

Delineating genetic management units of sambar deer (Rusa unicolor) in south-eastern Australia, using opportunistic tissue sampling and targeted scat collection
Christopher Davies A , Wendy Wright A , Faye Wedrowicz A , Carlo Pacioni C and Fiona E. Hogan B D
A , Wendy Wright A , Faye Wedrowicz A , Carlo Pacioni C and Fiona E. Hogan B D
A School of Science, Psychology and Sport, Federation University Australia, Gippsland Campus, Churchill, Vic. 3842, Australia.
B School of Science, Psychology and Sport, Federation University Australia, Berwick Campus, Berwick, Vic. 3806, Australia.
C Arthur Rylah Institute for Environmental Research, Department of Environment, Land Water and Planning, Heidelberg, Vic. 3084, Australia.
D Corresponding author. Email: fiona.hogan@federation.edu.au
Wildlife Research 49(2) 147-157 https://doi.org/10.1071/WR19235
Submitted: 16 December 2019 Accepted: 16 June 2021 Published: 20 October 2021
Journal Compilation © CSIRO 2022 Open Access CC BY-NC-ND
Abstract
Context: Invasive species are major drivers of biodiversity loss, requiring management to reduce their ecological impacts. Population genetics can be applied to delineate management units, providing information that can help plan and improve control strategies.
Aim: The present study aims to use a genetic approach to test the existence of three previously proposed sambar deer populations in south-eastern Australia. In doing so, the study aims to delineate management units of sambar deer in south-eastern Australia.
Methods: Sambar deer DNA was sourced opportunistically from tissue samples and targeted scat collection. Samples were collected from three areas in Victoria, south-eastern Australia: Mt Cole (MC), French Island (FI) and eastern Victoria (EV). Contemporary population structure was assessed using a suite of 11 polymorphic microsatellite markers. The number of maternal sambar deer lineages in south-eastern Australia was investigated through sequencing of the mitochondrial (mt)DNA control region.
Key results: Three distinct genetic clusters were identified. Differentiation among inferred clusters was found to be high, with FST ranging from 0.24 between EV and FI clusters and 0.48 between MC and FI clusters. Two mtDNA haplotypes were identified; R.u1 was found throughout EV and FI, and R.u2 was unique to MC. DNA isolated from scats provided reliable data and proved critical for sampling areas where hunting and culling of deer are not generally undertaken.
Conclusions: Three genetically distinct sambar deer management units in south-eastern Australia are defined – MC, FI and EV. Sambar deer control strategies should be applied to each management unit independently. This may be difficult or infeasible for the EV management unit, which is large and geographically complex. Further research may help identify additional fine-scale genetic structure in EV, allowing smaller, more practicable management units to be identified.
Implications: Genetic data can be used to identify management units for invasive species, which will be critical for the development of future management strategies and improving control operations. The approach outlined here could also be applied to improve the management of other introduced deer species in south-eastern Australia.
Keywords: Australia, DNA, invasive pest species, microsatellite, non-invasive sampling, opportunistic tissue sampling, Sambar deer, targeted scat collection.
Introduction
Invasive species are recognised globally as major drivers of biodiversity loss (Allendorf and Lundquist 2003). Australia has a long history of damaging invasive species introductions (Hoffmann and Broadhurst 2016), including accidental arrivals to the continent and those brought intentionally –either as domestic pets and livestock for recreational hunting opportunities, or as biological control agents (Phillips et al. 2007). Some examples of the most damaging invasive species include the feral cat (Felis catus), red fox (Vulpes vulpes) and cane toad (Bufo marinus). These species have been associated with major declines in small mammals across the Australian continent (Woinarski et al. 2015). Larger species, including feral goats (Carpa hircus) and deer, have also caused significant damage to Australian ecosystems (Bayne et al. 2004; Davis et al. 2016), including those already under threat from anthropogenic climate change (Department of Environment 2015). Invasive species can have pronounced economic impacts (Perrings et al. 2000; Pimentel et al. 2005). For example, in Australia during 2001, the financial costs associated with economic loss and control of invasive species was estimated to be over A$9 billion, rising to over A$13 billion in 2012 (Hoffmann and Broadhurst 2016).
Wild deer, particularly sambar deer (Rusa unicolor), are currently causing significant environmental damage across south-eastern Australia, and pressure exists on land managers to improve deer management practices. Sambar are large tropical deer. The species has a native range in south-east Asia, including areas of India, Sri Lanka, Thailand, Nepal and China (Leslie 2011). It was first introduced into Australia during the 1800s to provide game for hunting (Bentley 1957). Sambar deer are generalist browsers and have established self-sustaining wild populations in Victoria, New South Wales (NSW), the Australian Capital Territory (ACT) and Northern Territory (NT) (Moriarty 2004). Surveys based on deer harvest by recreational hunters provide evidence of rapidly increasing sambar deer numbers in Victoria over the last decade, with estimates of over 30 000 sambar deer harvested in 2009, rising to almost 90 000 in 2017 (Moloney and Turnbull 2018) and more than 100 000 in 2018 (Moloney and Powell 2019). Increased sambar deer numbers have raised concerns regarding their social, economic and ecological impacts.
The largest sambar deer populations in Australia are known to occur in Victoria, where the species has been identified as a potentially threatening process (Parliament of Victoria 2017), and have been implicated in damage to native ecosystems (Peel et al. 2005; Bilney 2013). Sambar deer have been shown to browse a wide selection of trees, shrubs, forbs and grasses, including threatened native species in the Yarra Ranges National Park (Forsyth and Davis 2011), and their potential to permanently change vegetation composition and structure has been documented. They also consume several weed species, so are likely to disperse environmental weeds across Victorian landscapes (Eyles 2002). Antler rubbing by sambar deer has damaged stands of yellow wood (Acronychia oblongifolia) and shiny nematolepsis (Nematolepis wilsonii) in the Yarra Ranges and in East Gippsland (Bennett and Coulson 2011; Bilney 2013), linking sambar to the loss of rare and threatened plant species. In the Alpine National Park (ANP) sambar create wallows and game trails, and can damage sensitive subalpine peat communities, which are listed as threatened ecosystems (Department of Environment 2015). Another particularly serious impact of wild deer is their ability to carry and transmit endemic and exotic diseases to livestock (Cripps et al. 2019), wildlife (Ryan and Power 2012) and people (Ng et al. 2011).
Based on sambar deer occurrence records, habitat connectivity and ecology, a recent study investigating sambar deer distribution proposed four reproductively isolated populations in Victoria: eastern Victoria; Mount Cole; Timboon; and French Island (Forsyth et al. 2015). Of these, the proposed population in eastern Victoria has the largest distribution, estimated to cover over 66 300 km2. This area encompasses most forested areas east of Melbourne through to the Victoria–NSW border and continues northwards into the ACT (Fig. 1; Forsyth et al. 2015). The eastern Victorian sambar deer population is thought to have been established by animals released by the Victorian Acclimatisation Society and by deer escaping from farms (Moriarty 2004). Known escapes and introductions occurred at Gembrook (undated), Kinglake (1863), Snake Island (1866) and Tooradin (1869–73) (Bentley 1967). The sambar deer populations at Mount Cole, French Island and Timboon (Fig. 1) are substantially smaller than the eastern Victorian population. Releases of sambar deer around Ercildoune in the 1870s and 1880s are thought to have founded the Mount Cole population (Forsyth et al. 2015). The origin of the French Island population is uncertain; it may have been established from a release during 1859 (Bentley 1967), or from animals originating from the Tooradin release swimming to the island (Forsyth et al. 2015). It is likely that the four sambar deer populations proposed by Forsyth et al. (2015) are genetically isolated from each other, because barriers to dispersal, including large expanses of cleared land, major roads and watercourses, exist among them (Forsyth et al. 2015). However, no previous studies have investigated the connectivity of sambar deer populations across Victoria.

|
Managing deer populations is a complex issue in Victoria. Despite clear evidence of the damage they cause to ecological and agricultural systems, they are also recognised as valuable game species (Davis et al. 2016). As such, recreational hunters expect healthy populations of deer to hunt. Some land managers have suggested that deer eradication is required where they are impacting sensitive vegetation communities (DEDJTR 2018). Current management strategies to reduce deer populations and their associated impacts in Australia include culling (DEDJTR 2018) and the use of fencing to exclude deer from sensitive areas (Bennett and Coulson 2008). A lack of understanding regarding deer distribution, abundance, connectivity and ability to spread to new areas makes the effective management of sambar deer in Victoria difficult. More research is required to investigate these aspects of sambar deer ecology and improve their management.
A molecular approach, where DNA is used to investigate the relatedness between sampled individuals, can be used to determine whether the putative populations are indeed genetically isolated and have the potential to be managed independently. Genetic data can be used to delineate population boundaries and assess connectivity between groups (Fraser et al. 2013). Investigating the genetic structure of animal populations, to identify individual management units, has previously been used to direct and improve eradication efforts for invasive species, including mink (Neovision vison), stoats (Mustela erminea) and feral pigs (Sus scrofa) (Hampton et al. 2004b; Veale et al. 2014; Mora et al. 2018). Small populations, with clear boundaries and no connectivity, represent the best opportunity for successful eradication because recruitment from other populations is unlikely to occur (Abdelkrim et al. 2005). Genetic data can also be used to detect dispersal pathways that could be subsequently targeted for ongoing control and surveillance (Adams et al. 2014). Subsequent to the implication of control actions, genetic data can also be used to determine the success of the program by identifying survivors and immigrants (Veale et al. 2013).
The ecology of sambar deer in Victoria is poorly understood, and no previous studies have incorporated a genetic approach for sambar deer research. Based on the ecology of sambar deer and habitat characteristics, a previous study has suggested that multiple, genetically isolated sambar deer populations exist in Victoria (Forsyth et al. 2015). Here, we aim to test this expectation using empirical genetic evidence. We used opportunistic sampling of tissue (sourced from hunters) and targeted non-invasive sampling of scats from three of the four proposed sambar deer populations (eastern Victoria, Mount Cole and French Island). Samples were not collected from Timboon due to difficulties accessing private properties and time constraints. Genetic samples were used to investigate the contemporary genetic structure of sambar deer in south-eastern Australia and attempt to delineate separate management units. This information will determine if sambar deer in Victoria form one large, homogenous population or are divided into distinct populations. Identifying genetically isolated populations will help land managers determine the feasibility of eradication and control efforts by determining the scale of connected sambar deer populations and likelihood of reinvasion after the implementation of management efforts.
Methods
Sample collection and DNA isolation
The collection of samples for population genetic studies from wild animals can be challenging. Deer are large but cryptic animals, inherently difficult and expensive to trap, which limits the ability to collect large numbers of tissue or blood samples for genetic analyses (Hampton et al. 2018). DNA from tissue can be provided by recreational hunters, but this requires a good relationship with hunters and restricts sampling to areas where hunting is permitted. Alternatively, deer DNA can be sourced non-invasively through the collection of faecal pellets, often referred to as scats (Davies et al. 2020). Because scats are continuously deposited in the environment, animals do not need to be culled or caught to be sampled. Scat collection therefore allows for targeted rather than opportunistic DNA collection.
Sambar deer tissue (ear or liver) and scat samples were collected from three of the four Victorian sambar populations proposed by Forsyth et al. (2015): eastern Victoria (EV); French Island (FI); and Mount Cole (MC) (Fig. 1). Sampling was conducted over a period of 3 years, from 2015 until 2018. All sampling was performed under the provisions of the Wildlife Act 1975 and National Parks Acts 1975 from the Department of Land Water and Planning (Permit no. 1000 7699).
All sambar deer tissue samples from EV (n = 56) were supplied by licenced recreational hunters and Parks Victoria. Deer scats, presumed to be individual sambar deer, were collected from EV (n = 33), FI (n = 23) and MC (n = 23). DNA was sampled from scats using a swabbing method described in Davies et al. (2020). Attempts were made to prevent swabbing scats from the same individual by employing an exclusion zone of ~100 m between scat samples. Geographic locations were recorded for scat samples using a handheld GPS device (Garmin, Schaffhausen, Switzerland), or georeferenced by recording the nearest road, track or town. DNA was isolated from tissue samples using DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany) and from scat samples using the Qiagen QIAamp DNA Mini kit, following protocols outlined in Davies et al. (2020).
Quality control of DNA isolates
DNA from the outer surface of scats, as targeted in the present study, can be degraded by environmental conditions such as rain. As such, DNA isolates from all samples collected were screened for DNA quality and quantity, as outlined in Davies et al. (2020).
Mitochondrial DNA amplification and sequencing
DNA isolates from EV (n = 69), FI (n = 5) and MC (n = 11) were amplified for a ~600-base-pair (bp) section of the mitochondrial (mt)DNA control region using primers CervCRH and CervtPro (Balakrishnan et al. 2003), following methods described in Davies et al. (2020). PCR products were sequenced using Sanger sequencing by the Australian Genome Research Facility (AGRF) in Melbourne, Australia. Sequences were trimmed using Sequence Scanner Software v2.0 (Applied Biosystems, Waltham, MA, USA) and the software MEGA7 (Kumar et al. 2016) was used to align sequences using CLUSTAL W (Thompson et al. 1994). To understand the number of sambar deer lineages introduced to Victoria and to identify mitochondrial differences that may contribute to population structure, aligned sequences were used to produce a haplotype network via the R packages pegas (Paradis 2010) and ape (Popescu et al. 2012). All new haplotypes were submitted to GenBank. Due to a lack of DNA template, mitochondrial DNA amplification and sequencing was not possible for all samples that were genotyped using microsatellite markers.
To gain information regarding the geographical origin of Victorian sambar deer, we compared sequence data produced by the present study with mt(DNA) control region sequence data reported by Gupta et al. (2015) and Martins et al. (2018), representing sambar deer from their native range in south-east Asia. We compared a 139-bp section of control region sequence in common between the present study and those of Gupta et al. (2015) and Martins et al. (2018). The 139-bp segment of control region sequence was also extracted from mitochondrial genome data (NC_020745.1, Hassanin et al. 2012) of the Javan rusa deer (Rusa timorensis), which was used as an outgroup. A rooted neighbour joining tree was produced using the R package ape (Paradis and Schliep 2019).
Microsatellite genotyping
DNA isolates from tissues (n = 56) and scats (n = 79) were genotyped using 11 fluorescent-dye-labelled cervid microsatellites: BL42; BM757; INRA121; IDVGA55; TGLA53; TGLA57 (Bonnet et al. 2002); Ca18; Ca43 (Gaur et al. 2003); CelJP38; OarFCB5; and RT7 (Pérez-Espona et al. 2008), previously described in Davies et al. (2020). The power of the microsatellite suite to identify individuals was high, with a probability of identity (PID) of 2.7 × 10−7 for unrelated individuals and 1.0 × 10−3 for full siblings (Davies et al. 2020). Genotyping was carried out on the Applied Biosystems 3730 DNA analyser and GENEMAPPER 3.7 software (Applied Biosystems) by AGRF. All DNA isolates derived from scat samples were genotyped in triplicate as recommended by Davies et al. (2020). Consensus genotypes from replicates were generated using ConGenR (Lonsinger and Waits 2015). The R package allelematch (Galpern et al. 2012) was used to identify identical genotypes, which were removed from the dataset.
Population structure analysis
Genetic structuring was assessed using three methods. First, the Bayesian clustering approach in the program STRUCTURE (Pritchard et al. 2000) was used to identify the most likely number of genetic clusters. The software assigns individuals to clusters by minimising deviations from Hardy–Weinberg (HW) proportions and Linkage Disequilibrium (LD). STRUCTURE was run with admixture and correlated allele frequencies, with 3 000 000 Markov Chain Monte Carlo (MCMC) iterations and a burn-in of 1 000 000 iterations for K from 1 to 10. The most likely number of clusters was chosen based on the value of K, with the highest posterior probability from values that have plateaued (Pritchard et al. 2000). To assess if missing genotype data influenced clustering, STRUCTURE analyses were repeated on the same set of individuals (n = 105) with a reduced number of loci (six in total), as well as a subset of individuals (n = 64) with complete genotypes (individuals missing data at more than one locus were omitted from analyses).
Second, GENELAND, which implements spatial Bayesian clustering methods (Guillot et al. 2005) was used to investigate fine-scale population structure for a subset of samples with coordinate data from across Victoria (n = 46) and EV alone (n = 27). GENELAND analyses were performed using the spatial model and run with both correlated and uncorrelated allele frequencies. The correlated allele frequency model is more powerful at detecting subtle population structure; however, it is more sensitive to departures from model assumptions, such as the presence of isolation by distance (Guillot 2008). For both correlated and uncorrelated allele frequency models, the maximum number of populations (K) was set to 10, with 1 000 000 iterations and a burn in of 50 000. The thinning parameter was set to 1000 and 10 independent runs were conducted. The most likely number of clusters inferred by GENELAND was chosen according to the run with the highest posterior probability. Convergence was assessed in line with the GENELAND manual (Guillot et al. 2005).
Third, Discriminant Analysis of Principal Components (DAPC) was used to assess population structure using the R package adegenet version 2.0.2 (Jombart 2008). DAPC is based on genetic distances rather than the minimisation of HW proportions (Jombart 2008). We used the ‘find.cluster’ function to infer groups. The optimal number of clusters was chosen according to the lowest BIC value. The number of principal components included in the DAPC analysis (n = 11) was determined using the ‘optim.a.score’ function.
Genetic statistics and isolation by distance analyses
GenAlEx version 6.5 (Peakall and Smouse 2006) was used to calculate the mean number of alleles (NA), mean expected (HE) and observed heterozygosity (HO) and number of private alleles (PA) across all loci for clusters assigned by STRUCTURE. Deviations from Hardy–Weinberg equilibrium (HWE) and genotypic disequilibrium for each cluster were calculated using GENEPOP 4.2 (Rousset 2008). The R package diveRsity (Keenan et al. 2013) was used to calculate allelic richness (AR, mean number of alleles corrected for differences in sample size), FST (Weir and Cockerham 1984) and Djost (Jost 2008) in order to estimate genetic differentiation between clusters. Mantel tests were performed in GenAlEx to test for correlation between geographical and genetic distances for all samples with coordinate data in defined clusters. We also tested for evidence of fine-scale isolation by distance, by conducting spatial autocorrelation analyses in GenAlEx for all individuals of each cluster, for distances of 15–150 km. MICRO-CHECKER 2.2.3 (Van Oosterhout et al. 2004) was used to check for the presence of null alleles, stuttering and allelic dropout for each defined cluster (EV, FI and MC).
Results
Quality control of DNA isolates
All DNA isolates from scats and tissue had DNA concentrations greater than 0.05 ng µL−1 and produced PCR products for the quality control amplification step, so were retained for genetic analysis, as recommended by Davies et al. (2020).
Mitochondrial DNA sequencing
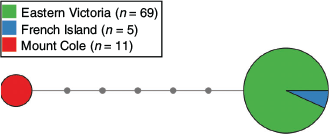
Mitochondrial control region sequencing of tissue (n = 56) and scat samples (n = 32) identified two different haplotypes (R.u1 and R.u2). The first haplotype (R.u1, GenBank accession number: MK473445) was found in individuals sampled from EV and FI, and has been previously reported by Davies et al. (2020). The second haplotype (R.u2, GenBank accession number: MK473444) was unique to sambar deer from MC (Fig. 2).
|
Phylogenetic analysis indicated that Victorian sambar deer are likely to have originated from Sri Lanka. The 139-bp Victorian mtDNA sequence from the present study was identical to that of sambar deer (n = 4) from Sri Lanka, whereas differences of 4–8 bp were observed between Victorian sambar deer and those sampled from different locations across south-east Asia (Supplementary material Fig. S1). This result is in agreement with historical records (Zoological and Acclimatisation Society of Victoria 1872) that indicate the importation of sambar deer from Sri Lanka to Victoria, Australia, during the 1800s.
Microsatellite genotyping
When collecting DNA from discarded sources, multiple scats of the same individual can be inadvertently sampled. Matching of genotypic data derived from scat samples (n = 79) revealed that 52 individual sambar deer had been sampled, with 27 individuals being sampled more than once (four individuals in EV, 12 at FI and 11 at MC). After removal of matching genotypes, 105 individual sambar deer (n = 52 originating from scats and n = 53 originating from tissues) were identified from the regions sampled, i.e. EV (n = 82), FI (n = 11) and MC (n = 12).
Population structure
Inspection of the mean log-likelihood inferred by STRUCTURE indicated the most likely number of populations to be K = 3 (Supplementary material Fig. S2). STRUCTURE clustered most individuals strongly (Q > 0.8) to their location of origin (Fig. 3). Two individuals were identified as not originating from their sampled region, one individual sampled in EV was assigned to the MC cluster and one individual from FI was assigned to the EV cluster. Cluster assignment for K = 3 appeared highly similar when only six loci were included and for the subset of individuals (n = 64) with complete genotypes (Supplementary material Fig. S3).

|
Using the uncorrelated allele frequency model across all regions, GENELAND detected K = 3, matching the results of STRUCTURE, whereas the correlated allele frequency model estimated K = 4, identifying an extra cluster (Kanumbra, n = 2) within the EV population (Fig. 4, Supplementary material Fig. S4). Using eastern Victorian samples alone, GENELAND estimated K = 1 using the uncorrelated allele frequency model and K = 3 using the correlated allele frequency model (Supplementary material, Fig. S5), possibly suggesting further substructure of sambar deer present within the EV population.

|
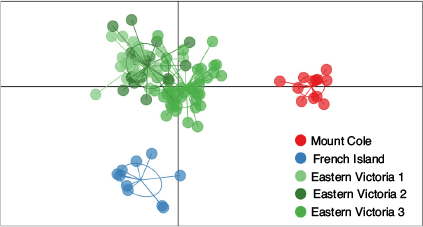
In total, five genetic clusters were inferred using DAPC (FI, MC and three clusters in EV), with all individuals grouping to their sampling location except one individual from FI that was assigned to EV (Fig. 5, Supplementary material Fig. S6).
|
Genetic statistics of inferred population clusters
MICRO-CHECKER indicated homozygote excess at locus BL42 in the EV region, consistent with the presence of null alleles or Wahlund effect. These findings were not consistent across all regions sampled – therefore BL42 was retained for further analysis. Mean observed heterozygosity (HO) and expected heterozygosity (HE) across all regions was 0.42 (s.d. = 0.14) and 0.43 (s.d. = 0.13) respectively (Table 1). Deviation from HWE (P < 0.05) was observed at locus RT7 within the EV cluster. Significant genotypic disequilibrium (P < 0.05) was detected between locus pairs CelJP38 and IDVGA55, BL42 and INRA121 in the EV cluster, and between Ca18 and OarFCB5 and BL42 and TGLA53 in the MC cluster. Both private alleles and genetic differentiation tests (FST and Djost) showed that the three clusters of sambar deer are likely to be separate, with little mixing between groups. Private alleles within a sampled group can provide a measure of genetic distinctiveness, but estimates are dependent upon sample size. The EV cluster had the highest number of private alleles (n = 15). Five private alleles were detected for the MC cluster, which is relatively high considering the small number of individuals sampled (n = 12); in comparison, for FI, where a similar number of individuals were sampled (n = 11), only one private allele was detected. All three population clusters were substantially differentiated using FST and Djost (Table 2). Genetic differentiation was high and ranged from 0.19 (FI–EV) to 0.44 (FI–MC) for Djost and from 0.24 (FI–EV) to 0.48 (FI–MC) for FST (Table 2). Mantel tests showed no correlation between geographical and genetic distance within each individual region (EV: r = 0.068; P = 0.220, FI: r = 0.006; P = 0.400, MC r = –0.002; P = 0.500).

|

|
Discussion
Sambar deer have established themselves as a problematic invasive species in south-eastern Australia, and a lack of understanding of the connectivity between populations has complicated their management. For the first time, we use microsatellite genotyping to investigate the population structure of sambar deer in Victoria and report three distinct population clusters (EV, FI and MC). Genetic clustering of individuals inferred in the pesent study agree with previous assumptions made by Forsyth et al. (2015), who suggested that sambar deer from EV, FI and MC are reproductively isolated. The FST and Djost values observed in the present study show that all clusters displayed high levels of genetic differentiation. The highest genetic differentiation between clusters was observed between FI and MC, and the lowest genetic differentiation between FI and EV. The MC cluster exhibited a separate mitochondrial lineage (haplotype, R.u2), whereas FI and EV shared a single haplotype (R.u1). This suggests that either (1) the FI sambar deer population was established from the same stock as other releases in EV, or (2) the area was colonised by animals swimming to the island from Tooradin, as suggested by Forsyth et al. (2015). The separate mitochondrial haplotype identified at MC also suggests a lack of gene flow between MC and other sambar deer populations (FI and EV).
Factors contributing to genetic differentiation
Several different factors may have contributed to the genetic differentiation observed in Victorian sambar deer, including barriers to dispersal, founder effect and maintenance of genetic differentiation from source populations. Although sambar deer are recognised as strong swimmers (Leslie 2011), the waters of Westernport Bay are likely to impede movement of individuals between the EV and FI populations. Sambar deer commonly display preferences for areas of thick vegetation cover (Bentley 1957), so large areas of cleared land are likely to be perceived as a barrier by this species. Expanses of cleared land exist between the forested areas inhabited by sambar deer in EV and FI, and are likely to have impeded movement of individual animals (and hence gene flow) between these areas. Likewise, large areas of inhospitable habitat exist between MC and the occupied range of sambar deer at EV and FI (Fig. 1). These areas would have also likely acted as a barrier to dispersal and gene flow. An additional element to consider is that even if the three populations were established from the same source, with limited or in the absence of gene flow, the MC and FI sambar populations are likely to be affected by high levels of genetic drift, driven by the low number of individuals in some of these populations.
Based on STRUCTURE results, one individual from EV showed ancestry from MC and one individual from FI displayed EV ancestry. The individual from EV displaying MC ancestry did not have the MC mitochondrial haplotype (R.u2) and had missing data at six of the 11 loci (Supplementary Data S1). Missing data can negatively affect population structure inference (Reeves et al. 2016) because Bayesian assignment, based on data from a small number of microsatellite loci genotyped in individuals with missing data, is potentially unreliable. The individual from FI displaying ancestry from EV had missing data at only one of the 11 loci and had alleles at six loci that were not present in other FI individuals. This finding potentially indicates the recent migration of individuals into the FI population; however, the small sample size obtained from FI limits confidence in the result, requiring further sampling to ensure that the different alleles identified in this individual are not present in deer located elsewhere on the island.
Mitochondrial data provides insights into the origin of Victorian sambar deer
Sequencing of the mitochondrial control region revealed two sambar deer haplotypes (Fig. 2), providing evidence of at least two female founders of the Victorian sambar deer population. One haplotype was shared among deer from EV and FI (R.u1), with the second unique to sambar deer from MC (Ru.2). The presence of the different haplotypes among regions may have resulted from a range of different scenarios. One explanation is that the individual animals originally brought to Australia were from a single population (likely from Ceylon, now Sri Lanka) containing a limited number of haplotypes, and by chance only individuals with the R.u2 haplotype were released at MC. This is in line with reports from Bentley (1967) and records from the Zoological and Acclimatisation Society of Victoria (1872), which indicate four adult sambar deer were imported from Ceylon in 1861. However, to decisively conclude that the Sri Lankan haplotype is not present elsewhere throughout the native range of sambar deer, given the lack of phylogeographic structure evident from the available data, more detailed sampling would be required. Alternatively, the sambar deer introduced to MC may have been sourced from a different area of the native range.
Opportunistic tissue sampling and non-invasive scat sampling
Sambar deer are inherently difficult to capture and sample for genetic studies. Our approach, which combined opportunistic sampling of tissue sourced from hunters or land management agencies (Parks Victoria) with targeted scat sampling, has enabled the collection of DNA from 105 individual sambar deer across a wide geographical area. Using this approach, we were able to describe the population structure of sambar deer across south-eastern Australia. DNA isolated from tissue (including blood) samples is typically higher in both yield and quality, and data derived from such samples are less prone to errors compared with those generated from degraded DNA. Although scat samples are relatively easy to collect, they are more likely to contain degraded DNA.
Although it is preferable to use tissue samples over scat samples for genetic studies, tissue sampling for deer can be limited to areas that allow hunting and/or locations where deer control efforts are undertaken. For example, such sampling would not usually be possible in national parks (where hunting is prohibited) unless control operations are being undertaken, where deer pose the greatest threat to biodiversity. Scat collection offers a valid alternative to tissue sampling in protected areas and, as in the present study, sampling can be focused in areas in which deer activity has been detected. Furthermore, deer scats can be easily differentiated from the scats of native mammals so non-experts, volunteers and citizen scientists could be utilised to conduct field sampling and increase sample sizes. Because differentiation among scats of individuals of a single species can be problematic, DNA profile matching is necessary to ensure that individuals are not represented more than once in a dataset.
Future directions
Increasing the sample size, geographical coverage of samples and number of microsatellite loci may help elucidate more subtle differences in population structure (Landguth et al. 2012) and possibly clarify the different estimate of K observed using GENELAND. However, the differences in K estimates we observed in the EV sambar population using the correlated and uncorrelated allele frequency models in GENELAND could have arisen for several reasons. The additional cluster inferred from the correlated allele frequency model may accurately represent further population structuring within the geographically large EV population. This explanation would also help explain deviations from HWE and genotypic disequilibrium observed in the EV cluster. Alternatively, the additional cluster may have been identified due to gaps in the sampling distribution (large areas of the EV population were not sampled). Previous studies have reported that the effect of isolation by distance (IBD) can cause spurious results when using GENELAND and other spatially explicit Bayesian clustering techniques (Frantz et al. 2009). Despite no evidence of IBD in the EV population, there may be other factors at play. For example, the large distribution and rapid expansion of sambar deer populations in Victoria may violate model assumptions.
During the present study, three of the four Victorian sambar deer populations identified by Forsyth et al. (2015) were sampled. Further collection of genetic data from the population not sampled for the present study (Timboon, shown in Fig. 1) would help determine whether deer within this area also form a distinct management unit. Using genetic data to determine whether eradication, control or containment is feasible at Timboon is a priority for future research due to the potential for this population to spread into neighbouring areas, including the Great Otway National Park.
Analyses of genetic data using a landscape genetic approach could be used to determine the influence of landscape features on gene flow and connectivity across Victoria. Such an approach could also identify leading edges of range expansion, which could be targeted for prioritised eradication or control (Rollins et al. 2009). Additionally, further genetic analyses will allow estimates of effective population size for each of the clusters identified in the present study. This will provide an indication of the size of each cluster and determine if each population is expanding or contracting. By collecting genetic information from scats before and after control operations, the effectiveness of the control operations can be assessed by estimating abundance of the target species using genetic mark–recapture models (Berry et al. 2012), or through the detection of genetic bottlenecks (Hampton et al. 2004a; Rollins et al. 2006). Incorporating genetic approaches into sambar deer research will provide important information regarding distribution and population dynamics and is therefore recommended.
It is important to note that habitat connectivity exists across the Victoria–NSW border, especially along densely forested expanses of the Great Dividing Range. It is therefore expected that geneflow would occur across State boundaries. The identification of sambar deer management units that occur across state boundaries would highlight the need for cooperation between Victorian and NSW management agencies and underscore the need to align deer management objectives across state borders.
Implications for sambar deer management
Current management strategies of deer in Victoria are often ad hoc, because little is known about the ecology of this invasive species (Davis et al. 2016). Sambar deer management interventions require more empirical data to help assign resources and improve management outcomes (Parliament of Victoria 2017). The findings of the present study have important implications for sambar deer management in Victoria. We have identified three genetically isolated sambar deer populations in Victoria, which should be applied as distinct management zones. Establishing deer management zones in Victoria will help guide where to conduct surveillance, monitoring, control and eradication efforts to meet localised deer management objectives (DEDJTR 2018).
Conclusion
The work of Hone et al. (2010) suggested that the removal of around 40% of sambar deer would be required to stop annual population growth. If sambar deer formed a single homogenous population across Victoria, removing this number of deer would be a daunting and unfeasible process, exacerbated by the rugged and inaccessible terrain preferred by the species. Here we provide evidence that multiple sambar deer populations exist in Victoria. With this information land managers can focus control and eradication efforts to the areas identified (EV, FI and MC) and make evidence-based decisions regarding whether control and eradication efforts are feasible and cost effective. We show sambar deer on French Island are genetically isolated from deer on the mainland, suggesting that eradication of sambar deer from French Island is possible and control operations undertaken on the island have a low risk of reinvasion. In contrast, the observed connectivity between sambar deer across eastern Victoria suggests that eradication attempts performed at geographical scales smaller than the entire EV region are not feasible because reinvasion is likely to occur quickly, unless further population structuring is shown by follow up studies with a higher resolution power as we suggest above. As such, an asset protection approach to deer impacts across EV that focuses on protecting vegetation communities assessed to be at the highest levels of risk (e.g. alpine peatlands and warm temperate rainforest communities) may be practical in the short-term. We suggest that incorporating a genetic approach into future studies will greatly improve the management of sambar deer in Victoria. The sampling methodology and genetic approach applied here could also be used to delineate management units for other introduced deer species in Victoria.
Conflicts of interest
The authors declare no conflicts of interest.
Data availability statement
The data that support the present study will be shared upon reasonable request to the corresponding author.
Declaration of funding
Christopher Davies was supported by an Australian Government Research Training Program (RTP) Stipend and RTP Fee-Offset Scholarship through Federation University Australia. This research was also funded by the Holsworth Wildlife Research Endowment – Equity Trustees Charitable Foundation and Federation University Australia’s School of Health and Life Sciences.
Acknowledgements
We thank those who helped collect deer tissue and scat samples, including Luke Treptow, Karl Sobott, Dwayne Needham, Dr Jordan Hampton and staff from Parks Victoria.
References
Abdelkrim, J., Pascal, M., Calmet, C., and Samadi, S. (2005). Importance of assessing population genetic structure before eradication of invasive species: examples from insular Norway rat populations. Conservation Biology 19, 1509–1518.| Importance of assessing population genetic structure before eradication of invasive species: examples from insular Norway rat populations.Crossref | GoogleScholarGoogle Scholar |
Adams, A. L., van Heezik, Y., Dickinson, K. J. M., and Robertson, B. C. (2014). Identifying eradication units in an invasive mammalian pest species. Biological Invasions 16, 1481–1496.
Allendorf, F. W., and Lundquist, L. L. (2003). Introduction: population biology, evolution, and control of invasive species. Conservation Biology 17, 24–30.
| Introduction: population biology, evolution, and control of invasive species.Crossref | GoogleScholarGoogle Scholar |
Atlas of Living Australia. (2019). Occurrence records download – Cervus unicolor. Available at https://doi.org/10.26197/5c62120762b48 [verified 12 February 2019]
Balakrishnan, C., Monfort, S., Gaur, A., Singh, L., and Sorenson, M. (2003). Phylogeography and conservation genetics of Eld’s deer (Cervus eldi). Molecular Ecology 12, 1–10.
| Phylogeography and conservation genetics of Eld’s deer (Cervus eldi).Crossref | GoogleScholarGoogle Scholar | 12492873PubMed |
Bayne, P., Harden, R., and Davies, I. (2004). Feral goats (Capra hircus) in the Macleay River gorge system, north-eastern New South Wales, Australia, impacts on soil erosion. Wildlife Research 31, 519–525.
| Feral goats (Capra hircus) in the Macleay River gorge system, north-eastern New South Wales, Australia, impacts on soil erosion.Crossref | GoogleScholarGoogle Scholar |
Bennett, A., and Coulson, G. (2008). Evaluation of an exclusion plot design for determining the impacts of native and exotic herbivores on forest understoreys. Australian Mammalogy 30, 83–87.
| Evaluation of an exclusion plot design for determining the impacts of native and exotic herbivores on forest understoreys.Crossref | GoogleScholarGoogle Scholar |
Bennett, A., and Coulson, G. (2011). The impacts of sambar (Cervus unicolor) on the threatened shiny nematolepis (Nematolepis wilsonii). Pacific Conservation Biology 16, 251–260.
| The impacts of sambar (Cervus unicolor) on the threatened shiny nematolepis (Nematolepis wilsonii).Crossref | GoogleScholarGoogle Scholar |
Bentley, A. (1957). A brief account of the deer in Australia. The Journal of Wildlife Management 21, 221–225.
| A brief account of the deer in Australia.Crossref | GoogleScholarGoogle Scholar |
Bentley, A. (1967). ‘An Introduction to the Deer of Australia with Special Reference to Victoria.’ (Australian Deer Research Foundation: Melbourne, Vic., Australia.)
Berry, O., Algar, D., Angus, J., Hamilton, N., Hilmer, S., and Sutherland, D. (2012). Genetic tagging reveals a significant impact of poison baiting on an invasive species. The Journal of Wildlife Management 76, 729–739.
| Genetic tagging reveals a significant impact of poison baiting on an invasive species.Crossref | GoogleScholarGoogle Scholar |
Bilney, R. J. (2013). Antler rubbing of yellow-wood by sambar in East Gippsland, Victoria. Victorian Naturalist 130, 68–74.
Bonnet, A., Thévenon, S., Maudet, F., and Maillard, J. C. (2002). Efficiency of semi-automated fluorescent multiplex PCRs with 11 microsatellite markers for genetic studies of deer populations. Animal Genetics 33, 343–350.
| Efficiency of semi-automated fluorescent multiplex PCRs with 11 microsatellite markers for genetic studies of deer populations.Crossref | GoogleScholarGoogle Scholar | 12354142PubMed |
Cripps, J. K., Pacioni, C., Scroggie, M. P., Woolnough, A. P., and Ramsey, D. S. L. (2019). Introduced deer and their potential role in disease transmission to livestock in Australia. Mammal Review 49, 60–77.
| Introduced deer and their potential role in disease transmission to livestock in Australia.Crossref | GoogleScholarGoogle Scholar |
Davies, C., Hogan, F. E., Wedrowicz, F., and Wright, W. (2020). A DNA toolbox for non-invasive genetic studies of sambar deer (Rusa unicolor). Australian Mammalogy 42, 58–66.
| A DNA toolbox for non-invasive genetic studies of sambar deer (Rusa unicolor).Crossref | GoogleScholarGoogle Scholar |
Davis, N. E., Bennett, A., Forsyth, D. M., Bowman, D. M. J. S., Lefroy, E. C., Wood, S. W., Woolnough, A. P., West, P., Hampton, J. O., and Johnson, C. N. (2016). A systematic review of the impacts and management of introduced deer (family Cervidae) in Australia. Wildlife Research 43, 515–532.
| A systematic review of the impacts and management of introduced deer (family Cervidae) in Australia.Crossref | GoogleScholarGoogle Scholar |
DEDJTR (2018). Draft deer management strategy. Victorian Department of Economic Development, Jobs, Transport and Resources, Melbourne, Vic., Australia.
Department of Environment (2015). National recovery plan for the alpine sphagnum bogs and associated fens ecological community. Australian Department of Environment, Canberra, ACT, Australia.
Eyles, D. (2002). Sambar deer (Cervus unicolor) as a potential seed vector for the spread of the environmental weed Himalayan honeysuckle (Leycesteria formosa) at Mount Buffalo National Park. B.Sc. (Hons) Thesis, University of Melbourne, Melbourne, Vic., Australia.
Forsyth, D. M., and Davis, N. E. (2011). Diets of non-native deer in Australia estimated by macroscopic versus microhistological rumen analysis. The Journal of Wildlife Management 75, 1488–1497.
| Diets of non-native deer in Australia estimated by macroscopic versus microhistological rumen analysis.Crossref | GoogleScholarGoogle Scholar |
Forsyth, D. M., Stamation, K., and Woodford, L. (2015). Distributions of sambar deer, rusa deer and sika deer in Victoria. Arthur Rylah Institute for Environmental Research, Melbourne, Vic., Australia.
Frantz, A. C., Cellina, S., Krier, A., Schley, L., and Burke, T. (2009). Using spatial Bayesian methods to determine the genetic structure of a continuously distributed population: clusters or isolation by distance? Journal of Applied Ecology 46, 493–505.
| Using spatial Bayesian methods to determine the genetic structure of a continuously distributed population: clusters or isolation by distance?Crossref | GoogleScholarGoogle Scholar |
Fraser, E. J., Macdonald, D. W., Oliver, M. K., Piertney, S., and Lambin, X. (2013). Using population genetic structure of an invasive mammal to target control efforts – an example of the American mink in Scotland. Biological Conservation 167, 35–42.
| Using population genetic structure of an invasive mammal to target control efforts – an example of the American mink in Scotland.Crossref | GoogleScholarGoogle Scholar |
Galpern, P., Manseau, M., Hettinga, P., Smith, K., and Wilson, P. (2012). Allelematch: an R package for identifying unique multilocus genotypes where genotyping error and missing data may be present. Molecular Ecology Resources 12, 771–778.
| Allelematch: an R package for identifying unique multilocus genotypes where genotyping error and missing data may be present.Crossref | GoogleScholarGoogle Scholar | 22463778PubMed |
Gaur, A., Singh, A., Arunabala, V., Umapathy, G., Shailaja, K., and Singh, L. (2003). Development and characterization of 10 novel microsatellite markers from chital deer (Cervus axis) and their cross-amplification in other related species. Molecular Ecology Notes 3, 607–609.
| Development and characterization of 10 novel microsatellite markers from chital deer (Cervus axis) and their cross-amplification in other related species.Crossref | GoogleScholarGoogle Scholar |
Guillot, G. (2008). Inference of structure in subdivided populations at low levels of genetic differentiation – the correlated allele frequencies model revisited. Bioinformatics 24, 2222–2228.
| Inference of structure in subdivided populations at low levels of genetic differentiation – the correlated allele frequencies model revisited.Crossref | GoogleScholarGoogle Scholar | 18710873PubMed |
Guillot, G., Mortier, F., and Estoup, A. (2005). GENELAND: a computer package for landscape genetics. Molecular Ecology Notes 5, 712–715.
| GENELAND: a computer package for landscape genetics.Crossref | GoogleScholarGoogle Scholar |
Gupta, S. K., Kumar, A., Gaur, A., and Hussain, S. A. (2015). Detection of 40 bp insertion–deletion in mitochondrial control region among sambar (Rusa unicolor) populations in India. BMC Research Notes 8, 581.
| Detection of 40 bp insertion–deletion in mitochondrial control region among sambar (Rusa unicolor) populations in India.Crossref | GoogleScholarGoogle Scholar | 26483190PubMed |
Hampton, J., Pluske, J. R., and Spencer, P. B. S. (2004a). A preliminary genetic study of the social biology of feral pigs in south-western Australia and the implications for management. Wildlife Research 31, 375–381.
| A preliminary genetic study of the social biology of feral pigs in south-western Australia and the implications for management.Crossref | GoogleScholarGoogle Scholar |
Hampton, J. O., Spencer, P. B. S., Alpers, D. L., Twigg, L. E., Woolnough, A. P., Doust, J., Higgs, T., and Pluske, J. (2004b). Molecular techniques, wildlife management and the importance of genetic population structure and dispersal: a case study with feral pigs. Journal of Applied Ecology 41, 735–743.
| Molecular techniques, wildlife management and the importance of genetic population structure and dispersal: a case study with feral pigs.Crossref | GoogleScholarGoogle Scholar |
Hampton, J. O., Finch, N. A., Watter, K., Amos, M., Pople, T., Moriarty, A., Jacotine, A., Panther, D., McGhie, C., Davies, C., Mitchell, J., and Forsyth, D. M. (2018). A review of methods used to capture and restrain introduced wild deer in Australia. Australian Mammalogy 41, 1–11.
Hassanin, A., Delsuc, F., Ropiquet, A., Hammer, C., Jansen van Vuuren, B., Matthee, C., Ruiz-Garcia, M., Catzeflis, F., Areskoug, V., Nguyen, T. T., and Couloux, A. (2012). Pattern and timing of diversification of Cetartiodactyla (Mammalia, Laurasiatheria), as revealed by a comprehensive analysis of mitochondrial genomes. Comptes Rendus Biologies 335, 32–50.
| Pattern and timing of diversification of Cetartiodactyla (Mammalia, Laurasiatheria), as revealed by a comprehensive analysis of mitochondrial genomes.Crossref | GoogleScholarGoogle Scholar | 22226162PubMed |
Hoffmann, B. D., and Broadhurst, L. M. (2016). The economic cost of managing invasive species in Australia. NeoBiota 31, 1–18.
| The economic cost of managing invasive species in Australia.Crossref | GoogleScholarGoogle Scholar |
Hone, J., Duncan, R. P., and Forsyth, D. M. (2010). Estimates of maximum annual population growth rates (rm) of mammals and their application in wildlife management. Journal of Applied Ecology 47, 507–514.
| Estimates of maximum annual population growth rates (rm) of mammals and their application in wildlife management.Crossref | GoogleScholarGoogle Scholar |
Jombart, T. (2008). adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24, 1403–1405.
| adegenet: a R package for the multivariate analysis of genetic markers.Crossref | GoogleScholarGoogle Scholar | 18397895PubMed |
Jost, L. (2008). GST and its relatives do not measure differentiation. Molecular Ecology 17, 4015–4026.
| GST and its relatives do not measure differentiation.Crossref | GoogleScholarGoogle Scholar | 19238703PubMed |
Keenan, K., McGinnity, P., Cross, T. F., Crozier, W. W., and Prodöhl, P. A. (2013). diveRsity: an R package for the estimation and exploration of population genetics parameters and their associated errors. Methods in Ecology and Evolution 4, 782–788.
| diveRsity: an R package for the estimation and exploration of population genetics parameters and their associated errors.Crossref | GoogleScholarGoogle Scholar |
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Molecular Biology and Evolution 33, 1870–1874.
| MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets.Crossref | GoogleScholarGoogle Scholar | 27004904PubMed |
Landguth, E. L., Fedy, B. C., Oyler-Mccance, S. J., Garey, A. L., Emel, S. L., Mumma, M., Wagner, H. H., Fortin, M.-J., and Cushman, S. A. (2012). Effects of sample size, number of markers, and allelic richness on the detection of spatial genetic pattern. Molecular Ecology Resources 12, 276–284.
| Effects of sample size, number of markers, and allelic richness on the detection of spatial genetic pattern.Crossref | GoogleScholarGoogle Scholar |
Leslie, D. M. (2011). Rusa unicolor (Artiodactyla: Cervidae). Mammalian Species 43, 1–30.
| Rusa unicolor (Artiodactyla: Cervidae).Crossref | GoogleScholarGoogle Scholar |
Lonsinger, R., and Waits, L. (2015). ConGenR: rapid determination of consensus genotypes and estimates of genotyping errors from replicated genetic samples. Conservation Genetics Resources 7, 841–843.
| ConGenR: rapid determination of consensus genotypes and estimates of genotyping errors from replicated genetic samples.Crossref | GoogleScholarGoogle Scholar |
Martins, R., Schmidt, A., Lenz, D., Wilting, A., and Fickel, J. (2018). Human-mediated introduction of introgressed deer across Wallace’s line: historical biogeography of Rusa unicolor and Rusa timorensis. Ecology and Evolution 8, 1465–1479.
| Human-mediated introduction of introgressed deer across Wallace’s line: historical biogeography of Rusa unicolor and Rusa timorensis.Crossref | GoogleScholarGoogle Scholar | 29435225PubMed |
Moloney, P. D., and Powell, Z. (2019). Estimates of the 2018 deer harvest in Victoria: results from surveys of Victorian Game Licence holders in 2018. Unpublished client report for the Game Management Authority, Arthur Rylah Institute for Environmental Research, Department of Environment, Land, Water and Planning, Melbourne, Vic., Australia.
Moloney, P. D., and Turnbull, J. D. (2018). Estimates of harvest for deer in Victoria: results from surveys of Victorian Game licence holders in 2016. Unpublished client report for the Game Management Authority, Arthur Rylah Institute for Environmental Research, Department of Environment, Land, Water and Planning, Melbourne, Vic., Australia.
Mora, M., Medina-Vogel, G., Sepúlveda, M. A., Noll, D., Álvarez-Varas, R., and Vianna, J. A. (2018). Genetic structure of introduced American mink (Neovison vison) in Patagonia: colonisation insights and implications for control and management strategies. Wildlife Research 45, 344–356.
| Genetic structure of introduced American mink (Neovison vison) in Patagonia: colonisation insights and implications for control and management strategies.Crossref | GoogleScholarGoogle Scholar |
Moriarty, A. (2004). The liberation, distribution, abundance and management of wild deer in Australia. Wildlife Research 31, 291–299.
| The liberation, distribution, abundance and management of wild deer in Australia.Crossref | GoogleScholarGoogle Scholar |
Ng, J., Yang, R., Whiffin, V., Cox, P., and Ryan, U. (2011). Identification of zoonotic Cryptosporidium and Giardia genotypes infecting animals in Sydney’s water catchments. Experimental Parasitology 128, 138–144.
| Identification of zoonotic Cryptosporidium and Giardia genotypes infecting animals in Sydney’s water catchments.Crossref | GoogleScholarGoogle Scholar | 21334325PubMed |
Paradis, E. (2010). pegas: an R package for population genetics with an integrated-modular approach. Bioinformatics 26, 419–420.
| pegas: an R package for population genetics with an integrated-modular approach.Crossref | GoogleScholarGoogle Scholar | 20080509PubMed |
Paradis, E., and Schliep, K. (2019). ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528.
| ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R.Crossref | GoogleScholarGoogle Scholar | 30016406PubMed |
Parliament of Victoria (2017). Inquiry into the control of invasive animals on Crown land. Environment Natural Resources and Regional Development Committee, Victorian Government, Melbourne, Vic., Australia.
Peakall, R., and Smouse, P. E. (2006). GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Molecular Ecology Notes 6, 288–295.
| GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research.Crossref | GoogleScholarGoogle Scholar |
Peel, B., Bilney, R. J., and Bilney, R. J. (2005). Observations of the ecological impacts of sambar (Cervus unicolor) in East Gippsland, Victoria, with reference to destruction of rainforest communities. Victorian Naturalist 122, 189–200.
Pérez-Espona, S., Pérez-Barbería, F. J., McLeod, J. E., Jiggins, C. D., Gordon, I. J., and Pemberton, J. M. (2008). Landscape features affect gene flow of Scottish Highland red deer (Cervus elaphus). Molecular Ecology 17, 981–996.
| Landscape features affect gene flow of Scottish Highland red deer (Cervus elaphus).Crossref | GoogleScholarGoogle Scholar | 18261043PubMed |
Perrings, C., Dalmazzone, S., and Williamson, M. H. (2000). ‘The Economics of Biological Invasions.’ (Edward Elgar Publishing: Cheltenham, UK.)
Phillips, B. L., Brown, G. P., Greenlees, M., Webb, J. K., and Shine, R. (2007). Rapid expansion of the cane toad (Bufo marinus) invasion front in tropical Australia. Austral Ecology 32, 169–176.
| Rapid expansion of the cane toad (Bufo marinus) invasion front in tropical Australia.Crossref | GoogleScholarGoogle Scholar |
Pimentel, D., Zuniga, R., and Morrison, D. (2005). Update on the environmental and economic costs associated with alien-invasive species in the United States. Ecological Economics 52, 273–288.
| Update on the environmental and economic costs associated with alien-invasive species in the United States.Crossref | GoogleScholarGoogle Scholar |
Popescu, A.-A., Huber, K., and Paradis, E. (2012). ape 3.0: new tools for distance-based phylogenetics and evolutionary analysis in R. Bioinformatics 28, 1536–1537.
| ape 3.0: new tools for distance-based phylogenetics and evolutionary analysis in R.Crossref | GoogleScholarGoogle Scholar | 22495750PubMed |
Pritchard, J. K., Stephens, M., and Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945–959.
| Inference of population structure using multilocus genotype data.Crossref | GoogleScholarGoogle Scholar | 10835412PubMed |
Reeves P. A.Bowker C. L.Fettig C. E.Tembrock L. R.Richards C. M. (2016 ). Effect of error and missing data on population structure inference using microsatellite data. bioRxiv
Rollins, L. A., Woolnough, A. P., and Sherwin, W. B. (2006). Population genetic tools for pest management: a review. Wildlife Research 33, 251–261.
| Population genetic tools for pest management: a review.Crossref | GoogleScholarGoogle Scholar |
Rollins, L. E. E., Woolnough, A., Wilton, A., Sinclair, R. O. N., and Sherwin, W. (2009). Invasive species can’t cover their tracks: using microsatellites to assist management of starling (Sturnus vulgaris) populations in Western Australia. Molecular Ecology 18, 1560–1573.
| Invasive species can’t cover their tracks: using microsatellites to assist management of starling (Sturnus vulgaris) populations in Western Australia.Crossref | GoogleScholarGoogle Scholar |
Rousset, F. (2008). genepop’007: a complete re-implementation of the genepop software for Windows and Linux. Molecular Ecology Resources 8, 103–106.
| genepop’007: a complete re-implementation of the genepop software for Windows and Linux.Crossref | GoogleScholarGoogle Scholar | 21585727PubMed |
Ryan, U., and Power, M. (2012). Cryptosporidium species in Australian wildlife and domestic animals. Parasitology 139, 1673–1688.
| Cryptosporidium species in Australian wildlife and domestic animals.Crossref | GoogleScholarGoogle Scholar | 22906836PubMed |
Thompson, J. D., Higgins, D. G., and Gibson, T. J. (1994). CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Research 22, 4673–4680.
| CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice.Crossref | GoogleScholarGoogle Scholar | 7984417PubMed |
Van Oosterhout, C., Hutchinson, W., Wills, D. P. M., and Shipley, P. (2004). micro-checker: software for identifying and correcting genotyping errors in microsatellite data. Molecular Ecology Notes 4, 535–538.
| micro-checker: software for identifying and correcting genotyping errors in microsatellite data.Crossref | GoogleScholarGoogle Scholar |
Veale, A. J., Edge, K. A., McMurtrie, P., Fewster, R. M., Clout, M. N., and Gleeson, D. M. (2013). Using genetic techniques to quantify reinvasion, survival and in situ breeding rates during control operations. Molecular Ecology 22, 5071–5083.
| Using genetic techniques to quantify reinvasion, survival and in situ breeding rates during control operations.Crossref | GoogleScholarGoogle Scholar | 24033616PubMed |
Veale, A. J., Gleeson, D. M., and Clout, M. N. (2014). Measuring connectivity of invasive stoat populations to inform conservation management. Wildlife Research 41, 395–406.
| Measuring connectivity of invasive stoat populations to inform conservation management.Crossref | GoogleScholarGoogle Scholar |
Weir, B. S., and Cockerham, C. C. (1984). Estimating F-statistics for the analysis of population structure. Evolution 38, 1358–1370.
| 28563791PubMed |
Woinarski, J. C. Z., Burbidge, A., and Harrison, P. L. (2015). Ongoing unraveling of a continental fauna: decline and extinction of Australian mammals since European settlement. Proceedings of the National Academy of Sciences of the United States of America 112, 4531–4540.
| Ongoing unraveling of a continental fauna: decline and extinction of Australian mammals since European settlement.Crossref | GoogleScholarGoogle Scholar |
Zoological and Acclimatisation Society of Victoria (1872). ‘Proceedings of the Zoological and Acclimitisation Society of Victoria,’ Vol. 1. (Stillwell and Knight: Melbourne, Vic., Australia.)