

Assessing the Applicability of the Geometric Counterpoise Correction in B2PLYP/Double-ζ Calculations for Thermochemistry, Kinetics, and Noncovalent Interactions*
Nisha Mehta A and Lars Goerigk A B
A and Lars Goerigk A B
A School of Chemistry, The University of Melbourne, Parkville, Vic. 3010, Australia.
B Corresponding author. Email: lars.goerigk@unimelb.edu.au
Australian Journal of Chemistry 74(11) 795-805 https://doi.org/10.1071/CH21133
Submitted: 1 June 2021 Accepted: 24 June 2021 Published: 19 July 2021
Abstract
We present a proof-of-concept study of the suitability of Kruse and Grimme’s geometric counterpoise correction (gCP) for basis set superposition errors (BSSEs) in double-hybrid density functional calculations with a double-ζ basis set. The gCP approach only requires geometrical information as an input and no orbital/density information is needed. Therefore, this correction is practically free of any additional cost. gCP is trained against the Boys and Bernardi counterpoise correction across a set of 528 noncovalently bound dimers. We investigate the suitability of the approach for the B2PLYP/def2-SVP level of theory, and reveal error compensation effects—missing London dispersion and the BSSE—associated with B2PLYP/def2-SVP calculations, and present B2PLYP-gCP-D3(BJ)/def2-SVP with the reparametrised DFT-D3(BJ) and gCP corrections as a more balanced alternative. Benchmarking results on the S66x8 benchmark set for noncovalent interactions and the GMTKN55 database for main-group thermochemistry, kinetics, and noncovalent interactions show a statistical improvement of the B2PLYP-gCP-D3(BJ) scheme over plain B2PLYP and B2PLYP-D3(BJ). B2PLYP-D3(BJ) shows significant overestimation of interaction energies, barrier heights with larger deviations from the reference values, and wrong relative stabilities in conformers, all of which can be associated with BSSE. We find that the gCP-corrected method represents a significant improvement over B2PLYP-D3(BJ), particularly for intramolecular noncovalent interactions. These findings encourage future developments of efficient double-hybrid DFT strategies that can be applied when double-hybrid calculations with large basis sets are not feasible due to system size.
Keywords: density functional theory, double-hybrid density functionals, noncovalent interactions, thermochemistry, London dispersion, atomic-orbital basis sets, computational chemistry, theoretical chemistry.
Introduction
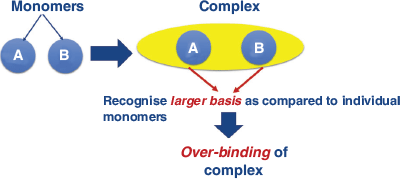
When using finite atomic orbital (AO) basis sets in an electronic structure calculation, the computed properties are subject to the basis set error.[1–4] The basis set error is classified into two types: the basis set superposition error (BSSE) and the basis set incompleteness error (BSIE).[1] Our present study focuses solely on the BSSE, for which several corrections have been proposed.[1,5–12] BSSE arises from an unbalanced basis set expansion of supramolecular complexes and their constituent monomers. In a dimer complex with subsystems A and B, the unoccupied basis functions of subsystem A can be used by subsystem B (and vice-versa) to lower its energy, which leads to the over-binding of complexes (Fig. 1). That artificial stabilisation can be removed by the counterpoise correction (CP) scheme, which was first introduced by Boys and Bernardi (the BB-CP scheme).[13] In this approach, the energy of each fragment is calculated with the basis functions of the full complex, but without electrons and nuclei of the other fragments; orbitals located at the positions of the other fragments are therefore also known as ‘ghost orbitals’. For example, the BB-CP correction for a dimer can be presented as:
|
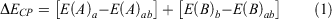
Here, a and b are the basis sets of subsystems A and B in their frozen complex geometries, with energies E(A) and E(B), respectively; the dimer’s basis set is represented by ab. The CP-corrected binding energy (BE) can then be obtained as:
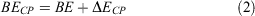
Similarly, the BB-CP correction can be applied to multimer systems beyond dimers.
The BB-CP scheme has two major shortcomings. The standard scheme can only calculate the intermolecular BSSE for noncovalently bound complexes. This is because it requires partitioning a system into all possible fragments. For some simple systems, this partitioning is straightforward, but for systems involving intramolecular noncovalent interactions (NCIs), this is not the case. The second problem is the computational cost, which increases with the number of fragments due to a larger number of separate calculations.
In 2006, Galao and Alvarez-Idaboy addressed intramolecular BSSE by introducing an ‘atom-by-atom’ scheme.[14] Jensen later generalised this into the ACP-n approach (atomic counterpoise method) where the total BSSE is calculated as the sum of atomic contributions.[15] A few empirical models for BSSE calculations—models that do not require additional quantum-chemical calculations, but rely on fitted statistical models—were introduced in Refs [16 and 17]. DiLabio and co-workers’ atom-centred potentials[18–20] compensate for basis set limitations in treating NCIs with dispersion-corrected Hartree-Fock (HF) and Density Functional Theory (DFT) methods, the latter of which we here refer to as density functional approximations (DFAs).
In 2012, Kruse and Grimme introduced a semi-empirical correction scheme called ‘geometrical counterpoise correction (gCP)’.[21] The gCP scheme is based on an atom pairwise approach. This scheme only requires geometric information as an input and no wave function data is needed. gCP has gained considerable attention in the computational chemistry community because of its efficiency, ease of use, and transferability. The gCP correction has to be parametrised for each basis set. For a given basis set, there is one set of parameters for HF and another set that applies to all GGA, meta-GGA, and hybrid DFT methods. In addition, the correction has also been used in specialised cases, such as HF-3c,[22] HSE-3c,[23] PBEh-3c,[24] B97-3c,[25] HFsol-3c,[26] PBEsol0-3c,[26] HSEsol-3c,[26] and r2SCAN-3c.[27] Those are DFT methods with specialised basis sets and other corrections to allow fast and accurate calculations. Regardless of the underlying DFT methods, the gCP correction has to be combined with a London-dispersion correction. Usually, this has been the DFT-D3[28,29] or DFT-D4[30,31] corrections.
In 2017, Head-Gordon and co-workers introduced the DFT-C[32] correction for BSSE, which is different from Kruse and Grimme’s gCP approach in a few critical areas; see Ref. [32] DFT-C for more details on those differences. Although DFT-C showed improvement over gCP, this approach is very specialised and implemented in fewer software packages, therefore, beyond the scope of this study.
DFT currently offers the highest cost-accuracy ratio for thermochemistry, kinetics, and NCIs. Among the various DFAs, double hybrid density functionals (DHDFs) stand out for their accuracy, reliability, and overall robustness.[33–40] DHDFs are a combination of conventional DFT and second-order Møller-Plesset perturbation (MP2) theory. It is known that conventional wave function electron correlation methods converge more slowly to the complete basis set (CBS) limit than HF or conventional DFT. Basis set convergence and BSSE for lower-rung DFT methods and double hybrids have been extensively studied for the S66x8[41] benchmark set for noncovalent interactions in Ref. [42]. While the basis set convergence of a DHDF calculation is faster than for wave function electron correlation methods,[43] there is a slower basis set convergence compared to conventional DFT due to the MP2 part. In electronic structure calculations, using a smaller basis set (e.g. a double-ζ AO basis) may become necessary for the treatment of large systems due to limited computing resources. Explicitly correlated approaches, such as R12 and F12 variants,[44–48] allow using smaller AO basis sets with faster convergence to the CBS limit, and this has also been demonstrated for DHDFs.[43] The other alternative to allow for faster DHDF calculations is a computationally efficient split-valence basis set named ‘DH-SVPD’,[49–51] which has been especially designed for DHDFs and the treatment of weakly bound systems.
We are motivated by the question if the gCP scheme is beneficial for double-hybrid DFT calculations, as they have so far been excluded from the gCP scheme, most likely due to MP2 having a different basis set convergence behaviour. Herein, we present a brief proof-of-concept study to see if any developments in such a direction are justified. For that reason we choose the B2PLYP[33] functional to test the impact of gCP. In the first step of our study, we analyse the major shortcomings of using B2PLYP with a small basis set (def2-SVP[52]), namely, BSSE and missing London dispersion. We will then present the B2PLYP-gCP-D3(BJ)[29]/def2-SVP scheme and compare it with B2PLYP-D3(BJ)/def2-SVP, B2PLYP/def2-SVP, and B2PLYP-D3(BJ)/quadruple-ζ results. The individual contributions from gCP and DFT-D3(BJ) will be analysed in all cases. This analysis will be carried out on the S66x8 set, followed by the GMTKN55[35] database for general main-group thermochemistry, kinetics, and NCIs.
This manuscript illustrates the benefits of using DHDF-gCP-D3(BJ)/double-ζ over DHDF/double-ζ and DHDF-D3(BJ)/double-ζ for electronic structure calculations. Our conclusions will reveal useful insights for new approaches, and enable potential future developments beyond B2PLYP.
Computational Details
Technical Details
Our starting point for determining improved electronic energies is a standard DHDF calculation (B2PLYP in this study) with a small basis set. Two terms are added to the B2PLYP energy (EB2PLY P) to account for BSSE and missing London dispersion forces. The total energy for the resulting B2PLYP-gCP-D3(BJ) method can be written as:
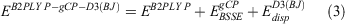
where 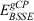 and
and 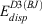 are the energy contributions from the gCP and DFT-D3(BJ) corrections, respectively, which will be described in more detail in the following two sections.
are the energy contributions from the gCP and DFT-D3(BJ) corrections, respectively, which will be described in more detail in the following two sections.
All electronic structure calculations were carried out with ORCA 4.0.0 and 4.0.1,[53,54] while the DFT-D3(BJ) and gCP energy corrections were obtained with the standalone programs by Grimme and co-workers.[55,56] The generally observed trends for B2PLYP were compared to BLYP[57–59] and B3LYP,[60,61] which are GGA and hybrid DFAs, respectively, with the same underlying DFT exchange and correlation components as B2PLYP. All BLYP calculations were treated with the resolution of the identity (RI-J) approximation for Coulomb integrals and appropriate auxiliary basis sets,[62,63] while hybrid, double-hybrid, and MP2-type calculations were done with the combined resolution-of-the-identity for Coulomb and the chain-of-sphere approximations for exchange integrals (RIJCOSX).[64] We also used the frozen core approximation for the second-order perturbative steps of the DHDF and MP2-type calculations to prevent core-core BSSE,[65] as well as the RI approximation with appropriate auxiliary basis sets to speed up those steps.[63] All calculations were carried out with ORCA’s quadrature grid ‘3’, followed by a non-iterative step with the larger grid ‘4’. The SCF convergence criterion was set to 10−7 Eh. The non-hydrogen atoms in the GMTKN55 sets G21EA,[66,67] AHB21,[68] and IL16[68] were augmented with diffuse s and p functions from the Dunning class of basis sets,[69] while hydrogens were subjected to diffuse s functions. Oxygens in WATER27[35,70] were also augmented with diffuse s and p functions. While the gCP correction in our case has not been fitted for diffuse functions, it was necessary to add them in the case of the aforementioned four benchmark sets to make our results commensurate with our previous GMTKN55 studies and to properly treat the negatively charged species[35,38,71–73] in those sets. Similar to our previous GMTKN55 papers, we use the prefix aug’ in front of a basis set name. For instance, “aug’-def2-SVP” indicates that diffuse functions were used for a specific benchmark set. If we have a more general discussion “(aug’)-def2-SVP” indicates that some results are based on basis sets with diffuse and others without diffuse functions.
The basis functions for core electrons of heavy elements were replaced with def2-ECP[52] effective core potentials in the GMTKN55 sets HAL59,[74,75] HEAVY28,[28] and HEAVYSB11.[28]
Fitting Procedure for gCP
The gCP correction is defined as:[21]
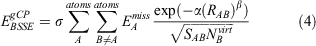
where 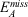 represents the difference in atomic energies between a large basis set and the target basis set for free atom A, SAB is the Slater-type overlap integral assessed over s-type orbitals,
represents the difference in atomic energies between a large basis set and the target basis set for free atom A, SAB is the Slater-type overlap integral assessed over s-type orbitals, 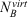 represents the number of virtual orbitals on atom B, and σ, α and β are scaling parameters. Slater exponents in SAB are weighted by a scaling parameter dubbed η. Those four scaling parameters need to be fitted for a given level of theory; in our case this meant that we fitted them for the B2PLYP/def2-SVP level of theory. The fitting procedure for the gCP correction uses the S66x8 benchmark set, which is an extension of the S66 dataset.[41] S66 contains 66 noncovalently bound dimers and is separated into systems that are dominated by electrostatics (hydrogen bonds), dispersion, or a mixture of both. To allow for an analysis of interaction energies in non-equilibrium situations, the S66x8 extension considers eight different intermolecular distances for each dimer, namely at 0.90re, 0.95re, re, 1.05re, 1.10re, 1.25re, 1.5re, and 2re, where re is the dimer-specific equilibrium distance. This means that S66x8 contains 528 dimers whose geometries we took from Ref. [41]. For our target level of theory (B2PLYP/def2-SVP), the standard BB-CP corrections were calculated for all 528 dimers. The four gCP scaling parameters were determined in a least-squares fit against those BB-CP values. For the dimers with the shortest intermolecular distances (i.e. 0.90re), the weights of the deviations from that correction were reduced to 0.5 during our fit, as recommended in the original gCP publication.[21]
represents the number of virtual orbitals on atom B, and σ, α and β are scaling parameters. Slater exponents in SAB are weighted by a scaling parameter dubbed η. Those four scaling parameters need to be fitted for a given level of theory; in our case this meant that we fitted them for the B2PLYP/def2-SVP level of theory. The fitting procedure for the gCP correction uses the S66x8 benchmark set, which is an extension of the S66 dataset.[41] S66 contains 66 noncovalently bound dimers and is separated into systems that are dominated by electrostatics (hydrogen bonds), dispersion, or a mixture of both. To allow for an analysis of interaction energies in non-equilibrium situations, the S66x8 extension considers eight different intermolecular distances for each dimer, namely at 0.90re, 0.95re, re, 1.05re, 1.10re, 1.25re, 1.5re, and 2re, where re is the dimer-specific equilibrium distance. This means that S66x8 contains 528 dimers whose geometries we took from Ref. [41]. For our target level of theory (B2PLYP/def2-SVP), the standard BB-CP corrections were calculated for all 528 dimers. The four gCP scaling parameters were determined in a least-squares fit against those BB-CP values. For the dimers with the shortest intermolecular distances (i.e. 0.90re), the weights of the deviations from that correction were reduced to 0.5 during our fit, as recommended in the original gCP publication.[21]
In passing, we would also like to mention that we additionally analysed a damped version of gCP, which was first used in the context of PBEh-3c.[24] The damped-gCP variant uses the basic form of gCP, but the short-range part of the correction is damped according to:
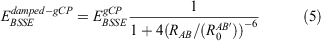
where 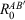 are radii introduced in the DFT-D3(0) correction with zero-damping.[28] During initial tests, we saw the statistical superiority of gCP over damped-gCP (see Table S4 in the Supplementary Material). Therefore, we decided to keep the basic form of gCP and the damped version will not be discussed any further.
are radii introduced in the DFT-D3(0) correction with zero-damping.[28] During initial tests, we saw the statistical superiority of gCP over damped-gCP (see Table S4 in the Supplementary Material). Therefore, we decided to keep the basic form of gCP and the damped version will not be discussed any further.
Fitting Procedure for DFT-D3(BJ)
The two-body London dispersion energy according to the DFT-D3(BJ) scheme is defined as:[29]
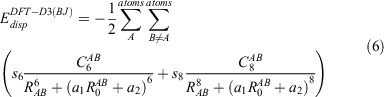
with chemical-environment-dependent[28] dispersion coefficients Cn (n = 6, 8), internuclear distances RAB, order-dependent scaling factors sn, cut-off radii 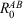 , and empirical parameters a1 and a2 that control the magnitude of added dispersion at short and medium interatomic distances. For more details on this correction, see Refs [29, 76, and 77]. As we will outline in the results section, we observed benefits of refitting DFT-D3(BJ) for our purposes over using the standard parametrisation, which is based on B2PLYP/def2-QZVP. In this section we cover briefly how we performed such refits.
, and empirical parameters a1 and a2 that control the magnitude of added dispersion at short and medium interatomic distances. For more details on this correction, see Refs [29, 76, and 77]. As we will outline in the results section, we observed benefits of refitting DFT-D3(BJ) for our purposes over using the standard parametrisation, which is based on B2PLYP/def2-QZVP. In this section we cover briefly how we performed such refits.
The s6 parameter is set to unity for most functionals to ensure the correct asymptotic behaviour of the dispersion energy. The exception are double hybrids, which have an additional long-range dispersion contribution from their MP2 part. Herein, we determined s6 for the B2PLYP/def2-SVP level of theory according to the procedure presented in Ref. [78]. For that purpose, we considered the Ne2, Ar2, and Kr2 dimers at interatomic separations of 7 Å, 10 Å, and 10 Å, respectively. The MP2 contributions to the interaction energies of the three dimers were calculated and compared to CCSD(T)[79]/aug-cc-pVTZ[69,80] dispersion energies. This allowed us to calculate the average amount of long-range dispersion recovered by B2PLYP/def2-SVP. This average was then subtracted from unity to get the value s6. Our resulting s6 is 0.830.
Having determined s6, the remaining three parameters s8, a1, and a2 in Eqn 6 were determined by a least-squares fit against the reference interaction energies of the combined S66x8, S22x5,[81] and NCIBLIND[82] benchmark sets. We carried out such fits for different BSSE-corrected B2PLYP/def2-SVP interaction energies, as outlined in the results section.
A Brief Overview of the GMTKN55 Database
To further assess the performance of B2PLYP/def2-SVP with the DFT-D3(BJ) and gCP corrections, we used the GMTKN55 database, which comprises 2462 total single-point energies distributed over 55 datasets. GMTKN55 is classified into five categories. The first category, ‘basic properties and reactions of small systems’, consists of 18 sets and covers reaction energies for small systems, total atomisation energies, ionisation potentials, electron affinities, and self-interaction-error-related problems. The second category offers nine test sets for isomerisation and reaction energies of larger systems. Barrier height related problems are dealt with in the third category (seven test sets in total). The fourth category comprises 12 benchmark sets dealing with intermolecular noncovalent interactions, while intramolecular interactions are described by the nine benchmark sets in the fifth category. A summary of the GMTKN55 database and the names of all individual 55 benchmark sets can be found in Table 1.

|
Results and Discussion
BSSE in Double-Hybrid DFT Calculations of the S66x8 Benchmark Set
First, we analyse the behaviour of BSSE for the S66x8 benchmark set. For def2-SVP, the average BB-CP corrections are reported for BLYP, B3LYP, and B2PLYP in Figs 2 and S1; the individual BB-CP values are shown in Table S1. The figures show the CP averages (CPavg) for the hydrogen-bonded, dispersion-dominated, and mixed complexes, as well as the CPavg values for the eight different intermolecular distances. Hydrogen-bonded complexes show the largest CPavg (BLYP = 3.10 kcal mol−1, B3LYP = 2.65 kcal mol−1, and B2PLYP = 2.76 kcal mol−1), followed by mixed (BLYP = 1.36 kcal mol−1, B3LYP = 1.16 kcal mol−1, and B2PLYP = 1.34 kcal mol−1), and dispersion-dominated complexes (BLYP = 1.22 kcal mol−1, B3LYP = 1.05 kcal mol−1, and B2PLYP = 1.31 kcal mol−1) (see Fig. 2 for details). The complexes with the shortest intermolecular distances show the largest CP corrections; e.g. the CPavg values for B2PLYP are 3.19, 2.79, 2.45, 2.16, 1.91, 1.30, 0.69, and 0.11 kcal mol−1 for the dimers for the eight distances 0.90re, 0.95re, re, 1.05re, 1.10re, 1.25re, 1.5re, and 2re, respectively (Fig. S1).
|
Upon closer inspection of our results, we found that GGAs have a larger BSSE than HF theory. This also explain why a hybrid, such as B3LYP has smaller BSSE than BLYP. We note that the same can be seen in the discussion of the original gCP paper in Ref. [21].
Furthermore, B2PLYP shows a tendency to yield larger CP corrections than the B3LYP hybrid DFA. For instance, the CPavg values for the entire S66x8 set are 1.82, 1.64, and 1.91 kcal mol−1 for B2PLYP, B3LYP, and BLYP, respectively. This increase in CPavg for B2PLYP is most likely due to its MP2 component. We validated this hypothesis by comparing our results for B2PLYP to those of other DHDFs with different degrees of MP2 contribution, as well as the MP2 method itself. The average CP correction for PBE0-2[134] (79 % Fock exchange and 50 % MP2 correlation) and PBE0-DH[135] (50 % Fock exchange and 12.5 % MP2 correlation) are 1.75 and 1.40 kcal mol−1. The CPavg values for MP2 and spin-component-scaled SCS-MP2[136] are 2.31 and 2.23 kcal mol−1, respectively. This shows that the amount of MP2 correlation is the major influencing factor for double hybrids. Our results in Figs 2 and S1 reveal that the BSSE of a double hybrid differs from that of the hybrids and pure DFAs. This justifies why the globally adjusted gCP parameters for DFT from Ref. [21] should not be used for double hybrids, which is why we refit them here for B2PLYP.
Adjustment of the DFT-D3 and gCP Corrections
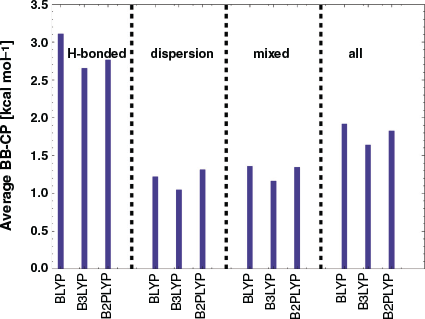
Although the BB-CP and gCP corrections were developed as independent procedures, London dispersion corrections play an important role in the DFT-based treatment of geometries, thermochemistry, kinetics, and noncovalent interactions.[29,35,71,72,133,137–146] Herein, we will study in detail if a refit of the DFT-D3(BJ) parameters has any positive effects. We find a significant improvement in performance when the DFT-D3(BJ) parameters s8, a1, and a2 in Eqn 6 are re-determined in the presence of the BB-CP correction with the procedure discussed earlier. This reparametrised version of B2PLYP-CP-D3(BJ) performs extremely well, with a decrease in the MAD from 0.73 to 0.26 kcal mol−1 for the S66x8 benchmark set when compared to a BB-CP based version that uses the original DFT-D3(BJ) parameters. Consequently, we decided to refit the DFT-D3(BJ) parameters for the gCP scheme as well.
When the damping parameters for the DFT-D3(BJ) correction are optimised in the presence of the gCP correction, we see a significant improvement in performance and the mean absolute deviation (MAD) is reduced by 0.39 kcal mol−1 compared to using the standard DFT-D3(BJ) parametrisation for B2PLYP (see Table 2 for details). Following that, we investigate whether we can apply the global gCP parameters implemented for DFT (Ref. [21]) to B2PLYP. As shown in Table 2, the accuracy obtained by re-fitting both the DFT-D3(BJ) and gCP parameters for B2PLYP is not negligible, which is why this is our chosen strategy for this study.

|
The various fitted parameters can be found in Tables S2 and S3 in the Supplementary Material. To avoid confusion, we would like to clarify that ‘B2PLYP-gCP-D3(BJ)’ in this manuscript always refers to the refitted gCP and DFT-D3(BJ) parameters, whereas ‘B2PLYP-D3(BJ)’ uses the original DFT-D3(BJ) damping parameters for B2PLYP regardless of the chosen basis set.
Impact of Reoptimised gCP and DFT-D3(BJ) on B2PLYP/def2-SVP Calculations
Analysis with the S66x8 Benchmark Set
In Fig. 3, we illustrate the effect of the gCP and dispersion corrections against the S66x8 reference interaction energies, which are based on high-level data at the CBS limit. Note that we use the reference values from the original publication[41] to make our analysis commensurate with previous studies,[21,147] particularly our GMTKN55 ones of the related S66.[35,38,71–73] For slightly changed reference values, which would not change our main conclusions, see Refs [42 and 148]. Dispersion and gCP contributions to B2PLYP-gCP-D3(BJ) are reported separately to demonstrate the energetic effects of the corrections. A positive mean deviation (MD) indicates an overestimation of the interaction energies. Starting from plain B2PLYP/def2-SVP [MD = −0.15 kcal mol−1 and MAD = 1.46 kcal mol−1], the inclusion of the dispersion correction is expected to make the dimers of S66x8 more stable, which is indeed the case with MD/MAD = 1.14/1.22 kcal mol−1 for B2PLYP-D3(BJ)/def2-SVP. These values are contaminated by BSSE. Additionally adding the gCP correction to the dispersion-corrected result induces the expected destabilising effect and brings the interaction energies closer to the reference values (MD/MAD = 0.11/0.56 kcal mol−1). The hydrogen-bonded systems in the S66x8 benchmark set show overestimation of the interaction energies for plain B2PLYP (MD = 1.74 kcal mol−1). Adding the dispersion correction [B2PLYP-D3(BJ)] leads to further over-stabilisation (MD = 2.47 kcal mol−1). This in turn is compensated for by the gCP correction and results in a final MD of 0.62 kcal mol−1 for B2PLYP-gCP-D3(BJ). The situation is different for the dispersion-driven systems of the S66x8 benchmark set, where B2PLYP yields interaction energies that are too low (MD = −1.72 kcal mol−1). Correcting the energies for dispersion and BSSE brings interaction energies closer to the reference interaction energies (MD = 0.39 kcal mol−1). A look at the third category of S66x8 shows that the gCP-D3(BJ)-corrected B2PLYP yields an MD that is better than the MD of plain B2PLYP (0.10 versus −0.51 kcal mol−1).
In conclusion, most of the results shown in this manuscript can be explained by the advantageous interplay of the opposing effects of the dispersion correction (stabilisation) and the addition of gCP correction (destabilisation). Our results in this section therefore validate small basis set calculations for NCIs. Although our herein presented approach is not competitive with the standard B2PLYP-D3(BJ) near the basis set limit [MAD(B2PLYP-D3(BJ)/def2-QZVP[52] = 0.16 kcal mol−1],[147] it certainly performs better than B2PLYP/def2-SVP and B2PLYP-D3(BJ)/def2-SVP without requiring additional computational resources.
Analysis with the GMTKN55 Benchmark Database
Next, we look at the GMTKN55 database and its categories. The benchmarking is done against the original GMTKN55 reference values which mostly consist of high-level data at the complete basis set limit. We focus mostly on the NCI benchmark sets but also discuss the other categories briefly. Individual statistics for all 55 sets are shown in Tables S5–S7 in the Supplementary Material.
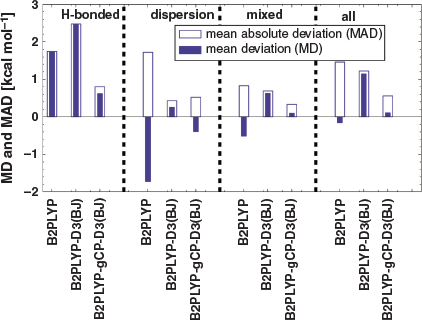
Fig. 4 shows the MDs of B2PLYP, B2PLYP-D3(BJ), and B2PLYP-gCP-D3(BJ) for the noncovalent interaction datasets of GMTKN55. Intermolecular NCIs are described by 12 benchmark sets (Table 1) and we discuss a few, select examples. The interaction energy trends for the n-alkane dimer set ADIM6[28,35] are similar to those of S66x8, meaning that interaction energies are insufficiently described by plain B2PLYP (MD = −2.88 kcal mol−1). The improvement by using gCP in conjunction with the reparametrised DFT-D3(BJ) is even better than for S66x8 and lowers the magnitude of the MD by 1.90 kcal mol−1. The interaction energy trends for the bound dimers in S22 and S66 are similar to those of ADIM6 and S66x8, meaning that the introduction of dispersion into the treatment leads to an over-stabilisation of dimers and yields MDs of 1.81 kcal mol−1 and 1.69 kcal mol−1, respectively. This in turn is mostly compensated by additionally adding the gCP correction to yield nearly perfect MDs of −0.01 and 0.21 kcal mol−1.
For RG18[35] (interaction energies in rare gas complexes) the gCP and dispersion corrections show the same trends as before, but both corrections have different impacts on the interaction energies and the overall destabilising effect of gCP is more prominent [MD(B2PLYP) = −0.09 kcal mol−1, MD[B2PLYP-D3(BJ)] = 0.42 kcal mol−1, and MD[B2PLYP-gCP-D3(BJ)] = −0.83 kcal mol−1]. The MD value of CARBHB12[35] (hydrogen-bonded complexes of carbene analogues with small molecules) shows that dispersion- and BSSE-uncorrected B2PLYP slightly overestimates the interaction energies (MD = 0.93 kcal mol−1). These interactions are overestimated even further when the dispersion correction is included with an MD value of 1.58 kcal mol−1, which is reduced to 1.16 kcal mol−1 when gCP and DFT-D3(BJ) are both applied. The halogen bonding interaction set HAL59[35,74,75] also yields good results when the gCP and DFT-D3(BJ) corrections are applied, with the MD of 1.51 kcal mol−1 for B2PLYP-D3(BJ) improving to 0.82 kcal mol−1 for B2PLYP-gCP-D3(BJ).
We remind the reader that def2-SVP was enhanced by diffuse functions to take anionic systems into account in benchmark sets such as WATER27[35,70] [binding energies in (H2O)n, H+(H2O)n, and OH–(H2O)n] or IL16[35,68] [interaction energies in anion-cation dimers] (see Computational Details section). Those diffuse functions were added to make our results consistent with previous GMTKN55 studies and to allow for an analysis of the entire database. We notice a strong underestimation of the interaction energies in WATER27 for B2PLYP-gCP-D3(BJ) (MD = −13.13 kcal mol−1) and IL 16 (MD = 5.13 kcal mol−1). In future work, reparametrising gCP for such a slightly changed version of def2-SVP might lead to improvements, but problems with the basis set cannot be ruled out either at this stage. That being said, the interaction energies of the cation-neutral dimers in CHB6[35,68] are overestimated by 2.64 kcal mol−1 for B2PLYP-gCP-D3(BJ) relative to B2PLYP. CHB6 does not require any diffuse functions and the fact that it is an outlier might also hint at basis set insufficiencies, which might require further study in the future.
When analysing intramolecular NCIs, we noticed that one of the largest deviations we observed for plain B2PLYP were for BUT14DIOL[35,132] (relative energies in butane-1,4-diol conformers) and SCONF[35,67,130] (sugar conformers) with MD values of 2.28 kcal mol−1 and 1.22 kcal mol−1, respectively. Both sets strongly benefit from the gCP-D3(BJ) combination (MDs = 0.89/0.02 kcal mol−1), which is not surprising as conformer stabilities in those sets are driven by intramolecular hydrogen bonding; we have seen in our discussion of S66x8 how hydrogen-bonded systems benefit from both corrections (see Fig. 3). For the intramolecular NCI category of GMTKN55, conformational energies are sometimes difficult to determine, but generally acceptable at the B2PLYP/large-basis level. B2PLYP-D3(BJ) with a small AO basis fails to establish the correct order. The benchmark reference of one of the systems in the SCONF test set, for example, predicts the two most stable conformers to be 5.54 kcal mol−1 apart from each other. B2PLYP-D3(BJ) predicts a difference of −1.25 kcal mol−1 between the two, where the minus sign indicates a reversed energetic order of the two conformers. The order is correct for B2PLYP-gCP-D3(BJ), with an energy difference of 3.96 kcal mol−1.
|
|
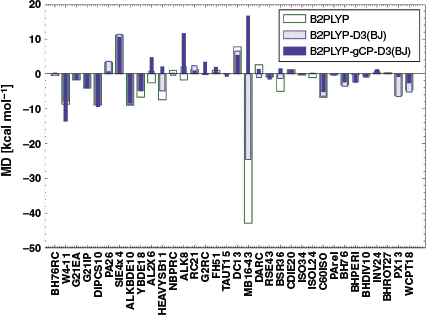
Having seen the impact of the refitted gCP and DFT-D3(BJ) corrections on the NCI test sets, we now turn our attention to the non-NCI subsets of GMTKN55 (see Fig. 5). We discuss the reaction barrier heights first (seven sets, see Table 1). In the PX13[116] test set (proton-exchange barriers in small-molecule clusters), B2PLYP and B2PLYP-D3(BJ) yield barriers that are on average too low (MDs of −6.19 and −6.51 kcal mol−1, respectively). Correcting for BSSE in B2PLYP-D3(BJ) raises the barriers significantly (MD = −0.81 kcal mol−1). For BHDIV10[35]—a test set for barrier heights of ‘diverse reactions’—the addition of the dispersion correction leads to a general underestimation of the barriers (MDs of −0.39 to −0.94 kcal mol−1, respectively), which is only partially compensated for by the gCP correction (MD = −0.89 kcal mol−1).
|
For BHROT27[35]—barrier heights for rotation around single bonds—there is little statistical difference between the tested methods. For BHPERI[67,111–113]—barrier heights of pericyclic reactions—B2PLYP shows better results than B2PLYP-D3(BJ) (MDs of −0.17 and −2.32 kcal mol−1, respectively) and this time the addition of gCP does not improve the results (MD = −2.46 kcal mol−1), which indicates that this set is more challenging then the previously discussed ones.
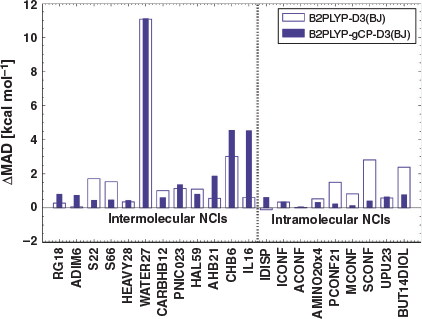
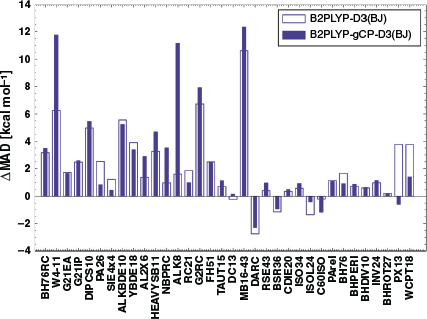
For several benchmark sets belonging to the remaining categories of GMTKN55, such as total atomisation energies for small systems (W4-11[83]), dissociation and other reactions of alkaline compounds (ALK8[35]), reaction energies of selected G2/97[149] systems (G2RC[35,67]), or oligomerisation and H2 fragmentations of NH3/BH3 systems (NBPRC[35,67,78,89]), B2PLYP or B2PLYP-D3(BJ) are statistically superior to B2PLYP-gCP-D3(BJ). A likely reason could be a seemingly better result due to error compensation between remaining errors of the method and the basis set incompleteness error. Alternatively, it can also mean that the reparametrised ‘gCP-D3(BJ)’ corrections might not be suitable for the properties covered by those benchmark sets. Additionally, atomisation energies in W4-11 are poorly described by a method relying on a small-basis MP2 contribution, such that it is not expected that either of the two corrections can improve the results. Having established the impact of the combined gCP and DFT-D3(BJ) corrections on B2PLYP/(aug’-)def2-SVP numbers, the remaining question is how close those numbers get to B2PLYP-D3(BJ) near the CBS limit, which for GMTKN55 is B2PLYP-D3(BJ)/(aug’)-def2-QZVP. In the following, we will collectively use the acronym ‘QZ’ for the larger basis set and ‘DZ’ for the smaller. In order to answer that remaining question, we calculated the differences in MADs between DZ-based levels and published[35] B2PLYP-D3(BJ)/QZ results; this difference is called ‘ΔMAD’. The resulting values are shown for each benchmark set in Figs 6 and 7.
|
|
A positive ΔMAD means that the investigated method gives a larger average error than B2PLYP-D3(BJ)/QZ. A negative value means that we observe fortuitous compensation between shortcomings of the tested method and the basis-set incompleteness error, which make the MADs at the DZ level smaller than at the QZ level.
As can be seen in Figs 6 and 7, most of the NCI benchmark sets of GMTKN55, as well as a significant number of non-NCI test sets, benefit from the inclusion of the gCP correction. For example, the ΔMADs for S22 and S66 improve by 1.26 kcal mol−1 and 1.06 kcal mol−1, respectively. As expected, however, most ΔMAD values are positive.
The first GMTKN55 study introduced so-called weighted total mean absolute deviation (WTMAD) schemes. Details on the two schemes called ‘WTMAD-1’ and ‘WTMAD-2’ can be found in Ref. [35]. For our purpose, it is only important to know that the idea behind a WTMAD is to combine the MADs of the 55 individual benchmark sets to into one number, which can be used to conveniently assess functional robustness and allows to establish a functional ranking. The interested reader can find WTMAD-2 values in Table S8 in the Supplementary Material. In Ref. [150], an article showing why the popular B3LYP/6-31G*[151] model chemistry should not be used, the authors introduced the WTΔMAD scheme for the GMTKN30[78] predecessor of GMTKN55. According to that idea the individual MADs are replaced with ΔMADs, but otherwise the WTMAD formalism stays the same. We did the same here for our GMTKN55 results based on GMTKN55’s WTMAD-2[35] scheme. Such WTΔMAD-2 values are listed in Table 3 for GMTKN55 and its individual categories. This allows us to compare the general applicability of B2PLYP-gCP-D3(BJ)/DZ relative to B2PLYP-D3(BJ)/QZ.

|
For the entire database, we obtain WTΔMAD-2 values of 8.28 kcal mol−1 for B2PLYP, 8.60 kcal mol−1 for B2PLYP-D3(BJ), and 6.17 kcal mol−1 for B2PLYP-gCP-D3(BJ), respectively, which shows an overall improvement for the latter. This is mostly due to the barrier height and NCI categories (see Table 3). We particularly notice a significant improvement when gCP is applied for intramolecular NCIs: B2PLYP-gCP-D3(BJ) has an WTΔMAD-2 of 7.59 kcal mol−1 compared to 16.33 and 21.87 kcal mol−1 for plain B2PLYP and B2PLYP-D3(BJ), respectively.
Overall, the accuracy of our developed scheme is limited due to the basis set incompleteness error associated with the small basis. That being said, gCP has been developed as a general-purpose tool and can be very easily adapted to different basis sets to limit that problem. Nevertheless, we have shown that the gCP scheme is in principle adaptable to double-hybrid DFT methods.
Conclusions
In this study, we investigated the applicability of Grimme’s geometric counterpoise correction (gCP) for the basis set superposition error (BSSE) in double-hybrid DFT calculations. This scheme only requires geometrical information as an input and no orbital/density information is needed. Therefore, this correction is almost free compared to the cost of the underlying electronic structure calculation. The choice of the functionals and basis sets was not the scope of this manuscript, and we exemplified our analysis with B2PLYP/def2-SVP. Our conclusions should be considered as a proof-of-concept study.
Our work revealed the error compensation between missing London dispersion and BSSE in B2PLYP/def2-SVP results for thermochemistry, kinetics, and noncovalent interactions. We suggest B2PLYP-gCP-D3(BJ) as the more reliable alternative, with both corrections explicitly parameterised for the chosen level of theory. In fact, reparametrising DFT-D3(BJ) for B2PLYP-gCP/def2-SVP was beneficial. Benchmark results on the S66x8 benchmark set and GMTKN55 database showed statistical improvements of B2PLYP-gCP-D3(BJ) over plain B2PLYP and B2PLYP-D3(BJ). B2PLYP just with dispersion correction [B2PLYP-D3(BJ)] showed a significant overestimation of interaction energies, barrier heights with larger deviations from reference values, and wrong relative stabilities in conformers. Those problems could be removed or their severity at least reduced by adding gCP. The error cancellation in plain B2PLYP is unsystematic and depends on the chemical nature of the test set. The improvement by gCP over B2PLYP-D3(BJ) is largest for noncovalent interactions (in particular intramolecular NCIs).
In summary, the addition of a specifically optimised gCP-D3(BJ) correction removes two major deficiencies (missing London dispersion and BSSE) and consequently shows a more robust and reliable picture of B2PLYP with a small basis set. Our observation of the opposing effects of the dispersion correction (stabilisation) and gCP correction (destabilisation) is commensurate with a similar picture drawn for the B3LYP/6-31G* level of theory in Ref. [150].
Despite the promising trends presented herein, a comparison of B2PLYP-gCP-D3(BJ)/def2-SVP and B2PLYP-D3(BJ)/def2-QZVP shows that the approach's accuracy is limited, mostly due to remaining basis set incompleteness errors, which again is something that can also be seen in Ref. [150]. However, if the application of larger basis sets is not feasible, we strongly recommend using the gCP-D3(BJ) variant. This study encourages the possibility of extending this method to other double hybrids and basis sets.
Supplementary Material
Additional information on BSSE in the S66x8 set, the optimised DFT-D3(BJ) and gCP parameters, an analysis of the damped-gCP variant, and all statistical results for GMTKN55 are available on the Journal’s website.
Data Availability Statement
The data that support this study are available in the article and accompanying online supplementary material. More detailed information on specific energies can be obtained from the corresponding author upon request.
Conflicts of Interest
The authors declare no conflicts of interest.
Declaration of Funding
This research did not receive any specific funding.
Acknowledgements
N. M. acknowledges a Melbourne International Engagement Award (MIEA) offered through the Melbourne India Postgraduate Program and a Melbourne Research Scholarship. L. G. is deeply grateful to the Royal Australian Chemical Institute for the award of a 2020 Rennie Memorial Medal. L. G. also acknowledges generous allocation of computational resources from the National Computational Infrastructure (NCI) Facility within the National Computational Merit Allocation Scheme (project fk5), and Research Platform Services (ResPlat) at The University of Melbourne (project punim0094).
References
[1] F. B. van Duijneveldt, J. G. van Duijneveldt van de Rijdt, J. H. van Lenthe, Chem. Rev. 1994, 94, 1873.| Crossref | GoogleScholarGoogle Scholar |
[2] B. Liu, A. McLean, J. Chem. Phys. 1973, 59, 4557.
| Crossref | GoogleScholarGoogle Scholar |
[3] N. R. Kestner, J. Chem. Phys. 1968, 48, 252.
| Crossref | GoogleScholarGoogle Scholar |
[4] H. Jansen, P. Ros, Chem. Phys. Lett. 1969, 3, 140.
| Crossref | GoogleScholarGoogle Scholar |
[5] D. W. Schwenke, D. G. Truhlar, J. Chem. Phys. 1985, 82, 2418.
| Crossref | GoogleScholarGoogle Scholar |
[6] J. Collins, G. Gallup, Chem. Phys. Lett. 1986, 123, 56.
| Crossref | GoogleScholarGoogle Scholar |
[7] M. J. Frisch, J. E. Del Bene, J. S. Binkley, H. F. Schaefer, J. Chem. Phys. 1986, 84, 2279.
| Crossref | GoogleScholarGoogle Scholar |
[8] D. Cook, J. Sordo, T. Sordo, Int. J. Quantum Chem. 1993, 48, 375.
| Crossref | GoogleScholarGoogle Scholar |
[9] L. Mentel, E. Baerends, J. Chem. Theory Comput. 2014, 10, 252.
| Crossref | GoogleScholarGoogle Scholar | 26579908PubMed |
[10] R. Kalescky, E. Kraka, D. Cremer, J. Chem. Phys. 2014, 140, 084315.
| Crossref | GoogleScholarGoogle Scholar | 24588177PubMed |
[11] E. Miliordos, S. S. Xantheas, J. Chem. Phys. 2015, 142, 094311.
| Crossref | GoogleScholarGoogle Scholar | 25747085PubMed |
[12] L. A. Burns, M. S. Marshall, C. D. Sherrill, J. Chem. Theory Comput. 2014, 10, 49.
| Crossref | GoogleScholarGoogle Scholar | 26579890PubMed |
[13] S. F. Boys, F. Bernardi, Mol. Phys. 1970, 19, 553.
| Crossref | GoogleScholarGoogle Scholar |
[14] A. Galano, J. R. Alvarez-Idaboy, J. Comput. Chem. 2006, 27, 1203.
| Crossref | GoogleScholarGoogle Scholar | 16752366PubMed |
[15] F. Jensen, J. Chem. Theory Comput. 2010, 6, 100.
| Crossref | GoogleScholarGoogle Scholar | 26614323PubMed |
[16] J. C. Faver, Z. Zheng, K. M. Merz, J. Chem. Phys. 2011, 135, 144110.
| Crossref | GoogleScholarGoogle Scholar | 22010701PubMed |
[17] J. C. Faver, Z. Zheng, K. M. Merz, Phys. Chem. Chem. Phys. 2012, 14, 7795.
| Crossref | GoogleScholarGoogle Scholar | 22377839PubMed |
[18] A. Otero-de-La-Roza, G. A. DiLabio, J. Chem. Theory Comput. 2017, 13, 3505.
| Crossref | GoogleScholarGoogle Scholar | 28636358PubMed |
[19] V. K. Prasad, A. Otero-de-la Roza, G. A. Di-Labio, J. Chem. Theory Comput. 2018, 14, 726.
| Crossref | GoogleScholarGoogle Scholar | 29262249PubMed |
[20] A. Otero-de-la-Roza, G. A. DiLabio, J. Chem. Theory Comput. 2020, 16, 4176.
| Crossref | GoogleScholarGoogle Scholar | 32470304PubMed |
[21] H. Kruse, S. Grimme, J. Chem. Phys. 2012, 136, 154101.
| Crossref | GoogleScholarGoogle Scholar | 22519309PubMed |
[22] R. Sure, S. Grimme, J. Comput. Chem. 2013, 34, 1672.
| Crossref | GoogleScholarGoogle Scholar | 23670872PubMed |
[23] J. G. Brandenburg, E. Caldeweyher, S. Grimme, Phys. Chem. Chem. Phys. 2016, 18, 15519.
| Crossref | GoogleScholarGoogle Scholar | 27240749PubMed |
[24] S. Grimme, J. G. Brandenburg, C. Bannwarth, A. Hansen, J. Chem. Phys. 2015, 143, 054107.
| Crossref | GoogleScholarGoogle Scholar | 26254642PubMed |
[25] J. G. Brandenburg, C. Bannwarth, A. Hansen, S. Grimme, J. Chem. Phys. 2018, 148, 064104.
| Crossref | GoogleScholarGoogle Scholar | 29448802PubMed |
[26] L. Doná, J. G. Brandenburg, B. Civalleri, J. Chem. Phys. 2019, 151, 121101.
| Crossref | GoogleScholarGoogle Scholar | 31575185PubMed |
[27] S. Grimme, A. Hansen, S. Ehlert, J.-M. Mewes, J. Chem. Phys. 2021, 154, 064103.
| Crossref | GoogleScholarGoogle Scholar | 33588555PubMed |
[28] S. Grimme, J. Antony, S. Ehrlich, H. Krieg, J. Chem. Phys. 2010, 132, 154104.
| Crossref | GoogleScholarGoogle Scholar | 20423165PubMed |
[29] S. Grimme, S. Ehrlich, L. Goerigk, J. Comput. Chem. 2011, 32, 1456.
| Crossref | GoogleScholarGoogle Scholar | 21370243PubMed |
[30] E. Caldeweyher, C. Bannwarth, S. Grimme, J. Chem. Phys. 2017, 147, 034112.
| Crossref | GoogleScholarGoogle Scholar | 28734285PubMed |
[31] E. Caldeweyher, S. Ehlert, A. Hansen, H. Neugebauer, S. Spicher, C. Bannwarth, S. Grimme, J. Chem. Phys. 2019, 150, 154122.
| Crossref | GoogleScholarGoogle Scholar | 31005066PubMed |
[32] J. Witte, J. B. Neaton, M. Head-Gordon, J. Chem. Phys. 2017, 146, 234105.
| Crossref | GoogleScholarGoogle Scholar | 28641421PubMed |
[33] S. Grimme, J. Chem. Phys. 2006, 124, 034108.
| Crossref | GoogleScholarGoogle Scholar | 16438568PubMed |
[34] L. Goerigk, S. Grimme, Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 576.
| Crossref | GoogleScholarGoogle Scholar |
[35] L. Goerigk, A. Hansen, C. Bauer, S. Ehrlich, A. Najibi, S. Grimme, Phys. Chem. Chem. Phys. 2017, 19, 32184.
| Crossref | GoogleScholarGoogle Scholar | 29110012PubMed |
[36] L. Goerigk, N. Mehta, Aust. J. Chem. 2019, 72, 563.
| Crossref | GoogleScholarGoogle Scholar |
[37] G. Santra, N. Sylvetsky, J. M. Martin, J. Phys. Chem. A 2019, 123, 5129.
| Crossref | GoogleScholarGoogle Scholar | 31136709PubMed |
[38] N. Mehta, M. Casanova-Páez, L. Goerigk, Phys. Chem. Chem. Phys. 2018, 20, 23175.
| Crossref | GoogleScholarGoogle Scholar | 30062343PubMed |
[39] J. M. L. Martin, G. Santra, Isr. J. Chem. 2020, 60, 787.
| Crossref | GoogleScholarGoogle Scholar |
[40] N. Mardirossian, M. Head-Gordon, J. Chem. Phys. 2018, 148, 241736.
| Crossref | GoogleScholarGoogle Scholar | 29960332PubMed |
[41] J. Rezác, K. E. Riley, P. Hobza, J. Chem. Theory Comput. 2011, 7, 2427.
| Crossref | GoogleScholarGoogle Scholar | 21836824PubMed |
[42] B. Brauer, M. K. Kesharwani, S. Kozuch, J. M. Martin, Phys. Chem. Chem. Phys. 2016, 18, 20905.
| Crossref | GoogleScholarGoogle Scholar | 26950084PubMed |
[43] A. Karton, J. M. Martin, J. Chem. Phys. 2011, 135, 144119.
| Crossref | GoogleScholarGoogle Scholar | 22010710PubMed |
[44] T. B. Adler, G. Knizia, H.-J. Werner, J. Chem. Phys. 2007, 127, 221106.
| Crossref | GoogleScholarGoogle Scholar | 18081383PubMed |
[45] H. Fliegl, W. Klopper, C. Hättig, J. Chem. Phys. 2005, 122, 084107.
| Crossref | GoogleScholarGoogle Scholar |
[46] G. Knizia, T. B. Adler, H.-J. Werner, J. Chem. Phys. 2009, 130, 054104.
| Crossref | GoogleScholarGoogle Scholar | 19206955PubMed |
[47] D. P. Tew, W. Klopper, C. Neiss, C. Hättig, Phys. Chem. Chem. Phys. 2007, 9, 1921.
| Crossref | GoogleScholarGoogle Scholar | 17431520PubMed |
[48] H.-J. Werner, G. Knizia, F. R. Manby, Mol. Phys. 2011, 109, 407.
| Crossref | GoogleScholarGoogle Scholar |
[49] J. S. García, E. Brémond, M. Campetella, I. Ciofini, C. Adamo, J. Chem. Theory Comput. 2019, 15, 2944.
| Crossref | GoogleScholarGoogle Scholar |
[50] E. Brémond, I. Ciofini, J. C. Sancho-García, C. Adamo, J. Phys. Chem. A 2019, 123, 10040.
| Crossref | GoogleScholarGoogle Scholar | 31596087PubMed |
[51] B. Tirri, I. Ciofini, J. C. Sancho-García, C. Adamo, E. Brémond, Int. J. Quantum Chem. 2020, 120, e26233.
| Crossref | GoogleScholarGoogle Scholar |
[52] F. Weigend, R. Ahlrichs, Phys. Chem. Chem. Phys. 2005, 7, 3297.
| Crossref | GoogleScholarGoogle Scholar | 16240044PubMed |
[53] F. Neese, Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73.
| Crossref | GoogleScholarGoogle Scholar |
[54] F. Neese, Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1327.
| Crossref | GoogleScholarGoogle Scholar |
[55] S. Grimme, DFT-D3 V3.1 2014 (University of Bonn).
[56] H. Kruse, J. G. Brandenburg, S. Grimme, A Geometrical Counterpoise Correction V2.0.2 2016.
[57] A. D. Becke, Phys. Rev. A 1988, 38, 3098.
| Crossref | GoogleScholarGoogle Scholar |
[58] C. Lee, W. Yang, R. G. Parr, Phys. Rev. B Condens. Matter 1988, 37, 785.
| Crossref | GoogleScholarGoogle Scholar | 9944570PubMed |
[59] B. Miehlich, A. Savin, H. Stoll, H. Preuss, Chem. Phys. Lett. 1989, 157, 200.
| Crossref | GoogleScholarGoogle Scholar |
[60] A. D. Becke, J. Chem. Phys. 1993, 98, 5648.
| Crossref | GoogleScholarGoogle Scholar |
[61] P. J. Stephens, F. J. Devlin, C. F. Chabalowski, M. J. Frisch, J. Phys. Chem. 1994, 98, 11623.
| Crossref | GoogleScholarGoogle Scholar |
[62] K. Eichkorn, F. Weigend, O. Treutler, R. Ahlrichs, Theor. Chem. Acc. 1997, 97, 119.
| Crossref | GoogleScholarGoogle Scholar |
[63] F. Weigend, M. Häser, H. Patzelt, R. Ahlrichs, Chem. Phys. Lett. 1998, 294, 143.
| Crossref | GoogleScholarGoogle Scholar |
[64] R. Izsák, F. Neese, J. Chem. Phys. 2011, 135, 144105.
| Crossref | GoogleScholarGoogle Scholar | 22010696PubMed |
[65] T. Schwabe, J. Phys. Chem. A 2013, 117, 2879.
| Crossref | GoogleScholarGoogle Scholar | 23473372PubMed |
[66] L. A. Curtiss, K. Raghavachari, G. W. Trucks, J. A. Pople, J. Chem. Phys. 1991, 94, 7221.
| Crossref | GoogleScholarGoogle Scholar |
[67] L. Goerigk, S. Grimme, J. Chem. Theory Comput. 2010, 6, 107.
| Crossref | GoogleScholarGoogle Scholar | 26614324PubMed |
[68] K. U. Lao, R. Schäffer, G. Jansen, J. M. Herbert, J. Chem. Theory Comput. 2015, 11, 2473.
| Crossref | GoogleScholarGoogle Scholar | 26575547PubMed |
[69] R. A. Kendall, T. H. Dunning, R. J. Harrison, J. Chem. Phys. 1992, 96, 6796.
| Crossref | GoogleScholarGoogle Scholar |
[70] V. S. Bryantsev, M. S. Diallo, A. C. T. van Duin, W. A. Goddard, J. Chem. Theory Comput. 2009, 5, 1016.
| Crossref | GoogleScholarGoogle Scholar | 26609610PubMed |
[71] A. Najibi, L. Goerigk, J. Chem. Theory Comput. 2018, 14, 5725.
| Crossref | GoogleScholarGoogle Scholar | 30299953PubMed |
[72] A. Najibi, L. Goerigk, J. Comput. Chem. 2020, 41, 2562.
| Crossref | GoogleScholarGoogle Scholar | 32870518PubMed |
[73] A. Najibi, M. Casanova-Páez, L. Goerigk, J. Phys. Chem. A 2021, 125, 4026.
| Crossref | GoogleScholarGoogle Scholar | 33938224PubMed |
[74] S. Kozuch, J. M. L. Martin, J. Chem. Theory Comput. 2013, 9, 1918.
| Crossref | GoogleScholarGoogle Scholar | 26583543PubMed |
[75] J. Rezac, K. E. Riley, P. Hobza, J. Chem. Theory Comput. 2012, 8, 4285.
| Crossref | GoogleScholarGoogle Scholar | 26605592PubMed |
[76] S. Grimme, A. Hansen, J. G. Brandenburg, C. Bannwarth, Chem. Rev. 2016, 116, 5105.
| Crossref | GoogleScholarGoogle Scholar | 27077966PubMed |
[77] L. Goerigk, in ‘Non-Covalent Interactions in Quantum Chemistry and Physics’, Otero de la Roza, A., DiLabio, G. A., Eds.; Elsevier: Amsterdam, 2017; pp. 195–219.
[78] L. Goerigk, S. Grimme, J. Chem. Theory Comput. 2011, 7, 291.
| Crossref | GoogleScholarGoogle Scholar | 26596152PubMed |
[79] K. Raghavachari, G. W. Trucks, J. A. Pople, M. Head-Gordon, Chem. Phys. Lett. 1989, 157, 479.
| Crossref | GoogleScholarGoogle Scholar |
[80] T. H. Dunning, J. Chem. Phys. 1989, 90, 1007.
| Crossref | GoogleScholarGoogle Scholar |
[81] L. Grafova, M. Pitonak, J. Rezac, P. Hobza, J. Chem. Theory Comput. 2010, 6, 2365.
| Crossref | GoogleScholarGoogle Scholar | 26613492PubMed |
[82] D. E. Taylor, J. G. Angyan, G. Galli, C. Zhang, F. Gygi, K. Hirao, J. W. Song, K. Rahul, O. A. von Lilienfeld, R. Podeszwa, et al. J. Chem. Phys. 2016, 145, 124105.
| Crossref | GoogleScholarGoogle Scholar | 27782652PubMed |
[83] A. Karton, S. Daon, J. M. L. Martin, B. Ruscic, Chem. Phys. Lett. 2011, 510, 165.
| Crossref | GoogleScholarGoogle Scholar |
[84] S. Parthiban, J. M. L. Martin, J. Chem. Phys. 2001, 114, 6014.
| Crossref | GoogleScholarGoogle Scholar |
[85] Y. Zhao, D. G. Truhlar, J. Phys. Chem. A 2006, 110, 10478.
| Crossref | GoogleScholarGoogle Scholar | 16942053PubMed |
[86] H. Yu, D. G. Truhlar, J. Chem. Theory Comput. 2015, 11, 2968.
| Crossref | GoogleScholarGoogle Scholar | 26575734PubMed |
[87] Y. Zhao, H. T. Ng, R. Peverati, D. G. Truhlar, J. Chem. Theory Comput. 2012, 8, 2824.
| Crossref | GoogleScholarGoogle Scholar | 26592123PubMed |
[88] E. R. Johnson, P. Mori-Sanchez, A. J. Cohen, W. Yang, J. Chem. Phys. 2008, 129, 204112.
| Crossref | GoogleScholarGoogle Scholar | 19045857PubMed |
[89] S. Grimme, H. Kruse, L. Goerigk, G. Erker, Angew. Chem. Int. Ed. 2010, 49, 1402.
| Crossref | GoogleScholarGoogle Scholar |
[90] Y. Zhao, B. J. Lynch, D. G. Truhlar, Phys. Chem. Chem. Phys. 2005, 7, 43.
| Crossref | GoogleScholarGoogle Scholar |
[91] Y. Zhao, N. Gonzalez-Garcia, D. G. Truhlar, J. Phys. Chem. A 2005, 109, 2012.
| Crossref | GoogleScholarGoogle Scholar | 16833536PubMed |
[92] J. Friedrich, J. Hänchen, J. Chem. Theory Comput. 2013, 9, 5381.
| Crossref | GoogleScholarGoogle Scholar | 26592276PubMed |
[93] J. Friedrich, J. Chem. Theory Comput. 2015, 11, 3596.
| Crossref | GoogleScholarGoogle Scholar | 26574443PubMed |
[94] S. Grimme, C. Mück-Lichtenfeld, E.-U. Würthwein, A. W. Ehlers, T. P. M. Goumans, K. Lammertsma, J. Phys. Chem. A 2006, 110, 2583.
| Crossref | GoogleScholarGoogle Scholar | 16494365PubMed |
[95] M. Piacenza, S. Grimme, J. Comput. Chem. 2004, 25, 83.
| Crossref | GoogleScholarGoogle Scholar | 14634996PubMed |
[96] H. L. Woodcock, H. F. Schaefer, P. R. Schreiner, J. Phys. Chem. A 2002, 106, 11923.
| Crossref | GoogleScholarGoogle Scholar |
[97] P. R. Schreiner, A. A. Fokin, R. A. Pascal, A. de Meijere, Org. Lett. 2006, 8, 3635.
| Crossref | GoogleScholarGoogle Scholar | 16898779PubMed |
[98] C. Lepetit, H. Chermette, M. Gicquel, J.-L. Heully, R. Chauvin, J. Phys. Chem. A 2007, 111, 136.
| Crossref | GoogleScholarGoogle Scholar | 17201396PubMed |
[99] J. S. Lee, J. Phys. Chem. A 2005, 109, 11927.
| Crossref | GoogleScholarGoogle Scholar | 16366644PubMed |
[100] A. Karton, J. M. Martin, Mol. Phys. 2012, 110, 2477.
| Crossref | GoogleScholarGoogle Scholar |
[101] Y. Zhao, O. Tishchenko, J. R. Gour, W. Li, J. J. Lutz, P. Piecuch, D. Truhlar, J. Phys. Chem. A 2009, 113, 5786.
| Crossref | GoogleScholarGoogle Scholar | 19374412PubMed |
[102] Y. Zhao, D. G. Truhlar, Theor. Chem. Acc. 2008, 120, 215.
| Crossref | GoogleScholarGoogle Scholar |
[103] D. Manna, J. M. L. Martin, J. Phys. Chem. A 2016, 120, 153.
| Crossref | GoogleScholarGoogle Scholar | 26654916PubMed |
[104] F. Neese, T. Schwabe, S. Kossmann, B. Schirmer, S. Grimme, J. Chem. Theory Comput. 2009, 5, 3060.
| Crossref | GoogleScholarGoogle Scholar | 26609985PubMed |
[105] S. N. Steinmann, G. Csonka, C. Carminboeuf, J. Chem. Theory Comput. 2009, 5, 2950.
| Crossref | GoogleScholarGoogle Scholar | 26609976PubMed |
[106] H. Krieg, S. Grimme, Mol. Phys. 2010, 108, 2655.
| Crossref | GoogleScholarGoogle Scholar |
[107] L.-J. Yu, A. Karton, Chem. Phys. 2014, 441, 166.
| Crossref | GoogleScholarGoogle Scholar |
[108] S. Grimme, M. Steinmetz, M. Korth, J. Org. Chem. 2007, 72, 2118.
| Crossref | GoogleScholarGoogle Scholar | 17286442PubMed |
[109] R. Huenerbein, B. Schirmer, J. Moellmann, S. Grimme, Phys. Chem. Chem. Phys. 2010, 12, 6940.
| Crossref | GoogleScholarGoogle Scholar | 20461239PubMed |
[110] R. Sure, A. Hansen, P. Schwerdtfeger, S. Grimme, Phys. Chem. Chem. Phys. 2017, 19, 14296.
| Crossref | GoogleScholarGoogle Scholar | 28537281PubMed |
[111] V. Guner, K. S. Khuong, A. G. Leach, P. S. Lee, M. D. Bartberger, K. N. Houk, J. Phys. Chem. A 2003, 107, 11445.
| Crossref | GoogleScholarGoogle Scholar |
[112] D. H. Ess, K. Houk, J. Phys. Chem. A 2005, 109, 9542.
| Crossref | GoogleScholarGoogle Scholar | 16866406PubMed |
[113] T. C. Dinadayalane, R. Vijaya, A. Smitha, G. N. Sastry, J. Phys. Chem. A 2002, 106, 1627.
| Crossref | GoogleScholarGoogle Scholar |
[114] A. Karton, L. Goerigk, J. Comput. Chem. 2015, 36, 622.
| Crossref | GoogleScholarGoogle Scholar | 25649643PubMed |
[115] L. Goerigk, R. Sharma, Can. J. Chem. 2016, 94, 1133.
| Crossref | GoogleScholarGoogle Scholar |
[116] A. Karton, R. J. O’Reilly, B. Chan, L. Radom, J. Chem. Theory Comput. 2012, 8, 3128.
| Crossref | GoogleScholarGoogle Scholar | 26605724PubMed |
[117] A. Karton, R. J. O’Reilly, L. Radom, J. Phys. Chem. A 2012, 116, 4211.
| Crossref | GoogleScholarGoogle Scholar | 22497287PubMed |
[118] P. Jurecka, J. Sponer, J. Cerny, P. Hobza, Phys. Chem. Chem. Phys. 2006, 8, 1985.
| Crossref | GoogleScholarGoogle Scholar | 16633685PubMed |
[119] M. S. Marshall, L. A. Burns, C. D. Sherrill, J. Chem. Phys. 2011, 135, 194102.
| Crossref | GoogleScholarGoogle Scholar | 22112061PubMed |
[120] J. Rezac, K. E. Riley, P. Hobza, J. Chem. Theory Comput. 2011, 7, 2427.
| Crossref | GoogleScholarGoogle Scholar | 21836824PubMed |
[121] D. Manna, M. K. Kesharwani, N. Sylvetsky, J. M. L. Martin, J. Chem. Theory Comput. 2017, 13, 3136.
| Crossref | GoogleScholarGoogle Scholar | 28530805PubMed |
[122] D. Setiawan, E. Kraka, D. Cremer, J. Phys. Chem. A 2015, 119, 1642.
| Crossref | GoogleScholarGoogle Scholar | 25325889PubMed |
[123] T. Schwabe, S. Grimme, Phys. Chem. Chem. Phys. 2007, 9, 3397.
| Crossref | GoogleScholarGoogle Scholar | 17664963PubMed |
[124] S. Grimme, Angew. Chem. Int. Ed. 2006, 45, 4460.
| Crossref | GoogleScholarGoogle Scholar |
[125] D. Gruzman, A. Karton, J. M. L. Martin, J. Phys. Chem. A 2009, 113, 11974.
| Crossref | GoogleScholarGoogle Scholar | 19795892PubMed |
[126] M. K. Kesharwani, A. Karton, J. M. L. Martin, J. Chem. Theory Comput. 2016, 12, 444.
| Crossref | GoogleScholarGoogle Scholar | 26653705PubMed |
[127] D. Reha, H. Valdes, J. Vondrasek, P. Hobza, A. Abu-Riziq, B. Crews, M. S. de Vries, Chemistry 2005, 11, 6803.
| Crossref | GoogleScholarGoogle Scholar | 16092140PubMed |
[128] L. Goerigk, A. Karton, J. M. L. Martin, L. Radom, Phys. Chem. Chem. Phys. 2013, 15, 7028.
| Crossref | GoogleScholarGoogle Scholar | 23403537PubMed |
[129] U. R. Fogueri, S. Kozuch, A. Karton, J. M. L. Martin, J. Phys. Chem. A 2013, 117, 2269.
| Crossref | GoogleScholarGoogle Scholar | 23379303PubMed |
[130] G. I. Csonka, A. D. French, G. P. Johnson, C. A. Stortz, J. Chem. Theory Comput. 2009, 5, 679.
| Crossref | GoogleScholarGoogle Scholar | 26609572PubMed |
[131] H. Kruse, A. Mladek, K. Gkionis, A. Hansen, S. Grimme, J. Sponer, J. Chem. Theory Comput. 2015, 11, 4972.
| Crossref | GoogleScholarGoogle Scholar | 26574283PubMed |
[132] S. Kozuch, S. M. Bachrach, J. M. Martin, J. Phys. Chem. A 2014, 118, 293.
| Crossref | GoogleScholarGoogle Scholar | 24328111PubMed |
[133] L. Goerigk, S. Grimme, Phys. Chem. Chem. Phys. 2011, 13, 6670.
| Crossref | GoogleScholarGoogle Scholar | 21384027PubMed |
[134] J.-D. Chai, S.-P. Mao, Chem. Phys. Lett. 2012, 538, 121.
| Crossref | GoogleScholarGoogle Scholar |
[135] E. Brémond, C. Adamo, J. Chem. Phys. 2011, 135, 024106.
| Crossref | GoogleScholarGoogle Scholar | 21766924PubMed |
[136] S. Grimme, J. Chem. Phys. 2003, 118, 9095.
| Crossref | GoogleScholarGoogle Scholar |
[137] S. Grimme, R. Huenerbein, S. Ehrlich, ChemPhysChem 2011, 12, 1258.
| Crossref | GoogleScholarGoogle Scholar | 21445954PubMed |
[138] S. Grimme, P. R. Schreiner, Angew. Chem. Int. Ed. 2011, 50, 12639.
| Crossref | GoogleScholarGoogle Scholar |
[139] S. Grimme, M. Steinmetz, Phys. Chem. Chem. Phys. 2013, 15, 16031.
| Crossref | GoogleScholarGoogle Scholar | 23963317PubMed |
[140] W. Hujo, S. Grimme, J. Chem. Theory Comput. 2013, 9, 308.
| Crossref | GoogleScholarGoogle Scholar | 26589033PubMed |
[141] S. Rösel, H. Quanz, C. Logemann, J. Becker, E. Mossou, L. Canadillas-Delgado, E. Caldeweyher, S. Grimme, P. R. Schreiner, J. Am. Chem. Soc. 2017, 139, 7428.
| Crossref | GoogleScholarGoogle Scholar | 28502175PubMed |
[142] A. A. Fokin, T. S. Zhuk, S. Blomeyer, C. Perez, L. V. Chernish, A. E. Pashenko, J. Antony, Y. V. Vishnevskiy, R. J. F. Berger, S. Grimme, et al. J. Am. Chem. Soc. 2017, 139, 16696.
| Crossref | GoogleScholarGoogle Scholar | 29037036PubMed |
[143] L. Goerigk, J. R. Reimers, J. Chem. Theory Comput. 2013, 9, 3240.
| Crossref | GoogleScholarGoogle Scholar | 26583999PubMed |
[144] L. Goerigk, C. A. Collyer, J. R. Reimers, J. Phys. Chem. B 2014, 118, 14612.
| Crossref | GoogleScholarGoogle Scholar | 25410613PubMed |
[145] P. Kraus, I. Frank, J. Phys. Chem. A 2018, 122, 4894.
| Crossref | GoogleScholarGoogle Scholar | 29750513PubMed |
[146] N. Mehta, T. Fellowes, J. M. White, L. Goerigk, J. Chem. Theory Comput. 2021, 17, 2783.
| Crossref | GoogleScholarGoogle Scholar | 33881869PubMed |
[147] L. Goerigk, H. Kruse, S. Grimme, ChemPhysChem 2011, 12, 3421.
| Crossref | GoogleScholarGoogle Scholar | 22113958PubMed |
[148] M. K. Kesharwani, A. Karton, N. Sylvetsky, J. M. Martin, Aust. J. Chem. 2018, 71, 238.
| Crossref | GoogleScholarGoogle Scholar |
[149] L. A. Curtiss, K. Raghavachari, P. C. Redfern, J. A. Pople, J. Chem. Phys. 1997, 106, 1063.
| Crossref | GoogleScholarGoogle Scholar |
[150] H. Kruse, L. Goerigk, S. Grimme, J. Org. Chem. 2012, 77, 10824.
| Crossref | GoogleScholarGoogle Scholar | 23153035PubMed |
[151] W. J. Hehre, R. Ditchfeld, J. A. Pople, J. Chem. Phys. 1972, 56, 2257.
| Crossref | GoogleScholarGoogle Scholar |
* Lars Goerigk is the recipient of a 2020 Rennie Memorial Medal from the Royal Australian Chemical Institute.