

The Challenge of Storage in the Hydrogen Energy Cycle: Nanostructured Hydrides as a Potential Solution
James M. Hanlon A , Hazel Reardon A , Nuria Tapia-Ruiz A and Duncan H. Gregory A BA WestCHEM, School of Chemistry, University of Glasgow, Glasgow G12 8QQ, UK.
B Corresponding author. Email: duncan.gregory@glasgow.ac.uk

James M. Hanlon graduated from the University of Glasgow in 2005 with a B.Sc. (Hons) in chemistry with medicinal chemistry. He then returned to the University of Glasgow in 2008 to undertake a Ph.D. with Professor Duncan H. Gregory on hydrogen storage materials under the ESPRC SUPERGEN program. His research interests include nanostructured hydrogen storage materials, hydrogen release systems, and ammonia storage materials. |

Hazel Reardon graduated from the University of Strathclyde with an M.Sc. in analytical chemistry in 2008. She then took up a position as a technical specialist in industry whilst undertaking a Royal Society of Chemistry accredited graduate program. In 2010, she began a Ph.D. at the University of Glasgow with Professor Duncan H. Gregory on the development of hydrogen storage materials. Her research interests are focused on new materials for modern energy and fuel solutions. |

Nuria Tapia-Ruiz graduated from the University of Barcelona with an honours degree in inorganic chemistry in 2009. She then moved to the University of Glasgow where she is currently studying for a Ph.D. in materials chemistry under the supervision of Professor Duncan H. Gregory. Her research interests are focused on developing new routes for the synthesis of nitrides with potential applications as energy storage materials. |

Duncan H. Gregory studied at the University of Southampton, completing his Ph.D. in 1993 under Professor Mark Weller. He was an EPSRC Advanced Fellow, Lecturer and Reader in Materials Chemistry at the University of Nottingham until 2006. He then took up the WestCHEM Chair in Inorganic Materials at the University of Glasgow and is Head of Inorganic Chemistry. His research interests centre on materials with potential applications in areas including sustainable energy. |
Australian Journal of Chemistry 65(6) 656-671 https://doi.org/10.1071/CH11437
Submitted: 16 November 2011 Accepted: 30 December 2011 Published: 20 February 2012
Abstract
Hydrogen has the capacity to provide society with the means to carry ‘green’ energy between the point of generation and the point of use. A sustainable energy society in which a hydrogen economy predominates will require renewable generation provided, for example, by artificial photosynthesis and clean, efficient energy conversion effected, for example, by hydrogen fuel cells. Vital in the hydrogen cycle is the ability to store hydrogen safely and effectively. Solid-state storage in hydrides enables this but no material yet satisfies all the demands associated with storage density and hydrogen release and uptake; particularly for mobile power. Nanochemical design methods present potential routes to overcome the thermodynamic and kinetic hurdles associated with solid state storage in hydrides. In this review we discuss strategies of nanosizing, nanoconfinement, morphological/dimensional control, and application of nanoadditives on the hydrogen storage performance of metal hydrides. We present recent examples of how such approaches can begin to address the challenges and an evaluation of prospects for further development.
Introduction
The continued increase in CO2 emissions and the finite reserves of fossil fuels demand a clean, renewable source of energy to be developed to meet the demands of the world’s population. The predominant candidate for an alternative energy source is solar and hydrogen is an ideal means by which converted solar energy can be stored and transported.
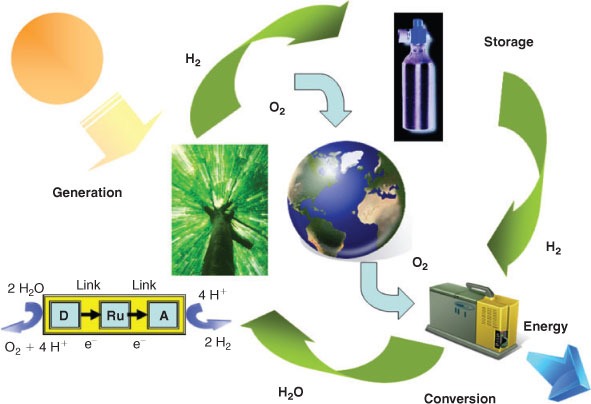
The two major issues before a hydrogen economy can replace a carbon economy based on fossil fuels are the production and storage of hydrogen (Fig. 1). Fuel cells are a relatively mature technology and permit clean power generation from hydrogen as a fuel. A potentially viable and sustainable method of producing hydrogen is by water splitting, giving H2 and O2 by artificial photosynthesis.[1–4] Hence, once such an environmentally friendly process for the production of hydrogen is fully functioning, then safe and efficient ubiquitous storage of hydrogen will be required. Containment of hydrogen in a solid state medium represents such a safe and efficient method of storage but demands a non-toxic and cheap storage material that can store hydrogen optimally in terms of weight and volume and with the minimum of energy requirements to charge and discharge the store.[5–7] Aside from these technical hurdles, the main challenge then lies in the acceptance of the hydrogen economy both socially and economically.
|
In terms of meeting the stringent technical criteria – especially demanding for mobile applications – among the best options in solid state storage materials available are the metal hydrides. The theoretical gravimetric storage capacities of the hydrides are relatively high (for example, LiH can theoretically store 12.8 wt % of H2). However, since the hydrogen in hydrides is bound in the solid by a chemical bond, dehydrogenation requires relatively high energies (and hence high release temperatures). Similarly, reversible storage and cycleabilty can be an issue. To overcome these kinetic and thermodynamic problems and to allow hydrides to become the basis of viable storage systems, new approaches are needed.
In this review, we focus on one such approach; that of nanostructuring metal and complex hydrides. Nanostructuring involves the reduction of the particle size and indeed tailoring of morphology at the nanometer scale by a variety of contrasting methods from physical size reduction through nanoconfinement in a host material or matrix to direct chemical synthesis of active nanometer-scale storage materials.[8–11] In parallel with the design of appropriate catalysts, nanostructuring of hydrides can result in major improvements in sorption properties compared with equivalent bulk materials and offers the potential to produce storage systems that meet the high demands of modern applications (notably, for example, in the transport sector, including automobiles, but also in stationary power generation).
Light Metal Hydrides
Nanometer-scale materials are becoming increasingly important in understanding how materials may be exploited to their maximum potential for hydrogen storage.[12] The synthesis of nanostructured materials in particular has extended the capability of light metal hydride systems as hydrogen stores and this work is dominated by the Mg–H system.[9,13] Here, we will focus on light metal hydride systems at the nanometer scale, the effects on the intrinsic chemical properties of the materials at this level, and the benefits of nanodesign for hydrogen storage.
Alkali Metal Hydrides
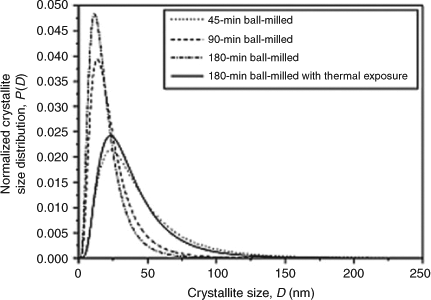
Advances in the hydrogen storage performance of alkali metal binary hydrides by nanostructuring have been overshadowed somewhat by the storage abilities of complex hydrides containing the respective component metals such as borohydrides, alanates, and amides. These complex hydrides are covered elsewhere in this review. Nevertheless, the simplicity of a single metal system remains appealing and at the very least represents a useful model to aid understanding of sorption at the nanometer scale. Ortiz et al. investigated the milling of LiH to establish how physical processing modified crystallite size (reduction to ~25 nm diameter) and subsequently studied the effects of heating to 558 K (for a period of 1 h) on the nanocrystals formed (Fig. 2).[14] They observed the formation of agglomerates as a result of the milling process that were thermally stable under the above conditions. This was ascribed to the porosity of the material. Although experimental studies on nanostructured lithium hydride for hydrogen storage, independent of composite matrices, are yet to be established in the literature, the stability of this material to moisture could present both technical and safety issues for storage. In a computational study, Gautam et al. studied various cluster sizes of LinHm, where n = 1–30 and m ≤ n, to probe the effect of varying the hydrogen ratio on the stability of clusters over this size range.[15] Their calculations predicted that the hydrogen content has a significant effect on the stability of the cluster. Irrespective of the cluster size, as m approached n (and particularly when m = n) the stabilization effect was marked. Experimental studies on the optimization of LiH particle size and morphology for hydrogen sorption to complement this work would be of significant value.
|
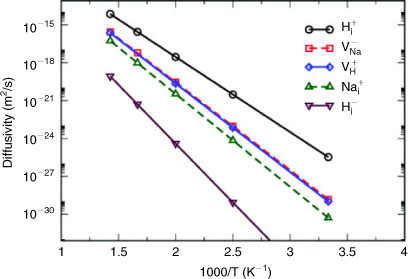
A recent theoretical study of the diffusion mechanisms in NaH by Hao et al. established that although Shottky defects (Na+ cation vacancy and H– anion vacancy pairs) were the most populous in NaH, it was charged H interstitials that were the dominant contributor to hydrogen diffusion (Fig. 3). Given this development in understanding, there is an opportunity to exploit the defect structure of NaH at the nanometer scale and although the influence of NaH as a component for hydrogen storage was highlighted recently by Pistidda et al. in their work on the NaH–MgB2 system, no evaluation of nanometer-scale properties of the hydride were pursued in this study.[16,17] In fact, directed nanostructuring of NaH has effectively been limited thus far to thin films. Films of 200 nm in thickness have been formed by sputtering, although subsequent analysis of properties is as yet rather limited.[18] Studies of the sorption behaviour of such NaH films in comparison to the bulk material should prove illuminating.
|
Alkaline Earth Metal Hydrides
If we consider the group 2 hydrides systematically, beryllium hydride has a theoretical hydrogen gravimetric capacity of 18.3 wt % but assessments of its suitability for hydrogen storage remain relatively pessimistic owing both to the difficulties associated with its preparation and concerns over safety.[19,20]
Magnesium, by contrast to beryllium, has been extensively studied for hydrogen storage and there exists a vast amount of information on nanostructured magnesium hydride. Here we will endeavour to highlight some of the most important developments and discuss only the most recent and significant advances in directed nanodesign in the Mg–H system.
Whisker-like structures of Mg have been known in the literature since the mid 1980s, but well controlled nanodesign of such configurations is now possible by growth by chemical vapour deposition (CVD) or other forms of solid–vapour reactions.[21,22] Zhu et al. demonstrate this very elegantly in their recent publication, which shows how variation in the evaporation temperature and hydrogen pressures at which CVD is conducted for MgH2 synthesis may generate a spectrum of morphologies in which the hydride may grow, including nanowires and nanoparticles.[23]
Physical processing of hydride nanoparticles by ball milling has been central to the development of hydrogen storage materials and is extremely valuable in improving uptake and release kinetics. Nowhere is this more evident than in the Mg–H system. Many materials have been introduced as catalysts or thermal propagators for hydrogen sorption as unique components, but it is emerging that synergistic effects from a mixture of additives bring advantages to the system, e.g., as shown with graphite and Nb2O5.[24] However, a major consideration when employing such additives during the milling process is the detrimental effect on gravimetric capacity; vital for potential mobile applications.
Mechanochemical synthesis of hydrogen storage materials by contrast to the purely physical effects of size reduction, is emerging as an attractive way to prepare hydrides without following more lengthy and energy-intensive milling procedures. The process results in a metal hydride or composite material within a salt or by-product matrix. In the case of light metal hydrides, nanoparticles of MgH2 have been synthesized using this method by Sheppard et al. with thermodynamic investigations indicating that a reduction in the size of the particles to ~7 nm had the effect of decreasing the equilibrium pressure, and hence the decomposition enthalpy and entropy.[25,26] This is promising for developing an enhanced knowledge of how MgH2 nanoparticles function and hence how they will perform as a hydrogen storage medium. Mechanochemical synthesis may become more important in the development of novel composite materials, in which the by-product may play a role in the sorption process, i.e., as a catalyst or as a thermal conductor for mediation of sorption conditions.
This leads us to the concept of encapsulation of light metal hydrides, where the encapsulation medium may play an important role in facilitating the sorption of hydrogen. Jeon et al. published research on the encapsulation of Mg within a poly(methyl methacrylate) (PMMA) polymer matrix, with the aim of creating an environment around the Mg which is selectively permeable to hydrogen only.[27] It should be noted here that the concept of magnesium nanoparticle encapsulation has been considered by others before this work; de Jongh et al. demonstrated the melt infiltration of magnesium into carbon nanosupports in 2007.[28] There were, however, several significant developments in the work by Jeon et al. From powder X-ray diffraction (XRD) data it was established that the nanoparticles were stable in air (after three days of exposure). The nanoparticles also exhibited a reversible storage capacity of up to ~6 wt % of hydrogen, which translated to ~4 wt % once the system mass was accounted for.
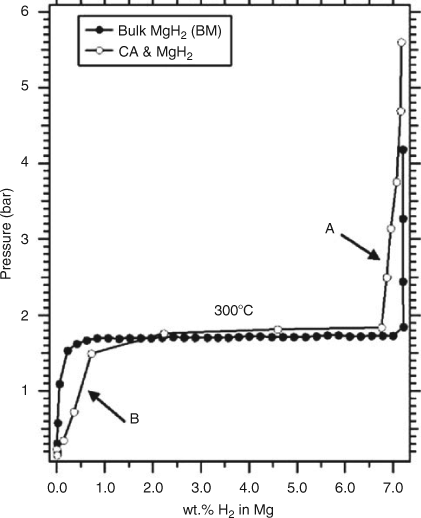
Furthermore, the mechanical flexibility of the polymer is important in extending the longevity of such a material, since expansion and contraction of the material during sorption cycling leads to decrepitation issues when powders are pressed into pellets or compacts.[29] Publications are now emerging on similarly themed nanosupports. For example, Paskevicius et al. were able to show how, from organic precursors, magnesium hydride could be encased within a carbon aerogel (CA).[30] The carbon nanostructure was not wholly effective in generating nanoparticles alone, however, since some bulk MgH2 was also identified. Furthermore, the hydride particles were susceptible to decomposition in air, since MgO is evident in their transmission electron microscopy (TEM) work after minimal air exposure, meaning that the aerogel only supports the material, and does not protect the hydride from oxidative decomposition. Nonetheless, these developments are important in establishing new designs for hydrogen storage materials, and novel synthesis routes, particularly since their results indicate full desorption of hydrogen from the supported material at 300°C, which is comparable with the bulk (Fig. 4).
|
From the above examples it is evident that organic–magnesium precursors are emerging in the literature for the synthesis of nanometer-scale magnesium owing to the added protective nature of the organic component of the mixture (e.g., bis(cyclopentadienyl)–magnesium, in the work of Jeon et al. and dibutyl magnesium from Paskevicius’ research) towards the active storage material.[27,30] In fact, this latter synthetic route to MgH2 has been known since the 1950s.[31] Similarly, preparation of Grignard reagents has traditionally been used in organic chemistry for the production of more reactive magnesium, and these methods have now become important in the development of novel routes to Mg for hydrogen storage.[32] One of the most difficult aspects of working with Mg and its hydride is the acute sensitivity to air and moisture, and so solvents used for its synthesis must be chosen carefully, as highlighted by Haas and Gedanken.[33] They used THF and dibutyldiglyme alongside Grignard (alkyl-magnesium chloride) precursors to synthesize Mg particles 4 nm in size by a sonoelectrochemistry technique.
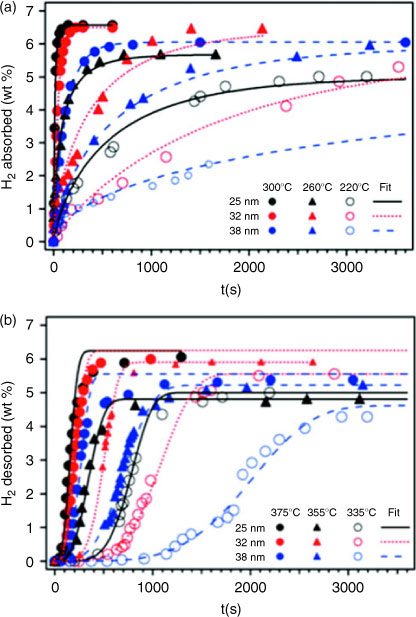
Organic surfactants have also been utilized for the synthesis and stabilization of magnesium nanoparticles with hydrogen storage and energy applications in mind. Aguey-Zinsou and Ares-Fernández used tetrabutylammonium bromide (TBA) to generate 5 nm colloids of Mg, with dehydrogenation beginning at 358 K, which is significantly lower than that of commercial, micrometer-scale MgH2 (~673 K).[34] Subsequently, magnesium-based colloids were prepared by Kalidindi and Jagirdar, involving the conversion of metal into organometallic colloids using THF, hexadecylamine (HDA), and HDA-toluene, by a solvated metal atom dispersion technique.[35] These colloids do not show selective permeability to hydrogen as in the work of Jeon et al. but developments in this field could yield important results for both stabilizing and protecting Mg in air. Moreover, solvent-based techniques for the production of nanostructured Mg and MgH2 highlight the search for a means to purify products whilst preventing reaction with air and moisture. For instance, attempts to remove LiCl generated by reactive milling by Paskevicius et al. were unsuccessful using THF alone, despite earlier work using THF (or diethyl ether) in analogous wet reactions to synthesize the metal.[26,36] Efforts have been made recently to establish controlled synthesis of Mg in various organo–potassium solutions.[37] Norberg et al. showed that the hydrogen absorption and desorption kinetics of the solvent-synthesized material are improved with respect to that of commercial micrometer-sized materials (Fig. 5a and 5b), with the reasons being attributed not only to the decrease in particle size but also to increased surface defects. This is not a new premise; ball milling is an easy way to achieve particle size reduction and surface defect enhancement. Therefore, the study by Norberg is important in establishing a novel, less energy intensive method for nanostructured Mg production. It should be noted, however, that the temperatures for evolution of the hydrogen are still relatively high (Fig. 5b), and it will be necessary to establish the onset temperature at which the hydrogen is first evolved.
|
Hence the synthesis of hydrogen storage materials, particularly magnesium, in solvents and with organic components is fast developing, with promising hydrogen sorption results and opportunities for morphology control. Refining the syntheses and demonstrating how these are competitive with solid state routes will be key to their future as economic, low energy routes to hydrogen storage materials.
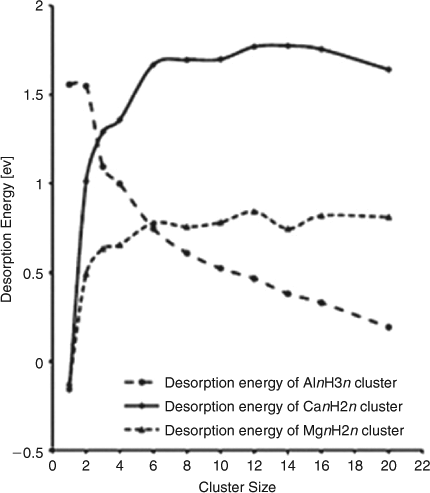
Of the heavier alkaline earth metals, Ca in clusters of CanH2n (where n = 1–4, 6, 8, 10, 12, 14, 16, 20) has been investigated by Density Functional Theory Vienna Ab-initio Simulation Package (DFT VASP) calculations to establish optimum cluster sizes for hydrogen storage, representing an important comparison to work on MgnH2n and AlnH3n clusters.[38] This work indicates that small CanH2n cluster sizes (n < 6) will have lower desorption energies than larger clusters, but that CaH2 alone shows less promise as a hydrogen storage medium than pure Mg and Al hydrides (Fig. 6).
|
Using similar computational techniques, calculations have also been conducted to establish how calcium may be embedded on graphene nanoribbons (GNRs) to give a theoretical hydrogen capacity of 5 wt % (Fig. 7).[39] Zigzag GNRs were shown to have a significantly stronger Ca binding energy in comparison to that of the armchair conformer, and so boron was used as a dopant in the case of the latter to control the sites at which Ca is embedded as it was predicted to prevent aggregates from forming. This work indicates that graphene nanostructures may be suitable hosts for Ca as a hydrogen store. Experimental work is now required to validate that this is indeed a practical storage method.
|
Sputtering has been used to form CaH2 thin films of 200 nm in thickness.[18] Results from optical spectrophotometry measurements did not show there to be any significant changes upon application of 1 bar of hydrogen to the hydride films at room temperature, but by measuring the transmittance through the thin films (i.e., by spectrophotometry) after exposure to air, the formation of Ca(OH)2 was indicated. Nanostructuring, in this instance, did not appear to have an effect on the subsequent reactions with MgB2 under hydrogen to establish if the calcium borohydride could be formed from the thin film. Catalysis, encapsulation, and formation of multinary phases incorporating Ca may represent the best approaches to the element’s utility for hydrogen storage.
Complex hydrides containing both alkali and alkaline earth metals may prove to be effective storage materials, but nanostructuring has yet to be developed in such systems. The high storage capacity of compounds such as NaMgH3 (theoretically 6 wt %), is particularly promising.[40,41] Wu et al. identified the mechanism by which hydride diffuses within NaMgH3 by a combination of theoretical approaches and experimental methods such as powder neutron diffraction (PND) and neutron vibrational spectroscopy (Fig. 8). Deuterated samples were analyzed at 5, 300, and 370 K by PND and Rietveld refinement was conducted to establish fundamental information regarding the temperature-dependant behaviour of the structure. Wu et al. explained that the mobility of H– in this ternary system was facilitated at low temperatures (5 K) by lattice distortion. This will undoubtedly become important in establishing the nanodesign criteria for hydrogen storage performance if this system is to be developed.
|
Of the light metal hydrides that have been synthesized at the nanometer scale to date, it is clear that most work has focussed on Mg-based materials. In addition, nanoscaffolds are now being readily pursued as a means by which the hydrides (or indeed the metals themselves) may be constrained or templated.[42] From porous carbon to complex polymers, the design of the nanoscaffold demands as much investigation as the active storage material itself.[43] As a result, porous materials are being designed specifically as host matrices for hydrogen storage and are developing rapidly in unison with metal hydride systems.[44]
Complex Hydrides
Borohydrides
Borohydrides possess some of the highest theoretical H2 gravimetric capacities and so have attracted considerable research interest.[45–47] LiBH4, for example, can theoretically store 18.5 wt % of H2 and dehydrides to lithium hydride and boron with the release of hydrogen. LiBH4 releases 13.8 wt % of H2 in this dehydrogenation step (Eqn 1) but proceeds no further under moderate conditions because of the high decomposition temperature of LiH (953 K):
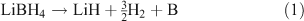
Mg(BH4)2 can theoretically store 14.9 wt % of H2 while Ca(BH4)2 has a theoretical gravimetric capacity of 11.6 wt % of H2. Hence, as with many simple (binary) light metal hydrides, the major disadvantage in the use of borohydrides is the temperature required for H2 release (dehydrogenation begins at 673 K).[48] Likewise, reversibility in these complex hydrides is also problematic with high temperature and pressure required for hydrogen uptake. For example, bulk LiBH4 rehydrogenation occurs under a pressure of 150 bar of H2 at 873 K.[49] Controlling the particle size of the borohydrides can begin to overcome some of these difficulties, as will be discussed below.
Physical Nanostructuring and Nanocatalysts
A strategy of ball milling with additives has been successful in destabilizing borohydrides, leading in turn to lower H2 release temperatures. Ball milling LiBH4 with MgH2 results in a reduction of the reaction enthalpy for hydrogenation and dehydrogenation of 25 kJ mol–1 of H2 when compared with the bulk material.[50] The maximum H2 release obtained was 10 wt %. Addition of metal halides to the milled mixture can have a marked effect on dehydrogenation just as in light metal hydride systems. Zhang et al. ball milled LiBH4 with halides of Ce and La to investigate the possible destabilization effect on LiBH4.[51] The authors found that the addition of CeCl3 to LiBH4 resulted in a dehydrogenation onset temperature of 493–553 K, which is much lower (by 120–180 K) than that of bulk LiBH4. A total of 3.05 wt % of H2 could be desorbed with respect to the total weight of the reactants. By contrast, for LaCl3 the dehydrogenation temperature decreased to 543 K (3.7 wt % of H2 was realised when the sample was rehydrogenated). Similarly, milling the dichlorides of Fe, Co, and Ni (FeCl2, CoCl2, and NiCl2) with LiBH4 produced materials with lower desorption temperatures.[52] By comparison, bulk LiBH4 released H2 in two steps: 1 % was desorbed at 583 K with the remainder released at 693 K. The transition metals destabilize LiBH4, which Zhang and Liu suggest is directly related to the relative instability of the Fe, Co, and Ni borohydrides themselves. The LiBH4–Fe(Co, Ni) ball milled samples also produced transition metal borides which then appear to catalyze subsequent H2 release from LiBH4. Similar effects are observed with MnCl2 with the resulting mixture (LiMn(BH4)3 + LiCl) desorbing 4.5 wt % of H2 at 473 K,[53] although rehydrogenation of the system was unsuccessful. Fluoride additives produce similar results and when Guo et al. ball milled LiBH4 with TiF3, H2 desorption commenced at 373 K with release of 5.0 wt % of H2 by 523 K.[54] Replacing metal halides with alanates (such as LiAlH4 as a source of Al) appears to improve kinetics in LiBH4 by similar mechanisms.[55] In the Al-containing system, dehydrogenation was complete at 673 K, with 2–2.4 wt % of H2 released in terms of the composite material (12–13.5 wt % when considering LiBH4 alone). A gradual loss of capacity was observed over multiple cycles (and attributed to diborane formation). As per the chlorides, the formation of AlB2 drives the dehydrogenation temperature lower.
Just as above with LiBH4, the dehydrogenation temperatures of group 2 borohydrides can be similarly reduced by milling with suitable additives and Mg(BH4)2 has been the subject of investigations with chloride additives by analogy to the lithium compound. Notably, improved kinetic performance for both the hydrogenation and dehydrogenation of Mg(BH4)2 is possible by milling with TiCl3 or SnCl3.[56] The addition of these chlorides results in a five-fold reduction in the time for the release of 95 % of the total H2. Adding the chlorides yielded a 9.7 wt % of H2 release compared with 7.7 wt % of H2 at 573 K for the unmodified material (in terms of the Mg(BH4)2 component only).
Nanoconfinement
Nanoconfinement of borohydrides is now a principal focus of contemporary research.[43] Nanoparticles of borohydride can be incorporated inside a porous scaffold host such as carbon and the particle size of the contained borohydride proves to be important; as the pore size of the host is reduced so the dehydrogenation temperature is depressed. For example, LiBH4 constrained in 13 nm diameter pores in carbon dehydrogenates up to 50 times faster than the native borohydride,[57] and Cahen et al. found that LiBH4 nanoconfined inside mesoporous carbon with a pore diameter of 4 nm resulted in 3.4 wt % of H2 being evolved at 573 K in 90 min. This compares with 0.2 wt % evolution of H2 from bulk LiBH4 at the same temperature (Fig. 9).[58]
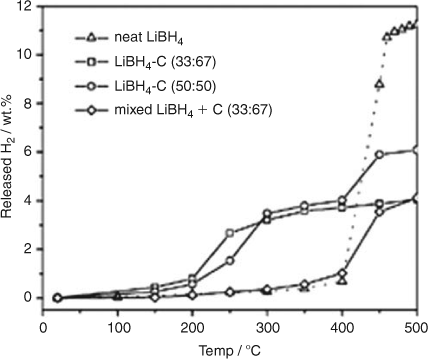
|
Other studies of LiBH4 in porous carbon have demonstrated that not only is the dehydrogenation temperature for LiBH4 reduced compared with the bulk material but so too is the reversibility of the hydrogenation/dehydrogenation process improved.[59,60] Hydrogenation of LiBH4 confined this way could occur under a pressure of 60 bar of H2 at 523 K. The nanoconfinement of LiBH4 also resulted in the suppression of an unwanted byproduct, diborane. By removing this byproduct, the borohydrides become a much more attractive prospect for H2 storage. Furthermore, when Ni is added as an additive, LiBH4 confined in mesoporous carbon can release ~3.5 wt % of H2 per gram (14 wt % of H2 per gram in terms of LiBH4 only) and is able to rehydrogenate at a higher rate. It was proposed that the combination of additive and nanoconfinement could alter the rehydrogenation reaction pathway.[61] By analogy to LiBH4, Mg(BH4)2 can be nanoconfined in activated carbon to also result in lower dehydrogenation temperatures compared with the respective bulk material.[62] The first H2 desorption step occurred at 530 K in the nanoconfined sample compared with 542 K in the bulk material with the second H2 desorption step peaking at 590 K for the nanoconfined sample as compared with 649 K for bulk Mg(BH4)2 with 6 wt % of H2 being released. Sartori et al. went on to study Mg(BH4)2 nanoconfined in an activated carbon scaffold by small angle X-ray and neutron scattering.[63] Although heating to 673 K resulted in no change to the size distribution of the confined particles (a distribution maintained at 1–6 nm in diameter), the thermal treatment enforced changes in the surface morphology; a change in surface roughness which might be directly related to the release of hydrogen. By comparison, heating bulk Mg(BH4)2 to 673 K resulted in both the particle size and morphology being affected (although the size distribution of the bulk particles is not discussed). From these observations, the nanoconfinement of the borohydride plays a vital role in the improvement of the dehydrogenation kinetics by the dimensional constraint of hydride particles.
Nanoconfinement in other porous hosts has thus far been somewhat limited. LiBH4 has been successfully constrained in oxides such as SiO2 and ZrO2.[64,65] When constrained in SiO2, the desorption of H2 commences at 423 K.[64] The nanoconfined LiBH4 cannot be rehydrided and the reaction is irreversible. This is a result of the decomposition products of LiBH4 and SiO2 reacting to form Li2SiO3 and Li4SiO4. Nanoconfinement in SiO2 and other oxides such as ZrO2 and Al2O3 were studied by both computational and experimental methods.[65] Thermogravimetric–mass spectrometry (TGA-MS) measurements showed that ZrO2 was the only oxide that did not react with LiBH4, hence the LiAlH4/ZrO2 system is reversible but the dehydrogenation temperature was not significantly lowered compared with bulk LiBH4.
Alanates
Alanates came to prominence as a hydrogen storage material in 1997 with the discovery made by Bogdanović et al. that doping with Ti resulted in the destabilization of NaAlH4.[66] This in turn led to improved properties of H2 uptake and release. NaAlH4 decomposes in two steps. In the first step the alanate dehydrogenates to Na3AlH6 releasing H2 (theoretically 3.7 wt %; Eqn 2). In the second step, Na3AlH6 decomposes to NaH, theoretically releasing 1.8 wt % of H2 (Eqn 3). The NaH then decomposes to release further H2, although because of the high temperatures required this step is not considered viable for a practical storage system (Eqn 4).
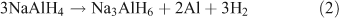
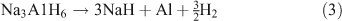

Similar to the borohydrides, the alanates suffer from high hydrogenation and dehydrogenation temperatures and poor reversibility is also an issue. Reducing the particle size of the alanates is seen as a potential method to overcome these problems as will be discussed below.
Physical Nanostructuring and Nanocatalysts
Following the work of Bogdanović,[66] TiCl3 has been studied as an additive with nanoparticles of NaAlH4. Vegge initially studied the potential role of Ti as an additive to NaAlH4 nanoparticles by DFT methods.[67] From these calculations, it was found that both the morphology and the presence of Ti are important factors in improving dehydrogenation. Indeed, experimentally when ball milling has been used to reduce the particle size of NaAlH4 in the presence of TiCl3, the latter acts as a catalyst for hydrogenation/dehydrogenation in a similar way to that in borohydride systems.[68] In fact, other sources of Ti appear to be at least as effective as the trichloride in catalysis (as is also observed for amides in the following section). Milling sodium alanate with micro- and nanosized TiO2 powders demonstrated that up to 5.1 wt % of H2 was released between 393 and 503 K for the latter; an improvement of 20–40 K over the former.[69] Also by analogy to the borohydrides, there is some recent evidence that rare earth metals can catalyze hydrogen sorption in alanates when milled.[70] Ce(SO4)2 and LaCl3 both improve H2 desorption compared with the bulk material; 5 % of the former yielded 3.97 wt % of H2 commencing at 378 K, while 5 % of the latter gave 4.3 wt % of H2 commencing at 428 K. Interestingly, the authors discovered that doping NaAlH4 with Ce(SO4)2 results in the formation of a porous microstructure. In a recent and promising extension to the approaches used to prepare nanometer-scale MgH2, Xiao et al. have used reactive ball milling (mechanochemical synthesis) to produce nanostructured NaAlH4.[71] These reactively milled samples absorbed 4 wt % of H2 in 10 min at 393 K and could desorb 95 % of the total H2 within 30 min at 423 K.
Ball milling has also been employed effectively to reduce the particle size of Mg(AlH4)2. Nanometer-sized Mg(AlH4)2 was synthesized by the reactive ball milling of AlH3 and MgH2. The resulting alanate had a decomposition enthalpy of approximately zero over two decomposition steps (although the first step has a rather high activation energy of 119 kJ mol–1).[72] Varin et al. had previously attempted to synthesize nanocrystalline Mg(AlH4)2 by the ball milling of Mg and Al in an atmosphere of H2 without success but succeeded when ball milling Na(AlH4)2 with MgCl2.[73] Nanocrystalline Mg(AlH4)2 was formed with an average particle size of 18 nm (with an NaCl by-product). The alanate decomposed to MgH2 and Al with the release of H2 between 398 and 453 K.[74] At 423 K, 2.24 wt % of H2 was released. The presence of these dehydrogenation products was confirmed by XRD analysis (Fig. 10).
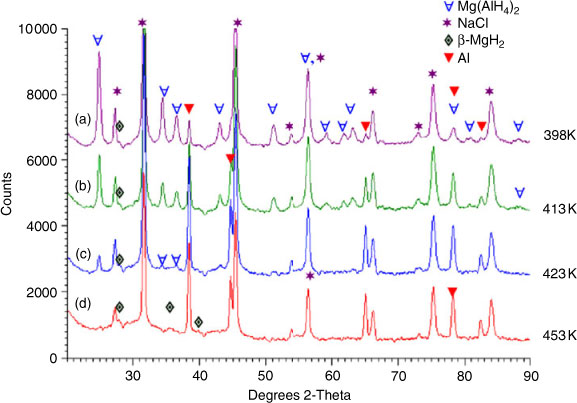
|
Nanoconfinement
Nanoconfinement of alanates, as with borohydrides, has been demonstrated to have a dramatic effect on storage performance. Notably, NaAlH4 nanoparticles confined in CAs with a pore size as low as 13 nm (Fig. 11) have led to a reduction in dehydrogenation temperature from 503 K in the bulk alanate to 413 K in the confined material.[75,76]

|
Furthermore, Neilsen et al. recently demonstrated that adding TiCl3 nanoparticles to CA-confined NaAlH4 promoted far faster dehydrogenation compared with both bulk and ball milled alanate.[77] The onset of H2 release was almost immediate in these confined materials (306 K) and the full extent of nanoconfinement and catalysis can be appreciated from the data in Table 1. The NaAlH4/aerogel/TiCl3 sample released 2.9 wt % of H2 (with respect to NaAlH4) during its first decomposition cycle, with 1.6 wt % of H2 being released during the fourth decomposition cycle.

|
If the pore size of the host structure is decreased below 13 nm, then the dehydrogenation temperature can be reduced further still (following a similar relationship to that observed in nanoconfined borohydrides). Thus Gao et al. found that NaAlH4 contained in 2 nm pores had the lowest H2 desorption temperature with H2 release commencing between 373 and 393 K.[78] Similarly, confinement of NaAlH4 in other carbons has been attempted with successful results. NaAlH4 confined in mesoporous carbon with a pore size of 4 nm (prepared using mesoporous silica as a template and sucrose as the carbon source) released 5.0 wt % of H2 at 453 K in 40 min compared with 0.5 wt % of H2 released from the bulk material in 90 min at the same temperature.[79] The cycling properties of NaAlH4 were also improved in this system over the bulk material. During cycling, the nanoconfined NaAlH4 releases 5 wt % of H2 during the first cycle, which decreases to 4.6 wt % for cycles 2–6 and then decreases further to 4.2 wt % for subsequent cycles. By comparison, bulk NaAlH4 releases 0.5 wt % of H2 during the first cycle which decreases to 0.2 wt % of H2 after five hydrogenation/dehydrogenation cycles under equivalent conditions. In a similar fashion, Baldè et al. studied the effect of constraining NaAlH4 within carbon fibres.[80] The onset of H2 release was possible below 343 K for particles 2–10 nm in size.
Mesoporous inorganic hosts can also be effectively utilized to confine alanates and as with borohydrides when so-confined, the dehydrogenation temperature is decreased.[81] H2 (1.4 wt %) is released from NaAlH4 confined in ordered mesoporous silica (OMS) at 423 K compared with a negligible amount in bulk, uncatalyzed NaAlH4 at the same temperature. Furthermore, raising the temperature to 453 K releases 3.0 wt % of H2 from the nanoconfined sample compared with 0.3 wt % for the bulk material under the same conditions. The reversibility of NaAlH4 is also improved when nanoconfined in OMS. When both bulk NaAlH4 and nanoconfined NaAlH4 were rehydrogenated at 398 K for 3 h and subsequently dehydrogenated, the nanoconfined sample released 0.6 wt % of H2 at 423 K compared with a negligible amount for the bulk material.
Amides, Imides, and Nitrides
Amides entered the hydrogen storage arena in 2002 when Chen et al. demonstrated that hydrogen could be reversibly stored within lithium nitride (Li3N).[82] The discovery of storage in this system, which involves the making and breaking of N–H bonds, has also facilitated the understanding of mechanism in other complex hydride systems.[83,84] The hydrogen uptake–release process involves two steps, theoretically storing a total of 10.4 wt % of H2 (Eqn 5):
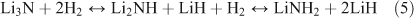
Practically, however, hydrogen release from imide to nitride requires high temperature conditions and so only the reaction involving H2 uptake–release between lithium amide (LiNH2) and lithium imide (Li2NH) is realistically reversible (yielding ~6.5 wt %)[81] and within sight of the system gravimetric and volumetric United States’ Department of Energy (US DOE) targets for 2015.[85] Among the challenges still to be met in amide systems, however, are high desorption temperatures, slow kinetics, and limited reversibility/cycle life. Nanodesign approaches potentially represent a route to overcome these barriers.
Physical Size Reduction by Ball Milling
Pinkerton et al. first studied the effects on the decomposition of one of the hydrogenated compounds in the Li–N–H system, LiNH2, by ball milling.[86] This is a competing reaction that occurs during the dehydrogenation of the LiNH2–LiH mixture (5) and it is of critical importance for hydrogen storage in the system since 1) the H2 capacity is negatively affected by the depletion of nitrogen because of ammonia emission, and 2) the ammonia produced can also poison the proton exchange membrane of a fuel cell (PEMFC). The above authors established that during LiNH2 decomposition the rate of ammonia evolution was dependent on the particle size. It was later shown that mechanical activation of as-received LiNH2 could decrease the particle size to 5.5 nm and that the subsequent increase in the surface area and defect density were responsible for the reduction of the onset temperature for dehydrogenation from 393 K to 298 K.[87] Furthermore, 6Li magic angle spinning nuclear magnetic resonance (MAS NMR) studies at room temperature showed a change in the local electronic structure around Li nuclei in milled LiNH2.[88] Importantly, temperature programmed desorption/mass spectrometry (TPD/MS) studies also illustrated that pre-milling LiNH2–LiH mixtures eliminated NH3 emission during dehydrogenation.[89] However, although milling can be an effective means to obtain Li–N–H nanoparticles with enhanced sorption properties, milling for prolonged periods (>100 h), can be detrimental to performance and lead to particle agglomeration.[90]
By analogy, the effect of mechanical milling has also been studied in the LiNH2–MgH2 system. Rohit et al. explored Li–Mg–N–H dehydrogenation kinetics in samples milled over different times.[91] They reported the in-situ formation of Mg(NH2)2 for mixtures milled for >15 h, which was responsible for enhanced dehydrogenation kinetics. Indeed, a rate increase of up to 49 % for 45 h ball-milled mixtures was observed. Scanning electron microscopy (SEM) images showed, however, that the particle sizes ranged from 2–6 μm and so agglomeration would appear to prevail at longer milling times just as in the Li–N–H system, although not necessarily to the great detriment of sorption kinetics.
Nanocatalysts
Addition of catalyst nanoparticles can address the slow dehydrogenation kinetics in complex hydride systems such as amides. Ichikawa et al. first reported improved desorption kinetics for a LiNH2 + LiH ball milled mixture by the addition of micrometer-size TiCl3.[92] The same authors examined the role of other catalysts in the Li–N–H system, including VCl3 and Ni, Fe, and Co nanoparticles (~10 nm diameter), commonly used for alanate systems.[93,94] They reported similarities between TiCl3 and VCl3 while discarding the use of the aforementioned metal nanoparticles because of ammonia production. No results for the catalyst-enhanced sample were reported after the third dehydrogenation cycle, suggesting perhaps a decrease in capacity over further cycles. This can be attributed perhaps to the formation of a chloride by-product during the hydrogenation/dehydrogenation process. In fact, nanosized Ti and TiO2 proved to be equally effective catalysts without unwanted by-product formation[95] and hydrogen desorption was dependent on additive particle size as seen in other complex hydride systems.[96] Furthermore, an increase in ammonia was also observed when increasing the particle size of Ti and TiO2 to the micrometer scale (Fig. 12). Several efforts have been made in order to understand the role of TiCl3 in the improved dehydrogenation properties of the LiNH2 + LiH mixture.[97] It has been claimed that TiCl3 acts as a grinding reagent, reducing the particle size and increasing the surface area of the hydrides, and preserves the active surface of LiNH2 improving the desorption kinetics. In addition, Isobe and coworkers used X-ray absorption spectroscopy (XAS) to propose that a synergistic effect exists between LiNH2 and TiCl3.[98] First principles calculations suggest that Ti occupies Li sites in the structure of milled LiNH2.[99]
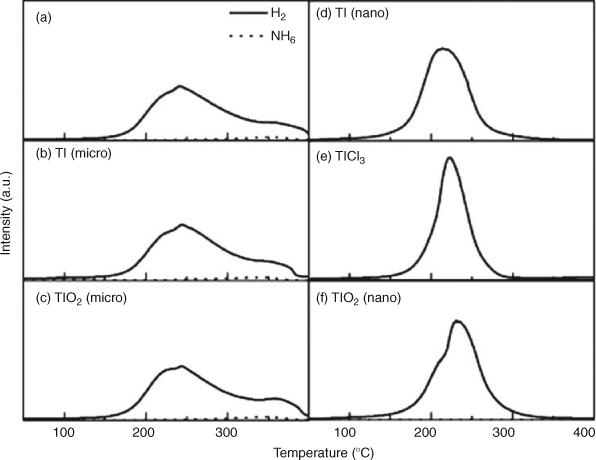
|
Despite the limited success of Ni as a nanocatalyst in the Li–N–H system, its addition to quaternary amide systems such as Li–Al–N–H and Li–B–N–H has proved beneficial.[100,101] In the former example, the addition of 5 % of 20 nm Ni particles contributed to the increased capacity of milled Li3AlH6–LiNH2 on cycling. Kojima et al. demonstrated that the reversible capacity of the Li–Al–N–H system (which desorbs 6.9 wt % of H2 in the first cycle and 1–2 wt % thereafter) was improved by the addition of Ni nanoparticles to 3–4 wt %; hence doubling the capacity of the pristine material. Similarly, adding 11 wt % 8 nm particles of NiCl2 to Li3(BH4)(NH2)2 caused a reduction in the dehydrogenation temperature onset of 112 K.
The quaternary Li–Mg–N–H system is in some respects more attractive than the Li–N–H system since depending on the Li : Mg ratio, the thermodynamics of uptake and release (and hence reversibility) become more favourable.[102] The kinetics in this system, however, remain problematic and nanocatalysts have been applied to try and address this. A hydrogen capacity of 5 wt % can be obtained for a LiNH2 : MgH2 molar ratio of 2.15 : 1 and the addition of single-walled carbon nanotubes (SWNTs) increases the dehydriding kinetics by a factor of 3 at 473 K.[103] More recently, studies on the use of nitrides (BN and TiN) as nanocatalysts have been described by Nayebossadri et al.[104] The dehydrogenation temperature in the Li–Mg–N–H system was investigated as a function of nanocatalyst concentration and particle size. Increasing the TiN nanoparticle concentration or reducing the particle size reduced the onset temperature whereas there was no such direct relationship for the nanometer-scale BN additive. It was proposed on this basis that BN acts so as to enhance diffusion across the interfaces in the system whereas TiN improves the surface reactivity of the LiNH2 particles. Thus TiN effectively behaves similarly to TiCl3 in the Li–N–H system. Indeed, the authors suggest the possibility of TiN formation by milling of LiNH2 and TiCl3 leading to the formation of the intermediate Ti(NH2)4, that would further decompose because of the high exothermic nature of the decomposition reaction observed in analogous systems.[105]
Complex Amides and Nanocomposites
Pinkerton et al. first reported the formation of the quaternary hydride Li3(BH4)(NH2)2 by direct reaction of LiNH2 and LiBH4 with a 2 : 1 molar ratio by ball milling or by heating.[106] This system is attractive given its high theoretical capacity (11.9 wt %; ~10 wt % at 523 K in practice) and minimal ammonia release. Dehydrogenation, however, yields Li3BN2 irreversibly. Wu and coworkers addressed the irreversibility problem by tuning the particle size of Li3(BH4)(NH2)2 by infiltration of the hydride into nanoporous carbon nanoscaffolds.[107] For example, on milling the hydride with activated carbon, AX-21, the dehydrogenation temperature was dramatically decreased and the samples infiltrated in AX-21 (2 nm pore diameter) were able to achieve partially reversibility (34 %) at 573 K under 50 atm of H2. However, improved reversibility occurs at the expense of ammonia formation, in contrast to the bulk material. This implies that perhaps a different (de)hydrogenation mechanism might occur at the nanometer-scale.
The LiNH2–MgH2–LiBH4 system also has a high gravimetric capacity (8–10 wt % experimentally) and it is believed that MgH2 acts to self-catalyze the material to release hydrogen.[108] Niemann et al. demonstrated that the crystallite size of MgH2 mediated the onset dehydrogenation temperature and that addition of MgH2 10 nm in size promoted H2 release (5–6 wt %) showing a main H2 evolution occurring at ~423 K whereas in particles with a crystallite size of 35–75 nm H2 was mainly released at 448 K.[109] The same authors also studied the catalytic effect of some nanoadditives (Ni, Co, Cu, Fe, and Mn) in the system with particle sizes ranging from 10–20 nm. Co and Ni reduced the temperature in the initial step to 348–373 K whereas other additives such as Fe and Mn increased the rate at which the hydrogen was released.[110]
Chemical Synthesis of Nanomaterials
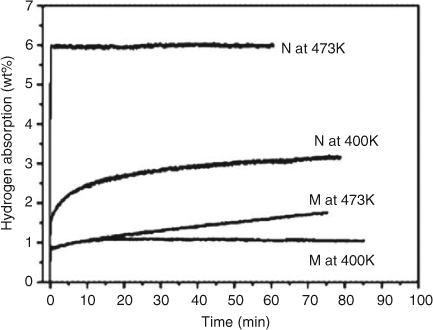
The chemical design of nanostructured amides offers opportunities to overcome thermodynamic and kinetic barriers but as yet the number of nanomaterials so-prepared is small. Xie et al. fabricated Li2NH hollow nanospheres by a plasma metal reaction[111] based on the Kirkendall effect. This effect promotes the formation of lithium defects which were proposed to be at the origin of the formation of the hollow nanostructure.[112] The majority of these nanospheres had diameters ranging from 100–200 nm with a shell thickness of 20 nm (Fig. 13a and 13b). The nanospheres exhibit improved hydrogen absorption/desorption properties compared with Li2NH microparticles synthesized by LiNH2 decomposition under vacuum. Li2NH nanospheres demonstrated outstanding kinetics and stored 6.0 wt % of H2 after 1 min at 473 K whereas the Li2NH microparticles could only uptake 1.6 wt % after 1 h at the same temperature (Fig. 14). It was also noted that the hollow nanospheres could remain intact after annealing at 573 K (Fig. 13c) and even after hydrogenation at 573 K under 35 bar (Fig. 13d). The activation energies (Ea) for hydrogen absorption were significantly different for the microparticles and nanospheres with values of 225 and 106 kJ mol–1, respectively. Moreover, a decrease in the desorption temperature by 115 K was observed for the Li2NH hollow nanospheres. These differences were attributed to the larger specific surface area, shorter Li+ diffusion pathways, and enhanced contact between LiNH2 and LiH at the nanometer-scale.

|
|
The same authors reported using similar methods for the formation of Mg(NH2)2 nanospheres.[113] Mg3N2 nanocubes were prepared by vaporizing bulk Mg in a NH3 atmosphere (in a plasma–metal reaction) and consequently treated with NH3 at 623 K for 12 h to form the amide nanospheres. An average particle size of 100 nm and a shell thickness of 10 nm were reported. In order to assess the dehydrogenation properties the hollow nanospheres were reacted with MgH2 nanoparticles and the results were compared with the performance of Mg(NH2)2 nanoparticles prepared by heating MgH2 nanoparticles under 4 bar of NH3 at 623 K for 12 h. Despite showing superior kinetics, the hollow nanospheres only released 2.5 wt % of H2 and absorption was not reported. Further studies by the same authors were conducted by ball milling LiH with the aforementioned Mg(NH2)2 hollow nanospheres (100 nm diameter) and Mg(NH2)2 particles with 500 and 2000 nm diameter to investigate the effects of particle size on desorption temperature.[114] The hollow Mg(NH2)2 nanospheres hydrogenated at the highest rate under 80 bar of H2 at 500 K and after 30 min the 100, 500, and 2000 nm samples took up 3.6, 3.2, and 3.0 wt % of H2, respectively. An analogous decrease in the dehydriding temperature was observed with diminishing particle diameter; 2000, 500, and 100 nm diameter particles/spheres desorbed at 539.8, 514.8, and 482.5 K, respectively, corresponding to activation energies of 182, 134.7, and 122.2 kJ mol–1, respectively.
Ammonia Borane
Ammonia borane (NH3BH3, AB) has attracted a great deal of attention as result of its low molecular weight and high hydrogen content (19.6 wt %).[115,116] AB decomposes at temperatures below 373 K and yields a polyaminoborane-like material ((NH2BH2)n, PAB) that further decomposes at 423 K to a polyiminoborane-like material ((NHBH)n, PIB). This can be expressed as follows:
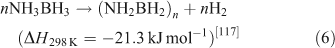
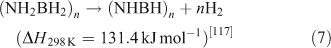
Overall, two equivalents of H2 can be released up to 473 K, which corresponds to a total capacity of 14.3 wt % of H2. At this temperature, however, small amounts of monomeric AB (NH2BH2), borazine (BHNH)3, and diborane (B2H6) are likely to be generated, with the effect of poisoning the hydrogen stream and consequently impairing PEM fuel cell performance.[118] Furthermore, the poor reversibility in the system makes AB regeneration impracticable on-board. Thus, the current challenges are to devise economic paths to fully re-charge the hydrogen storage material off-board and to prevent the formation of undesirable volatile impurities.
Gutowska et al. reported improvements in the thermodynamics and kinetics of AB by infiltration of the material within a mesoporous (7–8 nm diameter) silica scaffold (SBA-15).[119] TPD/MS studies of bulk AB and AB/SBA-15 demonstrated that for the latter, the onset temperature for H2 release was notably reduced. Moreover, the amount of borazine desorbed was diminished for the AB/SiO2 composite. 11B NMR spectroscopy confirmed the absence of borazine within the nanoscaffold suggesting a different dehydriding path. A difference by a factor of 20 in the calculated dehydrogenation enthalpy from differential scanning calorimetry (DSC) experiments for AB/SBA-15 over AB further supported this premise. Inelastic neutron spectroscopy also demonstrated different chemical properties in the bulk and the composite material; Paolone et al. observed that a phase transition in bulk AB at ~225 K was not found in the SBA-15 composite.[120] A similar concept involving embedding AB into a carbon cryogel (AB/C) (with pore diameters of 2–20 nm) (Fig. 15) was investigated by Feaver and Sepehri.[121] In this case, a one-step reaction rather than the predicted two-step process shown in Eqns 6 and 7 was observed. The composite showed a decrease in the dehydrogenation temperature to 363 K, which was attributed to (1) the nano-confinement of AB and (2) the catalytic effect of the carbon within the matrix. No borazine was detected on dehydrogenation of AB/C and 9 wt % of H2 was released with an enthalpy calculated as –120 kJ mol–1 (much more exothermic than bulk AB at –21 kJ mol–1). The exothermicity was rationalized as a possible reaction between the carboxylic groups of the carbon aerogel and the hydrogen released during the experiment.
|
Recently, Kurban et al. have used co-axial electrospinning to encapsulate AB in polystyrene (PS).[122] Polystyrene was chosen because of the thermal stability of the polymer during thermolysis of the AB and for its good permeability to H2. In the co-electrospinning process both the AB (core) and the PS (shell) were delivered by different routes through concentric nozzles. Both fluids are exposed to a high voltage that will eventually cause the expulsion of the fluids in the form of a jet. After a short time the jet bends, stretches, and thins forming nanofibres. The authors also explored the use of solvents with different miscibility towards AB showing that PS dissolved in semi-miscible (sMS) and immiscible (IS) solutions provided the best results. They demonstrated that porous sMS fibres decreased the dehydrogenation temperature of AB by 15–20 K whereas IS fibres (with minimal porosity) desorbed at similar temperatures to the bulk. In both cases no borazine was detected, perhaps due to trapping in the polymer.
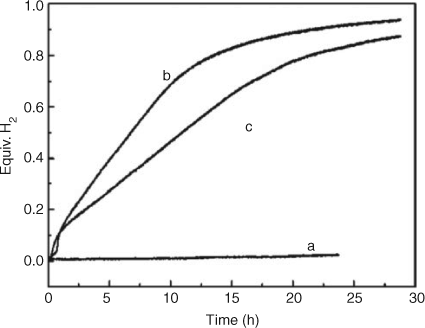
Inevitably, forming nanocomposites reduces the gravimetric capacity of AB and from this perspective nanocatalysts are a convenient alternative since the amount of catalyst added is usually ≤5 wt %. Neiner et al. demonstrated that by simple addition of BN nanoparticles to AB the onset temperature for hydrogen release could be reduced with a minimum onset at 344 K for an AB : BN mixture with a 1 : 4 weight ratio. Moreover the enthalpies for reactions (6) and (7) were –15 and 1.4 kJ mol–1, respectively.[123] However, during reaction (7) an increase in borazine over uncatalyzed AB was observed. It is not clear whether the second enthalpy value is therefore moderated by the positive vaporization enthalpy of borazine. Chen and coworkers demonstrated that the addition of 2 mol % of Co and Ni nanoparticles (3 nm in diameter) to AB by a ‘co-precipitation’ method decreased the onset temperature for H2 release to 323 K and detected no borazine.[124] Isothermal volumetric hydrogen release measurements also presented different features for the pristine and the catalytic enhanced sample; without additives, AB did not show any release of hydrogen when heating at 332 K for a day whereas for catalyzed AB, hydrogen was evolved vigorously at the same temperature. No hydrogen release was observed for AB without additives at this temperature for longer periods of time, suggesting that no initiation process involving the formation of the diammoniate of diborane (DADB) was required for the catalyzed samples (Fig. 16).
|
Concluding Remarks
Nano-approaches present considerable opportunities in hydrides, from simple binary systems to more complex bi and tri-metallic compounds with multistep dehydrogenation mechanisms. Whereas the landscape for light metal hydrides might have appeared limited following numerous studies considering nanoadditives as catalysts and physical size reduction methods (by ball milling), recent developments have illustrated that this is far from the case. Chemical synthesis has produced new morpholgies and new mechanisms to control size and shape and hence reactivity. Likewise, the development of nanocomposites, nanoencapsulation, and nanoconfinement has breathed new life into light metal hydride materials research. Indeed, the alkali metal and heavier alkaline earth metal hydrides would appear ripe for reappraisal in light of these developments.
Inevitably, ball milling and nanocatalyst approaches continue to be explored in complex hydrides where reaction ((de)hydrogenation) pathways are completely different to those in binary metal systems. In many cases, therefore, nanoadditives would appear to open up alternative, catalyzed reaction processes with reduced activation energies. Beyond this, however, nanostructuring of complex hydrides themselves can have profound consequences on both the thermodynamics of sorption and the kinetics of hydrogen uptake–release cycles. The nanometer-scaling of imides and amides, for example, induces rapid kinetics at reduced temperatures compared with micrometer-sized materials. Hugely encouraging gains in performance (reduced activation energies, diminished dehydrogenation temperatures) are also witnessed by the nanoconfinement of ammonia borane into mesoporous silica or encapsulation into PS.
There are evidently great opportunities for the design of hydrides at the nanometer scale and subsequent potential for step changes in hydrogen storage. While these advances also provide us with a track towards improved understanding of the storage of hydrogen in and on solids, they also, peversely, present us with additional challenges in constructing the systems that will ultimately act as a conduit between power generation and use. For example, how might one scale up production of the most exciting materials by orders of magnitude from milligrams to tonnes? How does the toxicology of such materials change over length scales and what are the consequences in terms of financial, ecological, and energy cost? These all assume that such materials can be succesfully integrated into storage and delivery systems – a significant engineering challenge. These are questions that, in time, will have to be answered, but inevitably the immediate issue is to push the envelope of materials performance and nanostructuring will play a leading role in this ambition.
Acknowledgements
DHG thanks the EPSRC for the funding of a studentship for JH under the SUPERGEN program (EP/E040071/1), the EPSRC, the Materials KTN and EADS IW for an Industrial CASE studentship for HR, and ScotCHEM for a SPIRIT studentship for NTR.
References
[1] J. Barber, Chem. Soc. Rev. 2008, 38, 185.| Crossref | GoogleScholarGoogle Scholar |
[2] N. D. McDaniel, S. Bernhard, Dalton Trans. 2010, 39, 10021.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtlSmsrrE&md5=14f930821cac3b8289bc7d54a3779504CAS |
[3] D. Gust, T. A. Moore, A. L. Moore, Acc. Chem. Res. 2009, 42, 1890.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtlyrtrnN&md5=31f8f2b6799f2b3c146caed304bb4212CAS |
[4] K. Kalyanasundaram, M. Graetz, Curr. Opin. Biotechnol. 2010, 21, 298.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXmslOht7w%3D&md5=0331f6b70ac7084cd92b8e321d959002CAS |
[5] (a) D. H. Gregory, J. Mater. Chem. 2008, 18, 2321.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXlsVOnsbk%3D&md5=f623028c8ecdfbe15f293672b10f9bb1CAS |
(b) T. K. Mandal, D. H. Gregory, Ann. Rep. Prog. Chem. Sect. A: Inorg. Chem. 2009, 105, 21.
| Crossref | GoogleScholarGoogle Scholar |
(c) T. K. Mandal, D. H. Gregory, Proc. IMechE, Part C: J. Mech. Eng. Sci. 2010, 224(C3), 539.J. M. Cameron, R. W. Hughes, Y. Zhao, D. H. Gregory, Chem. Soc. Rev. 2011, 40, 4099.
[6] A. W. C. Van den Berg, C. O. Arèan, Chem. Commun. 2008, 668.
| Crossref | GoogleScholarGoogle Scholar |
[7] S. Orimo, Y. Nakamori, J. R. Eliseo, A. Züttel, C. M. Jensen, Chem. Rev. 2007, 107, 4111.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhtVahsrbL&md5=69a6d615e973bb9760abe30b89c67dc8CAS |
[8] R. Schulz, J. Huot, G. Liang, S. Boily, G. Lalande, M. C. Denis, J. P. Dodelet, Mater. Sci. Eng. 1999, A267, 240.
| 1:CAS:528:DyaK1MXjslKit7c%3D&md5=ba57edb127cc7429eb5729c2faacbe98CAS |
[9] K.-F. Aguey-Zinsou, J.-R. Ares-Fernández, Energy Environ. Sci. 2010, 3, 526.
| 1:CAS:528:DC%2BC3cXnvVait7g%3D&md5=76d55ab049a4c6b98ad95e1ffbfcdc9dCAS |
[10] P. E. de Jongh, P. Adelhelm, ChemSusChem 2010, 3, 1332.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsFCkt77M&md5=f34ec3a7f8d7f065ad0a9e8660750d19CAS |
[11] T. K. Nielsen, F. Besenbacher, T. R. Jensen, Nanoscale 2011, 3, 2086.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXmt1yhtrw%3D&md5=9167774b92abbf88c84c9efd5a53a6e6CAS |
[12] C. J. Liu, L. Burghaus, F. Besenbacher, Z. L. Wang, ACS Nano 2010, 4, 5517.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtlWitbbJ&md5=854413399e2116c4658556efa3a08f88CAS |
[13] B. Peng, J. Liang, Z. Tao, J. Chen, J. Mater. Chem. 2009, 19, 2877.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXls12is7Y%3D&md5=5d1fa63cb96415e54c48b904ddd3503eCAS |
[14] A. L. Ortiz, W. Osborn, T. Markmaitree, L. L. Shaw, J. Alloy. Comp. 2008, 454, 297.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXjs12msbo%3D&md5=19d5ea0fbd30d0e285969b49d22eb79fCAS |
[15] S. Gautam, K. Dharamvir, N. Goel, J. Phys. Chem. A 2011, 115, 6383.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXmslOntbo%3D&md5=06a27ac5ac34a2b834e0ac76b51f7da0CAS |
[16] S. Hao, D. S. Sholl, J. Phys. Chem. Lett. 2010, 1, 2968.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtFygt7%2FJ&md5=91bfcfab75fb4e053e1960492a932ea8CAS |
[17] C. Pistidda, S. Garroni, C. B. Minella, F. Dolci, T. R. Jensen, P. Nolis, U. Bösenberg, Y. Cerenius, W. Lohstroh, M. Fichtner, M. D. Baró, R. Bormann, M. Dornheim, J. Phys. Chem. C 2010, 114, 21816.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsVais7bF&md5=395daddf9c37d866dc228d579ac255feCAS |
[18] M. Gonzalez-Silveira, R. Gremaud, H. Schreuders, M. J. van Setten, E. Batyrev, A. Rougier, L. Dupont, E. G. Bardají, W. Lohstroh, B. Dam, J. Phys. Chem. C 2010, 114, 13895.
| 1:CAS:528:DC%2BC3cXptFyjtbk%3D&md5=5540fad317d6edd942630689752f7282CAS |
[19] J. L. Maienschein, J. S. Bowers, J. S. Cantrell, T. A. Beiter, J. Alloy. Comp. 1992, 179, 157.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK38XhvVGjt70%3D&md5=dc8fa1517b10bd7df437cc72a9ffb547CAS |
[20] A. Zaluska, L. Zaluski, J. O. Strö-Olsen, J. Alloy. Comp. 2000, 307, 157.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXmtFOls7k%3D&md5=b7d95f45cab66310f89476c71ec31da0CAS |
[21] P. J. Herley, W. Jones, B. Vigeholm, J. Appl. Phys. 1985, 58, 292.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2MXksVSqtLs%3D&md5=fdc8c88e6fd27103dc50e1906feeac6eCAS |
[22] C. Zhu, S. Hosokai, I. Matsumoto, T. Akiyama, Cryst. Growth Des. 2010, 10, 5123.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtleiur3N&md5=1cb4bc229de4a6af7ea4b87f5c32bf47CAS |
[23] C. Zhu, S. Hosokai, T. Akiyama, Cryst. Growth Des. 2011, 11, 4166.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXpsFOhur8%3D&md5=e53331718066d1d7262c29954eeefb03CAS |
[24] C. Milanese, A. Girella, S. Garroni, G. Bruni, V. Berbenni, P. Matteazzi, A. Marini, Int. J. Hydrogen Energy 2010, 35, 9027.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtVGrurzM&md5=ba25e37a99226a071a3549df6386dbacCAS |
[25] D. A. Sheppard, M. Paskevicius, C. E. Buckley, J. Alloy. Comp. 2010, 492, L72.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXit1Crtb8%3D&md5=3e7562906f1f30ba9ce4899653e3d9acCAS |
[26] M. Paskevicius, D. A. Sheppard, C. E. Buckley, J. Am. Chem. Soc. 2010, 132, 5077.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXjs1Crsbg%3D&md5=e86b3072c7dcf5776062042f7d57dd1cCAS |
[27] K.-J. Jeon, H. R. Moon, A. M. Ruminski, B. Jiang, C. Kisielowski, R. Bardhan, J. J. Urban, Nat. Mater. 2011, 10, 286.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjvVGjtbw%3D&md5=ae2d2ca937b232c5745c25a804c31bc2CAS |
[28] P. E. de Jongh, R. P. Wagemans, T. M. Eggenhuisen, B. S. Dauvillier, P. B. Radstake, J. D. Meeldijk, J. W. Geus, K. P. de Jong, Chem. Mater. 2007, 19, 6052.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXht1aitrvF&md5=6596db9ef4fec60bdc0304c3cf1cd030CAS |
[29] A. Khandelwal, F. Agresti, G. Capurso, S. L. Russo, A. Maddalena, S. Gialanella, G. Principi, Int. J. Hydrogen Energy 2010, 35, 3565.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXktlShsrY%3D&md5=7553c94e0d63b74a35731ec28580ba27CAS |
[30] M. Paskevicius, H.-Y. Tian, D. A. Sheppard, C. J. Webb, M. P. Pitt, E. MacA. Gray, N. M. Kirby, C. E. Buckley, J. Phys. Chem. C 2011, 115, 1757.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXlsV2qug%3D%3D&md5=37dcf8b4c2cf3f9305d7d3ec6d82a603CAS |
[31] E. Wiberg, R. Bauer, Z. Naturforsch. B 1950, 5b, 396.
| 1:CAS:528:DyaG3MXis1CksA%3D%3D&md5=19449090b6ebfc5304d508564c931ae9CAS |
[32] T. P. Burns, R. D. Rieke, J. Org. Chem. 1987, 52, 3674.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2sXlsFelsL0%3D&md5=fba82d8e48d24a7e512ca466c0e8cfbaCAS |
[33] I. Haas, A. Gedanken, Chem. Commun. 2008, 1795.
| 1:CAS:528:DC%2BD1cXktVCitL8%3D&md5=ff311c44c42c2b3c8ded4e0e85834982CAS |
[34] K.-F. Aguey-Zinsou, J.-R. Ares-Fernández, Chem. Mater. 2008, 20, 376.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhsVOntbbL&md5=9978bbf872f2117ac4cd28321b0d542cCAS |
[35] S. B. Kalidindi, B. R. Jagirdar, Inorg. Chem. 2009, 48, 4524.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXjvF2gtrk%3D&md5=9962e4e63a3c6b54a42ce2faa2b49d7dCAS |
[36] E. C. Ashby, R. D. Schwartz, Inorg. Chem. 1971, 10, 355.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE3MXmtVarsA%3D%3D&md5=acb39c0e70b69cbd36532f0a977d8cdfCAS |
[37] N. S. Norberg, T. S. Arthur, S. J. Fredrick, A. L. Prieto, J. Am. Chem. Soc. 2011, 133, 10679.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXnvFagur4%3D&md5=e88a6e1965a2e7c02f83875e3cf15196CAS |
[38] D. K. Dixit, K. Gandhi, B. K. Dixit, Int. J. Hydrogen Energy 2011,
| Crossref | GoogleScholarGoogle Scholar |
[39] H. Lee, J. Ihm, M. L. Cohen, S. G. Louie, Nano Lett. 2010, 10, 793.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtFansLs%3D&md5=cd7faf9e063180a98e06109bdc90b74bCAS |
[40] H. Wu, W. Zhou, T. J. Udovic, J. J. Rush, T. Yildrim, Chem. Mater. 2008, 20, 2335.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXitVamtr8%3D&md5=9a947b5447ccae15289e170d23a07d83CAS |
[41] D. A. Sheppard, M. Paskevicius, C. E. Buckley, Chem. Mater. 2011, 23, 4298.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtFOit7zN&md5=aee8c526e81ccdfbd2ce241c483f8befCAS |
[42] A.-H. Lu, F. Schüth, Adv. Mater. 2006, 18, 1793.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28Xot1Kktr0%3D&md5=87a446921092307e3443b4c9ef3e3ee5CAS |
[43] J. J. Vajo, Curr. Opin. Solid State Mater. Sci. 2011, 15, 52.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjsVKmt78%3D&md5=eb2cd39f38077d7c2710bb658aa8fcfeCAS |
[44] H. Y. Tian, C. E. Buckley, M. Paskevicius, D. A. Sheppard, Int. J. Hydrogen Energy 2011, 36, 671.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXht1Gjt7o%3D&md5=8e5989b136cc54191967920c2766b23cCAS |
[45] H.-W. Li, Y. Yan, S. Orimo, A. Züttel, C. M. Jensen, Energies 2011, 4, 185.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhslals74%3D&md5=f0ce2bb531aa9c2e17b5eb621935c4b7CAS |
[46] E. Rönnebro, Curr. Opin. Solid State Mater. Sci. 2011, 15, 44.
| Crossref | GoogleScholarGoogle Scholar |
[47] A. J. Churchard, E. Banch, A. Borgschulte, A. J. Churchard, E. Banch, A. Borgschulte, Phys. Chem. Chem. Phys. 2011, 13, 16955.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXht1Slt7rF&md5=e106ae9c7f4931324370747f7d24dc8fCAS |
[48] E. M. Fedneva, V. L. Alpatova, V. I. Mikheeva, Russ. J. Inorg. Chem. 1964, 9, 826.
[49] P. Mauron, F. Buchter, O. Friederichs, A. Remhof, M. Bielmann, C. N. Zwicky, A. Züttel, J. Phys. Chem. B 2008, 112, 906.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhsVemtrvM&md5=0b155892c15f4f186f407aab0f0767a1CAS |
[50] J. J. Vajo, S. L. Skeith, J. Phys. Chem. B 2005, 109, 3719.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXht1aqu7s%3D&md5=24592414c13b4c18247e5a7d50ab73c6CAS |
[51] B. J. Zhang, B. H. Liu, Z. P. Li, J. Alloy. Comp. 2011, 509, 751.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsVCktbnP&md5=49b95eb6d364d5dd4fa44a7bc03b735fCAS |
[52] S. Barcelo, S. S. Mao, Int. J. Hydrogen Energy 2010, 35, 7228.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXos1ygsLs%3D&md5=5d6ac476c25da2c2b223d22ef2ea7b0bCAS |
[53] R. A. Varin, L. Zbroniec, Int. J. Hydrogen Energy 2010, 35, 3588.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXktlShs7w%3D&md5=5146efe4f3ddb227823e1bc46128cdc7CAS |
[54] Y. H. Guo, X. B. Yu, L. Gao, G. L. Xia, Z. P. Guo, H. K. Liu, Energy. Environ. Sci. 2010, 3, 465.
| 1:CAS:528:DC%2BC3cXnvVahsr4%3D&md5=594aaa24f458aeedc266e6bb96294106CAS |
[55] D. Blanchard, Q. Shi, C. B. Boothroyd, T. Vegge, J. Phys. Chem. C 2009, 113, 14059.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXotVejt74%3D&md5=13bc00ff8afa58637b5407786c62e9a8CAS |
[56] R. J. Newhouse, V. Stavila, S. J. Hwang, L. E. Klebanoff, J. Z. Zhang, J. Phys. Chem. C 2010, 114, 5224.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXisVaisbs%3D&md5=8c33c9f2d3295a5070f063fe46c95279CAS |
[57] A. F. Gross, J. J. Vago, S. L. Van Atta, G. L. Olson, J. Phys. Chem. C 2008, 112, 5651.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXjtlOisbw%3D&md5=b26646171e4a0c70b61961c34d744ed9CAS |
[58] S. Cahen, J.-B. Eymery, R. Janot, J.-M. Tarascon, J. Power Sources 2009, 189, 902.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXjsVChtb0%3D&md5=b92819f9351af145ef0d2caac974d5f0CAS |
[59] X. Liu, D. Peaslee, C. Z. Jost, E. H. Majzoub, J. Phys. Chem. C 2010, 114, 14036.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXpslWhu7o%3D&md5=678c8706e8a42ae2c87ccaf218c18a16CAS |
[60] X. Liu, D. Peaslee, C. Z. Jost, T. F. Baumann, E. H. Majzoub, Chem. Mater. 2011, 23, 1331.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhs1aiu7k%3D&md5=d4ff39c2e8f430ed7de7b4aa1ed9fc47CAS |
[61] P. Ngene, M. van Zwienen, P. E. de Jongh, Chem. Commun. 2010, 46, 8201.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtlentbfJ&md5=e26dca95e4e674c9df20156b895bdeb0CAS |
[62] M. Fichtner, Z. Zhao-Karger, J. Hu, A. Roth, P. Weider, Nanotechnology 2009, 20, 204029.
| Crossref | GoogleScholarGoogle Scholar |
[63] S. Sartori, K. D. Knudsen, Z. Zhao-Karger, E. G. Bardaji, J. Muller, M. Fichtner, B. C. Hauback, J. Phys. Chem. C 2010, 114, 18785.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXht12qt77I&md5=b87f7cf0bd0d100eadb4ae7d5a0de0a4CAS |
[64] P. Ngene, P. Adelhelm, A. M. Beale, K. P. de Jong, P. E. de Jongh, J. Phys. Chem. C 2010, 114, 6163.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXjtlajt7g%3D&md5=ff624f8adef699387ed7204feb4866e3CAS |
[65] S. M. Opalka, X. Tang, B. L. Laube, T. H. Vanserspurt, Nanotechnology 2009, 20, 204024.
| Crossref | GoogleScholarGoogle Scholar | 1:STN:280:DC%2BD1Mzis1KmsQ%3D%3D&md5=addff651d4ad2b210048aa42230125d8CAS |
[66] B. Bogdanović, M. Shwickardi, J. Alloy. Comp. 1997, 1, 253.
[67] T. Vegge, Phys. Chem. Chem. Phys. 2006, 8, 4853.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XhtFWnsbrI&md5=56fbb9aef124fe3a7f45c285a3d50397CAS |
[68] M. Fichtner, P. Canton, O. Kircher, A. Lèon, J. Alloy. Comp. 2005, 404–406, 732.
| Crossref | GoogleScholarGoogle Scholar |
[69] G. J. Lee, J. H. Shim, Y. W. Cho, K. S. Lee, Int. J. Hydrogen Energy 2008, 33, 3748.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXovF2gsbw%3D&md5=126a66a91ff4bc6ccdc10e4bce13f26eCAS |
[70] Z. Xueping, F. Xin, L. Shenglin, J. Alloy. Comp. 2011, 509, 5873.
| Crossref | GoogleScholarGoogle Scholar |
[71] X. Xiao, K. Yu, X. Fan, Z. Wu, X. Wang, C. Chen, Q. Wang, L. Chen, Int. J. Hydrogen Energy 2011, 36, 539.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXht1Gjsbg%3D&md5=a5674bb3137c12132b33db6eaaae19bfCAS |
[72] V. Iosub, T. Matsunaga, K. Tange, M. Ishikiriyama, Int. J. Hydrogen Energy 2009, 34, 906.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtVGjt7s%3D&md5=76a6745bb6330870b5468d2c0565f48fCAS |
[73] R. A. Varin, C. Chiu, T. Czujko, Z. Wronski, Nanotechnology 2005, 16, 2261.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXht1CiurvK&md5=3b2f4744f56325457207622890251322CAS |
[74] R. A. Varin, C. Chiu, T. Czujko, Z. Wronski, J. Alloy. Comp. 2007, 439, 302.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXlslOks7s%3D&md5=d76dce1e37725cd69a372bcb9d8fafceCAS |
[75] F. E. Pinkerton, J. Alloy. Comp. 2011, 509, 8958.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtVaju7vO&md5=5e39d6b09232af508336d9410e8a8693CAS |
[76] R. D. Stephens, A. F. Gross, S. L. Van Atta, J. J. Vajo, F. E. Pinkerton, Nanotechnology 2009, 20, 204018.
| Crossref | GoogleScholarGoogle Scholar |
[77] T. K. Nielsen, M. Polanski, D. Zasad, P. Javadian, F. Besenbacher, J. Bystrzycki, J. Skibsted, T. R. Jensen, ACS Nano 2011, 5, 4056.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXkt1Wntr8%3D&md5=5fa1cfd8a3c017014433694a473e17b7CAS |
[78] J. Gao, P. Adelhelm, M. H. W. Verkeuijlen, C. Rongeat, M. Herrich, P. J. M. van Bentum, O. Gutfleisch, A. P. M. Kentgens, K. P. de Jong, P. E. de Jongh, J. Phys. Chem. C 2010, 114, 4675.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXit1Oiu7s%3D&md5=e740cd7d3426ba819c8f615f0aad934cCAS |
[79] Y. Li, G. Zhou, F. Fang, X. Yu, Q. Zhang, L. Ouyang, M. Zhu, D. Sun, Acta Mater. 2011, 59, 1829.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXmtVKksg%3D%3D&md5=b88d8550e7c4a3b93042355e3f556dd6CAS |
[80] C. P. Baldé, B. P. C. Hereijgers, J. H. Bitter, K. P. de Jong, J. Am. Chem. Soc. 2008, 130, 6761.
| Crossref | GoogleScholarGoogle Scholar |
[81] S. Zheng, F. Fang, G. Zhou, G. Chen, L. Ouyang, M. Zhu, D. Sun, Chem. Mater. 2008, 20, 3954.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXmtVOgtLg%3D&md5=a20ce850fd2241a76b9c7aba43062b5cCAS |
[82] P. Chen, Z. Xiong, J. Luo, J. Lin, K. L. Tan, Nature 2002, 420, 302.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XovVektLg%3D&md5=97510779490575bca11081d4eb3b78ecCAS |
[83] D. H. Gregory, J. Mater. Chem. 2008, 18, 2321.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXlsVOnsbk%3D&md5=f623028c8ecdfbe15f293672b10f9bb1CAS |
[84] P. Chen, M. Zhu, Mater. Today 2008, 11, 36.
| Crossref | GoogleScholarGoogle Scholar |
[85] http://www.eere.energy.gov/hydrogenandfuelcells/pdfs/freedomcar_targets_explanations.pdf (accessed 15 December 2011).
[86] F. E. Pinkerton, J. Alloy. Comp. 2005, 400, 76.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXosVWqtbc%3D&md5=f2720c5f9b3cb947b91c0a2096e80eebCAS |
[87] T. Markmaitree, R. Ren, L. L. Shaw, J. Phys. Chem. B 2006, 110, 20710.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XpsFCqsr0%3D&md5=12aba3b2a20b0cca495cf24bd5b71901CAS |
[88] C. Lu, J. Hu, J. Kwak, Z. Yang, R. Ren, T. Markmaitree, L. Shaw, J. Power Sources 2007, 170, 419.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXmt1Grtbw%3D&md5=aae8e7bcca3c7e14609e7b4b455c47bfCAS |
[89] L. L. Shaw, R. Ren, T. Markmaitree, W. Osborn, J. Alloy. Comp. 2008, 448, 263.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXht1ylsrrM&md5=667da5d16d05498d1f33532b95a7aa41CAS |
[90] R. A. Varin, M. Jang, M. Polanski, J. Alloy. Comp. 2010, 491, 658.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtFejsLc%3D&md5=9c56d7d950fe456a25d358b1b0149168CAS |
[91] R. R. Shahi, T. P. Yadav, M. A. Shaz, O. N. Srivastava, Int. J. Hydrogen Energy 2008, 33, 6188.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXht12gurrM&md5=1021cf39dc1c905681d720cf18928a4eCAS |
[92] T. Ichikawa, I. Shigehito, N. Hanada, H. Fujii, J. Alloy. Comp. 2004, 365, 271.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXivVWjtw%3D%3D&md5=c43a348261e1856e4761272c85cce477CAS |
[93] D. Blanchard, H. W. Brinks, B. C. Hauback, P. Norby, Mater. Sci. Eng. 2004, 108, 54.
| Crossref | GoogleScholarGoogle Scholar |
[94] Z. Xueping, L. Shenglin, J. Alloy. Comp. 2009, 481, 761.
| Crossref | GoogleScholarGoogle Scholar |
[95] S. Isobe, T. Ichikawa, N. Hanada, H. Y. Leng, M. Fichtner, O. Fuhr, H. Fujii, J. Alloy. Comp. 2005, 404–406, 439.
| Crossref | GoogleScholarGoogle Scholar |
[96] M. Fichtner, O. Fuhr, O. Kircher, J. Rothe, Nanotechnology 2003, 14, 778.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXpsVOnsrg%3D&md5=6fe3f384b6d14aaa0fa963243bbf62eaCAS |
[97] M. Matsumoto, T. Haga, Y. Kawai, Y. Kojima, J. Alloy. Comp. 2007, 439, 358.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXlslOksLg%3D&md5=270b89cff836adbc883ee56297d6faedCAS |
[98] S. Isobe, T. Ichikawa, Y. Kojima, H. Fujii, J. Alloy. Comp. 2007, 360, 446.
[99] T. Tsumuraya, T. Shishidou, T. Oguchi, Phys. Rev. B 2008, 77, 235114.
| Crossref | GoogleScholarGoogle Scholar |
[100] Y. Kojima, M. Matsumoto, Y. Kawai, T. Haga, N. Ohba, K. Miwa, S. Towata, Y. Nakamori, S. Orimo, J. Phys. Chem. B 2006, 110, 9632.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XjslSlsrk%3D&md5=f6deb0203ff3db68a8112d3614407e43CAS |
[101] F. E. Pinkerton, M. S. Meyer, G. P. Meisner, M. P. Balogh, J. Alloy. Comp. 2007, 433, 282.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXjtFGmsrw%3D&md5=b77cd5412ad8f10f172e3efe9876fa30CAS |
[102] Z. Xiong, G. Wu, J. Hu, P. Chen, Adv. Mater. 2004, 16, 1522.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXovVymtrk%3D&md5=cc30ec8511b7a97fc5decee330758fbcCAS |
[103] Y. Chen, P. Wang, C. Liu, H. Cheng, Int. J. Hydrogen Energy 2007, 32, 1262.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXlvValtrs%3D&md5=73246f3ce3b757c32ed7f6bfc93ab7baCAS |
[104] S. Nayebossadri, K. F. Aguey-Zinsou, Z. Xiao, Int. J. Hydrogen Energy 2011, 36, 7920.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXmslSmtL4%3D&md5=e08c28325c8bcf2d7d492f923a35f61dCAS |
[105] X. Feng, Y. Bai, B. Lu, C. Wang, Y. Qi, Y. Liu, G. Geng, L. Li, Inorg. Chem. 2004, 43, 3558.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXjslOiu74%3D&md5=81277c1db052ceccb4b5bc3a3387811fCAS |
[106] F. E. Pinkerton, G. P. Meisner, M. S. Meyer, M. P. Balogh, M. D. Kundrat, J. Phys. Chem. B 2005, 109, 6.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXhtVOrur%2FI&md5=31ad91f3b75914dcf8bfedaa71023c2bCAS |
[107] H. Wu, W. Zhou, K. Wang, T. J. Udovic, J. J. Rush, T. Yildirim, L. A. Bendersky, A. F. Gross, S. L. Van Atta, J. J. Vajo, F. E. Pinkerton, M. S. Meyer, Nanotechnology 2009, 20, 204002.
| Crossref | GoogleScholarGoogle Scholar |
[108] J. Yang, A. Sudik, D. J. Siegel, D. Halliday, A. Drews, R. O. Carter , C. Wolverton, G. J. Lewis, J. W. A. Sachtler, J. J. Low, S. A. Faheem, D. A. Lesch, V. Ozolins, J. Alloy. Comp. 2007, 446–447, 345.
| Crossref | GoogleScholarGoogle Scholar |
[109] M. U. Niemann, S. S. Srinivasan, A. Kumar, E. K. Stefanakos, D. Y. Goswani, K. McGrath, Int. J. Hydrogen Energy 2009, 34, 8086.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtFOqurzE&md5=652d7044925fe49907c4ea67f0a9bd66CAS |
[110] S. S. Srinivasan, M. U. Niemann, J. R. Hattrick-Simpers, K. McGrath, P. C. Sharma, D. Y. Goswani, E. K. Stefanakos, Int. J. Hydrogen Energy 2010, 35, 9646.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtVyrur7M&md5=e40614a07527ee9ecf520d0b04501e2aCAS |
[111] L. Xie, J. Zheng, Y. Liu, Y. Li, X. Li, Chem. Mater. 2008, 20, 282.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhsVWqsL%2FF&md5=e02ee73d5cd91a87b03191081ad24e94CAS |
[112] Y. D. Yin, R. M. Rioux, C. K. Erdonmez, S. Hughes, G. A. Somorjai, A. P. Alivisatos, Science 2004, 304, 711.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXjsV2gt7g%3D&md5=210f485f6e3aa8e22284fb3adb4ae757CAS |
[113] L. Xie, Y. Li, R. Yang, Y. Liu, X. Li, Appl. Phys. Lett. 2008, 92, 231910.
| Crossref | GoogleScholarGoogle Scholar |
[114] L. Xie, Y. Liu, G. Li, X. Li, J. Phys. Chem. C 2009, 113, 14523.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXovVels7w%3D&md5=3fc6b26f1947d9f8b4ebcb3fb13ee9e1CAS |
[115] F. H. Stephens, V. Pons, R. T. Baker, Dalton Trans. 2007, 2613.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXmsFOisLo%3D&md5=c882b2628b6be61c78b3e08db5ba8979CAS |
[116] T. B. Marder, Angew. Chem. Int. Ed. 2007, 46, 8116.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhtlGju7rM&md5=f0e7653c16e87228c8fee25a4e33eaffCAS |
[117] D. A. Dixon, M. Gutowski, J. Phys. Chem. A 2005, 109, 5129.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXksVSqu7Y%3D&md5=a03137c0d07f17221b667eff8653edf5CAS |
[118] F. Baitalow, J. Baumann, G. Wolf, K. Jaenicke-Röbler, G. Leitner, Thermochim. Acta 2002, 391, 159.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38Xls12gsrY%3D&md5=a00fd7643c899ba7ae74922abc736276CAS |
[119] A. Gutowska, L. Li, Y. Shin, C. M. Wang, X. S. Li, J. C. Linehan, R. S. Smith, B. D. Kay, B. Schmid, W. Shaw, M. Gutowski, T. Autrey, Angew. Chem. Int. Ed. 2005, 44, 3578.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXlsVykt70%3D&md5=840b43ba945a5813c50914900ba05de7CAS |
[120] A. Paolone, O. Palumbo, P. Rispoli, R. Cantelli, T. Autrey, A. Karkamkar, J. Phys. Chem. C 2009, 113, 10319.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXmtlKit78%3D&md5=a54a99abb17d3080ace0d4abf428dae9CAS |
[121] A. Feaver, S. Sepehri, P. Shamberger, A. Stowe, T. Autrey, G. Cao, J. Phys. Chem. B 2007, 111, 7469.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXmtlOnsbY%3D&md5=8b043814a7b47225a42ebc7d48848b69CAS |
[122] Z. Kurban, A. Lovell, S. M. Bennington, D. W. K. Jenkins, K. R. Ryan, M. O. Jones, N. T. Skipper, W. I. F. David, J. Phys. Chem. C 2010, 114, 21201.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsVaisrrP&md5=27b69558e08002b7cd6865c926a204bdCAS |
[123] D. Neiner, A. Karkamkar, J. C. Lineham, B. Arey, T. Autrey, S. M. Kauzlarich, J. Phys. Chem. C 2009, 113, 1098.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhsFCmtLjI&md5=e02d89ffb66760751fd16d7631a79e4fCAS |
[124] T. He, Z. Xiong, G. Wu, H. Chu, C. Wu, T. Zhang, P. Chen, Chem. Mater. 2009, 21, 2315.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXltVKksLg%3D&md5=e543bad563c6408ffcb219846e057af7CAS |