

Preparation of Protein–Polymer Conjugates: Copolymerisation by RAFT
Fei Huang A , Judith A. Scoble B , John Chiefari A and Charlotte C. Williams B C
B C
A CSIRO Manufacturing, Bag 10, Bayview Avenue, Clayton, Vic. 3168, Australia.
B CSIRO Manufacturing, 343 Royal Parade, Parkville, Vic. 3052, Australia.
C Corresponding author. Email: Charlotte.Williams@csiro.au
Australian Journal of Chemistry 73(10) 1027-1033 https://doi.org/10.1071/CH19514
Submitted: 13 October 2019 Accepted: 28 February 2020 Published: 2 June 2020
Journal Compilation © CSIRO 2020 Open Access CC BY-NC-ND
Abstract
A method for the preparation of protein–polymer conjugates is presented that represents a different approach to current conjugation techniques. A protein–polymer conjugate was prepared by reversible addition–fragmentation chain-transfer (RAFT) copolymerisation, where one of the species of monomer used contains a protein. The enzyme horseradish peroxidase (HRP) was functionalised with an acrylate group via a polyethylene glycol (PEG) linker to the protein surface lysine residues. The PEG linker promoted aqueous solubility of the acrylate group, which led to an improved yield of HRP functionalisation. RAFT copolymerisation with N-acryloylmorpholine (NAM) resulted in synthesis of an HRP-RAFT copolymer, with full retention of the enzyme’s activity.
Introduction
Conjugation of a polymer to a therapeutic protein is known to modify and improve the biodistribution and pharmacokinetics of the protein of interest,[1–3] with several polymer–protein conjugates on the market for treatment of a range of disease states.[4] There is broad interest in straightforward synthetic methods to prepare protein–polymer conjugates and the use of reversible addition–fragmentation chain-transfer (RAFT)-derived polymers falls within the strategy,[5–8] and may serve as an alternative to modification with polyethylene glycol (PEGylation).[5,9] RAFT polymerisation is a versatile controlled, radical polymerisation method, that has been shown to prepare functional polymers with defined structures and molecular weights, and relatively low polydispersity (PDI) compared with other radical polymerisation methods.[10]
The preparation of protein–polymer conjugates is commonly reported by a method referred to as the grafting-to approach, where conjugates are prepared by reacting preformed polymers with a protein, often at a defined site or residue on the protein.[11–16] This can be achieved by installing a protein-reactive functional group onto the polymer (that will react with particular amino acid residues of the protein such as cysteine, lysine, or bridging disulfide bonds),[5,17–19] either by post-polymerisation RAFT end group modification, or the growing polymer itself contains an orthogonal and stable bio-reactive handle.[20] In some instances the protein may also be chemically modified or engineered to contain a site-selective reactive handle available for conjugation.[21,22] Disadvantages of this grafting-to method include difficulty in separating unreacted polymer and protein from a polymer–protein conjugate, especially if both the protein and polymer possess high molecular weight (MW).[23] Furthermore, the grafting-to approach is sterically demanding, and can lead to low yielding reactions due to the inherent low concentration of functional groups relative to the polymer and protein (bringing two large molecules together containing small single functional groups to react).[5,23] An excess of polymer is often used in these reactions in order to overcome steric limitations, which can also be wasteful.
Alternatively, there are reports that have demonstrated the ability to grow RAFT-derived polymers from proteins. This is referred to as the grafting-from method, which involves functionalisation (preferably in a site-selective manner) of a protein with a RAFT chain transfer agent (CTA) followed by polymerisation, resulting in polymer formation from the protein surface.[5] The proposed advantages are the simplification of the purification process (unreacted monomer should be easier to remove from the conjugate) and that, with steric constraints removed, high MW polymers can theoretically be prepared.[24]
Grafting a polymer from a protein by RAFT (or other controlled free radical techniques such as atom transfer radical polymerisation (ATRP)) must be performed under biologically relevant conditions, that is, conditions that allow for retention of the biological activity of the protein. With the right combination of reagents, RAFT can be performed in water, without the need for elevated temperatures.[13] Polymers have been grown, by RAFT or ATRP polymerisation methods, from the surface of proteins such as bovine serum albumin,[23,25–27] lysozyme,[26,28] and streptavidin.[28] However, these proteins are generally not amenable to biochemical assessment. In this work, we describe the use of an industrially relevant protein, horseradish peroxidase (HRP). HRP is an enzyme that is used widely by industry in biomarker assays such as immunohistochemistry.[29]
A third approach to making protein–polymer conjugates involves copolymerisation of a protein into a growing, RAFT-derived polymer; this is neither grafting-to nor grafting-from the protein but instead the protein is incorporated into the reaction as an activated monomer. In this work we demonstrate for the first time the copolymerisation, or incorporation, of an activated protein-functionalised monomer into a growing RAFT polymer. Here the protein-functionalised monomer is referred to as a protein-RAFT-monomer and is a protein species that can participate in a free radical polymerisation reaction, rather than referring to the state of the protein itself.
This work demonstrates an approach to synthesise protein–polymer conjugates, by making the protein one of the monomeric starting materials, through synthesis of a protein-RAFT-monomer. Not only does this approach give rise to high yielding conjugates, it ensures protein incorporation and demonstrates the biological tolerance of the RAFT polymerisation technique.
Results and Discussion
The use of RAFT as a method to prepare polymers allows for the introduction of a variety of monomer units that ultimately will determine the composition and structure of the polymer (Scheme 1). A range of copolymers can be prepared using the RAFT technique; by varying the type of monomer used, ratio of monomer, and the order of monomer additions, copolymers can be prepared as statistical or block copolymers, and polymers can span a range of structures and functions.[30]
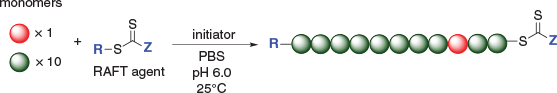
|
The active functional group of a monomer used in the RAFT reaction that is involved in the growing polymer is a vinylic group. Active monomers such as (meth)acrylates and (meth)acrylamides have their vinylic group conjugated to a carbonyl.[30] One can prepare a diverse and interesting range of monomers, by functionalising any molecule with these vinylic reactive groups, and that molecule can be used as a monomer for the polymerisation reaction. The Stayton group has elegantly demonstrated RAFT copolymerisation of antibiotic small molecule drugs (such as ciprofloxacin).[31] A monomer, referred to as a prodrug, was prepared whereby ciprofloxacin was activated by installation of a methacrylate functional group, which then could be used directly in copolymerisation reactions to yield ciprofloxacin-loaded polymers.
In this work an activated protein-RAFT-monomer is prepared by installation of the vinylic reactive group to the protein surface and submitting it to a protein-friendly RAFT polymerisation reaction in order to create a protein–polymer conjugate through copolymerisation. This work seeks to further understand the compatibility of RAFT polymerisation with biologically relevant functional proteins such as enzymes.
In order to copolymerise a protein-RAFT-monomer, the RAFT polymerisation reaction needs to be optimised to suit protein-friendly reaction conditions,[25,26,32,33] and the pH needs to maximise the stability of the enzyme; for HRP this is pH 6.0 (Scheme 1). The polymer selection was guided by monomers that exhibit high propagation rates and conversion, ultimately giving rise to high MW polymers, with retention of low polydispersity. The polymers that would constitute the backbone of these protein copolymer conjugates also need to be water soluble, stable at 25°C, and easy to prepare. To demonstrate this technique N-acryloylmorpholine (NAM) was used as the monomer. RAFT polymerisation reactions (Scheme 2) were performed in phosphate buffered saline (PBS), at 25°C and pH 6.0 with near 100 % monomer conversion being achieved, as determined by 1H NMR spectroscopy.

|
HRP is a 44 kDa glycoprotein with six lysine residues which are available for conjugation. Traditionally, conjugation methods to HRP have tended to focus on covalent attachment through these residues as there are only a small number of them and their modification does not adversely affect enzyme activity, indeed some chemical modification of the surface lysine residues can actually increase the stability of HRP.[34]
With protein-friendly RAFT conditions established, it was important to check the integrity of HRP after exposure to the RAFT polymerisation reaction. Although it has been shown that a cytotoxic small molecule, SN-38, maintains activity after exposure to RAFT polymerisation reactions,[35] there is evidence to suggest that some proteins may be susceptible to radical damage.[36] In order to assess the protein under these conditions, a RAFT polymerisation reaction was performed in the presence of a concentrated solution of HRP (1–2 mg mL−1). Poly(NAM) was synthesised using three different RAFT agents, with the water soluble and low-temperature-initiated RAFT initiator 2,2′-azobis[2-(2-imidazolin-2-yl)propane]dihydrochloride (VA-044), to give rise to water soluble RAFT polymers with high monomer incorporation (determined by 1H NMR analysis). The RAFT polymerisation reaction gave polymers with a high MW of 111 kDa (characterised by 1H NMR analysis) that were unhindered by the presence of a large protein at a reasonably high concentration. Furthermore, the enzyme itself retained activity after exposure to the radical catalysed polymerisation reaction (Table 1).

|
To determine HRP activity, a common assay was used that detects its interaction with 3,3′,5,5′-tetramethylbenzidine (TMB). TMB is a chromogenic small molecule that gives rise to a colour change in solution (λmax 450 nm) in the presence of both HRP and hydrogen peroxide (H2O2) with subsequent low pH treatment (Scheme 3).[37]

|
Conjugation reactions to install the acrylate/acrylamide (X = O and N respectively) onto HRP involved reaction with an N-hydroxysuccinimide (NHS) ester PEG crosslinker with the orthogonal vinylic polymerisable reactive group (i.e. acrylate/acrylamide) at the other end of the hetero-functional crosslinker (Scheme 4).

|
Acceptable PEG linker length was at least 5 kDa, for the NHS-PEG-acrylate (NHS-PEG-AC) when the PEG length was 1 kDa the crosslinker was not only insoluble in water but formed a gel in DMSO. The pH of the conjugation reaction (Scheme 4, pH 8.0) was in the optimal range for the NHS ester reaction with lysine amine residues[38] and was in the acceptable pH stability range for HRP.[39] The HRP-PEG-AC monomer (from here on referring to X = O) was isolated from the conjugation reaction mixture by size exclusion chromatography (SEC) in PBS, and was characterised by both SEC and by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) (Fig. 1). Lanes 2–9 are samples of fractions from the SEC moving from shorter to longer retention volume (i.e. higher to lower MW), lanes 6–9 show unreacted HRP; lanes 4 and 5 show an overlapping mixture of HRP and HRP-PEG-AC monomer; lanes 1–3 show clean HRP-PEG-AC monomer.

|
Importantly the PEGylated HRP conjugate demonstrated retention of enzymatic activity as the isolated HRP-PEG-AC monomer in solution as determined by TMB assay compared with untreated HRP at a similar concentration (Table 1).
With the acrylate functionalised-protein in hand, the HRP-PEG-AC monomer was incorporated as a comonomer into RAFT polymerisation reactions with NAM (Scheme 5). Three different RAFT agents were assessed, including dithiobenzoates and trithiocarbonates (Scheme 5). The RAFT agent was selected for water solubility, stability in water, and rate of monomer conversion.[30] Trithiocarbonate RAFT agent 1 was used due to its hydrophilicity. The dithiobenzoate RAFT agent 3 offers the option for selective aminolysis should RAFT end group removal be required for further conjugation.

|
In order to isolate the HRP-co-RAFT polymer conjugate from the copolymerisation reaction mixture, separation techniques including ion exchange chromatography and SEC were explored. The best method involved submitting the reaction mixture directly to a Concanavalin A (Con A) column before SEC. Con A is a tetrameric metalloprotein that binds to a range of proteins, it will bind to HRP through carbohydrates on the protein’s surface, e.g. high mannose-glycans. Con A is bound to the surface of the packing resin in order to allow for binding of HRP when solutions of protein flow across the resin. This allowed removal of any polymer alone that formed in the reaction without incorporated HRP. After washing the column with PBS, the HRP-co-RAFT polymer conjugate and any unreacted HRP-PEG-AC monomer was removed from the column by elution with a mannose solution, which competes for Con A binding and HRP containing bound materials are eluted from the column. Further isolation of HRP-co-RAFT polymer conjugate from unreacted HRP-PEG-AC monomer was achieved by SEC (Fig. 2a).

|
Analysis of the eluents from the Con A column by SEC (Fig. 2a) showed the clear presence of two materials containing protein, at 10 mL (higher MW) and at 14 mL. Detection at 280 nm indicates protein, 214 nm indicates presence of the NAM polymer (as well as protein and many other components of the reaction mixture), while absorbance at 403 nm is due to the heme group found in HRP. Samples of each fraction were taken across the whole SEC chromatogram and TMB assays were performed using the same volume of each fraction. The black histogram shown in Fig. 2 represents the TMB assay readout. Fig. 2b shows the SEC results of the same RAFT polymerisation reaction in the presence of unmodified HRP (not HRP-PEG-AC monomer), demonstrating that HRP is not non-specifically incorporated into the growing polymer unless a polymerisable reactive group is present on the protein. Fig. 2b shows unreacted HRP with retention of TMB activity at 16 mL, and the RAFT polymer alone, with no TMB activity at 10 mL, confirming no non-specific incorporation of the enzyme into the polymer.
Isolated HRP-co-RAFT polymer conjugate was further characterised by 1H NMR analysis (Fig. 3). Signals due to the enzyme were not visible, due to the higher concentration of NAM monomer, compared with protein, present in the final conjugate. The polymerisation reaction proceeded with high monomer conversion as determined by the loss of monomer signals in the 1H NMR spectrum.
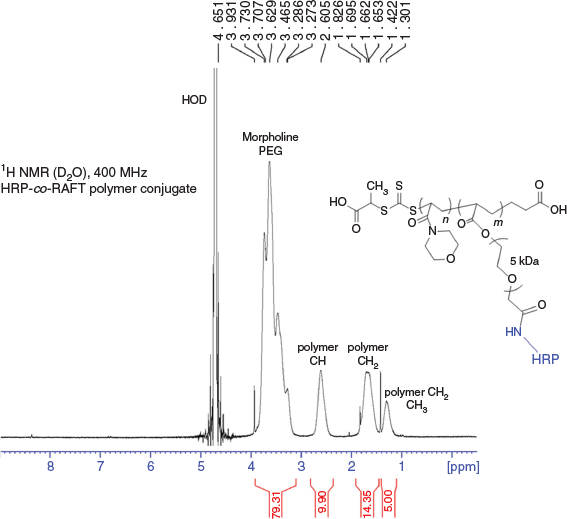
|
Furthermore, HRP functionalised with a short hydrocarbon linker and an acrylamide group (HRP-CO(CH2)2-ACA) was prepared in order to understand the importance of the PEG linker, and the acrylate versus acrylamide comonomer incorporation. An acrylamide NHS ester was prepared in situ and reacted in excess with HRP. The HRP-CO(CH2)2-ACA monomer was isolated by ultrafiltration using an Amicon centrifugal filter, MWCO 3 kDa, to remove excess acrylamide. The copolymerisation reaction with NAM (Scheme 6) showed some incorporation of HRP (by SEC and TMB overlay) into the polymer, but the result (not shown) was a broad mixture. This result demonstrates the importance of the PEG linker between the growing polymer and the enzyme; with only a short hydrocarbon linker, enzyme incorporation into the polymer is hindered.

|
Covalent conjugation of vinylic polymerisable functional groups, such as acrylates and acrylamides, to a functional protein, such as the enzyme HRP, can be performed without affecting the activity of the protein. Furthermore, a linker such as PEG between the site of protein conjugation and the polymerisable functional group is important to allow for that group to efficiently participate in a polymerisation reaction. This work represents an example of the creation of a protein-RAFT-monomer, and its participation in copolymerisation reactions using protein-friendly, radical initiated, RAFT polymerisation techniques to prepare defined protein–polymer conjugates. This method represents another mechanism by which protein–polymer conjugates can be prepared, adding to the current methodology alongside traditional graft-to and emerging graft-from techniques.
Conclusion
A functionally activated (acrylate), biologically active protein (HRP) has been copolymerised into a stable and water-soluble RAFT polymer using a low temperature, radical initiator to give rise to water soluble and stable protein–polymer conjugates.
The enzyme HRP demonstrates an example of a protein that can participate in a copolymerisation RAFT reaction under free radical polymerisation conditions without adverse effect on the polymerisation reaction or the activity of the protein itself. Although certain proteins may be susceptible to damage by small molecule radicals,[36] this was not the case with HRP. Using room temperature reaction conditions and aqueous buffer as a solvent allows for protein amenable conditions, although historically RAFT polymerisation reactions require organic solvents and higher temperatures.[10] Furthermore the protein-RAFT-monomer prepared here was stable and retained activity, even using simple amide coupling conjugation methods, through non-unique sites on the protein’s surface.
The copolymerisation technique reported here represents an example of another tool in the bioconjugation chemistry toolkit to prepare protein–polymer conjugates. The polymerisation reaction works well with high monomer conversion. This work demonstrates use with an industrially relevant enzyme, moving towards biomedical application.
Experimental
General
The water soluble initiator (2,2′-azobis[2-(2-imidazolin-2-yl)propane]dihydrochloride, VA-044) used in the RAFT polymerisation reactions was purchased from Wako Speciality Chemicals. The monomer, N-acryloylmorpholine (NAM), and the RAFT agents 2-(2-carboxyethylsulfanylthiocarbonylsulfanyl) propionic acid (1), 4-cyano-4-[(ethylsulfanylthiocarbonyl) sulfanyl]pentanoic acid (2), and 4-cyano-4-(phenyl-carbonothioylthio)pentanoic acid (3) were purchased from Sigma Aldrich. The NHS ester PEG acrylates and acrylamides were purchased from Creative PEGWorks. Horseradish peroxidase (HRP) was purchased from Sigma–Aldrich #P8375-25KU. 3,3′,5,5′-Tetramethylbenzidine (TMB) assay kits in solution for ELISA were purchased from Sigma–Aldrich. NMR spectra were performed on a Bruker 400 MHz Spectrometer. SEC were performed on a GE Healthcare Akta Express Purifier, the column used was a Superdex 200 Increase 10/300 GL, and SEC columns were eluted using PBS. The Con A columns were purchased from GE Healthcare. SDS-PAGE used 4–12 % BisTris pre-cast NuPAGE gels, MES running buffer, 200 V, 40 min.
Polymer Synthesis Reactions
In a sealable microwave vial was added 564 mg (4 mmol) NAM, 0.005 mmol RAFT agent (e.g. 1.3 mg RAFT agent 1), and 7.8 mg (0.025 mmol) of VA-044 followed by 2 mL of PBS (pH 6.0). The mixture was stirred at room temperature (note: < 25°C) until all reagents were dissolved. The vial was then sealed, needles inserted through a suba-seal lid, and the solution purged of oxygen by bubbling N2 through the reaction mixture for 40 min. After purging, needles were removed, and the vial placed into an oil bath at 25°C (activation temperature for VA-044 radical initiator) for 16 h. The reaction was stopped by opening the vial and exposing the reaction to air. The reaction mixture was then lyophilised before analysis by 1H NMR spectroscopy or SEC. δH (400 MHz, D2O) 1.2–1.4 (broad peak, RAFT CTA 1: CH3 + R-end polymer CH2), 1.6–2.0 (broad peak, NAM backbone polymeric CH2 + R-end polymeric CH2), 2.6–2.8 (broad peak, NAM backbone polymeric CH), 3.2–4.0 (broad peak, morpholine CH2 + Z-end polymer CH). Target DP = 800; MW from 1H NMR = 111 926 (MW determined by calculating actual DP from percentage monomer conversion by 1H NMR × monomer MW + CTA MW). SEC: broad peak detected at 214 nm eluted at 11.5 mL.
Polymer Synthesis in the Presence of HRP
Polymer synthesis in the presence of HRP followed the exact procedures as for the polymer synthesis alone, except the 2 mL of PBS (pH 6.0) contained 1 or 2 mg of dissolved HRP. After completion the reaction mixture was lyophilised before analysis by 1H NMR spectroscopy and SEC. 1H NMR results were as for the polymer alone, HRP could not be seen by 1H NMR analysis. Target DP = 800; MW from 1H NMR = 111 000 (MW determined by calculating actual DP from percentage monomer conversion by 1H NMR × monomer MW + CTA MW). Monomer conversion was near 100 % (by 1H NMR analysis). Fig. 2b shows the SEC analysis of the polymer formed in the presence of HRP.
Conjugation Reactions
HRP (2 mg, 4.545 × 10−5 mmol) was dissolved in 1 mL of PBS (10 mM (0.01M) phosphate at pH 8.0), and this solution was used to dissolve 11.4 mg (2.27 × 10−3 mmol, × 50 equivalents) of AC-PEG-SCM (Creative PEGWorks PHB-962; Acrylate-PEG-NHS (SCM – succinimidyl carboxy methyl ester)). The reaction mixture was placed on a slow rotating wheel for mixing, at room temperature, protected from light, for 16 h. The reaction mixture was submitted to SEC for purification and was characterised by both SEC and SDS-PAGE. SEC: overlaid absorbance wavelengths were detected at 214, 280 (protein), and 403 nm (UV max for HRP, due to heme group found in HRP), peak centred at 16.5 mL (due to HRP, characterised by SDS-PAGE); and peak centred at 14.44 mL (due to HRP-PEG-AC). SDS-PAGE: 4–12 % BisTris precast NuPAGE gel, SeeBlue 2 MW markers, MES running buffer, 200 V, 40 min (Fig. 1).
Note: HRP-PEG-AC with PEG ≥ 5 kDa could not be detected by mass spectrometry (MS), these samples contain broad MS peaks with a repeating PEG unit of 44 Da that masked detection of the protein components by MS. A control conjugation reaction using NHS-PEG4(PEG24)3 (Quanta Biodesign #10454, MW 4006.69) under the same reaction conditions was performed, the resulting HRP-PEG(4 kDa) was purified by SEC and submitted to liquid chromatography (LC)-MS detection using an ESI micrOTOF-Q, and showed a major mass of 47067 Da, which is the expected MW for 44174 Da HRP plus 4 kDa PEG-NHS.
Copolymerisation Reactions
In a sealable microwave vial was added 564 mg (4 mmol) of NAM, 0.005 mmol of RAFT agent (e.g. 1.3 mg of RAFT agent 1), and 7.8 mg (0.025 mmol) of VA-044 followed by a solution of HRP-PEG(5 kDa)-AC (Amax λ403 = 0.014; 1.25 µM) in 2 mL of PBS (pH 6.0) and the mixture was stirred at room temperature (note: < 25°C) until all reagents were dissolved. The vial was then sealed, needles inserted through a suba-seal lid, and the solution purged of oxygen by bubbling N2 through the reaction mixture for 40 min. After purging, needles were removed, and the vial placed into an oil bath at 25°C for 16 h. The reaction was stopped by opening the vial and exposing the reaction to air. The reaction mixture was loaded directly onto a Con A column for purification. The Con A column was equilibrated with 0.1 M NaOAc, 0.5 M NaCl, 1 mM CaCl2, and 1 mM MnCl2, at pH 6.0 at a flow rate of 0.1 mL min−1. The reaction mixture was loaded onto the Con A column, which was washed with excess buffer to remove protein-free polymer and low MW materials, and then the bound proteins were eluted with 0.5 M methyl-α-d-glucopyranoside in the binding buffer. The fractions from the Con A column were submitted to SEC for characterisation and separation of unreacted HRP-PEG-AC from the HRP-co-RAFT polymer conjugate. SEC: overlaid absorbance wavelengths detected at 214, 280, and 403 nm, a peak centred at 14.0 mL was due to HRP-PEG-AC and a peak centred at 10.2 mL was the HRP-co-RAFT polymer conjugate.
A TMB assay was performed across the SEC spectrum by taking aliquots of all fractions collected and placing them into a MaxiSorp plate and submitting each fraction to the TMB solution assay kit protocols (for ELISA), TMB assay readout detected at 450 nm on a FLUOstar Optima plate-reader.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgements
The authors thank Tom Nebl for the LC-MS result reported in the experimental. The authors are grateful to Mark York and Mischa Mueller for providing suggestions to improve this manuscript. This research did not receive any specific funding.
References
[1] S. A. Bhawani, A. Husaini, F. B. Ahmad, M. R. Asaruddin, Curr. Protein Pept. Sci. 2018, 19, 972.| Crossref | GoogleScholarGoogle Scholar | 28828988PubMed |
[2] I. Ekladious, Y. L. Colson, M. W. Grinstaff, Nat. Rev. Drug Discov. 2019, 18, 273.
| Crossref | GoogleScholarGoogle Scholar | 30542076PubMed |
[3] M. J. Vicent, H. Ringsdorf, R. Duncan, Adv. Drug Deliv. Rev. 2009, 61, 1117.
| Crossref | GoogleScholarGoogle Scholar | 19682516PubMed |
[4] R. Duncan, M. J. Vicent, F. Greco, R. I. Nicholson, Endocr. Relat. Cancer 2005, 12, S189.
| Crossref | GoogleScholarGoogle Scholar | 16113096PubMed |
[5] J. H. Ko, H. D. Maynard, Chem. Soc. Rev. 2018, 47, 8998.
| Crossref | GoogleScholarGoogle Scholar | 30443654PubMed |
[6] E. M. Pelegri-O’Day, E. W. Lin, H. D. Maynard, J. Am. Chem. Soc. 2014, 136, 14323.
| Crossref | GoogleScholarGoogle Scholar | 25216406PubMed |
[7] J. Y. Shu, B. Panganiban, T. Xu, Annu. Rev. Phys. Chem. 2013, 64, 631.
| Crossref | GoogleScholarGoogle Scholar | 23331303PubMed |
[8] W. Zhao, F. Liu, Y. Chen, J. Bai, W. Gao, Polymer 2015, 66, A1.
| Crossref | GoogleScholarGoogle Scholar |
[9] K. Knop, R. Hoogenboom, D. Fischer, U. S. Schubert, Angew. Chem. Int. Ed. 2010, 49, 6288.
| Crossref | GoogleScholarGoogle Scholar |
[10] J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo, S. H. Thang, Macromolecules 1998, 31, 5559.
| Crossref | GoogleScholarGoogle Scholar |
[11] E. Bays, L. Tao, C. W. Chang, H. D. Maynard, Biomacromolecules 2009, 10, 1777.
| 19505142PubMed |
[12] X. Huang, M. Li, D. C. Green, D. S. Williams, A. J. Patil, S. Mann, Nat. Commun. 2013, 4, 2239.
| Crossref | GoogleScholarGoogle Scholar | 23896993PubMed |
[13] I. Ozer, A. Chilkoti, Bioconjug. Chem. 2017, 28, 713.
| Crossref | GoogleScholarGoogle Scholar | 27998056PubMed |
[14] Y. Pang, J. Liu, Y. Qi, X. Li, A. Chilkoti, Angew. Chem. Int. Ed. 2016, 55, 10296.
| Crossref | GoogleScholarGoogle Scholar |
[15] Y. Xia, S. Tang, B. D. Olsen, Chem. Commun. 2013, 49, 2566.
| Crossref | GoogleScholarGoogle Scholar |
[16] J. S. Kim, A. R. Sirois, A. J. Vazquez Cegla, E. Jumai’an, N. Murata, M. E. Buck, S. J. Moore, Bioconjug. Chem. 2019, 30, 1220.
| Crossref | GoogleScholarGoogle Scholar | 30920802PubMed |
[17] T. A. Wright, R. C. Page, D. Konkolewicz, Polym. Chem. 2019, 10, 434.
| Crossref | GoogleScholarGoogle Scholar | 31249635PubMed |
[18] P. Wilson, A. Anastasaki, M. R. Owen, K. Kempe, D. M. Haddleton, S. K. Mann, A. P. Johnston, J. F. Quinn, M. R. Whittaker, P. J. Hogg, T. P. Davis, J. Am. Chem. Soc. 2015, 137, 4215.
| Crossref | GoogleScholarGoogle Scholar | 25794267PubMed |
[19] A. S. M. Wong, E. Czuba, M. Z. Chen, D. Yuen, K. I. Cupic, S. Yang, R. Y. Hodgetts, L. I. Selby, A. P. R. Johnston, G. K. Such, ACS Macro Lett. 2017, 6, 315.
| Crossref | GoogleScholarGoogle Scholar |
[20] G. N. Grover, H. D. Maynard, Curr. Opin. Chem. Biol. 2010, 14, 818.
| Crossref | GoogleScholarGoogle Scholar | 21071260PubMed |
[21] Y. Wang, C. Wu, Biomacromolecules 2018, 19, 1804.
| 29722971PubMed |
[22] M. P. Madej, G. Coia, C. C. Williams, J. M. Caine, L. A. Pearce, R. Attwood, N. A. Bartone, O. Dolezal, R. M. Nisbet, S. D. Nuttall, T. E. Adams, Biotechnol. Bioeng. 2012, 109, 1461.
| Crossref | GoogleScholarGoogle Scholar | 22170409PubMed |
[23] B. Sumerlin, ACS Macro Lett. 2012, 1, 141.
| Crossref | GoogleScholarGoogle Scholar |
[24] M. A. Gauthier, H. A. Klok, Chem. Commun. 2008, 23, 2591.
| Crossref | GoogleScholarGoogle Scholar |
[25] C. Boyer, V. Bulmus, J. Liu, T. P. Davis, M. H. Stenzel, C. Barner-Kowollik, J. Am. Chem. Soc. 2007, 129, 7145.
| Crossref | GoogleScholarGoogle Scholar | 17500523PubMed |
[26] J. Nicolas, V. San Miguel, G. Mantovani, D. M. Haddleton, Chem. Commun. 2006, 45, 4697.
| Crossref | GoogleScholarGoogle Scholar |
[27] N. Vanparijs, R. De Coen, D. Laplace, B. Louage, S. Maji, L. Lybaert, R. Hoogenboom, B. G. De Geest, Chem. Commun. 2015, 51, 13972.
| Crossref | GoogleScholarGoogle Scholar |
[28] K. L. Heredia, D. Bontempo, T. Ly, J. T. Byers, S. Halstenberg, H. D. Maynard, J. Am. Chem. Soc. 2005, 127, 16955.
| Crossref | GoogleScholarGoogle Scholar | 16316241PubMed |
[29] T. Nakata, N. Suzuki, J. Histochem. Cytochem. 2012, 60, 611.
| Crossref | GoogleScholarGoogle Scholar | 22610462PubMed |
[30] S. Perrier, Macromolecules 2017, 50, 7433.
| Crossref | GoogleScholarGoogle Scholar |
[31] D. Das, S. Srinivasan, A. M. Kelly, D. Y. Chiu, B. K. Daugherty, D. M. Ratner, P. S. Stayton, A. Convertine, Polym. Chem. 2016, 7, 826.
| Crossref | GoogleScholarGoogle Scholar |
[32] P. De, M. Li, S. R. Gondi, B. S. Sumerlin, J. Am. Chem. Soc. 2008, 130, 11288.
| Crossref | GoogleScholarGoogle Scholar | 18665597PubMed |
[33] M. Li, H. Li, P. De, B. S. Sumerlin, Macromol. Rapid Commun. 2011, 32, 354.
| Crossref | GoogleScholarGoogle Scholar | 21433183PubMed |
[34] O. Ryan, M. R. Smyth, C. O. Fagain, Enzyme Microb. Technol. 1994, 16, 501.
| Crossref | GoogleScholarGoogle Scholar | 7764889PubMed |
[35] C. C. Williams, S. H. Thang, T. Hantke, U. Vogel, P. H. Seeberger, J. Tsanaktsidis, B. Lepenies, ChemMedChem 2012, 7, 281.
| Crossref | GoogleScholarGoogle Scholar | 22144261PubMed |
[36] R. T. Dean, J. V. Hunt, A. J. Grant, Y. Yamamoto, E. Niki, Free Radic. Biol. Med. 1991, 11, 161.
| Crossref | GoogleScholarGoogle Scholar | 1937134PubMed |
[37] A. Frey, B. Meckelein, D. Externest, M. A. Schmidt, J. Immunol. Methods 2000, 233, 47.
| Crossref | GoogleScholarGoogle Scholar | 10648855PubMed |
[38] O. Koniev, A. Wagner, Chem. Soc. Rev. 2015, 44, 5495.
| Crossref | GoogleScholarGoogle Scholar | 26000775PubMed |
[39] See EC 1.11.1.7, pp. 1–6, in: Enzyme Handbook 7 (Eds D. Schomberg, M. Salzmann, D. Stephan) 1993 (Springer: Berlin).
[40] M. M. Kurfurst, Anal. Biochem. 1992, 200, 244.
| Crossref | GoogleScholarGoogle Scholar | 1378701PubMed |