmed paired-end reads were imported into CLC Genomics Workbench v. 20.0.2 (Qiagen) and de novo assembled (settings: length and similarity fraction = 0.9, minimum contig length = 300). The contigs generated from the de novo assemblies were then imported into Geneious v. 2022.0.12 (https://www.geneious.com). Each contig was then compared with a plant DNA reference database, which includes sequences of chloroplast gene regions from native and introduced flora found across South Australia (https://data.environment.sa.gov.au/Content/Publications/vascular-plants-bdbsa-taxonomy.xlsx), with all flora represented to the generic level. To identify matches within the database, BLASTn was used with an expect value of 1 × 10−20, and the top 10 hits for each matching contig were saved. In Geneious, the sequence classifier tool, ‘Classify Sequences’ (http://assets.geneious.com/documentation/geneious/Sequence_Classifier_Manual.pdf), was then used to compare the contigs with positive hits within the database of pooled BLASTn outputs obtained from the previous step. The Classify Sequences tool matches the query and database sequences using pairwise alignments and reports the identity for the proportion of overlap between the two sequences. The Classify Sequences tool uses a GenBank taxonomy string (https://www.ncbi.nlm.nih.gov/taxonomy/) attached to each database sequence to decide the appropriate levels of classification based upon user defined thresholds. Here, we used a 95% overlap identity threshold to record a match and a ≥98% identity overlap to classify a query sequence to genus. If there was a single match above this threshold, it was identified to species but for multiple near equal matches at ≥98% identity, the sequence was classified using the lowest common ancestor from the taxonomic string (e.g. two or more congeneric species with the same identity would be classified at the genus level, and species from different genera in the same family would be classified to a family level).
All plant species detected were then cross-referenced with the Australasian Virtual Herbarium (AVH; https://avh.chah.org.au) and the Atlas of Living Australia (ALA; https://www.ala.org.au) to assess the accuracy of sequencing results and determine their presence at either site. Where sequences were classified to a species level but were determined as absent from the site, and when multiple species from the same genus were present, they were each reclassified for ease of visualisation (represented as ‘Genus sp.’).
The proportion of sequencing reads per sample was determined by calculating the proportion of each plant species in relation to the total number of sequencing reads:
For ease of visualisation, species that had an abundance of <5% were grouped into their corresponding plant type, indicated in graphics as ‘other plant type’. A full list of each plant species which was detected for each individual YFRW is included as Supplementary material.
Results
Faecal microbiome
After removing ASVs that were below the threshold levels set to remove contaminants from samples (see Data availability for analysis code), the most commonly detected microbes across all YFRW faecal samples were from the phyla Bacteroidota and Firmicutes which constituted over 80% of the total ASVs detected in all samples (Fig. 3: Bacteriodota – browns; Firmicutes – greens). The phyla Verrucomicrobiota and Proteobacteria were also commonly detected but in much lower proportions (Fig. 3). Changes associated with microbial composition were evident when examining the relative abundance of microbes at Bimba Hill, where Bacteroidota slightly decreased and Firmicutes increased between spring 2021 and autumn 2022, but appeared stable at Italowie Gap throughout (Fig. 3).

Site and seasonal differences
Across all three sampling times, a total of 1065 ASVs were detected for Italowie Gap and 816 ASVs for Bimba Hill. Of these, 659 ASVs (54%) were shared between both sites, where Italowie Gap had 406 distinct ASVs and Bimba Hill had 157 distinct ASVs.
Location had a significant effect on the Alpha diversity metrics of microbial richness (ANOVA; P = 9.403 × 10−7), and microbial evenness (ANOVA; P = 0.01), where microbial richness was significantly higher in YFRW faecal samples collected from Italowie Gap compared to Bimba Hill (Fig. 4). Microbial composition was also statistically different depending on location when using Unweighted UniFrac (Adonis: P = 0.0001) and Weighted UniFrac (Adonis: P = 0.001) distances.
The faecal microbial Alpha diversity, ASV richness (top) and evenness (bottom), for yellow-footed rock-wallabies (Petrogale xanthopus xanthopus) at Italowie Gap (blue) and Bimba Hill (orange), South Australia, across three sampling times: autumn 2021, spring 2021, and autumn 2022.
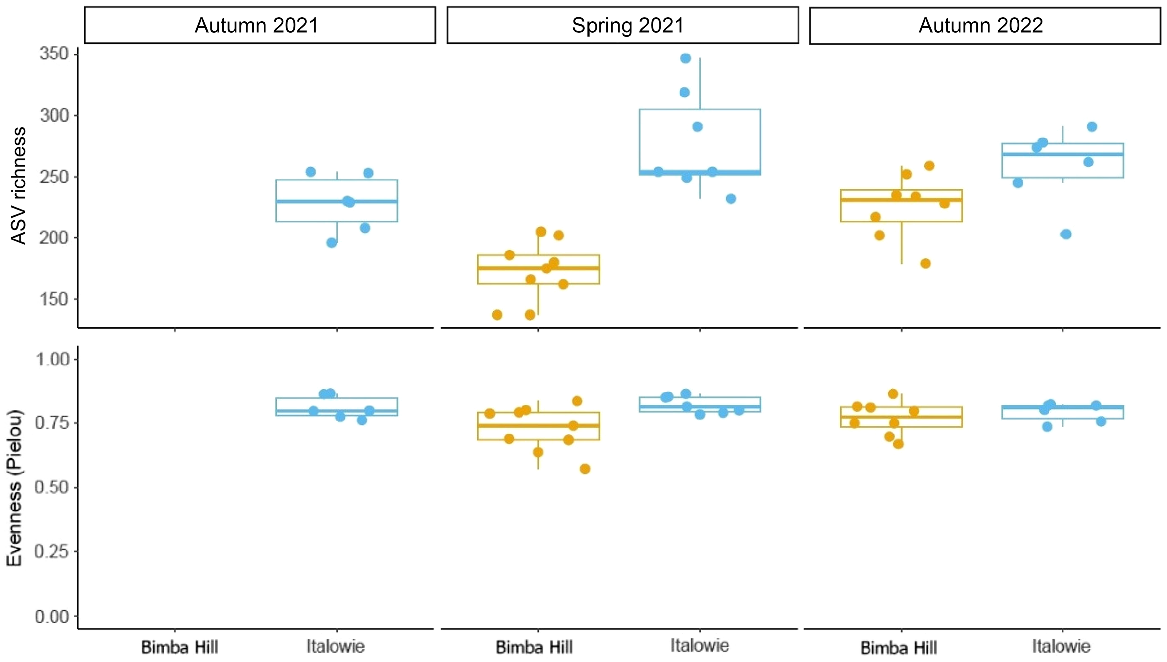
Season had a significant effect on microbial richness (ANOVA: P = 0.002) and also significantly affected microbial composition (Adonis: P = 0.0002) when using Unweighted UniFrac distances. Interestingly, the interaction between site and season also significantly affected microbial richness (ANOVA: P = 0.008).
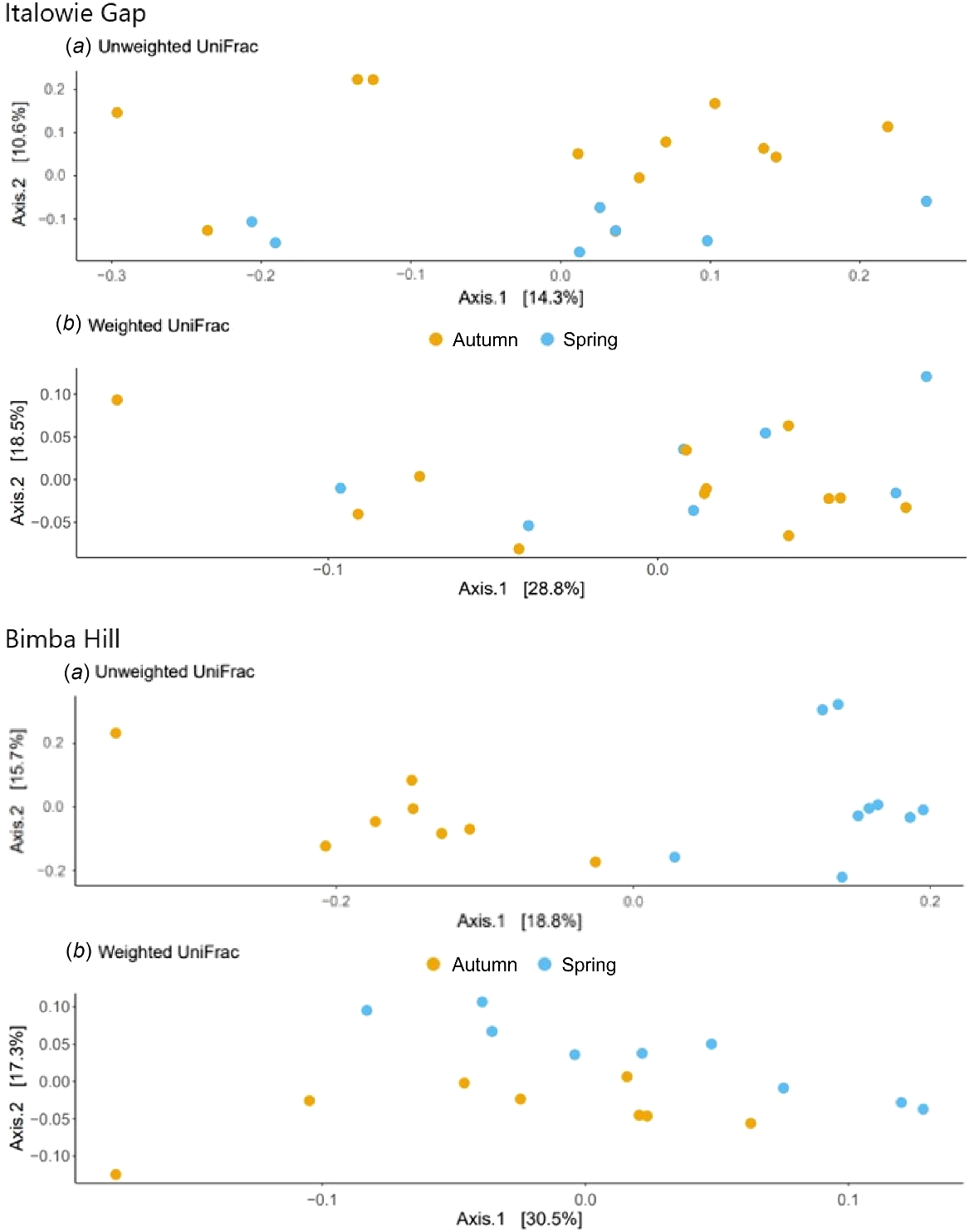
The differences in faecal microbial composition between sampling times was examined using a principal coordinates analysis (PCoA). Microbial composition estimated using Unweighted UniFrac for Italowie Gap accounted for approximately 14% of variance but did not differentiate between samples from spring 2021 and autumn 2022 (Fig. 5). In contrast the PCoA at Bimba Hill accounted for approximately 19% of variance and differentiated, although sparsely, in microbial composition between seasons (Fig. 5). The low levels of variation explained by these values indicates that other factors are contributing to changes in microbial composition. However, there does appear to be an effect of rainfall at Bimba Hill where microbial richness (Fig. 4) and composition (Fig. 5) have altered following high rainfall in autumn 2022 and a subsequent likely increase in vegetation abundance (Table 1; Fig. 2).
The Beta diversity of yellow-footed rock-wallaby (Petrogale xanthopus xanthopus) faecal microbes from two different sites, Italowie Gap (top) and Bimba Hill (bottom), sampled during spring 2021 (blue) and autumn 2022 (orange) using Unweighted UniFrac (top) and Weighted UniFrac (bottom) distances.
Intraindividual variation
Of all the individuals from which faecal microbial samples were collected in this study, nine were sampled across multiple periods (i.e. recaptured animals) (Italowie Gap n = 4; Bimba Hill n = 5), allowing for variations within individuals to be examined (Table 3). Intraindividual microbial composition at Bimba Hill appeared to be variable and included more variation than that observed in the Italowie Gap samples, where microbial composition was comparatively more stable between sampling periods (see Supplementary material, Fig. S1). Individuals from Bimba Hill showed a slight trend towards having a lower relative abundance of Bacteroidota and higher abundance of Firmicutes species during spring 2021 compared to autumn 2022 (see Supplementary material), as was also observed when examining the population as a whole (Fig. 3).
| Individual | Site | Autumn 2021 | Spring 2021 | Autumn 2022 | |
|---|---|---|---|---|---|
| Cecilia | Italowie | ✓ | ✓ | ||
| Eileen | Italowie | ✓ | ✓ | ✓ | |
| Pandora | Bimba | ✓ | ✓ | ||
| Parker | Bimba | ✓ | ✓ | ||
| Pickle | Bimba | ✓ | ✓ | ||
| Polly | Bimba | ✓ | ✓ | ||
| Pumpkin | Bimba | ✓ | ✓ | ||
| Ringo | Italowie | ✓ | ✓ | ||
| Zara | Italowie | ✓ | ✓ |
The trapping times where individual yellow-footed rock-wallabies (Petrogale xanthopus xanthopus) were resampled from multiple times for microbiome analyses. Times when microbiome samples were acquired are indicated with a tick.
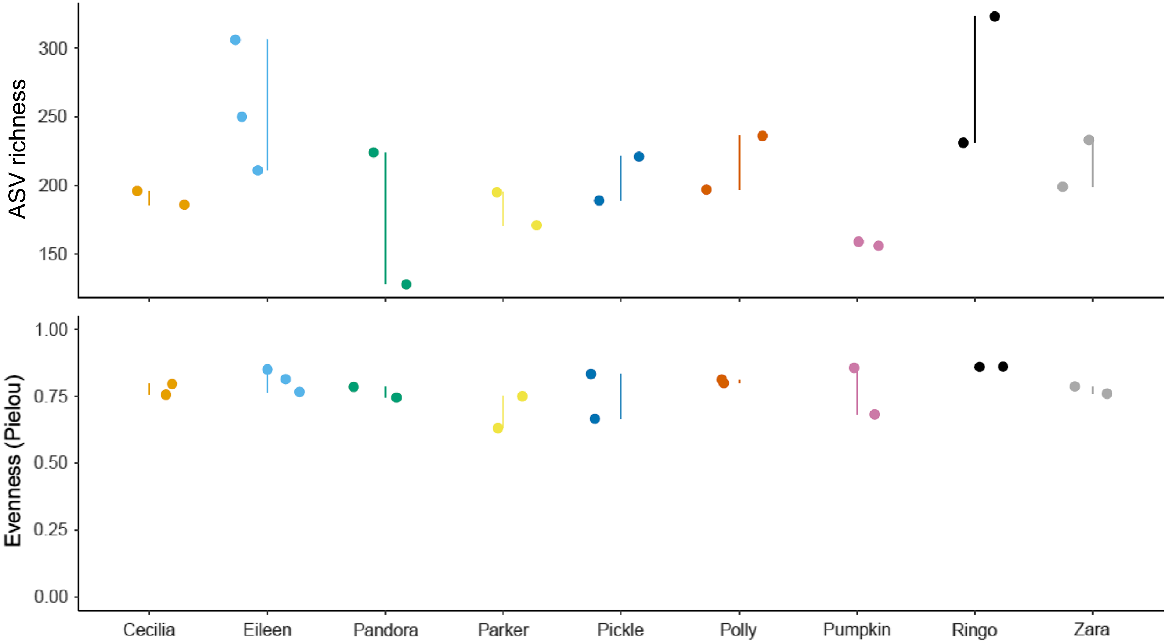
The only individual that showed a major variation in faecal microbial relative abundance was ‘Pumpkin’ at Bimba Hill (see Supplementary material), for which there was a significant decrease in Bacteroidota species during the second sampling (autumn 2022) compared to the first sampling (spring 2021). Due to the lack of change in Pumpkin’s microbial richness (Fig. 6), this suggests that microbial composition was different between sampling periods, with ‘Firmicutes – Other’ becoming more abundant in autumn 2022 (Figs 6, 7).
The differences in faecal microbial Alpha diversity within individual yellow-footed rock-wallabies (Petrogale xanthopus xanthopus) sampled across autumn 2021, spring 2021, and autumn 2022, in relation to microbial ASV richness (top) and evenness (bottom). Matching coloured points represent data from the same individual with the variation between sampling of that individual indicated with a vertical line.
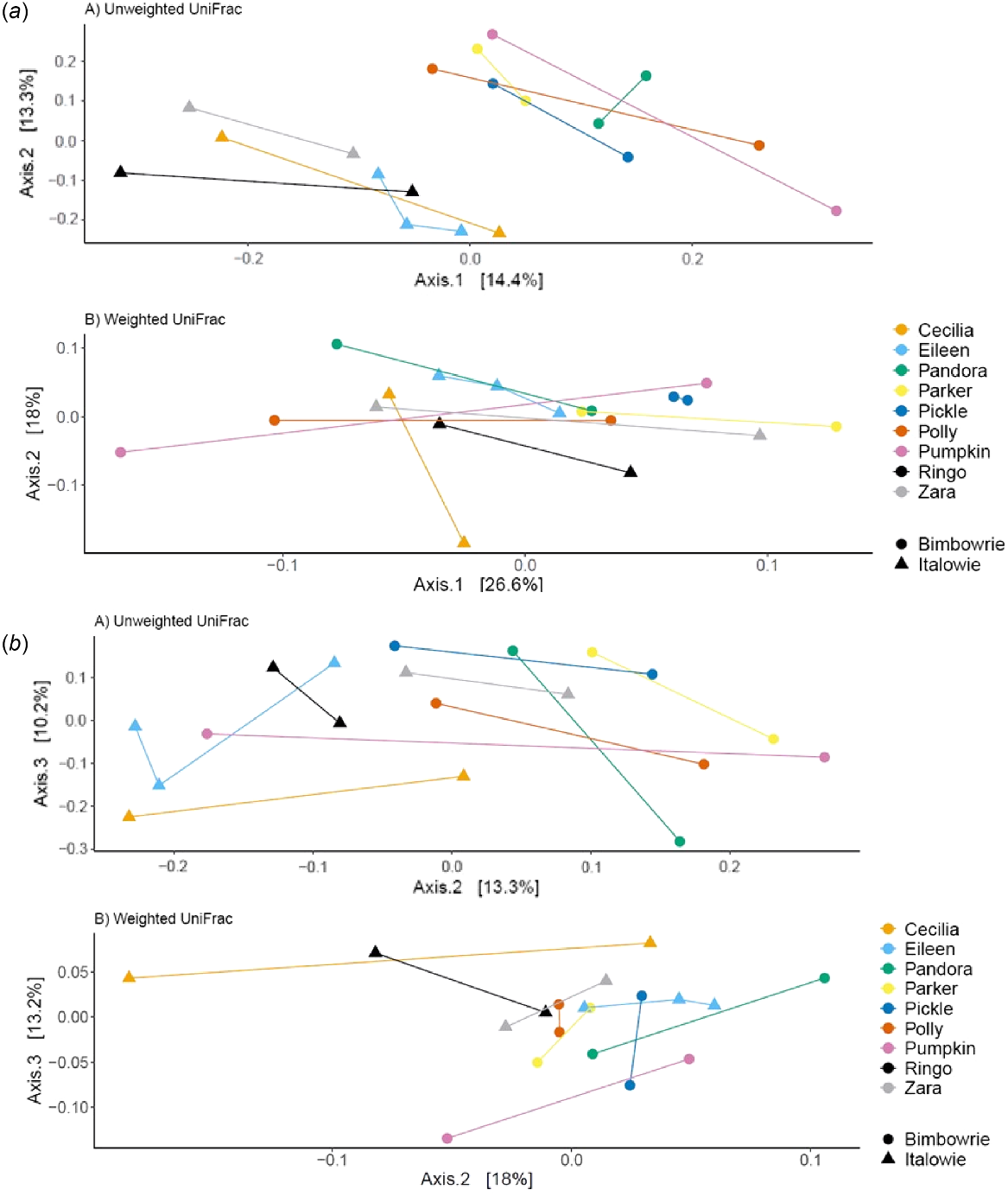
The differences in faecal microbial composition of yellow-footed rock-wallabies (Petrogale xanthopus xanthopus) that were sampled during autumn 2021, spring 2021, and autumn 2022 from Italowie Gap (Δ) or Bimba Hill (○) using Unweighted UniFrac (top) and Weighted UniFrac (bottom) distances. (a) Axis 1 versus 2, (b) Axis 2 versus 3. Each colour represents an individual, with differences in microbial composition between sampling indicated with a line.
Variation in microbial richness was also observed for other individuals across both study sites although they were not evident when examining relative abundance (Fig. 6; see Supplementary material). The three individuals that showed high variation in ASV richness throughout the sampling periods were ‘Eileen’ (Italowie Gap), ‘Pandora’ (Bimba Hill) and ‘Ringo’ (Italowie Gap) (Fig. 6).
Samples were clearly grouped by location when using Unweighted UniFrac distances along Axis 1 and Axis 2 (Fig. 7). However, along Axis 2 and Axis 3 samples were grouped by individual variation (Fig. 7). This further indicates that variation in microbial composition is driven by location.
No significant differences in microbial composition or diversity were detected between the sexes (see Supplementary material).
Dietary influences
Matched faecal microbiome and diet samples were collected from a total of 14 individuals at Italowie Gap; however, faecal microbiome sequencing of one individual (‘Ringo’) did not return any results, reducing the number of matched samples to 13. One individual (‘Eileen’) had matched samples collected on three occasions, and another individual (‘Zara’) had matched samples collected twice.
The average number of plant species detected across all individuals at each time point was nine in autumn 2021, eight in spring 2021, and nine in autumn 2022. Although the diet of individuals varied across the sampling period, Acacia species and Portulaca oleracea were detected regularly (Fig. 8). In general, Acacia species were detected at higher proportions during spring 2021, whereas P. oleracea typically occurred at higher proportions during autumn sampling periods (Fig. 8).
Top: the proportion (%) of genetic sequencing reads from plant species that were detected in yellow-footed rock-wallaby (Petrogale xanthopus xanthopus) scats from Italowie Gap during autumn 2021, spring 2021 and autumn 2022. Bottom: The relative abundance (%) of faecal microbes detected in scats from the same yellow-footed rock-wallabies (Petrogale xanthopus xanthopus), collected from the same location and sampling periods as above.
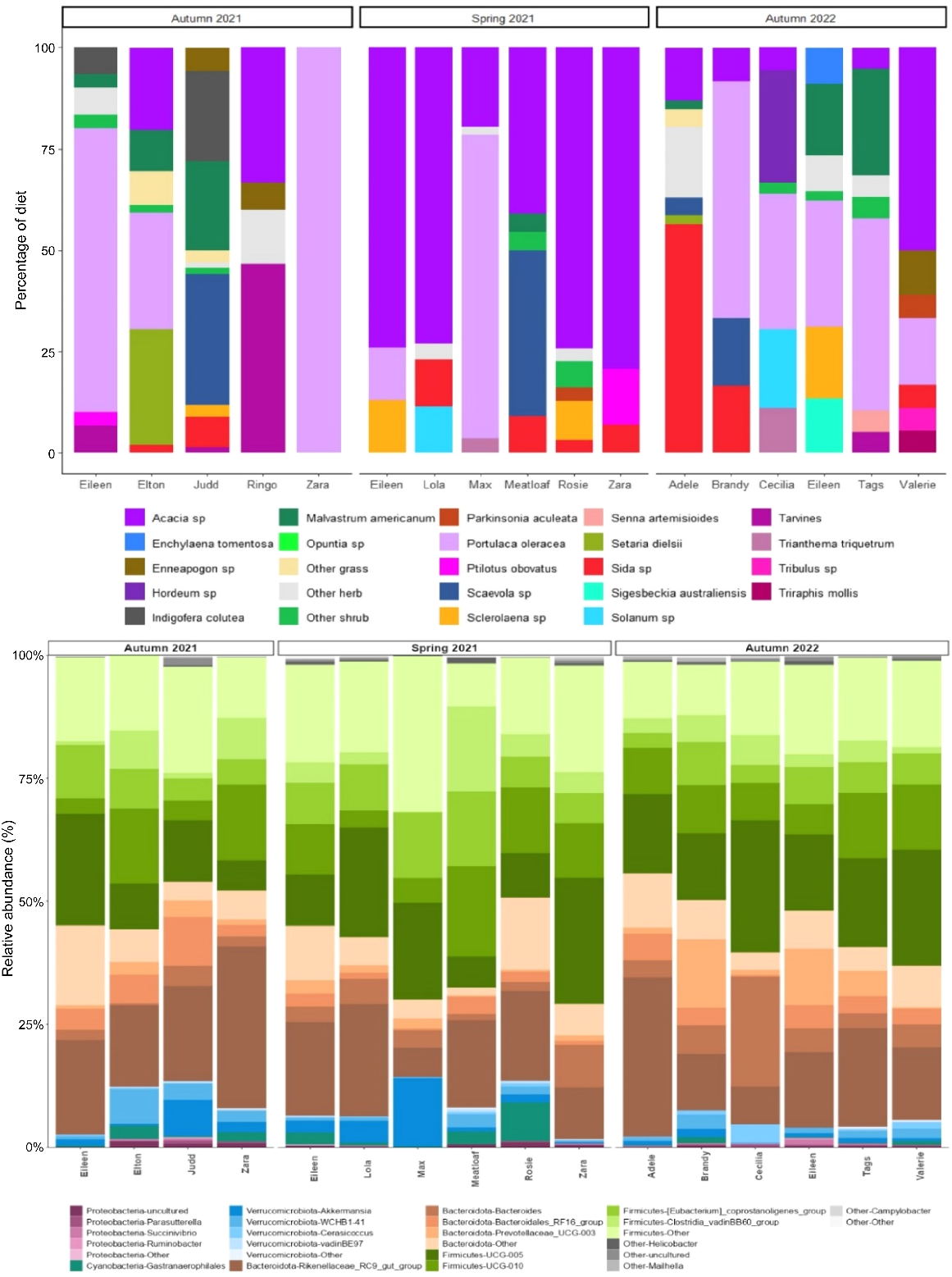
In the paired microbiome samples, the main phyla of microbiota detected were Bacteroidota and Firmicutes, with a small contribution from Verrucomicrobiota. During both autumn sampling periods (2021 and 2022), the proportion of Firmicutes seemed to decrease slightly in comparison to spring (2021) but this decline was not significant. This appeared to be related to increased rates of detection of Acacia species, suggesting there may be a relationship between increased Acacia intake and increased Firmicutes presence (Fig. 8), although the sample size is not great enough to support this observable trend statistically.
Matched faecal microbiome and diet samples (n = 8) were acquired from seven individuals at Bimba Hill, with one individual, ‘Pumpkin’, sampled during two periods.
During spring 2021, there was an average of nine plant species detected in scat samples in four samples, compared to seven plant species detected from another four samples in autumn 2022. This suggests that there was an increase in dietary diversity during spring 2021.
The detection of individual plant species from YFRWs at Bimba Hill was highly variable during spring 2021, but Calandrinia eremaea and Sclerolaena species were common. However, during autumn 2022 sequencing reads were dominated primarily by Acacia and Dysphania species (Fig. 9).
Top: the proportion (%) of genetic sequencing reads from plant species that were detected in yellow-footed rock-wallaby (Petrogale xanthopus xanthopus) scats from Bimba Hill during spring 2021 and autumn 2022. Bottom: The relative abundance (%) of faecal microbes detected in scats from the same yellow-footed rock-wallabies (Petrogale xanthopus xanthopus), collected from the same location and sampling periods as above.
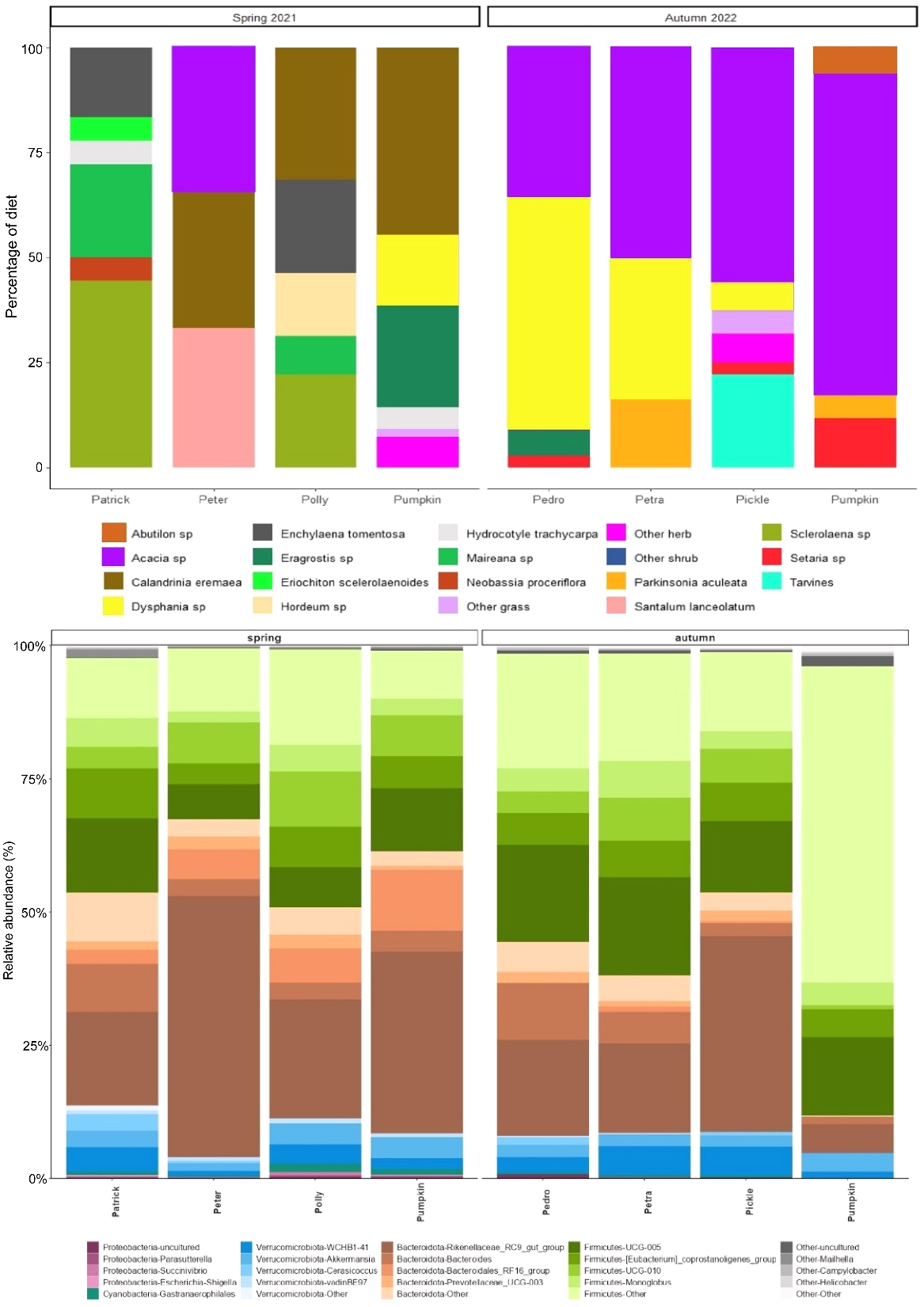
In general, the microbial composition of scats at Bimba Hill was similar to that observed at Italowie Gap where Bacteroidota and Firmicutes were the dominant phyla throughout sampling. However, the increased plant variability in diet samples during spring 2021 appeared to correspond to a higher abundance of Bacteroidota taxa, whereas the increase of Acacia species in autumn 2022 diet samples, appeared to coincide with an increase in Firmicutes microbes (Fig. 9).
Discussion
The importance of conserving the gut or faecal microbiome in wildlife has only recently been recognised (Banerjee et al. 2020), with few studies previously undertaken examining marsupials (Chhour et al. 2010; Cheng et al. 2015; Brice et al. 2019; Eisenhofer et al. 2021; Blyton et al. 2023; Eisenhofer et al. 2023). The faecal microbiome of the YFRW has not been characterised previously. Here we examined the faecal microbiome of this species across two geographically distinct populations, each with different habitat characteristics (Italowie Gap = connected, Bimba Hill = isolated). Due to their geographic separation, we also aimed to assess individual variation in microbial diversity and composition in response to season/rainfall, and how dietary composition might influence these parameters.
The dominant microbial phyla detected in all scat samples were Firmicutes and Bacteroidota, which is similar to that previously observed in other mammalian studies (Ley et al. 2008; Gulino et al. 2013; Brice et al. 2019; Eisenhofer et al. 2021; Awosile et al. 2023). The proportions of Firmicutes and Bacteroidota observed here for YFRW samples are similar to those reported in another microbiome study on three macropodid species (Macropus giganteus, Osphranter robustus, Osphranter rufus) (Gulino et al. 2013). In the SHNW, another semiarid-dwelling marsupial, the faecal microbiome consistently comprised >70% Firmicutes and Bacteroidota species, but also included microbial species absent in YFRW samples (Eisenhofer et al. 2021). SHNWs, however, utilise hindgut fermentation to access SCFAs, potentially explaining these differences. Another microbial phylum that was detected at low levels was Verrucomicrobiota. Although the role of Verrucomicrobiota is not well understood in the gut microbiome, it is commonly associated with soil (Zhang and Xu 2008; Bergmann et al. 2011), indicating this result may be a result of soil contact after deposition.
This study found that the greatest driver of microbiome diversity was location, where microbial richness was higher at Italowie Gap than at Bimba Hill, and microbial composition was also significantly different between sites. Habitat quality and type have previously been described as drivers of faecal microbial diversity in other species, including the black howler monkey (Alouatta pigra) (Amato et al. 2013) and African elephants (Loxodonta species) (Budd et al. 2020), and is often associated with differences in dietary intake. The Italowie Gap colony is situated in highly connected habitat, compared to the Bimba Hill colony which is in isolated outcrop habitat. Therefore, habitat connectivity may be acting as a proxy for habitat quality, facilitating the greater microbial richness at Italowie Gap, and the difference in microbial composition observed between sites.
Dietary differences between our study sites are likely driven by geographical variation in climate, rainfall, and soil type, altering vegetation community structure, which are likely influencing the location-based variation in faecal microbiome observed in this study. In a study conducted on koalas (Phascolarctos cinereus) (Brice et al. 2019), variation in faecal microbial composition was observed to be highly correlated with diet. While such a statistical correlation was not possible in the current study, and further research with a greater sample size is needed, a general trend was observed, where an increase in Firmicutes species occurred when Acacia species were detected in high proportions in diet samples. Acacia species, which are within the Family Leguminosae, utilise symbiotic bacteria within the root system to fix soil nitrogen, contributing to elevated protein accumulation within the plant tissue (Irlbeck and Hume 2003; Morton 2022). However, high levels of tannins are also present in the foliage, which blocks microbial fermentation of plant protein, restricting the availability of nutrients (Gartner and Hurwood 1976; McSweeney et al. 2008). For this reason, Acacia species are not a food source preferred by many macropodids, and typically is only eaten during drought when resource availability is low, where dead foliage with low tannin content is ingested (Irlbeck and Hume 2003). This is also reflected in this study as Acacia species were ingested in higher proportions during dry environmental conditions (refer to Fig. 1). As it has been previously found that a high carbohydrate or protein diet can increase the proportion of Firmicutes species present in the faecal microbiome (De Filippo et al. 2010; Zhang et al. 2020), the increased proportion of Firmicutes in this study observed at both sites during dry conditions may be attributed to the higher proportion of Acacia detected in diet samples.
One individual (‘Pumpkin’) from Bimba Hill had a markedly higher proportion of Firmicutes present during sampling in autumn 2022, compared to sampling in spring 2021. This also corresponded to a major dietary shift where in spring 2021 a variety of herbaceous and grass species (i.e. Calandrinia eremaea, Dysphania species, Eragrostis species) were detected in the sample, but during autumn 2022 Acacia species were the dominant plants detected. However, as this trend was not observed in all individuals that were sampled from multiple times, intraindividual variations may be impacting the results observed in ‘Pumpkin’ and would require further, more extensive sampling to support this conclusion.
‘Pumpkin’ was an adult female YFRW in above average body condition and had a young in pouch during both sampling times. No obvious pathologies or underlying health conditions were observed during either sampling time. Although a decrease in body weight, waist circumference, and body mass index has been correlated with an increase in Bacteroidetes in humans (Fava et al. 2013), ’Pumpkin’ experienced no notable weight change between samplings (<200 g difference). As microbial richness for this individual did not change markedly, the shift in microbial composition indicates that there was a change in the dominant microbes present between sampling times. This was further supported by the high detection rate of Acacia species in diet samples at Italowie Gap in spring 2021 also showing a slight increase in Firmicutes during this sampling period. This suggests that geographical variations in rainfall and the impact of habitat composition on vegetation structure are more than likely influencing the dietary composition of YFRWs, which also appears to be driving variation in faecal microbial composition.
An additional potential cause of this microbial variation may be neutral allopatric speciation. This process reduces the ability of microbes to be shared amongs populations if there is geographic separation due to random genetic mutations of the microbial genome and cospeciation with their host occurs (Groussin et al. 2020). The two populations in this study are separated by a large geographic distance (~180 km), and YFRWs are not known to migrate such large distances, with most migration events occurring over smaller distances (<13 km: Potter et al. 2020; Smith et al. 2023). As only approximately 54% of microbial ASVs are shared between populations, neutral allopatric speciation may well be influencing the microbial communities at these sites. However, to confirm this, further testing would be required regarding the unique microbial ASVs which were previously filtered from analysis, and would be better informed by sampling from additional YFRW populations.
If it were found that neutral allopatric speciation is occurring, may be particularly pertinent for future translocation programs conducted between regions of YFRW habitation, or if the historic link between the Flinders and Olary Ranges populations were ever to be re-established. Now the YFRWs faecal microbiome has begun to be characterised, there is potential for faecal transplants to be developed for use as a conservation tool for this species in broad-scale translocation or reintroduction programs, but the efficacy of faecal transplants and the spatial scale of microbial variation would need to be tested more thoroughly before implementation.
Conclusions
The results of this study suggest that the YFRW faecal microbiome is highly influenced by their geographical location. YFRWs from Italowie Gap had higher faecal microbial diversity than was observed at Bimba Hill, and microbial composition was markedly different between sites throughout. As only 54% of ASV were shared between sites, this suggests that microbial diversity and composition are mostly site-specific, likely influenced by geographical separation and local rainfall. This information may be an important consideration for conservation efforts, as there are underlying unique biological characteristics occurring within the species throughout its distribution.
Although diet appeared to have an effect on the YFRW faecal microbiome, a more in-depth investigation, including a higher sample size allowing for statistical analysis, would be needed to establish the correlation between faecal microbiome and diet. A trend between Acacia species and increased Firmicutes was observed (Italowie Gap in spring 2021, Bimba Hill in autumn 2022), and may be driven as a response to having a ‘protein-rich’ diet. This is worth further investigation, particularly if YFRWs are more reliant on Acacia species during dry/drought times. Additionally, how changes in microbial diversity and composition can affect YFRW health are unknown, and would require further monitoring and research.
This is particularly pertinent when, due to climate change, the frequency, severity and duration of droughts are predicted to increase, potentially altering, and decreasing, plant diversity and abundance, and thus altering the YFRW diet. On-going monitoring of YFRW faecal microbiome, diet, and health parameters will assist in informing the future conservation management strategies for this species.
Data availability
Raw microbial DNA sequencing data are publicly available at NCBI as bio-project PRJNA1110055. Raw plant DNA sequencing data are available on request to the corresponding author. Code for reproducing the microbiome analyses is available at https://github.com/EisenRa/2024_YFRW_faecal.
Declaration of funding
The authors acknowledge the generous funding provided for this project from the South Australia Arid Lands Landscape Board, Holsworth Wildlife Research Endowment and the Schultz Foundation.
Acknowledgements
We acknowledge the National Park and Wildlife Service rangers of the parks in which this study was conducted, Sian Johnson and Peter Watkins, for their commitment of knowledge and time to this project.
References
Adams-Hosking C, McAlpine C, Rhodes JR, Grantham HS, Moss PT (2012) Modelling changes in the distribution of the critical food resources of a specialist folivore in response to climate change. Diversity & Distributions 18, 847-860.
| Crossref | Google Scholar |
Amato KR, Yeoman CJ, Kent A, Righini N, Carbonero F, Estrada A, Gaskins HR, Stumpf RM, Yildirim S, Torralba M, Gillis M, Wilson BA, Nelson KE, White BA, Leigh SR (2013) Habitat degradation impacts black howler monkey (Alouatta pigra) gastrointestinal microbiomes. The ISME Journal 7, 1344-1353.
| Crossref | Google Scholar | PubMed |
Amato KR, Arrieta M-C, Azad MB, Bailey MT, Broussard JL, Bruggeling CE, Claud EC, Costello EK, Davenport ER, Dutilh BE, Swain Ewald HA, Ewald P, Hanlon EC, Julion W, Keshavarzian A, Maurice CF, Miller GE, Preidis GA, Segurel L, Singer B, Subramanian S, Zhao L, Kuzawa CW (2021) The human gut microbiome and health inequities. Proceedings of the National Academy of Sciences - PNAS 118, e2017947118.
| Crossref | Google Scholar |
Amir A, McDonald D, Navas-Molina JA, Kopylova E, Morton JT, Zech Xu Z, Kightley EP, Thompson LR, Hyde ER, Gonzalez A, Knight R (2017) Deblur rapidly resolves single-nucleotide community sequence patterns. mSystems 2, e00191-16.
| Crossref | Google Scholar | PubMed |
Arumugam R, Ravichandran P, Yeap SK, Sharma RSK, Zulkifly SB, Yawah D, Annavi G (2023) Application of high-throughput sequencing (HTS) to enhance the well-being of an endangered species (Malayan tapir): characterization of gut microbiome using MG-RAST. Metagenomic Data Analysis 2649, 175-194.
| Crossref | Google Scholar |
Awosile B, Crasto C, Rahman MK, Daniel I, Boggan S, Steuer A, Fritzler J (2023) Fecal microbial diversity of coyotes and wild hogs in Texas Panhandle, USA. Microorganisms 11, 1137.
| Crossref | Google Scholar |
Banerjee A, Cornejo J, Bandopadhyay R (2020) Emergent climate change impact throughout the world: call for “Microbiome Conservation” before it’s too late. Biodiversity and Conservation 29, 345-348.
| Crossref | Google Scholar |
Bates D, Mäechler M, Bolker B, Walker S (2015) Fitting linear mixed-effects models using lme4. Journal of Statistical Software 67, 1-48.
| Crossref | Google Scholar |
Bergmann GT, Bates ST, Eilers KG, Lauber CL, Caporaso JG, Walters WA, Knight R, Fierer N (2011) The under-recognized dominance of Verrucomicrobia in soil bacterial communities. Soil Biology & Biochemistry 43, 1450-1455.
| Crossref | Google Scholar | PubMed |
Blyton MDJ, Pascoe J, Hynes E, Soo RM, Hugenholtz P, Moore BD (2023) The koala gut microbiome is largely unaffected by host translocation but rather influences host diet. Frontiers in Microbiology 14, e1085090.
| Crossref | Google Scholar |
Bokulich NA, Kaehler BD, Rideout JR, Dillon M, Bolyen E, Knight R, Huttley GA, Gregory Caporaso J (2018) Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6, e90.
| Crossref | Google Scholar |
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, Bai Y, Bittinger K, Brejnrod A, Brislawn CJ, Brown CT, Callahan BJ, Caraballo-Rodríguez AM, Chase J, Cope EK, Silva RD, Diener C, Dorrestein PC, Douglas GM, Durall DM, Duvallet C, Edwardson CF, Estaki M, Fouquier J, Gauglitz JM, Gibbons SM, Gibson DL, Gonzalez A, Gorlick K, Guo J, Hillmann B, Holmes S, Holste H, Huttenhower C, Huttley GA, Janssen S, Jarmusch AK, Jiang L, Kaehler BD, Kang KB, Keefe CR, Keim P, Kelley ST, Knights D, Koester I, Kosciolek T, Kreps J, Langille MGI, Lee J, Ley R, Liu Y-X, Loftfield E, Lozupone C, Maher M, Marotz C, Martin BD, McDonald D, McIver LJ, Melnik AV, Metcalf JL, Morgan SC, Morton JT, Naimey AT, Navas-Molina JA, Nothias LF, Orchanian SB, Pearson T, Peoples SL, Petras D, Preuss ML, Pruesse E, Rasmussen LB, Rivers A, Robeson MS, II, Rosenthal P, Segata N, Shaffer M, Shiffer A, Sinha R, Song SJ, Spear JR, Swafford AD, Thompson LR, Torres PJ, Trinh P, Tripathi A, Turnbaugh PJ, Ul-Hasan S, van der Hooft JJJ, Vargas F, Vázquez-Baeza Y, Vogtmann E, von Hippel M, Walters W, Wan Y, Wang M, Warren J, Weber KC, Williamson CHD, Willis AD, Xu ZZ, Zaneveld JR, Zhang Y, Zhu Q, Knight R, Caporaso JG (2019) Author Correction: Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nature Biotechnology 37, 1091.
| Crossref | Google Scholar | PubMed |
Brice KL, Trivedi P, Jeffries TC, Blyton MDJ, Mitchell C, Singh BK, Moore BD (2019) The koala (Phascolarctos cinereus) faecal microbiome differs with diet in a wild population. PeerJ 7, e6534.
| Crossref | Google Scholar |
Budd K, Gunn JC, Finch T, Klymus K, Sitati N, Eggert LS (2020) Effects of diet, habitat, and phylogeny on the fecal microbiome of wild African savanna (Loxodonta africana) and forest elephants (L. cyclotis). Ecology and Evolution 10, 5637-5650.
| Crossref | Google Scholar | PubMed |
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R (2011) Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proceedings of the National Academy of Sciences 108, 4516-4522.
| Crossref | Google Scholar |
Chen S (2023) Ultrafast one-pass FASTQ data preprocessing, quality control, and deduplication using fastp. iMeta 2, e107.
| Crossref | Google Scholar | PubMed |
Chen J, He X, Huang J (2014) Diet effects in gut microbiome and obesity. Journal of Food Science 79, 442-451.
| Crossref | Google Scholar |
Cheng Y, Fox S, Pemberton D, Hogg C, Papenfuss AT, Belov K (2015) The Tasmanian devil microbiome-implications for conservation and management. Microbiome 3, e76.
| Crossref | Google Scholar |
Chhour K-L, Hinds LA, Jacques NA, Deane EM (2010) An observational study of the microbiome of the maternal pouch and saliva of the tammar wallaby, Macropus eugenii, and of the gastrointestinal tract of the pouch young. Microbiology 156, 798-808.
| Crossref | Google Scholar |
Copley PB (1983) Studies on the yellow-footed rock-wallaby, Petrogale xanthopus Gray (Marsupialia: Macropodidae) I. Distribution in South Australia. Australian Wildlife Research 10, 47-61.
| Crossref | Google Scholar |
Correa F, Torti V, Spiezio C, Checcucci A, Modesto M, Borruso L, Cavani L, Mimmo T, Cesco S, Luise D, Randrianarison RM, Gamba M, Rarojoson NJ, Sanguinetti M, Di Vito M, Bugli F, Mattarelli P, Trevisi P, Giacoma C, Sandri C (2021) Disentangling the possible drivers of Indri indri Microbiome: a threatened lemur species of Madagascar. Frontiers in Microbiology 12, 668274.
| Crossref | Google Scholar |
Davis MB, Shaw RG, Etterson JR (2005) Evolutionary responses to changing climate. Ecology 86, 1704-1714.
| Crossref | Google Scholar |
De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, Collini S, Pieraccini G, Lionetti P, Hartl DL (2010) Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proceedings of the National Academy of Sciences 107, 14691-14696.
| Crossref | Google Scholar |
Donohue ME, Asangba AE, Ralainirina J, Weisrock DW, Stumpf RM, Wright PC (2019) Extensive variability in the gut microbiome of a highly-specialized and critically endangered lemur species across sites. American Journal of Primatology 81, e23046.
| Crossref | Google Scholar | PubMed |
Eisenhofer R, Helgen KM, Taggart D (2021) Signatures of landscape and captivity in the gut microbiota of southern hairy-nosed wombats (Lasiorhinus latifrons). Animal Microbiome 3, 4.
| Crossref | Google Scholar |
Eisenhofer R, Brice KL, Blyton MD, Bevins SE, Leigh K, Singh BK, Helgen KM, Hough I, Daniels CB, Speight N, Moore BD (2023) Individuality and stability of the koala (Phascolarctos cinereus) faecal microbiota through time. PeerJ 11, e14598.
| Crossref | Google Scholar |
Fava F, Gitau R, Griffin BA, Gibson GR, Tuohy KM, Lovegrove JA (2013) The type and quantity of dietary fat and carbohydrate alter faecal microbiome and short-chain fatty acid excretion in a metabolic syndrome ‘at-risk’ population. International Journal of Obesity 37, 216-223.
| Crossref | Google Scholar | PubMed |
Gartner RJW, Hurwood IS (1976) The tannin and oxalic acid content of Acacia aneura (mulga). Australian Veterinary Journal 52, 194-196.
| Crossref | Google Scholar | PubMed |
Gibson RK, Broome L, Hutchinson MF (2018) Susceptibility to climate change via effects on food resources: the feeding ecology of the endangered mountain pygmy-possum (Burramys parvus). Wildlife Research 45, 539-550.
| Crossref | Google Scholar |
Groussin M, Mazel F, Alm EJ (2020) Co-evolution and co-speciation of host-gut bacteria systems. Cell Host & Microbe 28, 12-22.
| Crossref | Google Scholar | PubMed |
Gulino L-M, Ouwerkerk D, Kang AYH, Maguire AJ, Kienzle M, Klieve AV (2013) Shedding light on the microbial community of the macropod foregut using 454-amplicon pyrosequencing. PLoS ONE 8, e61463.
| Crossref | Google Scholar | PubMed |
Höglund J, Laurila A, Rödin-Mörch P (2019) Population genomics and wildlife adaptation in the face of climate change. In ‘Population genomics: wildlife’. (Eds PA Hohenlohe, OP Rajora) pp. 333–355. (Springer International Publishing: USA) 10.1007/13836_2019_69
Huang Z, Liu K, Ma W, Li D, Mo T, Liu Q (2022) The gut microbiome in human health and disease – Where are we and where are we going? A bibliometric analysis. Frontiers in Microbiology 13, e1018594.
| Crossref | Google Scholar |
Irlbeck NA, Hume ID (2003) The role of acacia in the diets of Australian marsupials? A review. Australian Mammalogy 25, 121-134.
| Crossref | Google Scholar |
Kau AL, Ahern PP, Griffin NW, Goodman AL, Gordon JI (2011) Human nutrition, the gut microbiome and the immune system. Nature 474, 327-336.
| Crossref | Google Scholar | PubMed |
Klamt M, Thompson R, Davis J (2011) Early response of the platypus to climate warming. Global Change Biology 17, 3011-3018.
| Crossref | Google Scholar |
Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS, Schlegel ML, Tucker TA, Schrenzel MD, Knight R, Gordon JI (2008) Evolution of mammals and their gut microbes. Science 320, 1647-1651.
| Crossref | Google Scholar |
Lozupone C, Knight R (2005) UniFrac: a new phylogenetic method for comparing microbial communities. Applied and Environmental Microbiology 71, 8228-8235.
| Crossref | Google Scholar | PubMed |
Lozupone CA, Hamady M, Kelley ST, Knight R (2007) Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Applied and Environmental Microbiology 73, 1576-1585.
| Crossref | Google Scholar | PubMed |
Marshall VM, Taggart DA, Ostendorf B (2018) Scale-dependent habitat analysis and implications for climate change risk for the southern hairy-nosed wombat. Australian Mammalogy 40, 162-172.
| Crossref | Google Scholar |
McMurdie PJ, Holmes S (2013) phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8, e61217.
| Crossref | Google Scholar | PubMed |
McSweeney CS, Collins EMC, Blackall LL, Seawright AA (2008) A review of anti-nutritive factors limiting potential use of Acacia angustissima as a ruminant feed. Animal Feed Science and Technology 147, 158-171.
| Crossref | Google Scholar |
Murray KD, Borevitz JO (2018) Axe: rapid, competitive sequence read demultiplexing using a trie. Bioinformatics 34, 3924-3925.
| Crossref | Google Scholar | PubMed |
Oksanen J, Simpson G, Blanchet F, Kindt R, Legendre P, Minchin P, O’Hara R, Solymos P, Stevens MHM, Szoecs E, Wagner H, Barbour M, Bedward M, Bolker B, Borcard D, Carvalho G, Chirico M, De Caceres M, Durand S, Evangelista HBA, FitzJohn R, Friendly M, Furneaux B, Hannigan G, Hill MO, Lahti L, McGlinn D, Ouellette M, Ribeiro Cunha E, Smith T, Stier A, Ter Braak CJF, Weedon J, Borman T (2022) vegan: Community Ecology Package. R package version 2.6-4. Available at https://CRAN.R-project.org/package=vegan
Pannoni SB, Proffitt KM, Holben WE (2022) Non-invasive monitoring of multiple wildlife health factors by fecal microbiome analysis. Ecology and Evolution 12, e8564.
| Crossref | Google Scholar | PubMed |
Potter S, Neaves LE, Lethbridge M, Eldridge MDB (2020) Understand historical demographic processes to inform contemporary conservation of an arid zone specialist: the yellow-footed rock-wallaby. Genes 11, 154-178.
| Crossref | Google Scholar | PubMed |
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO (2013) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Research 41, D590-D596.
| Crossref | Google Scholar | PubMed |
Redford KH, Segre JA, Salafsky N, del Rio CM, McAloose D (2012) Conservation and the microbiome. Conservation Biology 26, 195-197.
| Crossref | Google Scholar | PubMed |
Ribas MP, García-Ulloa M, Espunyes J, Cabezón O (2023) Improving the assessment of ecosystem and wildlife health: microbiome as an early indicator. Current Opinion in Biotechnology 81, 102923.
| Crossref | Google Scholar | PubMed |
Rohland N, Reich D (2012) Cost-effective, high-throughput DNA sequencing libraries for multiplexed target capture. Genome Research 22, 939-946.
| Crossref | Google Scholar | PubMed |
Round JL, Mazmanian SK (2009) The gut microbiota shapes intestinal immune responses during health and disease. Nature Reviews Immunology 9, 313-323.
| Crossref | Google Scholar | PubMed |
Ruykys L (2017) Multi-scale habitat associations of the black-footed rock-wallaby in north-western South Australia. Wildlife Research 44, 207-218.
| Crossref | Google Scholar |
Short J (1982) Habitat requirements of the brush-tailed rock-wallaby, Petrogale penicillata, in New South Wales. Australian Wildlife Research 9, 239-246.
| Crossref | Google Scholar |
Smith DA, Lethbridge MR, Allen BL, Andrew RL (2023) Inferring inter-colony movement within metapopulations of yellow-footed rock-wallabies using estimates of kinship. Conservation Genetics 24, 265-278.
| Crossref | Google Scholar |
Sommer F, Bäckhed F (2013) The gut microbiota – masters of host development and physiology. Nature Reviews Microbiology 11, 227-238.
| Crossref | Google Scholar | PubMed |
Waycott M, van Dijk KJ, Biffin E (2021) A hybrid capture RNA bait set for resolving genetic and evolutionary relationships in angiosperms from deep phylogeny to intraspecific lineage hybridization. BioRxiv 1-10.
| Crossref | Google Scholar |
West AG, Waite DW, Deines P, Bourne DG, Digby A, McKenzie VJ, Taylor MW (2019) The microbiome in threatened species conservation. Biological Conservation 229, 85-98.
| Crossref | Google Scholar |
Zhang L, Xu Z (2008) Assessing bacterial diversity in soil. Journal of Soils and Sediments 8, 379-388.
| Crossref | Google Scholar |
Zhang H, van der Wielen N, van der Hee B, Wang J, Hendriks W, Gilbert M (2020) Impact of fermentable protein, by feeding high protein diets, on microbial composition, microbial catabolic activity, gut health and beyond in pigs. Microorganisms 8, 1735.
| Crossref | Google Scholar |