

Synthesis and antibacterial activity of 6″-decanesulfonylacetamide-functionalised amphiphilic derivatives of amikacin and kanamycin
Dylan C. Farr A , Lendl Tan B , Juanelle Furness B , I. Darren Grice A C , Nicholas P. West B and Todd A. Houston A *
A C , Nicholas P. West B and Todd A. Houston A *
A
B
C
Abstract
Aminoglycoside antibiotics represent the first class of successful drugs in the treatment of tuberculosis; however, mycobacteria and other bacterial species possess several drug resistance mechanisms to inactivate these natural products. In the past 15 years, a variety of amphiphilic aminoglycosides have been shown to have improved activity against infectious microorganisms and to subvert resistance mechanisms. Here, we report on four novel synthetic compounds derived from two existing potent antitubercular compounds and describe their activity against both Mycobacterium tuberculosis and Staphylococcus aureus. It was found that a decanesulfonylacetamide-based conjugate of amikacin displayed promising preliminary antitubercular activities, warranting further investigation to assess the therapeutic potential of these unique antimicrobials.
Keywords: amide–triazole conjugates, amikacin, amphiphilic aminoglycosides, antibiotics, antimicrobial agents, kanamycin, Mycobacterium tuberculosis, n-decanesulfonylacetamide.
Introduction
Structural modification of the aminoglycoside (AG) scaffold has historically been a successful approach towards creating novel antibiotics. This approach led to the discovery of amikacin (AMI), a potent AG that is used for the treatment of tuberculosis (TB) today.1 Recently, a new class of antibiotics derived from the polycationic AG scaffold has produced several promising lead compounds that have revived activities against drug-resistant bacterial species of clinical importance. This new class of antimicrobial agent is derived from conjugation of hydrophobic groups such as amphiphilic alkyl chains, amino acids, peptides and aromatics to AG scaffolds, creating semi-synthetic amphiphilic AGs (AAGs). AAGs have shown promising preliminary results against several drug-resistant bacterial strains by evading common mechanisms of resistance and exerting novel mechanisms of action.2–5
Polycationic anti-bacterials (PAs) are a structurally diverse class of antibacterial agents having broad-spectrum activities and multiple modes of action. Amphiphilic PAs such as cationic antimicrobial peptides act on bacterial membranes, whereas non-amphiphilic PAs such as AGs act by binding to 16S rRNA (ribosomal RNA).6 However, mode of action studies reveal AAGs predominately target bacterial membranes, thereby leading to depolarisation and increased permeability.7–10 Amphiphilic cationic bacterial membrane disrupters interact with negatively charged components of the bacterial cell membrane such as phosphatidylglycerol, lipopolysaccharides and teichoic acids orders of magnitude more tightly than the bivalent cations that hold these negatively charged components together.11,12 Through displacement of bivalent cations, disruption of the outer membrane occurs, leading to uncontrolled cell permeability and cell death. Bacterial anionic lipids are attractive targets in the design of antibacterial agents as they can exert broad-spectrum effects while displaying potent activities towards resistant bacterial strains. Furthermore, owing to their mode of action, which results in the depolarisation of bacterial membranes through the formation of pores, in vitro resistance to cationic amphiphiles is rarely observed.13 AAGs have also demonstrated the potential to boost innate immune responses, inducing immunomodulatory responses, offering new perspectives for the treatment of persistent and dormant infections.14
In this work, we have chemically conjugated an amphiphilic sulfonylacetamide-based moiety to the scaffolds of AMI and kanamycin (KAN) (Fig. 1). Long alkyl chained sulfonylacetamides have been previously reported to display potent antitubercular activities.15,16 The most active of these, n-decanesulfonylacetamide (DSA), has a reported minimum inhibitory concentration (MIC) of 0.75–1.5 µg mL–1 towards Mycobacterium tuberculosis, which is comparable with first-line agents used to treat TB.15 These simple alkyl sulfonylacetamides have been shown to significantly decrease mycolic acid levels in mycobacteria, providing promising drug leads due to the importance of lipid biosynthesis to the virulence and intracellular survival of M. tuberculosis.16,17 However, simple alkyl sulfonylacetamides displayed poor pharmacokinetics within an in vivo model, resulting in suboptimal bioavailability. In addition, we have previously shown conversion of the amide in DSA into a thioamide afforded reduced antitubercular activity and improved activity against Staphylococcus aureus.18
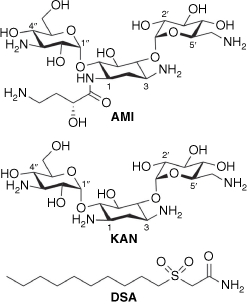
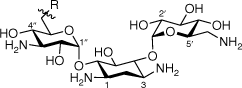
Within this new work, a series of AAGs were synthesised from the scaffolds of AMI and KAN utilising the active functionality of the sulfonylacetamide DSA. We were interested in investigating the conversion of AGs into antibiotic hybrids possessing sulfonylacetamide functionality to then explore potential structural candidates for further development of antimicrobial agents. Amphiphilic derivatisation of the AMI and KAN scaffold at 6″ was achieved through straightforward single-site modifications as outlined in Scheme 1. Triazole AAG conjugates were prepared by copper-catalysed click chemistry, while amide linkers were synthesised utilising carbodiimide coupling protocols. The resulting conjugates were evaluated for their antimicrobial activities towards M. tuberculosis and S. aureus.
(a) Synthesis of 6″ AMI and KAN sulfonylacetamide-based amide and triazole conjugates 9, 10, 17 and 18; (b) synthesis of structural building blocks 20 and 21. Reagents and conditions: (a) Boc2O, triethylamine (TEA), H2O, MeOH, 50°C, 24 h; (b) 2,4,6-TPSCl, pyridine, room temperature (rt), 18 h; (c) NaN3, DMF, 70°C, 18 h; (d) 20, sodium ascorbate, CuSO4, H2O, i-PrOH, 40°C, 2–4 h; (e) TFA, DCM, rt, 1–2 h; (f) H2(g), Pd(OH)2/C, MeOH, rt, 18 h; (g) 21, 1‐ethyl‐3‐(3‐dimethylaminopropyl)carbodiimide (EDC), N‐hydroxysuccinimide (NHS), THF, H2O, rt to 50°C, 15–24 h; (h) KHSO5, MeOH, H2O, 18 h; (i) TFA, DCM, rt, 1–2 h; (j) propargyl bromide, K2CO3, acetone, rt, 24 h; (k) KHSO5, MeOH, H2O, rt, 18 h; (l) bromoacetic acid, K2CO3, MeCN, 50°C, 24 h. (Boc, t-butyloxycarbonyl)
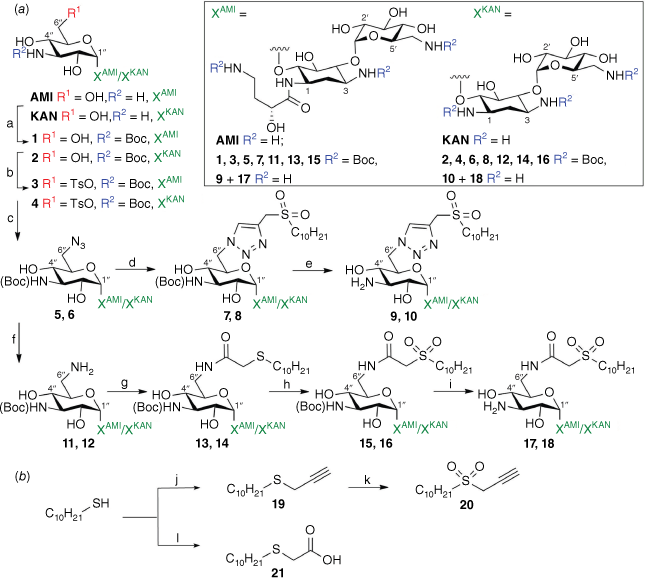
Results and discussion
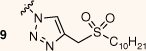
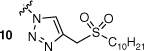
The synthesis of amphiphilic AMI and KAN-based sulfonylacetamide conjugates is described in Scheme 1. To determine the importance and durability of the amide linkage, the decanesulfonyl group was also added by a triazole linker. Derivatisation of the 6″ position of the aminoglycoside scaffolds was achieved following a straightforward synthetic approach involving subsequent functionalisation of the 6″ position, as outlined in Scheme 1. The 6″-azido-functionalised AG derivatives were prepared following synthetic strategies previously described for neomycin.19 Briefly, the amines of the AG scaffolds had Boc protecting groups installed to allow selective tripsylation of the 6″ primary alcohol. Subsequent displacement of the tripsyl leaving group allowed installation of an azide at the 6″ position, affording azide–alkyne cycloaddition with a previously prepared lipid alkyne 20. Finally, Boc functionality was removed to afford the final targets 9 and 10 as TFA salts. Synthesis of the amide conjugates was achieved by reduction of the 6″ azide to an amine by hydrogenation, followed by 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) coupling with a previously prepared carboxylic acid derivative of DSA (21). Following conjugation, the sulfide was oxidised to a sulfone and Boc functionality was removed to afford 17 and 18 as TFA salts.
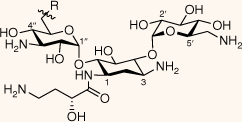
The synthesised AAGs were evaluated for their antitubercular and antistaphylococcal activities (Table 1). In general, conjugation of DSA lipid moieties to the AG scaffold reduced antibacterial activity when compared with the parent scaffold. The most active compound was the amide-conjugated AMI derivative 17, which displayed an MIC90 of 22 µg mL–1 towards M. tuberculosis. Although less efficient when compared with the parent scaffold, 17 displayed a modest level of inhibition, suggesting DSA derivatives of AGs with increased lipophilicity can attain antitubercular activities (Fig. 2).
| Compound | R | H37Rv MIC90 (µg mL–1) | ATCC25923 MIC (µg mL–1) | |
|---|---|---|---|---|
| AMI | – | 0.6 | – | |
| KAN | – | 1.3 | – | |
 |  | >50 | 100–200 | |
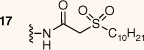 | 22 | 200 | ||
 |  | >50 | 100–200 | |
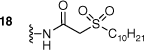 | >50 | >200 |
Percentage growth of M. tuberculosis following exposure to AMI alkylsulfonyl conjugates 9 and 17, and KAN alkylsulfonyl conjugates 10 and 18, tested at a serial dilution of 50–0.098 µg mL–1 (log of concentration in micrograms per millilitre). AMI and KAN controls serial diluted at a concentration of 10–0.020 µg mL–1.
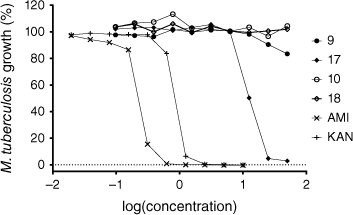
Although the amide derivative 17 demonstrated antitubercular activities, the corresponding triazole conjugate 9 displayed low inhibitory efficiency, suggesting the amide linker utilised in 17 influences antitubercular activity. Interestingly, membrane-targeting AAG derivatives reported by Herzog et al.20 demonstrated lipid conjugates utilising both amide and triazole linkers exhibited similar antimicrobial activities across several bacterial species. Although it has been reported that the length and nature of the amphiphilic moiety can greatly influence antibacterial inhibition,21 the polycationic scaffold utilised can also affect antimicrobial activities.2 The latter falls in line with data presented here: the AMI conjugate 17 displays antitubercular activities whereas the corresponding KAN derivative 18 has an abrupt loss of killing efficiency while maintaining the same amphiphilic functionality. This could imply that 17 is exerting an inhibitory effect by hitting a specific molecular target(s), rather than a broad interaction with the bacterial membrane, particularly considering the importance of lipid metabolism in M. tuberculosis.
As AAGs can act through binding of bacterial membranes as opposed to targeting rRNA, lipophilicity has a significant influence on the membrane permeabilisation and killing efficiency of the therapeutic.10,21–24 Lipophilicity may be an influential determinant towards the activity of the AMI amide conjugate 17 compared with KAN corresponding derivative 18. The AMI derivative 17 is the most active within this study, whereas the corresponding KAN derivative 18 displayed no antitubercular activity (MIC90 > 50 µg mL–1). AMI is a semi-synthetic derivative of KAN, structurally differing by an (S)-4-amino-2-hydroxybutanoyl (AHB) substituent at C1. Although AMI is more active towards M. tuberculosis compared with KAN, the difference in activities between 17 and 18 is unexpected given their structural similarities in terms of their lipophilic moieties, AG scaffold and net positive charge.
Although amphiphilic AMI and KAN-based lipid conjugates have reported antistaphylococcal activities, the small series reported here displays low inhibitory efficiency. AMI and KAN-derived polycarbamates and polyethers reported by Bera et al.2 were assessed against a spectrum of bacterial species, including for antistaphylococcal activities and were found to have a broad spectrum effect. When compared with the antistaphylococcal activities of 9, 10, 17 and 18 reported here, the influence of amphiphilicity is apparent, as most poly-modified conjugates provided enhanced antibacterial activities towards S. aureus. Results reported by Zhang et al.25 also found amphiphilicity to be influential as di-conjugates of KAN were demonstrated to be more active compared with mono-conjugates, which often displayed no activity towards S. aureus. A KAN 4″,6″-di-O-nonyl derivative was the most active towards S. aureus (MIC 8 µg mL–1) whereas single-site modification of O-nonyl at either 4″ or 6″ resulted in inactive compounds (≥250 µg mL–1). Polyconjugates have also demonstrated inhibitory efficiency towards S. aureus and other bacterial pathogens when constructed from the scaffolds of other AGs, such as tobramycin and neomycin.2,26 Mechanistic studies suggest the improved activities of di-conjugates in comparison with mono-conjugates is due to the complementary molecular shape allowing insertion within bacterial membranes.8
Few AMI-based AAGs have been reported and to our knowledge, none have been assessed for their inhibitory effect towards M. tuberculosis. Most of the functionalised AGs assessed and reported for antimycobacterial efficacy are conjugates of the tobramycin scaffold, along with KAN, netilmicin and sisomicin.27–29 The activities of a series of 6″-thioether and sulfone-linked alkyl chained conjugates of KAN towards M. smegmatis has been previously described.27 The optimal chain length found for mycobacterial inhibition was C11–C13, in line with the lipid length of conjugates reported here. Interestingly, oxidation of the thioether linkage to a sulfone decreased inhibitory effect towards S. aureus, while improving mycobacterial activity.27 This suggests that sulfonylacetamide-based conjugates may provide superior Gram-negative activities with unoxidised lipid moieties as opposed to the sulfone utilised in the inhibitor’s design. Additionally, the structural position of AHB appendages on the scaffolds of AMI and KAN have been previously shown to influence mycobacterial activities against M. smegmatis.29 In line with results presented here, the most active AMI derivative reported in the study towards mycobacterial species displayed no effectiveness towards Gram-positive species, including S. aureus.29
It has been found that amphiphilic derivatisation is capable of reducing the effect of AG modifying enzymes (AMEs).29 Enzymatic modification of AGs is the most prevalent mechanism of bacterial resistance. Owing to structural changes resulting from amphiphilic conjugation of the AG scaffold, AAGs have the ability to reduce affinity and evade the action of AMEs.27,29,30 A putative benefit of the DSA-conjugated AGs may be a means to circumvent structural changes from AMEs resulting in restored activity towards resistant bacterial strains. Alkyl chain 6″ conjugates of tobramycin have demonstrated antibacterial activities across an array of organisms including resistant strains through membrane interruption.30 Most notable were activities against bacterial strains expressing Eis, an acetyltransferase that causes high levels of resistance to AGs in extensively drug-resistant TB.31–33 Within the reported library, sulfur-linked alkyl conjugates were found to be poor substrates for several AMEs, including Eis, compared with tobramycin. It has been established that AAGs circumvent resistant mechanisms and exert antimicrobial inhibition through cell wall disruption; therefore, exploring activities towards resistant strains in addition to mechanistic studies would provide further insight into how valuable these derivatives are as lead compounds for further drug development.
Conclusion
In this work, a series of AAGs were synthesised utilising various synthetic strategies to chemically conjugate lipophilic moieties with previously reported antitubercular activities to the 6″ position of AMI and KAN by amide or triazole functionality. The antibacterial activities of the small series of amphiphilic AMI and KAN-based conjugates is described. Notably, the amide-linked sulfonylacetamide conjugate of AMI (17) was identified to have promising preliminary activity against M. tuberculosis (MIC90 22 µg mL–1). The activities of the AAGs synthesised here was influenced by the AG scaffold, lipid moiety and structural composition of the linker utilised. Although 17 displayed antitubercular efficiency, the target of action remains unknown and it could be exerting its effect by a membranolytic interaction or exhibiting a prodrug-dependent mode of action following the release of free AMI and a lipid moiety post-cleavage of the amide linker. Mechanistic studies would assess the therapeutic potential of these unique antimicrobials while providing further insight into how valuable these derivatives are as lead compounds for further, more refined, drug design and development.
Experimental procedures
Determination of minimum inhibitory concentration against Mycobacterium tuberculosis and Staphylococcus aureus
M. tuberculosis (H37Rv) was grown in Middlebrook 7H9S media, with 10% albumin dextrose catalase (ADC) supplementation, 1% tryptone, 0.05% glycerol and 0.05% Tween 80. Freshly seeded cultures were grown at 37°C for ~14 days to mid-exponential phase (optical density measured at a wavelength of 600 nm, OD600 = 0.4–0.8) for the inhibitory assay. The microbial culture was diluted to OD600 = 0.001 in 7H9S media and 100 µL (2 × 104 colony forming units mL–1) was added to each well, as well as the serial-diluted compound. Each 96-well plate prepared for the assay included rifampicin at a range of 0.4–200 nM as a positive control. The plates were incubated for 5 days at 37°C in a humidified incubator prior to the addition of resazurin (30 µL of a 0.02% solution) and 12.5 µL of Tween-80 to each well. Following 24-h incubation, the fluorescence was measured on a Fluorostar Omega fluorescent plate reader with an excitation wavelength of 530 nm and emission read at 590 nm.
S. aureus (ATCC 25923) was grown in lysogeny broth (LB) at 37°C with 160 rpm shaking. The MIC was determined using S. aureus cultures grown overnight in LB media and subcultured 1:100 the following morning. When cells were in mid-log phase, 105 cells were added to a microtitre plate for MIC determination containing the appropriate therapeutic that was serial-diluted. Bacterial cells were grown overnight for 18 h at 37°C in aerobic conditions and cell viability was determined by measuring absorbance at 600 nm on a Tecan Infinite 200 Pro plate reader.
General synthetic procedures
Chemicals for which synthesis is not reported were obtained from commercial suppliers. All solvents used were either reagent grade, distilled or dried and distilled prior to use. 1H and distortionless enhancement by polarisation transfer 13C NMR spectra were obtained using a Bruker Avance III HD Ascend 400 MHz spectrometer operating at 400 and 100 MHz respectively, or a Bruker Avance III HD 600 MHz operating at 600and 150 MHz. Signals are recorded to two decimal places in terms of chemical shifts (δ, ppm) relative to MeOD and D2O. The abbreviations for multiplicities used are: s, singlet; d, doublet; t, triplet and m, multiplet. Low-resolution mass spectrometry (LRMS) spectra were recorded with a Bruker Esquire 3000, and high-resolution (HR) MS with a Bruker maXis II ETD. Flash chromatography was conducted using flash chromatography grade silica, 200–400 mesh, purchased from Sigma–Aldrich with distilled solvents. Thin layer chromatography (TLC) was used to monitor reactions using Merck TLC plates. TLC plates were visualised under UV light and developed with appropriate TLC stains such as H2SO4 and charring with heat.
To a stirred solution of amikacin sulfate (1.50 g, 2.60 mmol) dissolved in 1:1 H2O:MeOH (50 mL) was added TEA (15 equiv.) followed by di-tert-butyldicarbonate (10 equiv.). The resulting reaction mixture was stirred at 50°C for 24 h. On completion, the MeOH was evaporated, and the residue was partitioned between EtOAc (100 mL) and H2O (50 mL), extracted with EtOAc (3 × 50 mL), dried over Na2SO4 and concentrated under vacuum. Purification by flash chromatography with 9:1 DCM:MeOH afforded 1 as white solid in a yield of 63% (1.62 g, 1.60 mmol). LRMS (electrospray ionisation, ESI): m/z calculated for [M + Na]+ C42H75N5NaO21, 1009.1; found 1009.1.
A solution of kanamycin sulfate (2.00 g, 3.43 mmol) was reacted following the synthetic conditions outlined for the synthesis of compound 1. Compound 2 was afforded as a white solid in a yield of 72% (2.13 g, 2.40 mmol). LRMS (ESI): m/z calculated for [M + Na]+ C38H68N4NaO19, 907.4; found 907.8.
Compound 1 (650 mg, 0.66 mmol) was dissolved in anhydrous pyridine (30 mL) and reacted with 2,4,6-TPSCl (28 equiv.) under an atmosphere of argon for 18 h. Following completion, pyridine was removed under vacuum by co-evaporation with toluene and the remaining residue was partitioned between H2O (100 mL) and EtOAc (100 mL), then extracted with EtOAc (3 × 50 mL). The organic layers were combined, dried over Na2SO4 and concentrated under reduced pressure. Product 3 was purified on flash silica in a gradient of isocratic DCM → 98:2 DCM:MeOH affording 3 as a white solid in a yield of 78% (640 mg, 0.51 mmol). LRMS (ESI): m/z calculated for [M + Na]+ C57H97N5NaO23, 1274.6; found 1275.2.
A solution of 2 (2.13 g, 2.40 mmol) was reacted following the same methodology outlined for the synthesis of 3. Purification of 4 was achieved by a gradient elution with isocratic DCM → 98:2 → 95:5 DCM:MeOH on flash silica, affording 4 as a white solid with a yield of 48% (1.31 g, 1.14 mmol). LRMS (ESI): m/z calculated for [M + Na]+ C53H90N4NaO21S, 1173.6; found 1174.2.
A solution of 3 (640 mg, 0.51 mmol) was dissolved in anhydrous DMF (10 mL), to which NaN3 (4 equiv.) was added; the resulting mixture was heated to 70°C and reacted for 18 h under an atmosphere of argon. On completion, the sample was partitioned between EtOAc (100 mL) and NaHCO3 (50 mL) and extracted into EtOAc (3 × 50 mL). The organic fractions were combined, dried over Na2SO4 and concentrated under vacuum. The resulting residue was further purified by flash chromatography; purification of 5 in 12:1 DCM:MeOH afforded the target as an amorphous white solid with a yield of 29% (148 mg, 0.15 mmol). LRMS (ESI): m/z calculated for [M + Na]+ C42H74N8NaO20, 1033.5; found 1033.5.
Compound 4 (1.31 g, 1.14 mmol) was reacted following the synthetic conditions reported for the synthesis of 5. Compound 6 was purified by flash chromatography with a gradient solvent system of DCM:MeOH 95:5 → 12:1, affording a white solid with a yield of 81% (840 mg, 0.92 mmol). LRMS (ESI): m/z calculated for [M + Na]+ C38H67N7NaO18, 932.4; found 932.9.
A solution of 5 (100 mg, 0.10 mmol) was stirred with the lipid acetylene 20 (1.2 equiv.) dissolved in a 1:1 solution of H2O and i-PrOH (5 mL). To the resulting reaction mixture were added sodium ascorbate (0.2 equiv.) and CuSO4 (0.1 equiv.). The resulting solution was heated to 40°C and stirred for 2–4 h. Following completion, as indicated by TLC, the crude reaction mixture was concentrated under vacuum and the crude residue was eluted with 10:1 DCM:MeOH, affording 7 as a white solid at a yield of 40% (50 mg, 0.04 mmol). LRMS (ESI): m/z calculated for [M + Na]+ C55H98N8NaO22S, 1277.6; found 1277.9.
Synthesis of 8 was achieved by reacting 6 (100 mg, 0.11 mmol) with the reaction conditions reported for the synthesis of 7. Compound 8 was obtained as a white solid with a yield of 60% (76 mg, 0.07 mmol). LRMS (ESI): m/z calculated for [M + Na]+ C51H91N7NaO20S, 1176.6; found 1176.9.
The conjugated intermediate 7 (50 mg, 0.04 mmol) was dissolved in DCM (1 mL) and stirred at 0°C, to which TFA (0.5 mL per 50 mg (w/v%)) was added dropwise. The reaction was carried out for 1–2 h at ambient temperature, concentrated under reduced pressure and azeotroped with toluene to afford the final deprotected conjugate 9 as a white solid at a yield of 97% (45 mg, 0.04 mmol). 1H NMR (400 MHz, D2O) δ 8.24 (s, 1H), 5.46 (s, 1H), 5.14 (s, 1H), 4.64–4.54 (m, 2H), 4.33–4.22 (m, 2H), 4.18 (dd, J = 10.4, 2.6 Hz, 1H), 4.14–3.98 (m, 2H), 3.90–3.57 (m, 6H), 3.56–3.37 (m, 3H), 3.37–3.21 (m, 2H), 3.23–3.09 (m, 2H), 2.32–2.06 (m, 3H), 2.07–1.66 (m, 2H), 1.58–1.38 (m, 2H), 1.42–1.07 (m, 12H), 0.86 (t, J = 6.5 Hz, 3H). 13C NMR (100 MHz, D2O) δ 175.4, 163.1, 162.8, 97.3, 96.8, 86.9, 79.8, 78.5, 72.6, 72.2, 70.8, 70.7, 69.6, 69.5, 68.7, 67.8, 66.9, 55.1, 52.1, 50.6, 48.9, 48.8, 48.0, 40.3, 36.9, 31.2, 30.8, 30.2, 28.6, 28.4, 28.4, 28.1, 27.4, 22.0, 21.2, 13.4. HRMS 9 (ESI): m/z calculated for [M + 2H]2+ C35H68N8O14S, 428.2288; found 428.2284.
Synthesis of 10 was achieved by reacting 8 (76 mg, 0.07 mmol) with the synthetic methodology reported for compound 9. Compound 10 was isolated as a white solid with a yield of 99% (73 mg, 0.07 mmol). 1H NMR (600 MHz, MeOD) δ 8.13 (s, 1H), 5.38 (d, J = 3.9 Hz, 1H), 5.09 (d, 1H), 4.62–4.55 (m, 3H), 4.30–4.25 (m, 1H), 4.10–4.04 (m, 1H), 3.85–3.74 (m, 2H), 3.73–3.67 (m, 2H), 3.65–3.54 (m, 3H), 3.49–3.39 (m, 4H), 3.23 (t, J = 9.4 Hz, 1H), 3.18 (t, 2H), 3.06 (dd, J = 13.0, 8.9 Hz, 1H), 2.48 (s, 1H), 1.89–1.82 (m, 2H), 1.51–1.44 (m, 2H), 1.40–1.25 (m, 12H), 0.90 (t, J = 6.9 Hz, 3H). 13C NMR (150 MHz, MeOD) δ 129.9, 129.2, 102.1, 96.9, 85.5, 81.1, 74.4, 73.2, 73.1, 73.0, 72.8, 70.4, 69.9, 68.7, 56.5, 53.3, 52.1, 51.7, 50.4, 48.7, 42.3, 33.0, 30.6, 30.5, 30.4, 30.2, 29.5, 28.9, 23.7, 22.9, 14.4. HRMS (ESI): m/z calculated for [M + 2H]2+ C31H61N7O12S, 377.7050; found 377.7027.
To a solution of 5 (148 mg, 0.15 mmol) stirring in MeOH (3–10 mL) was added Pd(OH)2/C 20 wt-% (1.2 equiv.). The reaction system was degassed and flushed with H2(g) several times. Hydrogenation was then carried out under 1 atm (101.3 kPa) H2(g) overnight for 18 h. The reaction was filtered over Celite 545 and concentrated under vacuum, affording 11 as a white solid with a mass yield of 78% (113 mg, 0.11 mmol). LRMS (ESI): m/z calculated for [M + Na]+ C42H76N6NaO20, 1007.5; found 1007.7.
Synthesis of 12 was achieved by reacting 6 (639 mg, 0.70 mmol) with the synthetic methodology reported for 11. Compound 12 was isolated as a white solid with a yield of 71% (438 mg, 0.50 mmol). LRMS (ESI): m/z calculated for [M + Na]+ C38H69N5NaO18, 906.5; found 905.9.
To a solution of 11 (113 mg, 0.11 mmol) dissolved in 9:1 THF:H2O (5–10 mL) were added the decyl acid 21 (1.5 equiv.), EDC (1.5 equiv.) and N‐hydroxysuccinimide (NHS) (1.5 equiv.). The reaction was carried out at 50°C for 24 h. Once it was complete, the solvent was removed under reduced pressure and the resulting residue was purified by flash chromatography. Elution of the crude material in DCM:MeOH 15:1 → 9:1 afforded 13 in a yield of 27% (37 mg, 0.03 mmol) as a white solid. LRMS (ESI): m/z calculated for [M + Na]+ C54H98N6NaO21S, 1221.6; found 1222.4.
Synthesis of 14 was achieved by reacting 12 (438 mg, 0.50 mmol) with the synthetic conditions reported for the synthesis of 13. Following a reaction time of 15 h at ambient temperature, the crude reaction mixture was eluted in DCM:MeOH 95:5 → 12:1, isolating 14 as a white solid in a yield of 43% (233 mg, 0.21 mmol). LRMS (ESI): m/z calculated for [M + Na]+ C50H91N5NaO19S, 1120.6; found 1120.9.
Compound 13 (37 mg, 0.03 mmol) was dissolved in MeOH:H2O (8:3) and reacted with KHSO5 (3 equiv.). The reaction was carried out for 18 h, and on completion, the reaction mixture was partitioned in EtOAc (50 mL) and H2O (50 mL) and extracted in EtOAc (5 × 30 mL). The organic layers were combined, dried over Na2SO4 and concentrated. Purification of 15 in a gradient elution of 95:5 → 9:1 DCM:MeOH yielded the target as a white solid at 42% (16 mg, 0.01 mmol). LRMS (ESI): m/z calculated for [M + Na]+ C54H98N6NaO23S, 1253.6; found 1254.8.
Synthesis of 16 was achieved by reacting 14 (233 mg, 0.21 mmol) with the synthetic conditions reported for the synthesis of 15. Elution of 16 in 12:1 DCM:MeOH afforded the target as a white solid at 53% (127 mg, 0.11 mmol). LRMS (ESI): m/z calculated for [M] C50H91N5O20S, 1129.6; found 1128.7.
Compound 15 (16 mg, 0.01 mmol) was reacted following the TFA deprotection conditions reported for the synthesis of compound 9. Compound 17 was isolated as a white solid at a yield of 95% (14 mg, 0.01 mmol). 1H NMR (400 MHz, D2O) δ 5.64 (d, J = 3.8 Hz, 1H), 5.23 (d, J = 3.9 Hz, 1H), 4.39–4.27 (m, 4H), 4.25–4.08 (m, 1H), 3.99–3.94 (m, 3H), 3.92–3.81 (m, 2H), 3.77 (dd, J = 9.9, 3.7 Hz, 1H), 3.73–3.59 (m, 4H), 3.58–3.42 (m, 5H), 3.33–3.21 (m, 3H), 2.36–2.21 (m, 3H), 2.11–1.97 (m, 1H), 1.97–1.83 (m, 3H), 1.63–1.49 (m, 2H), 1.48–1.25 (m, 12H), 0.94 (t, J = 6.7 Hz, 3H). 13C NMR (150 MHz, MeOD) δ 177.4, 164.8, 99.6, 96.4, 81.9, 81.4, 74.6, 73.7, 73.0, 73.0, 71.9, 71.1, 70.4, 70.1, 69.2, 56.9, 54.8, 50.4, 42.2, 38.3, 33.1, 30.7, 30.7, 30.6, 30.5, 30.4, 30.3, 30.2, 29.8, 29.4, 23.7, 22.9, 14.4. HRMS (ESI): m/z calculated for [M + 2H]2+ C34H68N6O15S, 416.2232; found 416.2209.
Compound 16 (127 mg, 0.11 mmol) was reacted following the TFA deprotection conditions reported for the synthesis of compound 9. Compound 18 was isolated as a white solid in a yield of 98% (111 mg, 0.11 mmol). 1H NMR (600 MHz, MeOD) δ 5.46 (d, J = 3.9 Hz, 1H), 5.07 (d, J = 3.6 Hz, 1H), 4.06 (td, J = 9.5, 3.0 Hz, 1H), 4.03–3.98 (m, 1H), 3.88–3.79 (m, 3H), 3.77–3.72 (m, 1H), 3.70 (t, J = 9.2 Hz, 1H), 3.64–3.55 (m, 3H), 3.49–3.43 (m, 2H), 3.44––3.39 (m, 2H), 3.37 (t, J = 10.3 Hz, 1H), 3.21 (dd, J = 9.4, 0.8 Hz, 1H), 3.02 (dd, J = 13.0, 9.2 Hz, 1H), 2.50 (dt, J = 12.3, 4.2 Hz, 1H), 1.99–1.91 (m, 1H), 1.87–1.79 (m, 2H), 1.51–1.43 (m, 2H), 1.41–1.23 (m, 12H), 0.90 (t, J = 7.0 Hz, 3H). 13C NMR (150 MHz, MeOD) δ 165.0, 102.2, 97.0, 85.4, 81.2, 74.4, 73.5, 73.0, 73.0, 72.6, 70.5, 70.1, 68.8, 56.4, 54.7, 51.9, 48.9, 42.2, 41.5, 33.0, 30.6, 30.5, 30.4, 30.2, 29.4, 29.2, 23.7, 22.8, 14.4. HRMS (ESI): m/z calculated for [M + 2H]2+ C30H61N5O13S, 365.6994; found 365.7003.
To a solution of 1-decanethiol (1.03 mL, 5.00 mmol) and K2CO3 (2.07 g, 15.0 mmol) dissolved in anhydrous acetone (10 mL) was added 80% propargyl bromide in toluene (0.67 mL, 7.50 mmol). The reaction proceeded under an atmosphere of argon at ambient temperature for 24 h. Following completion, the reaction was quenched with H2O and the mixture partitioned between 1 M HCl (100 mL) and EtOAc (100 mL), then extracted with EtOAc (2 × 50 mL). The organic layers were combined and washed with 1 M HCl (3 × 30 mL), H2O (30 mL) and brine (30 mL), dried over Na2SO4 and concentrated under vacuum to afford 19 as an amber oil in a yield of 70% (1.09 g, 5.13 mmol). 1H NMR (400 MHz, CDCl3) δ 3.23 (d, J = 2.6 Hz, 2H), 2.67 (t, J = 7.4 Hz, 2H), 2.21 (t, J = 2.6 Hz, 1H), 1.61 (p, J = 7.4 Hz, 2H), 1.47–1.34 (m, 2H), 1.34–1.18 (m, 12H), 0.87 (t, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 80.3, 70.9, 32.0, 31.8, 29.7, 29.6, 29.4, 29.3, 29.1, 29.0, 22.8, 19.3, 14.2. LRMS (ESI): m/z calculated for [M + H]+ C13H25S, 213.2; found 213.2.
Compound 19 (1.09 g, 5.13 mmol) was dissolved in MeOH (30 mL), the solution cooled to 0°C, and KHSO5 (4.75 g, 15.4 mmol) dissolved in H2O (15 mL) was added dropwise. The reaction was carried out at ambient temperature for 18 h. On completion, the MeOH solution was partially concentrated under vacuum and the residue was partitioned between H2O (100 mL) and DCM (100 mL) and extracted into DCM (3 × 30 mL). The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure to afford 20 as a white solid in a yield of 98% (1.23 g, 5.03 mmol). 1H NMR (400 MHz, CDCl3) δ 3.83 (d, J = 2.7 Hz, 2H), 3.19 (t, J = 7.9 Hz, 2H), 2.50 (t, J = 2.7 Hz, 1H), 1.93–1.80 (m, 2H), 1.50–1.40 (m, 2H), 1.39–1.20 (m, 12H), 0.88 (t, J = 6.6 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 76.5, 71.8, 51.5, 45.0, 32.7, 29.6, 29.4, 29.4, 29.1, 28.5, 22.8, 22.1, 14.2. LRMS (ESI): m/z calculated for [M + Na]+ C13H24NaO2S, 267.1; found 267.2.
To a solution of 1-decanethiol (5 mL, 24.3 mmol) and K2CO3 (10.0 g, 72.4 mmol) dissolved in MeCN (40 mL) was added bromoacetic acid (4.17 g, 30.0 mmol). The resulting solution was stirred at 50°C for 24 h under an atmosphere of argon. Following reaction completion, the sample was dried under reduced pressure and partitioned between 1 M HCl (100 mL) and EtOAc (100 mL), extracted into EtOAc (3 × 50 mL), and washed with 1 M HCl (1 × 30 mL) and brine (1 × 30 mL). The combined organic layers were dried over Na2SO4 and concentrated under vacuum. Compound 21 was isolated as a white solid in a yield of 94% (5.31 g, 22.9 mmol). 1H NMR (400 MHz, CDCl3) δ 3.25 (s, 2H), 2.65 (t, J = 7.42 Hz, 2H), 1.59 (p, J = 7.7 Hz, 2H), 1.42–1.27 (m, 2H), 1.27 (s, 12H), 0.87 (t, J = 6.81 Hz 3H). 13C NMR (100 MHz, CDCl3) δ 174.9, 33.6, 33.0, 32.0, 29.7, 29.6, 29.4, 29.3, 29.1, 28.9, 22.4, 13.8. LRMS 21 (ESI): m/z calculated for [M–H]− C12H23O2S, 231.1; found 231.1.
Data availability
The data that support this study will be shared upon reasonable request to the corresponding author.
References
1 Kawaguchi H, Naito T, Nakagawa S, Fujisawa K. BB-K8, a new semisynthetic aminoglycoside antibiotic. J Antibiot 1972; 25(12): 695-708.
| Crossref | Google Scholar | PubMed |
2 Bera S, Zhanel GG, Schweizer F. Antibacterial activities of aminoglycoside antibiotics-derived cationic amphiphiles. Polyol-modified neomycin B-, kanamycin A-, amikacin-, and neamine-based amphiphiles with potent broad spectrum antibacterial activity. J Med Chem 2010; 53(9): 3626-3631.
| Crossref | Google Scholar | PubMed |
4 Zhang J, Chiang F-I, Wu L, Czyryca PG, Li D, Chang C-WT. Surprising alteration of antibacterial activity of 5′′-modified neomycin against resistant bacteria. J Med Chem 2008; 51(23): 7563-7573.
| Crossref | Google Scholar | PubMed |
5 Dezanet C, Kempf J, Mingeot-Leclercq M-P, Décout J-L. Amphiphilic aminoglycosides as medicinal agents. Int J Mol Sci 2020; 21(19): 7411.
| Crossref | Google Scholar | PubMed |
6 François B, Russell RJ, Murray JB, Aboul-ela F, Masquida B, Vicens Q, Westhof E. Crystal structures of complexes between aminoglycosides and decoding A site oligonucleotides: role of the number of rings and positive charges in the specific binding leading to miscoding. Nucleic Acids Res 2005; 33(17): 5677-5690.
| Crossref | Google Scholar | PubMed |
7 Baussanne I, Bussière A, Halder S, Ganem-Elbaz C, Ouberai M, Riou M, Paris J-M, Ennifar E, Mingeot-Leclercq M-P, Décout J-L. Synthesis and antimicrobial evaluation of amphiphilic neamine derivatives. J Med Chem 2010; 53(1): 119-127.
| Crossref | Google Scholar | PubMed |
8 Sautrey G, Zimmermann L, Deleu M, Delbar A, Machado LS, Jeannot K, Van Bambeke F, Buyck JM, Decout J-L, Mingeot-Leclercq M-P. New amphiphilic neamine derivatives active against resistant Pseudomonas aeruginosa and their interactions with lipopolysaccharides. Antimicrob Agents Chemother 2014; 58(8): 4420-4430.
| Crossref | Google Scholar | PubMed |
9 Ouberai M, El Garch F, Bussiere A, Riou M, Alsteens D, Lins L, Baussanne I, Dufrêne YF, Brasseur R, Decout J-L, Mingeot-Leclercq MP. The Pseudomonas aeruginosa membranes: a target for a new amphiphilic aminoglycoside derivative? Biochim Biophys Acta 2011; 1808(6): 1716-1727.
| Crossref | Google Scholar | PubMed |
10 Udumula V, Ham YW, Fosso MY, Chan KY, Rai R, Zhang J, Li J, Chang C-WT. Investigation of antibacterial mode of action for traditional and amphiphilic aminoglycosides. Bioorg Med Chem Lett 2013; 23(6): 1671-1675.
| Crossref | Google Scholar | PubMed |
11 Hancock RE. Peptide antibiotics. Lancet 1997; 349(9049): 418-422.
| Crossref | Google Scholar | PubMed |
12 Shai Y. Mode of action of membrane active antimicrobial peptides. Biopolymers 2002; 66(4): 236-248.
| Crossref | Google Scholar | PubMed |
13 Lazaridis T, He Y, Prieto L. Membrane interactions and pore formation by the antimicrobial peptide protegrin. Biophys J 2013; 104(3): 633-642.
| Crossref | Google Scholar | PubMed |
14 Guchhait G, Altieri A, Gorityala B, Yang X, Findlay B, Zhanel GG, Mookherjee N, Schweizer F. Amphiphilic tobramycins with immunomodulatory properties. Angew Chem Int Ed Engl 2015; 54(21): 6278-6282.
| Crossref | Google Scholar | PubMed |
15 Jones PB, Parrish NM, Houston TA, Stapon A, Bansal NP, Dick JD, Townsend CA. A new class of antituberculosis agents. J Med Chem 2000; 43(17): 3304-3314.
| Crossref | Google Scholar | PubMed |
16 Parrish NM, Houston T, Jones PB, Townsend C, Dick JD. In vitro activity of a novel antimycobacterial compound, N-octanesulfonylacetamide, and its effects on lipid and mycolic acid synthesis. Antimicrob Agents Chemother 2001; 45(4): 1143-1150.
| Crossref | Google Scholar | PubMed |
17 Parrish NM, Ko CG, Hughes MA, Townsend CA, Dick JD. Effect of n-octanesulphonylacetamide (OSA) on ATP and protein expression in Mycobacterium bovis BCG. J Antimicrob Chemother 2004; 54(4): 722-729.
| Crossref | Google Scholar | PubMed |
18 Sun H-K, Pang A, Farr DC, Mosaiab T, Britton WJ, Anoopkumar-Dukie S, Grice ID, Kiefel MJ, West NP, Grant GD, Houston TA. Thioamide derivative of the potent antitubercular 2-(decylsulfonyl) acetamide is less active against Mycobacterium tuberculosis, but a more potent antistaphylococcal agent. Aust J Chem 2018; 71(9): 716-719.
| Crossref | Google Scholar |
19 Quader S, Boyd SE, Jenkins ID, Houston TA. Multisite modification of neomycin B: combined Mitsunobu and click chemistry approach. J Org Chem 2007; 72(6): 1962-1979.
| Crossref | Google Scholar | PubMed |
20 Herzog IM, Feldman M, Eldar-Boock A, Satchi-Fainaro R, Fridman M. Design of membrane targeting tobramycin-based cationic amphiphiles with reduced hemolytic activity. MedChemComm 2013; 4(1): 120-124.
| Crossref | Google Scholar |
21 Dhondikubeer R, Bera S, Zhanel GG, Schweizer F. Antibacterial activity of amphiphilic tobramycin. J Antibiot 2012; 65(10): 495-498.
| Crossref | Google Scholar | PubMed |
22 Mingeot-Leclercq M-P, Décout J-L. Bacterial lipid membranes as promising targets to fight antimicrobial resistance, molecular foundations and illustration through the renewal of aminoglycoside antibiotics and emergence of amphiphilic aminoglycosides. MedChemComm 2016; 7(4): 586-611.
| Crossref | Google Scholar |
23 Benhamou RI, Shaul P, Herzog IM, Fridman M. Di‐N‐methylation of anti‐Gram‐positive aminoglycoside‐derived membrane disruptors improves antimicrobial potency and broadens spectrum to Gram‐negative bacteria. Angew Chem 2015; 127(46): 13821-13825.
| Crossref | Google Scholar |
24 Berkov-Zrihen Y, Herzog IM, Benhamou RI, Feldman M, Steinbuch KB, Shaul P, Lerer S, Eldar A, Fridman M. Tobramycin and nebramine as pseudo‐oligosaccharide scaffolds for the development of antimicrobial cationic amphiphiles. Chem Eur J 2015; 21(11): 4340-4349.
| Crossref | Google Scholar | PubMed |
25 Zhang Q, Alfindee MN, Shrestha JP, Nziko VdPN, Kawasaki Y, Peng X, Takemoto JY, Chang C-WT. Divergent synthesis of three classes of antifungal amphiphilic kanamycin derivatives. J Org Chem 2016; 81(22): 10651-10663.
| Crossref | Google Scholar | PubMed |
26 Berkov-Zrihen Y, Herzog IM, Feldman M, Fridman M. Site-selective displacement of tobramycin hydroxyls for preparation of antimicrobial cationic amphiphiles. Org Lett 2013; 15(24): 6144-6147.
| Crossref | Google Scholar | PubMed |
27 Fosso MY, Shrestha SK, Green KD, Garneau-Tsodikova S. Synthesis and bioactivities of kanamycin B-derived cationic amphiphiles. J Med Chem 2015; 58(23): 9124-9132.
| Crossref | Google Scholar | PubMed |
28 Fosso MY, Zhu H, Green KD, Garneau-Tsodikova S, Fredrick K. Tobramycin variants with enhanced ribosome‐targeting activity. ChemBioChem 2015; 16(11): 1565-1570.
| Crossref | Google Scholar | PubMed |
29 Thamban Chandrika N, Green KD, Houghton JL, Garneau-Tsodikova S. Synthesis and biological activity of mono-and di-N-acylated aminoglycosides. ACS Med Chem Lett 2015; 6(11): 1134-1139.
| Crossref | Google Scholar | PubMed |
30 Herzog IM, Green KD, Berkov‐Zrihen Y, Feldman M, Vidavski RR, Eldar‐Boock A, Satchi‐Fainaro R, Eldar A, Garneau‐Tsodikova S, Fridman M. 6′′‐Thioether tobramycin analogues: towards selective targeting of bacterial membranes. Angew Chem 2012; 124(23): 5750-5754.
| Crossref | Google Scholar |
31 Chen W, Biswas T, Porter VR, Tsodikov OV, Garneau-Tsodikova S. Unusual regioversatility of acetyltransferase Eis, a cause of drug resistance in XDR-TB. Proc Natl Acad Sci 2011; 108(24): 9804-9808.
| Crossref | Google Scholar | PubMed |
32 Green KD, Chen W, Garneau-Tsodikova S. Identification and characterization of inhibitors of the aminoglycoside resistance acetyltransferase Eis from Mycobacterium tuberculosis. ChemMedChem 2012; 7(1): 73-77.
| Crossref | Google Scholar | PubMed |
33 Zaunbrecher MA, Sikes RD, Metchock B, Shinnick TM, Posey JE. Overexpression of the chromosomally encoded aminoglycoside acetyltransferase Eis confers kanamycin resistance in Mycobacterium tuberculosis. Proc Natl Acad Sci 2009; 106(47): 20004-20009.
| Crossref | Google Scholar | PubMed |