

Copper mediated in situ nucleophilic addition of polyalcohols to dicyanonitrosomethanide†
Mohd. R. Razali A B , Aron Urbatsch A , Boujemaa Moubaraki A , Keith S. Murray A , Glen B. Deacon A and Stuart R. Batten A *
A *
A School of Chemistry, 19 Rainforest Walk, Monash University, Vic. 3800, Australia.
B School of Chemical Sciences, Universiti Sains Malaysia, Penang, Malaysia.
Australian Journal of Chemistry 75(9) 725-731 https://doi.org/10.1071/CH21323
Submitted: 6 December 2021 Accepted: 7 February 2022 Published: 22 March 2022
© 2022 The Author(s) (or their employer(s)). Published by CSIRO Publishing. This is an open access article distributed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License (CC BY-NC-ND)
Abstract
The transition metal-promoted in situ nucleophilic addition of triethanolamine (teaH3) and N-methyldiethanolamine (mdeaH2) to the dicyanonitrosomethanide (dcnm) anion results in the formation of [Cu(hbnm)]·MeOH (1) and [Cu(mbnm)]·2MeCN (2), (hbnm = hydroxyethylaminobis(ethoxy(imino)methyl(cyano)nitrosomethanide) and mbnm = methylaminobis(ethoxy(imino)methyl(cyano)nitrosomethanide). Complexes 1 and 2 are coordination polymers, each containing the addition products of two alcohol arms of teaH3 and mdeaH2 to dcnm anions.
Keywords: coordination polymer, copper, crystal structure, dicyanonitrosomethanide, in situ reactions, magnetic properties, nucleophilic addition, small cyano anions.
Introduction
Small cyano anions (SCAs) are ligands of significant interest due to their coordination diversity and their ability to form a wide variety of compounds, including coordination polymers and metal clusters.[1–3] SCAs can be very adept at forming transition metal complexes as they show a large range of functionalities and binding modes. The dicyanomethanide (dca, [N(CN)2−]) and tricyanomethanide (tcm, [C(CN)3−])) ligands, for example, have been widely used in the synthesis of complexes of various geometries and dimensionalities, owing to their ability to display a variety of coordination modes.[4] This in turn leads to long-range magnetic ordering in MII(dca)2 species.[4]
One SCA, dicyanonitrosomethanide (dcnm), has demonstrated its propensity to undergo nucleophilic addition with water, alcohols and amines to either one (usually) or both nitrile arms.[5–16] Nucleophilic addition to the nitrile groups of the dcnm anion affords new ligands with polynucleating binding modes which can lead to the formation of metal clusters with potential single molecule magnet (SMM) behaviour.[6–11] With the addition of alcohols to dcnm, the nitrile arm is converted to an imine and an ether functionality, however the addition of alcohol moieties has also been shown to deactivate the β-carbon of the second nitrile arm of dcnm, inhibiting the addition of a second nucleophile.[12]
While the in situ nucleophilic addition of small alcohols such as methanol and to some extent ethanol is readily achieved in the presence of a transition metal, the addition of longer chain alcohols is limited and only a few have been reported.[5,16] The addition of 2-methoxyethanol represents the largest known alcohol added to dcnm and led to the formation of cyano(imino(2-methoxyethoxy)methyl)nitrosomethanide (cgnm).[12]
In order to further explore the general robustness and versatility of these in situ nucleophilic addition reactions, reactions of dcnm with alcohols that were larger and contained multiple alcohol functionalities were of interest. In particular, the addition of species with multiple alcohol functionalities may facilitate the formation of dianionic or trianionic polydentate bridging ligands. Reactions of dcnm with species such as triethanolamine (teaH3) in the presence of metals, and the subsequent formation and crystallisation of metal complexes, was also of particular interest given the propensity of both teaH3[10,17–19] and dcnm derivatives[6–12,20–23] to form polynuclear metal complexes. Many of these, in the triethanolamine series, display single molecule magnetic (SMM) properties.[24] In situ decomposition of the teaH3 ligand, to give a {Fe6Dy3} SMM cluster containing 1,1,2,2-tetrahydroxyethane, has been reported previously,[25] however in this work we were expecting larger rather than smaller ligands.
Results and discussion
Reaction of copper chloride with Me4N(dcnm) and teaH3 (mole ratio 1:3:1) in a mixture of acetonitrile and methanol gave the double-stranded 1D polymer [Cu(hbnm)]·MeOH (1) (hbnm = hydroxyethylaminobis(ethoxy(imino)methyl(cyano)nitrosomethanide)) (Scheme 1) as dark brown crystals. Hbnm is the bis-dcnm addition-product of teaH3; two of the teaH3 arms added to one nitrile arm of each of two dcnm anions, and notably, the last –CH2CH2OH arm remains unreacted (Scheme 1), as supported by the ν(OH) band at 3404 cm−1 in the IR spectrum.
|
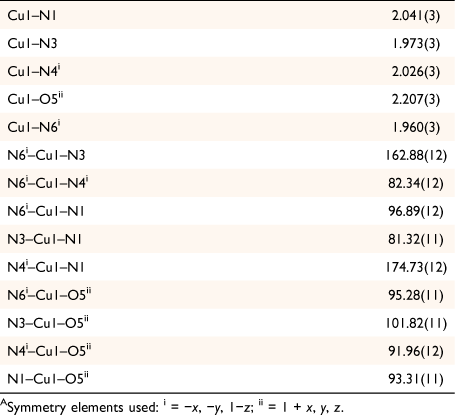
Complex 1 crystallises in the triclinic space group P1̄ with the asymmetric unit containing one crystallographically unique [Cu(hbnm)] moiety and one disordered methanol molecule in the lattice. The copper atom has a five-coordinate square pyramidal geometry and each dianionic hbnm ligand displays a µ3-κ2(N,N′)Cu:κ2(N″,N‴)Cu′:κ1(O)Cu″ coordination mode (Fig. 1a), coordinating to three equivalent copper ions. Each of the two substituted arms of the ligand chelates a copper ion with its nitroso and imine nitrogen atoms, while the third, unreacted hydroxyethyl arm coordinates to a third copper ion via its oxygen atom, facilitating the formation of the 1D network shown in Fig. 1b. The copper ion displays a Jahn–Teller distortion at the apical bond from the hydroxy group of hbnm (2.207(3) Å vs 1.960(3) Å–2.041(3) Å for the equatorial bonds, Table 1).

|
|
The tertiary amine (N7) at the centre of hbnm is not involved in any coordination to a metal. Instead, it acts as intramolecular hydrogen-bond acceptor for an imine group (N6). Another imine group (N3) forms intermolecular hydrogen bonds with the disordered methanol molecules in the lattice (Fig. 1c). One of the hbnm nitroso oxygen atoms (O1) acts as a bifurcated hydrogen bond acceptor to the hydroxyethyl group and to the disordered methanol molecule in the lattice. Attempts to synthesise the mono- and tris-analogous of hbnm were unsuccessful. Dcnm to teaH3 ratios of 4:1 and 5:1 resulted only in the formation of hbnm with the percentage yield depending on the amount of CuCl2 used. A 1:1 reaction gave no precipitate.
Since the third hydroxyethyl group of teaH3 did not undergo nucleophilic addition to dcnm, removal of this group should maintain the same level of nucleophilic addition as in hbnm formation. However, as the unreacted hydroxyethyl group was observed to coordinate to the copper ion in 1, removal of this group will nonetheless affect the overall structure. Thus, the addition product between N-methyldiethanolamine (mdeaH2) and dcnm, methylaminobis(ethoxy(imino)methyl(cyano)nitrosomethanide (mbnm)) (Scheme 2), was isolated as a copper complex from a synthetic procedure similar to that which led to the formation of 1, but with acetonitrile rather than a mixture of acetonitrile/methanol as the solvent. Crystallisation of [Cu(mbnm)]·2MeCN (2) was achieved from a reaction with a 2:1 dcnm to mdeaH2 ratio. As for hbnm, nucleophilic addition of two arms of mdeaH2 occurred, each to only one nitrile arm of dcnm, giving the bis-substituted product, mbnm.
|
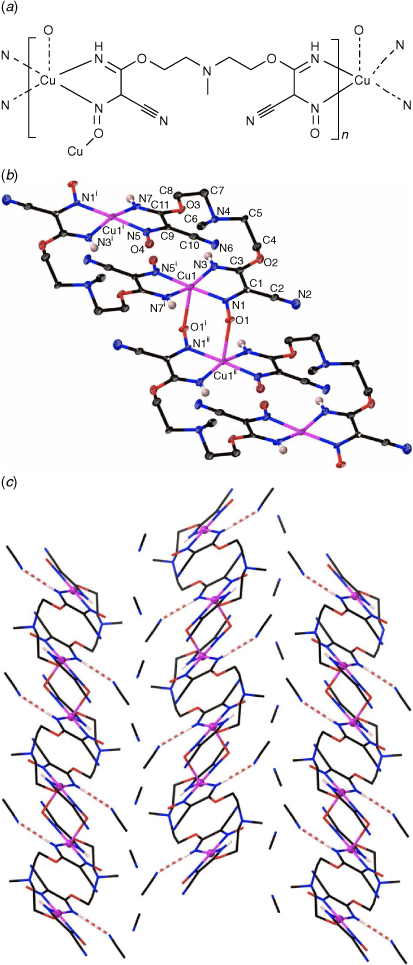
The 1D polymer 2 crystallises in the monoclinic space group P21/c with the asymmetric unit containing one [Cu(mbnm)] moiety and two lattice acetonitrile molecules. The dianionic ligand in 2 displayed a µ3-κ2(N,N′)Cu:κ2(N″,N‴)Cu′:κ1(O)Cu″ coordination mode, which is similar to that of hbnm in 1. Coordination of the hydroxyethyl arm of hbnm to a copper ion in 1 is, however, replaced by binding of a nitroso oxygen atom (O1) of a cyanonitrosomethanide moiety to the copper ion in 2. The second nitroso oxygen atom O4 in mbnm is not involved in coordination (Fig. 2a).
|
The structure contains approximately planar [Cu2(mbnm)2] rings in which the two bridging ligands chelate the metal atoms at either end through the imine and nitroso nitrogen atoms. These rings are then crosslinked into 1D chains via coordination of a nitroso oxygen atom to the apical position of a five-coordinate copper atom of an adjoining ring, and vice versa. This creates a second type of ring in which pairs of copper atoms are linked by pairs of bridging nitroso groups (Fig. 2b). The distance between the nitroso-bridged Cu1 and Cu1ii is 4.07 Å, whereas Cu⋯Cu distance across the larger ring is 5.53 Å. The copper atoms show Jahn–Teller distortion, with the longest bond being between Cu1 and the nitroso oxygen atom O1ii (2.334(6) Å, Table 2). The imine group N3 in mbnm acts as a hydrogen bond donor to the tertiary amine nitrogen N4, while the second imine group N7 forms an intermolecular hydrogen bond with a lattice acetonitrile (Fig. 2c).
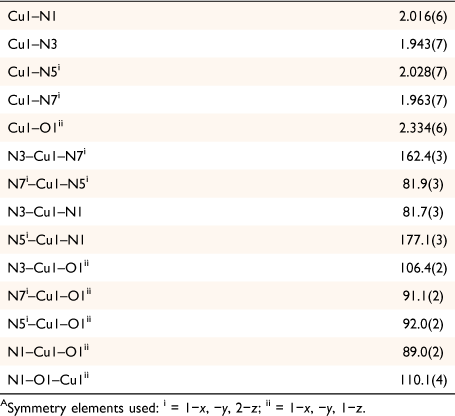
|
With the coordination of the nitroso oxygen in 2, induced by the removal of the coordinating ethanol arm of hbnm seen in 1, short Cu–NO–Cu bridges were created which were not present in 1. This is significant as these nitroso bridging motifs have been shown in previous metal clusters of dcnm derivatives to create efficient pathways for magnetic exchange.[6–12,20,21] Thus the magnetic properties of complex 2 were investigated.
Fig. 3 shows a χMT vs T plot per copper atom for complex 2. The χMT value remains constant at 0.45 cm−3 mol−1 K between 300 and 40 K, then decreases below 40 K to reach a minimum of 0.38 cm−3 mol−1 K at 4 K. The corresponding molar susceptibility data, χM, follow a Curie–Weiss law dependence, χM = C/(T − θ) where C = Curie constant and θ = Weiss constant. (C = 0.45 cm3 mol−1 K; θ = −0.59 K; Fig. 3, right). This indicates that the antiferromagnetic coupling within the nitroso-bridged Cu2 dinuclear moieties is weak. This is not surprising because the bridging is through the Cu1–N1{equatorial}O1{apical}–Cu1ii pathways (Fig. 2b) that involve Cu(dx2−y2)–O,N(p)–Cu(dz2) overlap. Fitting to a S = 1/2 dimer (−2JS1·S2) model[26] shown in Eqn 1 and Fig. 3, left, gave best-fit values of g = 2.19, 2J = −2.02 cm−1, P = 0.001 and Nα = 6.0 × 10−5 cm3 mol−1. J is the exchange coupling constant, g the Lande factor, β the Bohr Magneton, Nα the temperature independent paramagnetic susceptibility per Cu(II), and P is the fraction of S = ½ monomer.
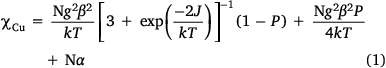

|
Conclusions
In summary, two Cu(II) coordination polymers were successfully synthesised by nucleophilic addition of primary alcohols to a nitrile arm of the dcnm anion. Furthermore, both ligands, hbnm in 1 and mbnm in 2, result from the addition reactions of multiple alcohol moieties on the one ligand with dcnm anions. To our knowledge, this is the first report of such a reaction. Although the yields were moderate the reactions were reproducible. Notably, no products containing ligands derived from addition reactions with a third arm of teaH3 were observed in the precipitated materials, however this may be driven by the fact that formation of the hbnm ligand leads to insoluble polymeric products before these additional reactions can occur. We can also not rule out other species in solution containing fully reacted teaH3 derivatives. A magnetic susceptibility study of complex 2 revealed weak antiferromagnetic coupling within nitroso-bridged Cu2 dinuclear moieties, due to the Cu1–N1{equatorial}O1{apical}–Cu1ii bridging pathways, which involve Cu(dx2−y2)–O,N(p)–Cu(dz2) overlap.
Experimental
Laboratory reagents and solvents were used without further purification. Elemental analyses (C, H, N) were performed by the Campbell Analytical Laboratory, University of Otago, New Zealand. ATR-IR spectra were recorded with a Bruker Equinox 55 series FTIR spectrometer in the range 4000–500 cm−1 with a resolution of 4 cm−1. DC magnetic susceptibility measurements were carried out on a Quantum Design MPMS 5 SQUID magnetometer calibrated by use of a standard palladium sample (Quantum Design) of accurately known magnetisation and checked from time to time by use of magnetochemical calibrants such as CuSO4·5H2O. Powder samples of mass ca. 20 mg were accurately weighed and dispersed in Vaseline in order to avoid torquing of the crystallites. The sample mulls were contained in a calibrated gelatine capsule held at the centre of a drinking straw that was fixed at the end of the sample rod. Magnetisation isotherm measurements were made in fields of between 0 and 5 T. Diamagnetic corrections for the ligands were obtained using Pascal’s table.
Me4N(dcnm) was prepared by a metathesis reaction between Ag(dcnm) and Me4NCl in water. Ag(dcnm) was prepared according to a literature procedure.[27]
Synthesis of [Cu(hbnm)]·MeOH (1)
Me4N(dcnm) (150 mg, 892 µmol) dissolved in acetonitrile (6 mL) was added to CuCl2 (38 mg, 282 µmol) in methanol (2 mL). Triethanolamine (44 mg, 295 µmol) in 3 mL acetonitrile was added, turning the solution to dark blue. Dark blue plate crystals of 1 formed in 2 weeks and were then filtered and washed with methanol and diethyl ether. Yield: 28 mg, 65 µmol, 23% (based on Cu). Anal. calcd for C13H19CuN7O6: C 36.12, H 4.17, N 22.69. Found: C 36.05, H 4.11, N 22.66. ATR-IR (ν/cm−1): 3404m, 3251m, 3036w, 2215m, 1620s, 1399s, 1313m, 1139m, 893w, 731w. The use of further excess Me4N(dcnm), either 200 mg or 300 mg, also resulted the formation of 1, as determined by IR spectra and unit cell determination.
Synthesis of [Cu(hbnm)]·2MeCN (2)
Me4N(dcnm) (100 mg, 595 µmol) dissolved in acetonitrile (6 mL) was added to Cu(ClO4)2·6H2O (55 mg, 148 µmol) in acetonitrile (2 mL). N-Methyldiethanolamine (35 mg, 294 µmol) in 3 mL acetonitrile was added, turning the solution to dark blue. Red block crystals of 2 formed in a month and were then filtered and washed with methanol and ether. Yield: 25 mg, 55 µmol, 37% (based on Cu). Anal. calcd for C15H19CuN9O4: C 39.74, H 4.20, N 27.82. Found: C 39.65, H 4.12, N 27.75. ATR-IR (ν/cm−1): 3152w, 2862m, 2213m, 1639m, 1605s, 1405s, 1290m, 1164m, 1121m, 975w, 929w, 804w, 756w.
Crystallography details
Data for complex 1 were collected at 100 K at the Australian Synchrotron MX1 beamline (synchrotron radiation, λ = 0.70846).[28] The data collection and integration were performed within the Blu-Ice[29] and XDS[30] software programs. Data completeness (92.4%) was slightly lower than ideal due to the constraints of the goniometer on the MX1 beamline. Data for 2 were collected using a Bruker Smart Apex X8 diffractometer with Mo Kα radiation λ = 0.71073 Å. Data were collected at 123 K, maintained using an open flow of nitrogen gas from an Oxford Cryostreams cryostat. The data collection and integration was performed within the SMART and SAINT+ software programs, and corrected for Lorentz-polarisation and absorption effects using the Apex II program Suite.[31] Complexes 1 and 2 were solved by direct methods (SHELXS-97), and refined (SHELXL-97) by full matrix least-squares on all F2 data.[32] The program OLEX2 was used as a graphical SHELXL interface.[33] Data for 1 and 2 have been deposited with the Cambridge Crystallographic Database with CCDC numbers 2126333–2126334.
Crystallographic details of [Cu(hbnm)]·MeOH (1)
C13H19CuN7O6: M = 432.90, dark blue plate, 0.05 × 0.01 × 0.01 mm3, triclinic, space group P1̄, a = 8.7280(17) Å, b = 10.204(2) Å, c = 10.430(2) Å, α = 101.00(3)°, β = 104.67(3)°, γ = 90.28(3)°, V = 880.7(3) Å3, Z = 2, ρcalc = 1.617 g cm−3, F000 = 438, 6421 reflections collected, 2890 unique (Rint = 0.0380), GooF = 1.073, R1 = 0.0439, wR2 = 0.1172, R indices based on 2605 reflections with I > 2σ(I), synchrotron radiation, λ = 0.70846, 257 parameters, 0 restraints, µ = 1.288. The methyl group of the methanol molecule in the lattice is disordered over two positions with occupancies refined as 50:50. The hydrogen atoms of the methanol were not located nor assigned due to this disorder.
Crystallographic details of [Cu(mbnm)]·2MeCN (2)
C15H19CuN9O4: M = 452.93, dark brown block, 0.10 × 0.10 × 0.05 mm3, monoclinic, space group P21/c, a = 11.564(3) Å, b = 19.853(5) Å, c = 9.139(2) Å, β = 108.247(6)°, V = 1992.7(8) Å3, Z = 4, ρcalc = 1.510 g cm−3, F000 = 932, 12 763 reflections collected, 4533 unique (Rint = 0.1520), GooF = 1.053, R1 = 0.0964, wR2 = 0.1979, R indices based on 2160 reflections with I > 2σ(I), 266 parameters, 14 restraints, µ = 1.138. C9 and C11 restrained using ISOR commands.
Data availability
Data for 1 and 2 have been deposited with the Cambridge Crystallographic Database with CCDC numbers 2126333–2126334.
Conflicts of interest
The authors declare no conflicts of interest.
Declaration of funding
We thank the Australian Research Council for funding (DP1097198, FT0991840, LE100100109, LE100100197). MRR thanks USM and Monash University for a Postgraduate Publication Award.
Acknowledgements
Part of this research was undertaken on the MX1 beamline at the Australian Synchrotron, part of ANSTO.
References
[1] DR Turner, ASR Chesman, KS Murray, GB Deacon, SR Batten, Chem Commun 2011, 47, 10189.| Crossref | GoogleScholarGoogle Scholar |
[2] N Gerasimchuk, Dalton Trans 2019, 48, 7985.
| Crossref | GoogleScholarGoogle Scholar | 31090771PubMed |
[3] S Benmansour, C Atmani, F Setifi, S Triki, M Marchivie, CJ Gómez-García, Coord Chem Rev 2010, 254, 1468.
| Crossref | GoogleScholarGoogle Scholar |
[4] SR Batten, KS Murray, Coord Chem Rev 2003, 246, 103.
| Crossref | GoogleScholarGoogle Scholar |
[5] M Hvastijová, J Kohout, JW Buchler, R Boča, J Kožı’šek, L Jäger, Coord Chem Rev 1998, 175, 17.
| Crossref | GoogleScholarGoogle Scholar |
[6] ASR Chesman, DR Turner, B Moubaraki, KS Murray, GB Deacon, SR Batten, Dalton Trans 2012, 41, 3751.
| Crossref | GoogleScholarGoogle Scholar |
[7] MR Razali, NF Chilton, A Urbatsch, B Moubaraki, SK Langley, KS Murray, GB Deacon, SR Batten, Polyhedron 2013, 52, 797.
| Crossref | GoogleScholarGoogle Scholar |
[8] DJ Price, SR Batten, KJ Berry, B Moubaraki, KS Murray, Polyhedron 2003, 22, 165.
| Crossref | GoogleScholarGoogle Scholar |
[9] ASR Chesman, DR Turner, B Moubaraki, KS Murray, GB Deacon, SR Batten, Inorg Chim Acta 2012, 389, 99.
| Crossref | GoogleScholarGoogle Scholar |
[10] MR Razali, A Urbatsch, SK Langley, JG MacLellan, GB Deacon, B Moubaraki, KS Murray, SR Batten, Aust J Chem 2012, 65, 918.
| Crossref | GoogleScholarGoogle Scholar |
[11] ASR Chesman, DR Turner, KJ Berry, NF Chilton, B Moubaraki, KS Murray, GB Deacon, SR Batten, Dalton Trans 2012, 41, 11402.
| Crossref | GoogleScholarGoogle Scholar |
[12] ASR Chesman, DR Turner, B Moubaraki, KS Murray, GB Deacon, SR Batten, Eur J Inorg Chem 2010, 2010, 59.
| Crossref | GoogleScholarGoogle Scholar |
[13] EI Izgorodina, ASR Chesman, DR Turner, GB Deacon, SR Batten, J Phys Chem B 2010, 114, 16517.
| Crossref | GoogleScholarGoogle Scholar | 21086972PubMed |
[14] ASR Chesman, DR Turner, DJ Price, B Moubaraki, KS Murray, GB Deacon, SR Batten, Chem Commun 2007, 3541.
| Crossref | GoogleScholarGoogle Scholar |
[15] ASR Chesman, DR Turner, GB Deacon, SR Batten, Chem Asian J 2009, 4, 761.
| Crossref | GoogleScholarGoogle Scholar |
[16] MR Razali, A Urbatsch, GB Deacon, SR Batten, Inorg Chim Acta 2013, 403, 120.
| Crossref | GoogleScholarGoogle Scholar |
[17] SK Langley, NF Chilton, B Moubaraki, KS Murray, Dalton Trans 2012, 41, 1033.
| Crossref | GoogleScholarGoogle Scholar | 22113523PubMed |
[18] SK Langley, L Ungur, NF Chilton, B Moubaraki, LF Chibotaru, KS Murray, Chem Eur J 2011, 17, 9209.
| Crossref | GoogleScholarGoogle Scholar | 21732432PubMed |
[19] S-J Liu, S-D Han, J-M Jia, L Xue, Y Cui, S-M Zhang, Z Chang, CrystEngComm 2014, 16, 5212.
| Crossref | GoogleScholarGoogle Scholar |
[20] MR Razali, ASR Chesman, NF Chilton, SK Langley, B Moubaraki, KS Murray, GB Deacon, SR Batten, Dalton Trans 2013, 42, 1400.
| Crossref | GoogleScholarGoogle Scholar | 23160602PubMed |
[21] ASR Chesman, DR Turner, B Moubaraki, KS Murray, GB Deacon, SR Batten, Aust J Chem 2009, 62, 1137.
| Crossref | GoogleScholarGoogle Scholar |
[22] AA Opalade, CJ Gomez-Garcia, N Gerasimchuk, Cryst Growth Des 2019, 19, 678.
| Crossref | GoogleScholarGoogle Scholar |
[23] AA Opalade, A Karmakar, GMDM Rúbio, AJL Pombeiro, N Gerasimchuk, Inorg Chem 2017, 56, 13962.
| Crossref | GoogleScholarGoogle Scholar | 29120177PubMed |
[24] LM Wittick, KS Murray, B Moubaraki, SR Batten, L Spiccia, KJ Berry, Dalton Trans 2004, 1003.
| Crossref | GoogleScholarGoogle Scholar | 15252679PubMed |
[25] S Schmidt, D Prodius, V Mereacre, GE Kostakis, AK Powell, Chem Commun 2013, 49, 1696.
| Crossref | GoogleScholarGoogle Scholar |
[26] B Bleaney, KD Bowers, Proc R Soc London, Sect A 1952, 214, 451.
| Crossref | GoogleScholarGoogle Scholar |
[27] G Longo, Gazz, Chim Ital 1931, 61, 575.
[28] NP Cowieson, D Aragao, M Clift, DJ Ericsson, C Gee, SJ Harrop, N Mudie, S Panjikar, JR Price, A Riboldi-Tunnicliffe, R Williamson, T Caradoc-Davies, J Synchrotron Rad 2015, 22, 187.
| Crossref | GoogleScholarGoogle Scholar |
[29] TM McPhillips, SE McPhillips, HJ Chiu, AE Cohen, AM Deacon, PJ Ellis, E Garman, A Gonzalez, NK Sauter, RP Phizackerley, SM Soltis, P Kuhn, J Synchrotron Rad 2002, 9, 401.
| Crossref | GoogleScholarGoogle Scholar |
[30] W Kabsch, J Appl Crystallogr 1993, 26, 795.
| Crossref | GoogleScholarGoogle Scholar |
[31] APEXII, v2.1.0. Madison, Wisconsin: Bruker AXS Ltd; 2005.
[32] GM Sheldrick, Acta Crystallogr, Sect A 2008, 64, 112.
| Crossref | GoogleScholarGoogle Scholar | 18156677PubMed |
[33] OV Dolomanov, LJ Bourhis, RJ Gildea, JAK Howard, H Puschmann, J Appl Cryst 2009, 42, 339.
| Crossref | GoogleScholarGoogle Scholar |
† Dedicated to Prof. Peter Junk, friend and colleague, on the occasion of his 60th birthday. Dedicated also to Prof. Glen Deacon, friend, colleague, co-author, and 85 years young.