

Unraveling the genetic diversity and evolutionary lineages of Catharanthus roseus cultivars through plastome analysis and DNA barcoding
Abeer Al-Andal A *
A *
A
Abstract
This investigation elucidates the genetic heterogeneity and phylogenetic affinities among eight cultivars of Catharanthus roseus, focusing on petal color and morphological variations.
The primary objective was to elucidate the genetic disparities and evolutionary trajectories among these cultivars, thereby augmenting our comprehension of their genomic architecture and phylogenetic lineages.
The genomic DNA of the cultivars underwent sequencing, assembly, and annotation utilizing the bioinformatic tools NOVOPlasty and GeSeq.
Results showed minimal plastome size variation among cultivars (154,928 bp to 155,066 bp). Group 1 cultivars (1, 6, 8) had elongated petals, whereas Group 2 (2, 3, 4, 5, 7) had broader, orbicular petals. Sequence analysis showed significant variations in photosynthesis-related genes, with distinct single nucleotide polymorphism (SNP) frequencies and insertion/deletion (Indel) patterns between groups. The examination of codon usage and simple sequence repeat (SSR) biomarkers did not yield significant contributions to understanding the speciation process. Phylogenetic relationships were determined using DNA barcoding and key plastid markers (matK, rbcL, trnL). The trnL gene effectively clustered cultivars by petal morphology. Phylogenetic trees showed close genetic relationships within the same tribe, with C. roseus being genetically distinct from other species.
This study has provided comprehensive chloroplast genome assemblies for C. roseus cultivars, advancing our understanding of their genetic diversity and phylogenetic relationships.
The findings enhance our comprehension of speciation mechanisms within the Apocynaceae family and offer important insights for the refinement of taxonomic frameworks, contributing to a deeper evolutionary perspective on the diversification of C. roseus and related species.
Keywords: codon usage, Indels, matK, rbcL, SNPs, speciation, SSRs, trnL.
Introduction
Catharanthus roseus, commonly known as the Madagascar periwinkle, is an evergreen perennial shrub classified within the Rauvolfieae tribe of the Apocynaceae family (order Gentianales, clade Angiosperms) (Endress and Bruyns 2000; Wang et al. 2023a). This plant is renowned for its medicinal properties and pharmacological potential, particularly in the production of alkaloids used in cancer treatment (Goswami et al. 2024). The alkaloids vincristine and vinblastine, isolated from C. roseus, have been integral in the development of chemotherapy drugs for treating leukemia and Hodgkin’s lymphoma (Torka et al. 2022; Banyal et al. 2023). The species is also known for its antioxidant, anti-inflammatory, and antimicrobial properties, making it a valuable resource for pharmaceutical and cosmetic industries (Pham et al. 2019). This plant is also valued for its ornamental appeal, because it is grown globally for its diverse range of vibrant flowers (Goswami et al. 2024; Ravikumar and Dhatt 2024). The plant has a long history of therapeutic use in traditional medicine, where it is employed to treat a variety of ailments, including cancer, diabetes, and hypertension (Chaturvedi et al. 2022). The eight C. roseus cultivars utilized in the present study represent a diverse range of morphological traits, particularly in petal color, shape, and pigmentation (Samiyarsih et al. 2019; Susanto and Dwiati 2024). They were selected on the basis of these distinct characteristics, which are typical of regions with warm climates where C. roseus is commonly cultivated for its ornamental and medicinal value (Aruna et al. 2015). These cultivars are not specific to any single region, but are widely used in landscaping and horticulture across various regions, including the United States, Europe, Africa, and Asia.
The nuclear genome of C. roseus has been subjected to comprehensive scrutiny, with investigations focusing on its architectural configuration and overall magnitude. This species exhibits a diploid chromosome count of 2n = 16 and an estimated genome size of approximately 738 Mbp (Guimarães et al. 2012). Recent advancements in genomic analyses have facilitated the assembly of a nearly complete genome, comprising 89 scaffolds that span 561.7 Mbp and are organized into eight pseudochromosomes (Xu et al. 2023). Notably, five of these chromosomes have been extensively mapped to extend towards the telomeres at one or both termini. Prior studies on the nuclear genome of C. roseus have employed molecular markers such as inter simple sequence repeats (ISSRs) and simple sequence repeats (SSRs) to evaluate genetic diversity among cultivars, showing pronounced polymorphism and distinct groupings, while also elucidating the role of transcription factors and gene expression in secondary metabolism, thereby contributing to our understanding of its evolutionary and taxonomic frameworks (Guimaraes et al. 2012; She et al. 2019). However, considering the chloroplast genome in the context of diversity, evolution, and taxonomy offers several advantages. Chloroplast DNA, being maternally inherited, provides a clear lineage and simplifies genetic analysis by mitigating the complexity associated with biparental inheritance of nuclear DNA (Yang et al. 2017). Chloroplast DNA markers, including matK, rbcL, and trnL genes, are extensively utilized for species and subspecies delineation because of their high variability and conservation across diverse plant species (Yang et al. 2017; Meena et al. 2020; Li et al. 2021; Matiz-Ceron et al. 2022; Wang et al. 2022a).
Investigating the structural variations of chloroplast variability in C. roseus also provides insights into the evolutionary trajectories and genetic diversity of this plant species, which is valuable for both scientific understanding and practical applications. By analyzing chloroplast genomes, researchers can elucidate phylogenetic relationships, identify molecular barcodes for species delineation, and inform conservation strategies (Caron et al. 2019). This knowledge can enhance our comprehension of plant evolution and adaptation, contributing to the sustainable management of genetic resources. Additionally, understanding chloroplast variability can have practical implications for improving crop traits and enhancing the biosynthesis of valuable secondary metabolites, such as those used in cancer treatments, thereby supporting advancements in biotechnology and pharmaceutical industries (Kulagina et al. 2022; Jamal and Ahmad 2024).
The molecular composition of plant chloroplast genomes exhibits remarkable conservation, typically featuring a quadripartite arrangement consisting of a large single-copy (LSC) region, a small single-copy (SSC) region, and two inverted repeat (IR) regions (Jiang et al. 2023; Sebastin et al. 2024). Within plastid genomes, single nucleotide polymorphisms (SNPs), insertions and deletions (Indels), and simple sequence repeats (SSRs) serve as key genetic markers, offering valuable insights into plant evolution, genetic diversity, and ecological adaptation (Ahmad et al. 2023; Frazão et al. 2023). These markers display significant variability, particularly in non-coding regions such as intergenic spacers and introns, owing to reduced selective pressures, despite their important role in regulating numerous functional genes (Leypold and Speicher 2021; Frazão et al. 2023). In coding sequences, frameshift Indels may lead to non-functional proteins, whereas in-frame Indels typically cause less severe alterations by adding or removing a few amino acids. The high polymorphism observed in SSRs, together with SNPs and Indels, enhances their applicability in DNA barcoding, population genetics studies, and phylogenetic analyses. Moreover, these genetic markers play a crucial role in determining conservation priorities for plant species, including C. roseus, contributing to our comprehension of plant evolution, adaptation, and biodiversity (Leypold and Speicher 2021).
This investigation endeavors to conduct a comprehensive analysis of the chloroplast genomes of eight C. roseus cultivars, juxtaposing their plastomic compositions against the reference plastome (KC561139.1) of the ‘Pacifica Punch’ cultivar. The study on C. roseus and its allied species within the Apocynaceae family aim to address several research gaps, where current phylogenetic analyses often struggle to resolve relationships at the species level within the Apocynaceae family, particularly because of incomplete lineage sorting or hybridization events (Endress and Bruyns 2000; Nazar et al. 2013, 2019; Wang et al. 2023a). In addition, there is a need for effective conservation and breeding strategies that leverage genetic diversity within C. roseus to enhance desirable traits (Kumar et al. 2014; Salama et al. 2020). The biosynthesis pathways of valuable secondary metabolites in C. roseus are not fully understood, and the evolutionary history and ecological adaptation of C. roseus are not well-documented, particularly in relation to its plastome (Sharma et al. 2021). It also seeks to provide a comprehensive genetic characterization of C. roseus cultivars by elucidating their phylogenetic inter-relationships by using chloroplast DNA markers, which will shed light on evolutionary trajectories and genetic diversity. Additionally, the research explores plastome variability among cultivars, which is crucial for informing biotechnological applications. The study also aims to establish molecular barcodes by using chloroplast genes (matK, rbcL, and trnL) for species and subspecies delineation, contributing to the authentication and identification of medicinal herbs. Furthermore, by providing foundational data on the plastome, this work supports biotechnological advancements in enhancing the production of valuable secondary metabolites and informs breeding programs aimed at improving desirable traits and conserving genetic resources.
Materials and methods
Plant material
This study focused on eight distinct cultivars of C. roseus, chosen for their diverse floral characteristics, which included variations in petal coloration, eye and central pigmentation, and petal morphology, as previously described (El-Domyati et al. 2012). Fresh, healthy, uniform-sized young leaf samples, free from any observable disease or pest infestations, were systematically collected from the Macca region in south-western Saudi Arabia, by following the standard protocols (El-Domyati et al. 2012). To minimize starch and polysaccharide content, the leaves were enclosed in opaque plastic bags for 24 h before being excised. The harvested leaves were subsequently sterilized with 70% ethanol, immediately frozen in liquid nitrogen, and stored at −80°C until further processing.
DNA isolation
Leaf genomic DNA was isolated from the eight different cultivars by using the DNeasy Plant Mini Kit (QIAGEN, Hilden, Germany) and quantified using both the Qubit (Invitrogen, Waltham, MA, USA) and Nanodrop (Nanodrop, Waltham, MA, USA) systems. To eliminate RNA contaminants, the DNA samples were subjected to enzymatic treatment using ribonuclease A (RNase A) at a concentration of 10 mg/mL (Sigma, USA). This process involved incubating the samples with the enzyme at 37°C for a duration of 30 min. Subsequently, the concentration of DNA in the various samples was visualized by agarose gel electrophoresis and quantified through spectrophotometric approach. The first method was performed to verify the integrity of isolated DNA, and the latter method exploits the principle of nucleic acid absorption of ultraviolet light, specifically at a wavelength of 260 nm. The DNA concentration was calculated using the following equation: DNA concentration (ug/mL) = OD260 × 50× dilution factor.
Whole-genome shotgun sequencing and plastome analysis
The extracted DNA was then shipped to BGI (Shenzhen, China) for whole-genome shotgun sequencing on the Illumina HiSeq 2000 platform, yielding >6 Gb of 100-bp paired-end reads. The raw sequencing data for the eight genomes (labeled isolates 1–8, Supplementary Fig. S1) were submitted to the NCBI repository, where they were allocated Bioproject ID PRJNA1191663. The cleaned Fastq files from different C. roseus cultivars were assembled using a reference-guided approach, with the published chloroplast genome sequence (Accession no. KC561139.1) serving as the reference plastome. Subsequently, the de novo assembler NOVOPlasty (ver. 4.3.5) was utilized to circularize the plastomes under default settings. This is a specialized tool designed to efficiently extract organelle genomes from whole-genome sequencing data that operates by initiating with a seed sequence and then extending it in both directions until a complete circular genome is assembled (Dierckxsens et al. 2017). Each plastome was thoroughly examined for structural completeness, and the genomes were re-oriented to match the reference plastome’s configuration to ensure consistency during subsequent analyses. To maintain the structural integrity of the plastomes, the two inverted repeat (IR) regions in the assembled genomes were adjusted to align with the orientation found in the reference. The eight circularized plastid genomes (coded 1–8, Fig. S1) were subsequently archived in the NCBI repository, where they were assigned accession IDs PQ677102, PQ677100, PQ677101, PQ677099, PQ677097, PQ677098, PQ677103, and PQ677096 respectively. These eight plastomes were annotated using GeSeq, an accurate and versatile tool for annotating organelle genomes (Tillich et al. 2017), which identifies genes, exons, introns, and other functional regions of the chloroplast genomes. The annotated genomes were aligned using multiple alignment using fast fourier transform (MAFFT) (ver. 7; Katoh and Standley 2013), a well-established software for performing multiple sequence alignments. This alignment enabled the assessment of sequence similarity and divergence across the chloroplast genomes of the eight C. roseus cultivars, the reference plastome, and additional species from the Apocynaceae family.
Molecular marker analysis
Single nucleotide polymorphisms (SNPs) and insertions/deletions (Indels) between each of the eight cultivars and those of the reference plastome (Accession no. KC561139.1) in the genic regions were identified using Bowtie2 (ver. 2.5.4). This recent version is an update to the ultrafast and memory-efficient tool for aligning sequencing reads to long reference sequences. Variant calling was then performed using the genome analysis toolkit (GATK) to detect base substitutions, including synonymous and non-synonymous variants (DePristo et al. 2011; Van der Auwera et al. 2013). The functional consequences of the identified SNPs and Indels were annotated using SnpEff (Cingolani et al. 2012). SnpEff is a powerful and versatile bioinformatics tool designed for annotating and predicting the effects of genetic variants. Codon usage frequencies for the different amino acids in the coding sequences (CDS) of the eight plastomes were calculated using the sequence manipulation suite (ver. 2; Stothard 2000). This version is a comprehensive collection of JavaScript programs designed for generating, formatting, and analyzing short DNA and protein sequences. Simple sequence repeats (SSRs) were identified in the plastomes of the eight cultivars by using the MegaSSR tool (Mokhtar et al. 2023), with a particular focus on the most common SSR motifs, such as mono A, mono T, and di/tri-nucleotide repeats. MegaSSR is a standalone pipeline designed for large-scale identification, classification, and development of SSR markers in genomic data.
Phylogenetic relationships
For phylogenetic analysis, sequences from the biomarker genes matK, rbcL, and trnL were extracted from the chloroplast genomes of the eight C. roseus cultivars, as well as the reference genome, to create molecular barcodes for the different cultivars. These sequences were then aligned against the corresponding sequences of the three genes in 22 other species from the Apocynaceae family. The alignments for these three genes were repeated, with the C. roseus cultivar ‘Cooler Orchid’ (coded 5) serving as a representative model for the eight cultivars to facilitate clustering within species of the same or different tribes. The comparative analyses aimed to evaluate sequence conservation and variation across species in the Apocynaceae family. The aligned sequences were visualized, and phylogenetic trees were constructed using the maximum likelihood estimation (MLE) method (Yang 1994), with the tree-building parameters and model selection based on preliminary analyses that identified the best fit for the data. This phylogenetic analysis provided insights into the evolutionary relationships among the studied C. roseus cultivars, as well as between these cultivars and other species from the Apocynaceae family.
Results
Structures of plastomes of the eight C. roseus cultivars
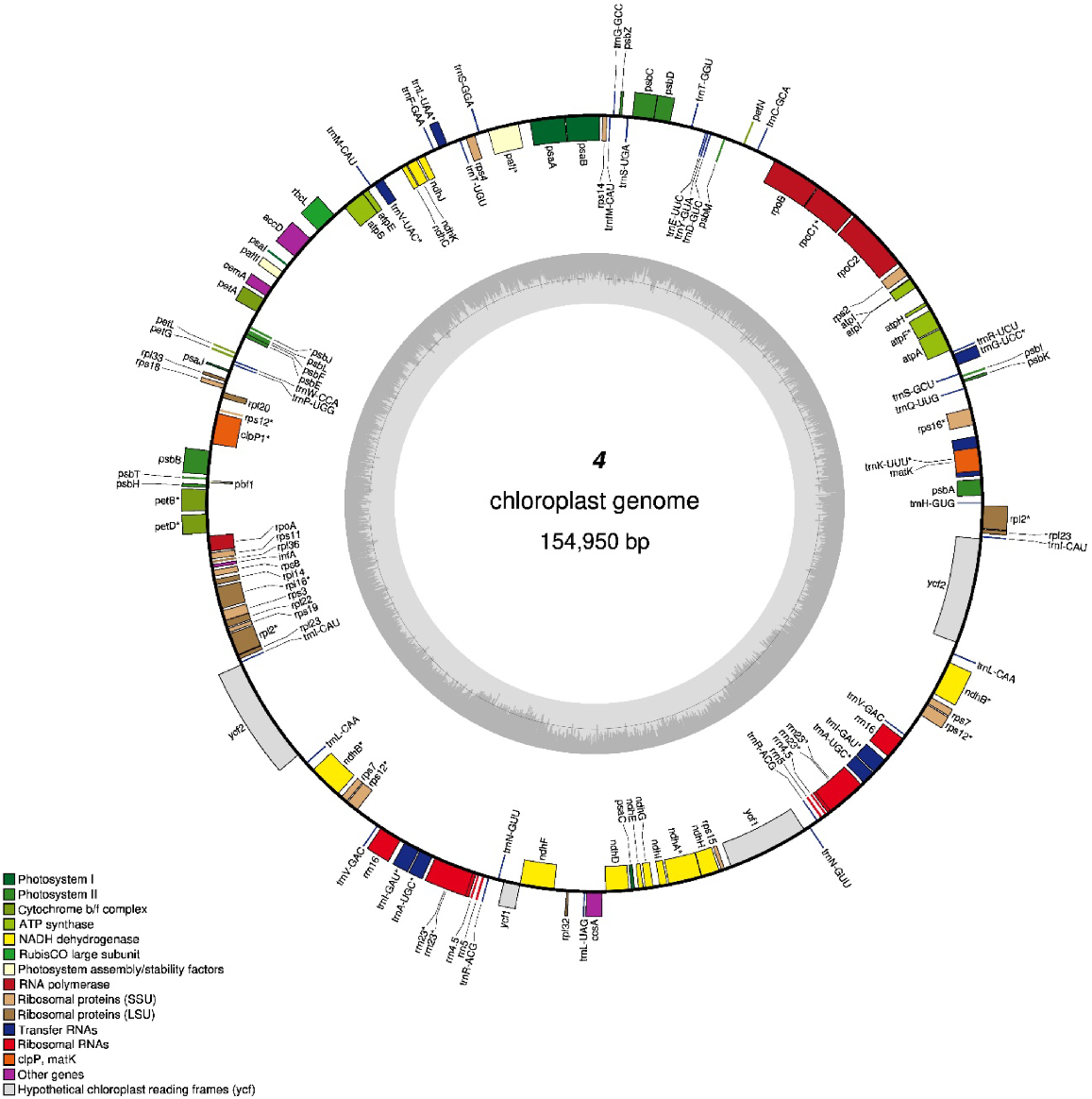
This study examined eight cultivars of C. roseus, each displaying distinct variations in petal color, eye, and center pigmentation (Fig. S1). Cultivars coded 1, 6, and 8 (Group 1) exhibit elongated, flatter petals, while cultivars coded 2, 3, 4, 5, and 7 (Group 2), along with the cultivar of the reference plastome, e.g. ‘Pacifica Punch Halo’ (KC561139.1), feature broad, rounded petals. Notably, the ‘Experimental Rose Pink’ cultivar (coded 4) presents a petal morphology strikingly similar to that of the reference plastome, albeit with a more delicate and refined appearance, complemented by a subtler color palette. Both of these cultivars display a pale pink hue, highlighted by a red radiating eye and a central red spot. The plastome sizes among the eight C. roseus cultivars vary, ranging from 154,928 bp in cultivar coded 5 to 155,066 bp in cultivars coded 6 and 8. In contrast, cultivars coded 2, 3, and 4 exhibit plastome sizes comparable to that of the reference plastome, measuring 154,950 bp (Figs 1, S2–S10). All chloroplast genomes of C. roseus displayed the characteristic quadripartite structure, consisting of the large single-copy (LSC) region, the small single-copy (SSC) region, and two inverted repeat regions (IRa and IRb), with the IR regions typically demarcating the separation between the SSC and LSC regions. As indicated in Table 1, the photosynthesis-related genes pafI, pafII, and pbfI were uniquely present in the chloroplasts of the eight C. roseus cultivars. In contrast, the reference plastome harbors the photosynthesis-related genes ycf3, ycf4, and psbN at the same loci. Notably, the ycf15 gene, associated with photosynthesis in the reference plastome, was absent in all eight cultivars (Fig. S10, Table 1).
The circular chloroplast genome (154,950 bp) of the Catharanthus roseus cultivar ‘Experimental Rose Pink’ (coded 4) serving as a representative model for the eight cultivars examined in this study. The map delineates the loci of various genes along with their transcriptional orientations; genes situated external to the circle are transcribed in a counterclockwise manner, whereas those positioned internally are transcribed clockwise. Genes are distinctly color-coded according to their functional categories. The inner circle graphically represents GC content in a dark gray hue and AT content in a light gray hue. Additionally, the map delineates three pivotal structural regions: the large single copy (LSC), the small single copy (SSC), and the inverted repeat (IR). The extent of the inverted repeat regions (IRa and IRb) is indicated by thick lines. The circular chloroplast genomes of the remaining seven cultivars are depicted in Figs S2–S8 in the supplementary material.
| Gene category | Gene group | Gene name | |
|---|---|---|---|
| Self-replication | Large subunit of ribosomal proteins | rpl2, rpl3, rpl14, rpl16, rpl20, rpl22, rpl23, rpl32, rpl33, rpl36 | |
| Small subunit of ribosomal proteins | rps2, rps3, rps4, rps7, rps8, rps11, rps12, rps14, rps15, rps16, rps18, rps19 | ||
| DNA-dependent RNA polymerase | rpoA, rpoB, rpoC1, rpoC2 | ||
| rRNA genes | rrn16, rrn23, rrn4.5, rrn5 | ||
| tRNA genes | trnA-UGC, trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnfM-CAU, trnG-GCC, trnG-UCC, trnH-GUG, trnI-CAU, trnI-GAU, trnK-UUU, trnL-CAA, trnL-UAA, trnL-UAG, trnM-CAU, trnN-GUU, trnP-GGG trnP-UGG, trnQ-UUG, trnR-ACG, trnR-UCU, trnS-GGA, trnS-GCU, trnS-UGA, trnT-GGU, trnT-UGU, trnV-GAC, trnV-UAC, trnW-CCA, trnY-GUA | ||
| Photosynthesis | Photosystem I | psaA, psaB, psaC, psaI, psaJ | |
| Photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ | ||
| Cytochrome b6/f complex | petA, petB, petD, petG, petL, petN | ||
| ATP synthase | atpA, atpB, atpE, atpF, atpH, atpI | ||
| NADH dehydrogenase | ndhA, ndhB, ndhC, ndhD, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | ||
| Rubisco | rbcL | ||
| Conserved open reading frames | pafI, pafII, pbfI, ycf1, ycf2, ycf3, ycf4, ycf15, ycf68 | ||
| Other genes | Maturase | matK | |
| Protease | clpP1 | ||
| Envelop membrane protein | cemA | ||
| Subunit acetyl- CoA-carboxylate | accD | ||
| c-type cytochrome synthesis gene | ccsA |
Genes highlighted in blue signify those that are exclusive to the eight plastomes, whereas genes marked in red indicate those unique to the reference chloroplast genome (KC561139.1). Additionally, genes depicted in green represent the chloroplast genes frequently employed in barcode detection among plants, which facilitated the retrieval of barcodes and the assessment of genetic distances among the eight cultivars, with the reference plastome serving as the comparative baseline. The trnL–UAA gene region, has been standardized in plant barcoding systems becauseo of its effectiveness in resolving relationships at various taxonomic levels, from species identification to higher-level phylogenetic analysis.
Illustrative data from sequence alignment of chloroplasts of the eight cultivars in comparison to that of the reference plastome is displayed in Fig. S11. The sequence alignment showed the occurrence of two prominent events involving Indel segments that are exclusively present in either Group 1 or Group 2 (Fig. S12). Notably, this finding lends further support to the genetic distinction between the two groups of C. roseus cultivars, which correlates with variations in petal morphology. The reference plastome, which exhibits a petal shape analogous to that of Group 2 cultivars, clustered with those cultivars in the analysis. The phylogenetic tree constructed from the plastome sequencing data of the eight cultivars, along with the reference plastome, corroborated previous findings, particularly in terms of the observed genetic distances among the cultivars under investigation (Fig. 2). Consequently, we hypothesize that petal morphology may serve as a pivotal evolutionary biomarker for speciation within C. roseus, potentially driving genetic differentiation between these groups. Subsequent molecular analyses were conducted to either corroborate or refute our hypothesis regarding speciation.
Phylogenetic tree constructed to assess the genetic distances among the plastomes of various Catharanthus roseus cultivars, as compared with the reference plastome (KC561139.1), by utilizing the maximum likelihood (ML) algorithm. The varying lengths of the branches, along with the numbers at the nodes representing bootstrap values, offer valuable insights into the relationships among the analyzed germplasm. Cultivars positioned at the ends of the branches exhibit greater genetic divergence, and higher bootstrap values indicate stronger support for the inferred groupings. Two genetically distinct groups were delineated, with cultivars coded 1, 6, and 8 forming one cluster (Group), and cultivars coded 2, 3, 4, 5, and 7 constituted the second. The reference plastome appears to exhibit a closer genetic affinity to the cultivars in Group 2.

Molecular and genetic analysis of the genic regions of the eight plastomes
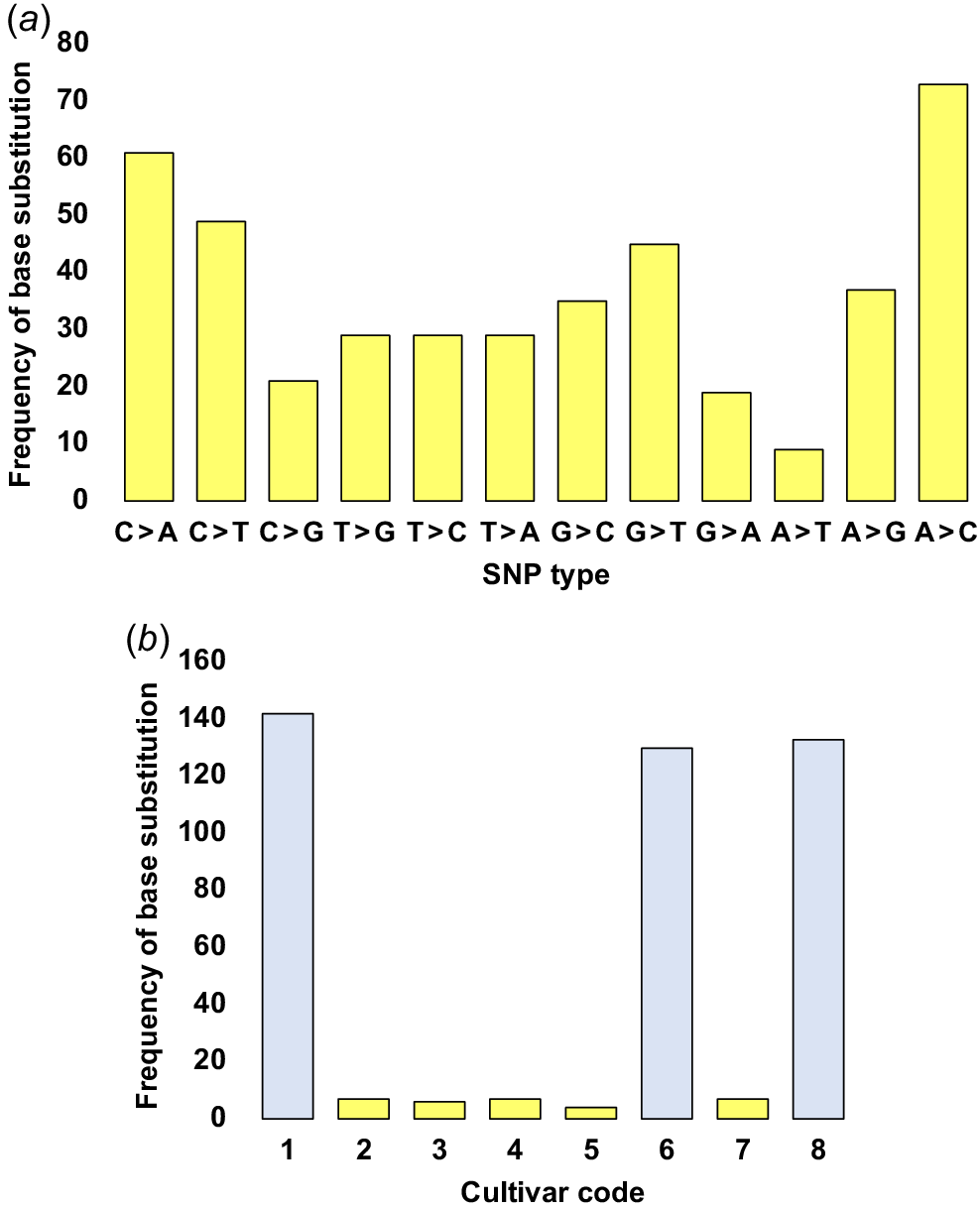
Thus, we systematically examined the variations in sequencing data across genic regions of the plastome among the different cultivars of the two groups, juxtaposing these findings with those of the reference plastome (Supplementary Tables S1–S8). The results for the frequency of base substitution at the level of SNPs showed an average of 9–73 variants, of which A > C, followed by C > A, C > T, and G > T showed the highest records, while A > T, followed by G > A, G > A, and T > G/C/A showed the lowest records (Fig. 3a). Expectedly, the highest records of base substitution were shown for cultivars of Group 1, where cultivar coded 1 exhibited 142 variants, whereas those coded 6, and 8 exhibited 130, and 133 variants, and base substitution recodes for cultivars of Group 2 showed an average of four to seven variants (Fig. 3b). The analysis also focused on the consequences of SNPs and in-frame insertions and deletions (Indels) in the genic regions. The first exhibited two distinct outcomes, and the second manifested as a single consequence. Consequences of SNP were classified into two categories, namely, synonymous mutations, which result in no alteration of the amino acid sequence, and non-synonymous mutations, which encompass both missense alterations and the introduction of premature stop codons within the translated protein sequences. In contrast, in-frame Indels lead to the insertion or deletion of one or more amino acids within the synthesized proteins.
(a) Alteration in genic sequences regarding the diverse types of single nucleotide polymorphisms (SNPs) across plastomes of the eight Catharanthus roseus cultivars (designated codes 1–8) and (b) the frequency of base substitutions observed within individual cultivars in comparison to the reference plastome of ‘Pacifica Punch Halo’ (KC561139.1) cultivar. Codes 1–8 correspond to cultivars ‘Patricia White’, ‘First Kiss Polka Dot’, ‘First Kiss Peach’, ‘Experimental Rose Pink’, ‘Cooler Orchid’, ‘Experimental Deep Pink’, ‘Victory Red’, and ‘Blue Pearl’ respectively. Additional details are available in Tables S1–S8.
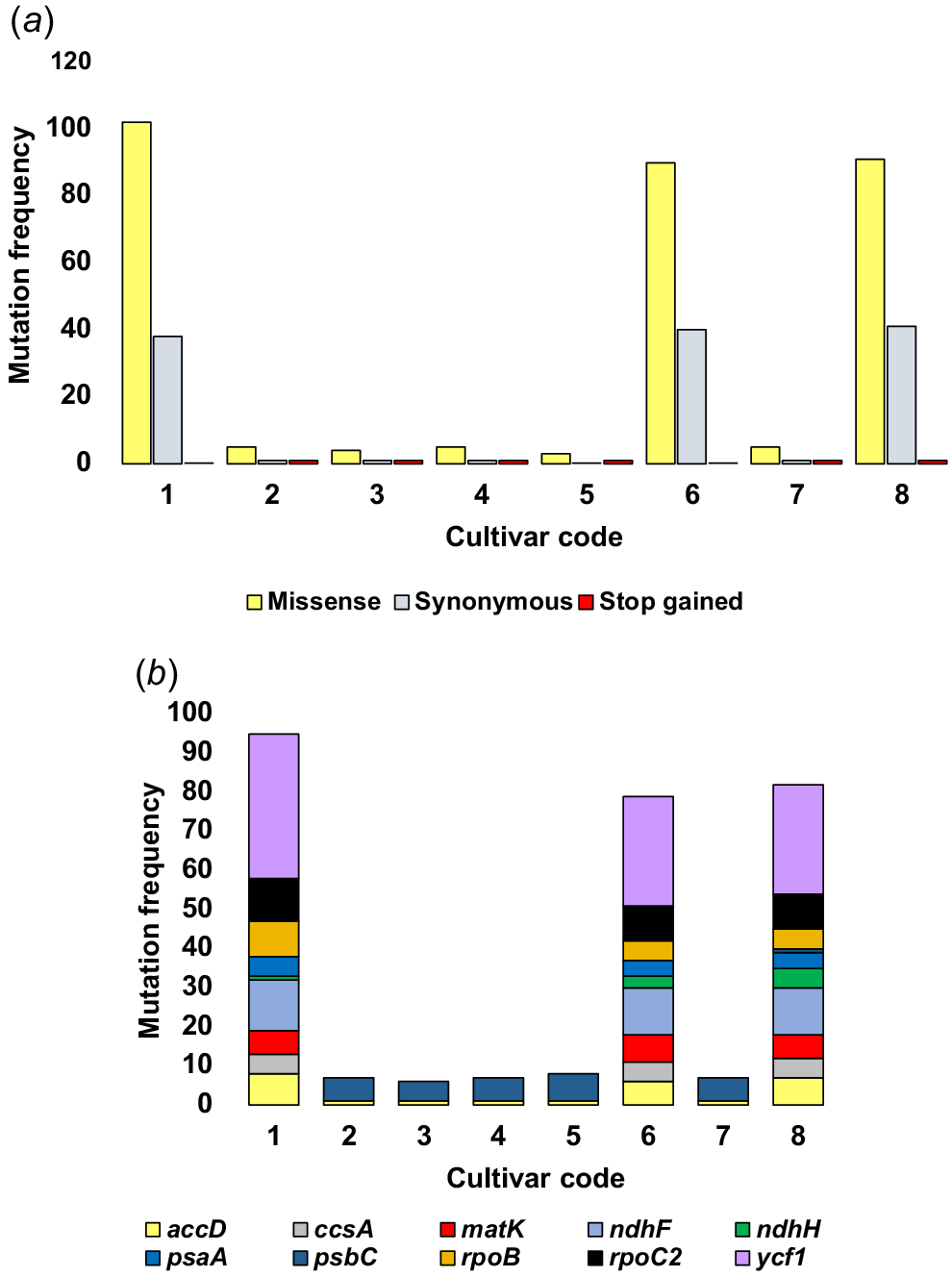
The results in Tables S1–S8 also supported our prior hypothesis for the close genetic relationships among different cultivars on the basis of petal shape, where alteration frequency in plastomes of Group 2 cultivars was few as compared with those of the reference plastome manifesting an average of as little as three to five missense variants, one stop gained variant, and one of synonymous variant took place in as few as two chloroplast genes, namely psbC and accD (Fig. 4a, b, Tables S2–S5, S7). In contrast, sequence alterations of Group 1 cultivars in comparison with the reference plastome reached an average of as high as 90–102 for missense variants, and 38–41 for synonymous variants took place in as high as 35–39 genes (Fig. 4a, Tables S1, S6, S8). The top 10 chloroplast genes in terms of the number of variants are ycf1, ndhf, rpoC2, and accD (Fig. S4b, Tables S1, S6, S8).
The top mutation frequencies in genic sequences regarding (a) the synonymous and non-synonymous (missense and stop-gained) plastome variant types, as well as (b) the most prevalent events occurring in plastome genes within the eight Catharanthus roseus cultivars (designated codes 1–8). Codes 1–8 correspond to cultivars ‘Patricia White’, ‘First Kiss Polka Dot’, ‘First Kiss Peach’, ‘Experimental Rose Pink’, ‘Cooler Orchid’, ‘Experimental Deep Pink’, ‘Victory Red’, and ‘Blue Pearl’ respectively. Further information is available in Tables S1–S8.
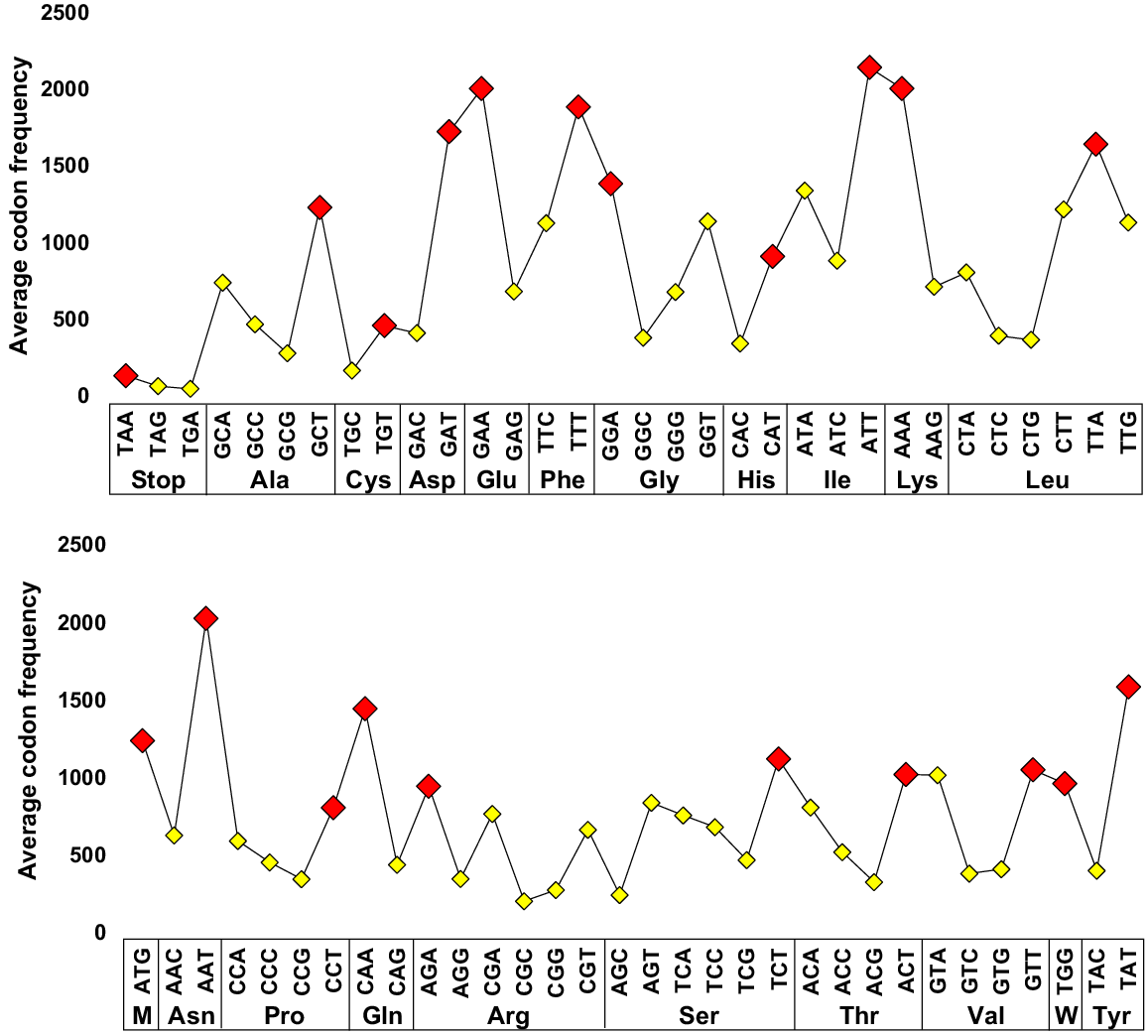
The codon frequency analysis conducted within the genic regions of the chloroplast genomes across the eight cultivars of C. roseus showed a clear bias in codon usage. The most prevalent codons were ATT, encoding isoleucine, AAT, encoding asparagine, GAA, encoding glutamic acid, and AAA, encoding lysine. Conversely, the least utilized codons encompassed the three stop codons, namely, TGA, TAG, and TAA, alongside TGC, which encodes cysteine (Fig. 5, Table 2). Further scrutiny of codon utilization patterns indicated a dominant presence of nucleotides T followed by A in the third position across codons encoding 18 of the 20 amino acids, with these patterns being exemplified by the four most abundant codons within the plastome genic regions of the eight cultivars. In stark contrast, the third codon position predominantly exhibited the least frequency of occurrence for nucleotide pairs such as CA and GA, as evidenced by the four least frequent codons observed.
The average codon frequency for the 20 amino acids in the genic region of the chloroplast sequence across the eight cultivars of Catharanthus roseus. The most frequently observed codon for each amino acid is marked with a red rhombus. For the common codons associated with amino acids that have 2-, 3-, 4-, and 6-fold degeneracy, as well as stop codons, the third nucleotide is either thiamine (T) or adenine (A). In contrast, for the two non-degenerate codons, i.e. methionine (M) and tryptophan (W), the third nucleotide is guanine (G). Additional information is available in Table 2.
| Codon | aa | Cultivar code | Codon | aa | Cultivar code | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |||||
| TAA | * | 131 | 141 | 141 | 141 | 136 | 132 | 141 | 132 | ATG | Met | 1234 | 1243 | 1243 | 1243 | 1243 | 1245 | 1243 | 1245 | |
| TAG | * | 59 | 76 | 76 | 76 | 67 | 60 | 76 | 60 | AAC | Asn | 629 | 634 | 634 | 634 | 634 | 630 | 634 | 630 | |
| TGA | * | 49 | 54 | 54 | 54 | 54 | 49 | 54 | 49 | AAT | 2020 | 2037 | 2037 | 2037 | 2037 | 2019 | 2037 | 2019 | ||
| GCA | Ala | 749 | 743 | 743 | 743 | 743 | 742 | 743 | 742 | CCA | Pro | 592 | 597 | 597 | 597 | 595 | 596 | 597 | 596 | |
| GCC | 471 | 470 | 470 | 470 | 470 | 474 | 470 | 474 | CCC | 455 | 456 | 456 | 456 | 456 | 463 | 456 | 463 | |||
| GCG | 279 | 284 | 284 | 284 | 284 | 285 | 284 | 285 | CCG | 349 | 352 | 352 | 352 | 352 | 349 | 352 | 349 | |||
| GCT | 1236 | 1230 | 1230 | 1230 | 1230 | 1242 | 1230 | 1242 | CCT | 812 | 810 | 810 | 810 | 810 | 808 | 810 | 808 | |||
| TGC | Cys | 172 | 169 | 169 | 169 | 169 | 169 | 169 | 169 | CAA | Gln | 1454 | 1450 | 1450 | 1450 | 1452 | 1442 | 1450 | 1440 | |
| TGT | 463 | 463 | 463 | 463 | 463 | 462 | 463 | 462 | CAG | 437 | 449 | 449 | 449 | 440 | 436 | 449 | 436 | |||
| GAC | Asp | 417 | 415 | 415 | 415 | 415 | 414 | 415 | 414 | AGA | Arg | 947 | 948 | 948 | 948 | 948 | 947 | 948 | 947 | |
| GAT | 1730 | 1728 | 1728 | 1728 | 1728 | 1726 | 1728 | 1726 | AGG | 351 | 354 | 354 | 354 | 353 | 350 | 354 | 350 | |||
| GAA | Glu | 2011 | 2009 | 2009 | 2009 | 2009 | 2011 | 2009 | 2011 | CGA | 766 | 775 | 775 | 775 | 770 | 768 | 775 | 768 | ||
| GAG | 687 | 683 | 683 | 683 | 683 | 692 | 683 | 692 | CGC | 206 | 208 | 208 | 208 | 208 | 206 | 208 | 206 | |||
| TTC | Phe | 1124 | 1138 | 1138 | 1138 | 1131 | 1123 | 1138 | 1123 | CGG | 285 | 278 | 278 | 278 | 278 | 285 | 278 | 285 | ||
| TTT | 1897 | 1887 | 1887 | 1887 | 1887 | 1893 | 1887 | 1893 | CGT | 668 | 668 | 668 | 668 | 668 | 669 | 668 | 669 | |||
| GGA | Gly | 1392 | 1389 | 1389 | 1389 | 1390 | 1386 | 1389 | 1386 | AGC | Ser | 248 | 247 | 247 | 247 | 247 | 251 | 247 | 251 | |
| GGC | 381 | 385 | 385 | 385 | 385 | 383 | 385 | 383 | AGT | 845 | 840 | 840 | 840 | 843 | 845 | 840 | 845 | |||
| GGG | 686 | 679 | 679 | 679 | 682 | 684 | 679 | 684 | TCA | 761 | 763 | 763 | 763 | 763 | 759 | 763 | 759 | |||
| GGT | 1155 | 1139 | 1139 | 1139 | 1139 | 1149 | 1139 | 1149 | TCC | 692 | 683 | 683 | 683 | 683 | 690 | 683 | 688 | |||
| CAC | His | 341 | 347 | 347 | 347 | 345 | 347 | 347 | 345 | TCG | 472 | 476 | 476 | 476 | 476 | 470 | 476 | 470 | ||
| CAT | 918 | 918 | 918 | 918 | 918 | 904 | 918 | 904 | TCT | 1138 | 1116 | 1116 | 1116 | 1116 | 1136 | 1116 | 1136 | |||
| ATA | Ile | 1358 | 1337 | 1337 | 1337 | 1337 | 1355 | 1337 | 1353 | ACA | Thr | 819 | 812 | 812 | 812 | 812 | 815 | 812 | 815 | |
| ATC | 881 | 891 | 891 | 891 | 891 | 879 | 891 | 879 | ACC | 526 | 525 | 525 | 525 | 526 | 522 | 525 | 522 | |||
| ATT | 2157 | 2143 | 2143 | 2143 | 2143 | 2149 | 2143 | 2149 | ACG | 329 | 331 | 331 | 331 | 338 | 328 | 331 | 328 | |||
| AAA | Lys | 2010 | 2011 | 2011 | 2011 | 2011 | 2006 | 2011 | 2006 | ACT | 1029 | 1021 | 1021 | 1021 | 1027 | 1027 | 1021 | 1027 | ||
| AAG | 705 | 723 | 723 | 723 | 723 | 703 | 723 | 703 | GTA | Val | 1022 | 1021 | 1021 | 1021 | 1021 | 1024 | 1021 | 1024 | ||
| CTA | Leu | 815 | 804 | 804 | 804 | 810 | 823 | 804 | 823 | GTC | 384 | 388 | 388 | 388 | 388 | 385 | 388 | 385 | ||
| CTC | 394 | 398 | 398 | 398 | 396 | 396 | 398 | 396 | GTG | 411 | 417 | 417 | 417 | 415 | 411 | 415 | 409 | |||
| CTG | 367 | 375 | 375 | 375 | 371 | 363 | 375 | 363 | GTT | 1049 | 1053 | 1053 | 1053 | 1050 | 1059 | 1053 | 1059 | |||
| CTT | 1225 | 1218 | 1218 | 1218 | 1221 | 1224 | 1218 | 1224 | TGG | Trp | 967 | 964 | 964 | 964 | 965 | 971 | 964 | 969 | ||
| TTA | 1652 | 1641 | 1641 | 1641 | 1646 | 1658 | 1641 | 1658 | TAC | Tyr | 399 | 410 | 410 | 410 | 410 | 399 | 410 | 399 | ||
| TTG | 1124 | 1147 | 1147 | 1147 | 1137 | 1120 | 1147 | 1120 | TAT | 1576 | 1593 | 1593 | 1593 | 1593 | 1585 | 1591 | 1583 | |||
Codons highlighted in red denote the most prevalent codons for specific amino acids (aa) within a plastome, whereas those marked in blue signify unique codons. Further details can be found in Fig. 5.
It is noteworthy that the overwhelming majority of amino acids (18 of 20) were encoded by codons exhibiting two-, three-, four-, or six-fold degeneracy. Among these codons, the third nucleotide position displayed the greatest degree of variability, surpassing the variability observed at the first and second nucleotide positions. This pattern highlights the inherent flexibility of the genetic code, particularly at the third codon position, where synonymous substitutions are often tolerated without altering the encoded amino acid, thereby contributing to a higher level of nucleotide variation in this region. Comparative analysis between the two groups of C. roseus cultivars showed absence of any discernible preferential patterns in codon usage for the 18 amino acids, in addition to the three stop codons (TGA, TAG, TAA) (Fig. 5, Table 2). This lack of divergence in codon preference between the cultivars of the two groups suggests that the level of genetic differentiation, or speciation, within C. roseus was insufficient to induce significant alterations in codon usage bias. Consequently, it can be inferred that the molecular divergence between these cultivars is not pronounced enough to provoke selective pressures that would lead to distinguishable shifts in codon preference, thus supporting the notion of the reduced degree of speciation at the plastomic level within this species.
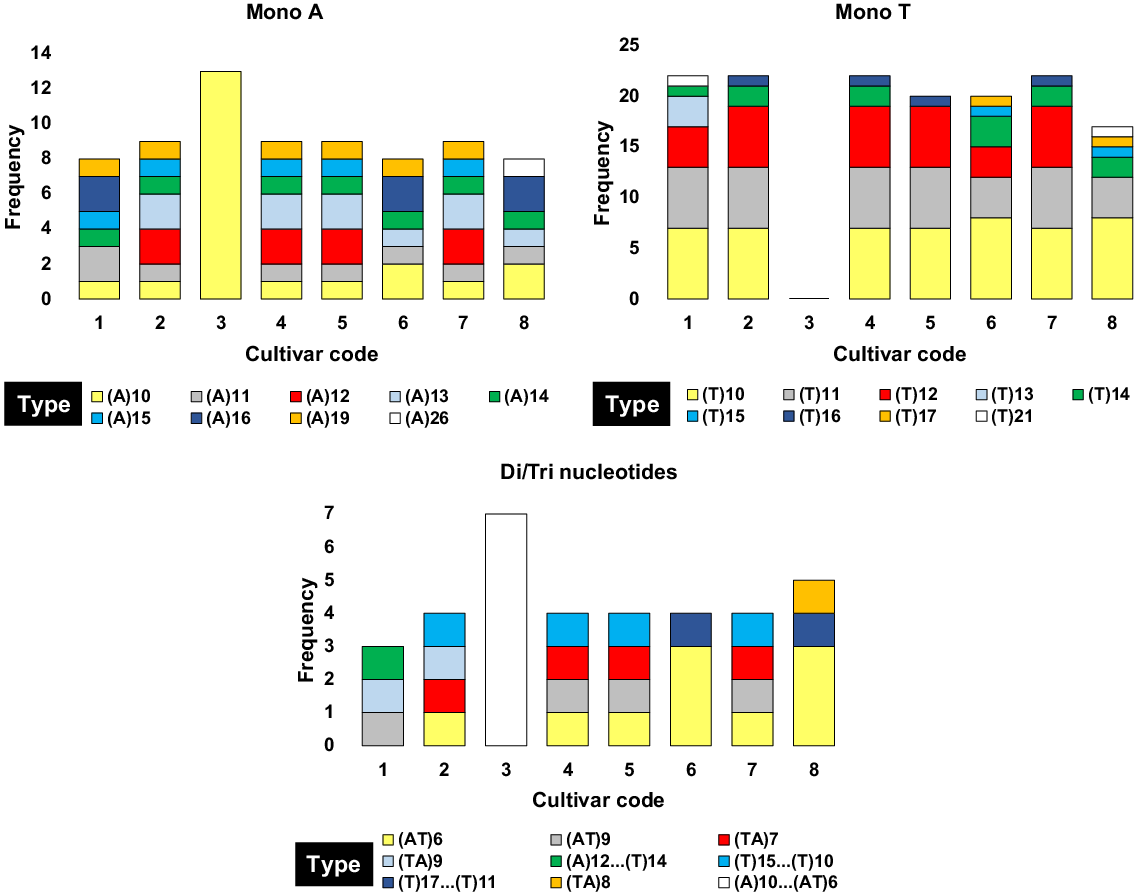
In the context of simple sequence repeat (SSR) biomarkers across plastomes of the eight C. roseus cultivars, we observed inconsistent results concerning the three key SSR events, namely, mono ‘A’, mono ‘T’, and Di/Tri nucleotides, particularly for the cultivar ‘First Kiss Peach’ (coded 3). Because of these unreliable findings, we opted to exclude this cultivar from SSR analysis regarding the genetic divergence between the two groups of C. roseus cultivars (Fig. 6, Table S11). In total, nine distinct SSR types were identified for each of the three SSR categories (Fig. 6, Tables S9–S10, S12–S16). When examining the SSR profiles of the cultivars from both groups, we detected varying degrees of similarity in the frequency distribution of different SSR types across the three SSR events. Notably, cultivars coded 4 and 7 exhibited a remarkable homogeneity in their SSR type frequencies, with complete concordance observed across all three SSR events (Fig. 6, Tables S12, S15). The observed variability in SSR frequencies and types within C. roseus was shown to be inadequate as a definitive metric for inferring speciation rates. This constraint likely stems from the limited availability of SSR loci in plastome research, compounded by the shallow evolutionary significance of these biomarkers for assessing genetic divergence at the population level. Although SSRs possess potential for elucidating genomic variability, their efficacy in precisely quantifying speciation events remains equivocal, necessitating further refinement in both methodological approaches and evolutionary interpretations.
Frequencies of the nine types of simple sequence repeat (SSR) types for the three SSR events, e.g. mono ‘A’, mono ‘T’, and Di/Tri nucleotides observed in the plastomes of the eight Catharanthus roseus cultivars (designated codes 1–8). Codes 1–8 correspond to cultivars ‘Patricia White’, ‘First Kiss Polka Dot’, ‘First Kiss Peach’, ‘Experimental Rose Pink’, ‘Cooler Orchid’, ‘Experimental Deep Pink’, ‘Victory Red’, and ‘Blue Pearl’ respectively. Additional details are provided in Tables S9–S16.
Barcoding and phylogenetic relationships
Further analysis incorporated three well-established molecular biomarkers, namely matK, rbcL, and trnL, to investigate the barcoding potential and elucidate the phylogenetic relationships among various C. roseus cultivars and closely related species within the Apocynaceae family. The barcodes derived for these three genes in the eight C. roseus cultivars are presented in Figs S13, S15 and 7 respectively. Additionally, multiple sequence alignments of these genes across the eight cultivars, compared with the reference plastome and analog sequences from 22 other species spanning seven tribes within the Apocynaceae family, are displayed in Figs S14, S16 and S17. The accession numbers and specific gene locations in the plastomes of the species within this family are detailed in Table S17. Notably, the barcodes for the three genes were able to partially distinguish between the two groups of C. roseus cultivars, with the trnL gene, demonstrating the greatest resolution and ability to differentiate between the groups (Fig. 7).
Barcodes of the trnL gene for the eight Catharanthus roseus cultivars (designated codes 1–8) as well as that of the reference plastome of ‘Pacifica Punch Halo’ (KC561139.1) cultivar. Codes 1–8 correspond to cultivars ‘Patricia White’, ‘First Kiss Polka Dot’, ‘First Kiss Peach’, ‘Experimental Rose Pink’, ‘Cooler Orchid’, ‘Experimental Deep Pink’, ‘Victory Red’, and ‘Blue Pearl’ respectively. Multiple sequence alignment including the eight cultivars as well as various species of the family Apocynaceae is presented in Fig. S17 (a Jalview file).

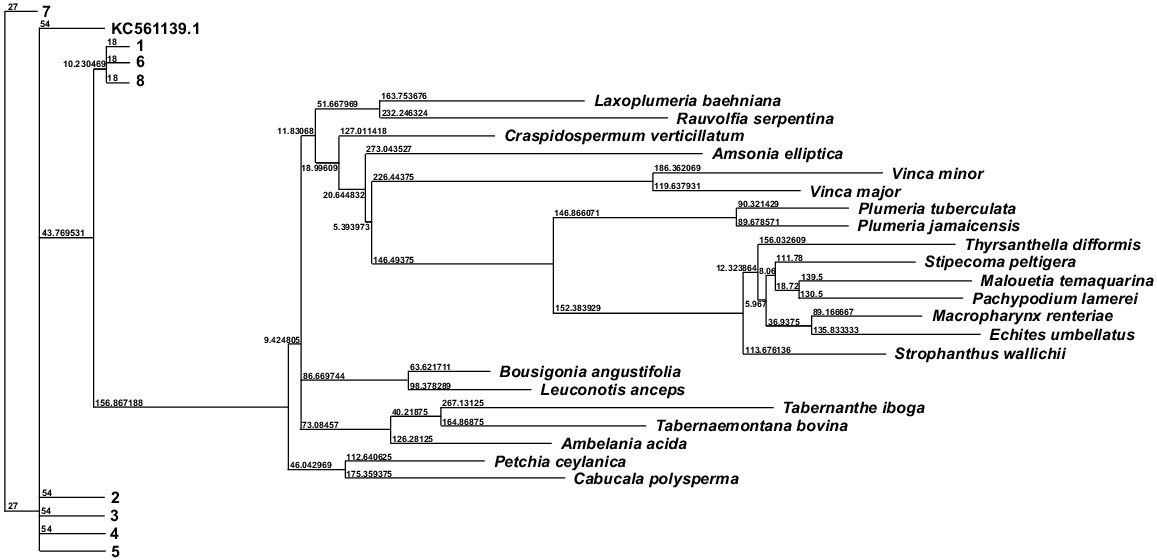
Three distinct phylogenetic trees were constructed on the basis of the sequence variations in the three well-established molecular biomarkers, i.e. matK, rbcL, and trnL, employing MLE to elucidate the evolutionary relationships among the eight C. roseus cultivars, the reference plastome, on the one hand, and between C. roseus and 22 species spanning seven tribes within the Apocynaceae family (Figs S18, S19 and 8 respectively). In the various phylogenetic trees, longer branches indicate a higher degree of genetic divergence between the groups. This means that the samples or cultivars at the ends of these branches are more genetically distinct from each other. On the other hand, shorter branches suggest closer genetic relationships, indicating that the samples share more genetic similarities. Each node in a dendrogram typically has a number that represents a specific point of divergence. Numbers can also represent statistical support values, such as bootstrap values, which indicate the confidence level in the branching pattern. Higher numbers often suggest stronger support for the grouping, whereas lower numbers may indicate less confidence.
Phylogenetic tree based on the trnL biomarker gene illustrating genetic distances among Catharanthus roseus cultivars, their reference cultivar ‘Pacifica Punch Halo’ (KC561139.1), and various species within the Apocynaceae family, reconstructed using the maximum likelihood (ML) algorithm. The differential branch lengths, along with the bootstrap values at the nodes, provide key insights into the phylogenetic relationships of the examined germplasm. Species at the terminal ends of the branches exhibit greater genetic divergence, whereas higher bootstrap values indicate stronger statistical support for the inferred clades. Notably, the rbcL gene biomarker successfully resolves the separation between two distinct groups of Catharanthus roseus cultivars, as determined by full plastome sequencing; Cultivars 1, 6, and 8 are grouped in Cluster 1, and Cultivars 2, 3, 4, 5, and 7 form Cluster 2. The construction of the tree was based on a multiple sequence alignment of the trnL gene, incorporating data from the eight cultivars along with various species from the Apocynaceae family. The tribes for these plant species are provided in Table S17.
In concordance with the barcoding data, the phylogenetic tree constructed from the trnL gene displayed the most pronounced discriminatory power in segregating the two cultivar groups, manifesting the highest level of resolution. This tree distinctly clustered cultivars coded 1, 6, and 8 into a singular group, whereas cultivars coded 2, 3, 4, and 5 were segregated into a separate, well-defined clade (Fig. 8). Cultivar 7, along with the reference plastome, deviated from the expected grouping on the basis of prior assumptions. Neither matK nor rbcL were able to effectively distinguish between the two cultivar groups, although matK highlighted a closer genetic relationship between cultivars coded 6 and 8 (Figs S18 and S19 respectively).
As the phylogenetic relationships between C. roseus and other species within the Apocynaceae family became less discernible owing to the presence of the nine C. roseus cultivars, we opted to use the barcode sequences of the ‘Experimental Rose Pink’ cultivar (coded 4) as a representative of the species. Consequently, three additional phylogenetic trees were generated on the basis of the sequences of genes matK, rbcL, and trnL for 23 species, including C. roseus, to improve the resolution of the genetic distances among species in the Apocynaceae family (Figs S20–S22). These phylogenetic trees revealed a clear clustering of species within the same tribe, except in three instances where species from different tribes exhibited close genetic relationships. Specifically, Bousigonia angustifolia from the tribe Apocynae showed a close relationship with Leuconotis anceps from the tribe Tabernaemontaneae; Cabucala polysperma from the tribe Apocynae clustered with Petchia ceylanica from the tribe Rauvolfieae; and Laxoplumeria baehniana from the tribe Plumerieae grouped closely with Rauvolfia serpentina from the tribe Rauvolfieae.
Interestingly, C. roseus appears to be genetically distinct from other species, both within its own tribe and across the broader Apocynaceae family. This observation underscores the need for further comprehensive phylogenetic analyses of C. roseus and other Apocynaceae species to refine their classification and resolve their accurate positioning within the evolutionary tree of the family. This will facilitate a clearer understanding of the taxonomic relationships and evolutionary trajectories within the family, as well as its inter-relationships with other families across the plant kingdom.
Discussion
Significance of plastomes in molecular profiling
The chloroplast genome is a useful tool for demonstrating variability among C. roseus cultivars because, despite the reduced structural variability among cultivars of the same species, chloroplast DNA exhibits high intraspecific genetic diversity because of its maternally inherited nature and the presence of variable markers such as SNPs, Indels, and SSRs. This makes it an ideal marker for distinguishing subtle genetic differences among cultivars. Furthermore, the plastome functions as an indispensable organelle in plant metabolic processes, serving as the locus for a multitude of quintessential biochemical pathways, encompassing photosynthesis, the biosynthesis of amino acids and fatty acids, as well as the genesis of chlorophyll and carotenoid pigments (Lubna et al. 2024). Although datasets derived from nuclear genomes have attained considerable prominence in phylogenetic and plant genomic investigations, plastomes remain crucial for elucidating the maternal evolutionary trajectories of angiosperm taxa, furnishing indispensable insights that augment and enrich the understanding of nuclear data and differences within genomic compartments (Dong et al. 2023). Plastomes have long been recognized for their evolutionary conservation and exhibit a moderate rate of molecular sequence evolution compared with the nuclear and mitochondrial genomes in plants (Wu et al. 2024). Maternal genetic contributions and evolutionary patterns potentially overlooked by nuclear genome studies can be elucidated through the implementation of a pan-plastome-based methodology, which undoubtedly offers a sophisticated avenue for investigating the intricacies of plant lineages and their phylogenetic relationships (Kan et al. 2024; Wang et al. 2024a).
In the majority of terrestrial plants, plastomes exhibit a remarkable degree of conservation in their structural configuration, gene order, and overall gene composition (Ravi et al. 2008; Cao et al. 2022). Chloroplasts are particularly valuable in plant phylogenetic and biotechnological studies because of their high abundance, moderate nucleotide evolution rate, and limited genetic recombination (Li et al. 2023). Throughout the evolutionary trajectory of chloroplasts, introns were integrated, and post-transcriptional modifications emerged as fundamental eukaryotic features that were assimilated into the plastid genome (Zhang et al. 2023). This assimilation prompted the recruitment of a diverse array of nucleus-encoded RNA-binding proteins, thereby facilitating the interplay between the nucleus and chloroplast in the context of co-evolution (Zoschke and Bock 2018; Forsythe et al. 2021). These molecular adaptations significantly enhance the versatility and evolutionary potential of chloroplast biogenesis and functional diversification (Zoschke and Bock 2018; Zupok et al. 2021).
Genetic differentiation and morphological divergence
NOVOPlasty, employed in the present study, is a specialized de novo assembler tailored for extracting and assembling organelle genomes, such as chloroplasts, from whole-genome sequencing data (Dierckxsens et al. 2017; Dierckxsens et al. 2020). It efficiently constructs circular genomes into a single, high-quality contig, which is vital for pinpointing genetic variations. Although NOVOPlasty is primarily intended for assembling organelle genomes, it can also identify genetic variations by utilizing short sequences surrounding mutations as starting points to assemble adjacent sequences. This approach allows for the detection of polymorphic sites and their surrounding genetic context (Dierckxsens et al. 2020). By ensuring accurate assembly without introducing mismatches, NOVOPlasty is well-suited for detecting subtle intraspecific variations (Sun et al. 2020; Shang et al. 2022).
The results of this study indicate that the eight C. roseus cultivars exhibit distinct genetic profiles, which correlate with observable differences in petal morphology (Fig. S1). The cultivars were divided into two groups as indicated earlier. These morphological differences are striking and suggest the potential role of selection in shaping these traits, possibly driven by environmental factors or pollinator preferences. In many plant species, flower morphology is subject to strong selective pressures, especially in species with specialized pollinators (Fenster et al. 2004). The correlation between petal morphology and genetic divergence in C. roseus supports the hypothesis that distinct ecological or evolutionary pressures may underlie the observed phenotypic differentiation (Stelkens and Seehausen 2009).
Although the cultivars exhibited notable morphological differences, the size of their plastomes showed only slight variation, ranging from 154,928 bp to 155,066 bp. This minimal variation indicates that the genetic differences at the plastome level are quite subtle. Nonetheless, the chloroplast genome remains vital for plant development and physiological processes, especially in photosynthesis and the production of secondary metabolites (Chen et al. 2018; Liu et al. 2020). This minor variation in plastome size among cultivars is consistent with findings from other studies on plastome stability, where plastomes tend to remain highly conserved within species or closely related taxa, as highlighted in clade Angiosperms, exemplified by C. roseus (Shaw et al. 2014). Given that plastomes typically evolve at a slower rate than do nuclear genomes (Ruhlman and Jansen 2014), the lack of significant plastome size differences among these cultivars suggests that the speciation process within C. roseus may be ongoing but not yet fully realized at the plastomic level.
Interestingly, even though the phylogenetic trees showed some degree of genetic differentiation, the reference plastome exhibited a somewhat unexpected clustering with Group 2 cultivars. This suggests that the reference plastome, which exhibits petal morphology similar to Group 2 cultivars, may share closer genetic relationships with these cultivars than was initially hypothesized. This finding raises questions about the true taxonomic boundaries within C. roseus and the potential for hybridization or introgression between cultivars. Hybridization events can lead to phenotypic convergence, even in the presence of genetic divergence (Arnold 2004, 2016), and this phenomenon may help explain the similarities between the reference plastome and Group 2 cultivars, despite their possible divergent evolutionary histories.
The presence of Indels exclusive to either Group 1 or Group 2 cultivars further corroborates the genetic divergence between the two groups. Indels in chloroplast genomes are often associated with functional variation and can be considered evolutionary biomarkers that reflect adaptive divergence (Liu et al. 2022). However, although the two distinct Indels observed in this study provide evidence of genetic differentiation, the overall plastomic structure remains largely intact across cultivars, limiting their ability to act as definitive biomarkers of speciation. Prior research has indicated that plastomes can provide valuable insights into evolutionary relationships at the family or genus level, but they are not suitable for tracking fine-scale speciation within C. roseus, underscoring the need for alternative molecular biomarkers, such as nuclear genes or microsatellites, to better resolve recent genetic divergence (Nazar et al. 2019; Zhang et al. 2024).
Phylogenetic resolution by using molecular biomarkers
Plastome-derived phylogenetic methodologies represent formidable instruments in the realm of plant phylogenetics and evolutionary investigations (Yang et al. 2024). Notwithstanding certain constraints, including the complexities associated with resolving incomplete lineage sorting (ILS) and interspecific hybridization events, plastids, which are characterized by their highly conserved structural integrity and minimal recombinatory propensity, continue to serve as invaluable genomic entities for sequencing endeavors and phylogenetic reconstructions within the realm of angiosperm systematics (Wang et al. 2024b; Xiang et al. 2024). The application of matK, rbcL, and trnL genes for DNA barcoding and subsequent phylogenetic analysis yielded a partial resolution of the genetic relationships among the cultivars of C. roseus and their corresponding reference plastome (Figs 7, 8, S13, S15, S18–S22). The three genes were chosen because of their considerable sequence variability and evolutionary importance, particularly within the context of plant phylogenetics (Kress and Erickson 2007; Chase and Fay 2009), and more specifically within the Apocynaceae family (Cabelin and Alejandro 2016). Phylogenetic trees generated from these biomarkers exhibited a differential capacity to resolve the genetic divergence between the two groups of C. roseus cultivars, with the trnL gene, followed by the matK gene, offering superior resolution over the rbcL gene. Previous studies have consistently underscored the efficacy of the matK biomarker gene as the pre-eminent molecular barcode for species authentication within the Apocynaceae family, particularly when paired with the trnL–F biomarker genes (Cabelin and Alejandro 2016). Other research has posited that matK and trnL represent formidable candidate biomarkers, with resolution down to the genus level, surpassing the discriminatory power of other commonly used biomarkers, including rbcL (Schneider et al. 2005; Müller et al. 2006; de Groot et al. 2011; Matiz-Ceron et al. 2022). The findings concerning the trnL gene can be explained by its elevated mutation rate and significant evolutionary role, particularly attributed to the marked variability in the length and sequence composition of its spacer region (Hao et al. 2009). Nevertheless, the stem-loop regions P6, P8, and P9 within the trnL intron may be subject to neutral evolutionary dynamics, evading the functional constraints typically imposed on more conserved regions, thus facilitating greater sequence divergence without compromising the structural integrity of the gene (Hao et al. 2009). Other studies have suggested that the trnL gene is a more robust DNA biomarker compared to rbcL, with the potential for its use in quantitatively assessing inter- and intra-specific dietary differences, further reinforcing its utility in molecular ecology (Mallott et al. 2018). Nonetheless, the most promising emerging research trend appears to be super-barcoding, the approach that examines the variability of entire plastid genomes (Krawczyk et al. 2023; Wang et al. 2023b).
Codon usage patterns and their implications for speciation
The prevalence of synonymous codons exhibits substantial variability across genes and organisms, resulting from the intricate interplay of mutational processes, selective pressures, and stochastic genetic drift (Gao et al. 2024; Shi et al. 2024). Investigating codon usage patterns serves as a pivotal foundation for elucidating phylogenetic relationships and evolutionary trajectories in plant species, while simultaneously providing a robust framework for the refinement of expression vectors in genetic engineering endeavors, thereby augmenting the expression efficacy of target genes Yuan (Shi et al. 2024). The codon frequency analysis conducted on the chloroplast genomes of the eight C. roseus cultivars showed a distinct bias in codon usage across the eight cultivars, which is a common phenomenon observed in plant plastomes (Suzuki and Morton 2016). This bias in codon usage is indicative of selective pressures acting on the translational machinery to optimize protein synthesis efficiency, particularly with respect to the amino acid composition and the overall stability of the genome (Duret 2002). The most prevalent codons in our study were those that encode isoleucine (ATT), asparagine (AAT), glutamic acid (GAA), and lysine (AAA), which completely aligns with findings of a very recent report in other plant species where codons encoding these amino acids are frequently being utilized (Zhou et al. 2024). The dominance of the nucleotides T and A at the third codon position is consistent with the well-established pattern of codon usage in plant plastomes, where a bias toward T or A at this position is typically observed (Dong et al. 2020; Hu et al. 2024). This might explain why chloroplast genomes are A/T rich (Turudić et al. 2021). This phenomenon has also been attributed to the stability of the A/T pair in the double-stranded structure of the plastome owing to the low melting temperature compared with the G/C pair, which is thought to contribute to the efficiency of gene expression. In contrast, the low frequency of codons with C/G pairs, such as CA and GA, at the third position further underscores this preferential usage, highlighting the selective constraints acting on codon placement (Wang et al. 2018).
Unlike sense codons of a given genetic code, the stop codons (TGA, TAG, and TAA), which are the least utilized in the plastomes of the cultivars studied, should be introduced once, whereas sense codons can globally be utilized at varying rations on the basis of the targeted amino acid sequence. Expectedly, stop codons are typically less prone to evolutionary pressures because they play a critical role in translation termination, and any deviation from their expected usage could be detrimental to the translational process (Belinky et al. 2018). Alterations in stop codons exhibit a marked correlation with an elevated frequency of substitutions immediately following the stop codon. These regions, which typically exhibit greater evolutionary conservation than do more distal loci, appear to undergo these substitutions as a compensatory mechanism. This suggests that the genetic modifications occurring in these proximal regions may serve to counterbalance or mitigate the functional impacts of changes occurring within the stop codon itself, thereby preserving the overall integrity and functionality of the encoded protein sequence across evolutionary timeframes (Belinky et al. 2018). In summary, the codon frequency patterns observed in the C. roseus cultivars are consistent with those reported in other plants, suggesting that selective pressures to maintain functional efficiency in protein synthesis drive codon usage preferences in the chloroplast genome. The dominance of A/T-rich codons, especially at the third position, reflects the inherent stability and functional requirements of the plastome, which are further reinforced by the low occurrence of codons with C/G pairs at the same position. These findings provide valuable insights into the molecular evolution of plastomes and underscore the importance of understanding codon usage patterns in the context of plastome evolution and gene function.
The analysis of codon usage across the chloroplast genomes of the eight C. roseus cultivars also provided valuable insights into the underlying genetic structure of the species. Despite substantial genetic variation, particularly in terms of SNPs and Indels, the study found no significant differences in codon preference between the two groups of cultivars. This lack of divergence in codon usage is indicative of a low degree of molecular differentiation at the plastomic level, reinforcing the idea that C. roseus cultivars are still in the early stages of speciation. Bias in codon usage has been shown to correlate with functional constraints in genome evolution (Duret 2002); however, in this case, the absence of significant changes in codon preference suggests that there are no major selective pressures acting on the chloroplast genome that would lead to the establishment of distinct codon usage patterns. This observation aligns with findings from other studies that have suggested that plastomic evolution is often constrained by functional requirements, such as the need for efficient protein synthesis and stability of the genome (Robbins and Kelly 2023). The observed lack of divergence in codon usage patterns further underscores the idea that plastomes evolve slowly compared with nuclear genomes and that significant molecular divergence at the plastomic level may take much longer to manifest (Drouin et al. 2008; Charboneau et al. 2021).
The lack of codon preference divergence between the two groups of cultivars also has important implications for speciation research. Codon usage has been widely employed as a biomarker for assessing evolutionary rates and speciation in many organisms, including plants (Zhou et al. 2016). However, in the case of C. roseus, it appears that the plastomic divergence is not pronounced enough to induce significant shifts in codon usage. This suggests that plastomic data, including codon usage analysis, may not be reliable indicators of speciation in closely related cultivars, at least within the short time frame of divergence observed in this study.
SSR biomarkers and their role in assessing speciation
Due to the high polymorphism and extensive applicability of chloroplast SSRs in population genetics and speciation research, they offer significant contributions to evolutionary studies (Wang et al. 2022b). SSRs play a pivotal role in driving genome recombination and structural rearrangements, thereby enhancing genetic diversity and influencing biogeographical distributions (Triest 2008). Notwithstanding, the analysis of SSR biomarkers across the plastomes of eight C. roseus cultivars showed inconsistent results, particularly with the cultivar ‘First Kiss Peach’ (coded 3), which exhibited unreliable results and was subsequently excluded from SSR analysis. In total, nine distinct SSR types were identified across three key categories (mono ‘A’, mono ‘T’, and Di/Tri nucleotides), with cultivars coded 4 and 7 showing remarkable homogeneity in their SSR profiles, suggesting low genetic divergence between them. Consistent with the findings of this investigation, a recent study has elucidated that the repetitive sequence units predominantly comprise adenine (A) and thymine (T) nucleotides, with a notable preponderance of A/T mononucleotide repeats (Gu et al. 2024). The findings of the present study highlighted the limitations of plastid SSR biomarkers in distinguishing fine-scale genetic variation within closely related cultivars, because they tend to evolve slowly and exhibit low polymorphism. While plastid SSRs can be useful for broader phylogenetic studies, their utility in detecting subtle intraspecific differences in C. roseus is constrained. Further studies incorporating additional molecular biomarkers, such as nuclear SSR biomarkers or whole-genome sequencing, are recommended for more accurate speciation analyses and a deeper understanding of genetic diversity within C. roseus and related species.
The study on C. roseus chloroplast genomes highlighted several connections between the results and potential applications in conservation and breeding programs. The study showed minimal plastome size variation among C. roseus cultivars, suggesting that these plants are still in the early stages of speciation at the plastomic level. This information is crucial for conservation efforts, as it indicates that genetic diversity within the species is not yet fully realized at the plastome level. The genetic variations detected among cultivars can be leveraged to create molecular markers, which are invaluable for marker-assisted selection (MAS) in breeding programs (Collard and Mackill 2008). This approach allows breeders to select for desirable traits more efficiently, such as improved petal morphology or enhanced secondary metabolite production.
In summary, a substantial body of research has emphasized the critical significance of chloroplast genome sequencing and genetic markers in unraveling the complex inter-relationships among species and shedding light on their evolutionary trajectories (Zeb et al. 2022). These studies collectively underscore the profound utility of chloroplast genomic data in advancing our comprehension of phylogenetic connections and evolutionary dynamics across diverse plant lineages.
Conclusions
In conclusion, the results of this study suggest that although there is some degree of genetic differentiation between the two groups of C. roseus cultivars, the speciation process is not yet fully realized at the plastomic level. The lack of significant divergence in key molecular biomarkers, such as codon usage and SSR profiles, indicates that the genetic differences observed among cultivars are shallow. The observed genetic distinctions may reflect early stages of divergence or ongoing gene flow between cultivars, potentially being facilitated by hybridization or introgression. The partial resolution of phylogenetic relationships using the trnL biomarker emphasizes the need for more refined genetic biomarkers to capture the fine-scale differentiation that may be occurring within C. roseus. The integration of these biomarkers, alongside a more extensive phylogenetic sampling, holds the potential to clarify the complex evolutionary dynamics governing C. roseus and its phylogenetic associations with other taxa within the Apocynaceae family previously explored (Stevens and Davis 2001; Stevens and Davis 2005; Kalwij 2012; Struwe 2014; The Angiosperm Phylogeny Group et al. 2016; Hosni and Shamso 2022; Shamso et al. 2023; Tiernan et al. 2023; Wang et al. 2023a). Ultimately, a more detailed understanding of the speciation processes within this species could have important implications for conservation biology, particularly in the context of biodiversity preservation and the management of plant genetic resources.
Data availability
The raw sequencing data for the eight C. roseus genomes (identified as Isolates 1–8) were assigned Bioproject ID PRJNA1191663 in the NCBI repository. The corresponding circularized plastid genomes of these cultivars were then assembled, annotated, and deposited in the NCBI repository, where they were assigned accession numbers PQ677102, PQ677100, PQ677101, PQ677099, PQ677097, PQ677098, PQ677103, and PQ677096 respectively.
Declaration of funding
The authors extend their appreciation to the Deanship of Research and Graduate Studies at King Khalid University for funding this work through large Research Project under Grant number RGP2/56/45.
References
Ahmad W, Asaf S, Al-Rawahi A, Al-Harrasi A, Khan AL (2023) Comparative plastome genomics, taxonomic delimitation and evolutionary divergences of Tetraena hamiensis var. qatarensis and Tetraena simplex (Zygophyllaceae). Scientific Reports 13, 7436.
| Crossref | Google Scholar |
Arnold ML (2004) Transfer and origin of adaptations through natural hybridization: were Anderson and Stebbins right? The Plant Cell 16, 562-570.
| Crossref | Google Scholar |
Aruna MS, Prabha MS, Priya NS, Nadendla R (2015) Catharanthus Roseus: ornamental plant is now medicinal boutique. Journal of Drug Delivery and Therapeutics 5, 1-4.
| Google Scholar |
Banyal A, Tiwari S, Sharma A, Chanana I, Patel SKS, Kulshrestha S, Kumar P (2023) Vinca alkaloids as a potential cancer therapeutics: recent update and future challenges. 3 Biotech 13, 211.
| Crossref | Google Scholar |
Belinky F, Babenko VN, Rogozin IB, Koonin EV (2018) Purifying and positive selection in the evolution of stop codons. Scientific Reports 8, 9260.
| Crossref | Google Scholar |
Cabelin VLD, Alejandro GJD (2016) Efficiency of matK, rbcL, trnHpsbA, and trnL–F (cpDNA) to molecularly authenticate Philippine ethnomedicinal Apocynaceae through DNA barcoding. Pharmacognosy Magazine 12, S384-S388.
| Crossref | Google Scholar |
Cao Q, Gao Q, Ma X, Zhang F, Xing R, Chi X, Chen S (2022) Plastome structure, phylogenomics and evolution of plastid genes in Swertia (Gentianaceae) in the Qing-Tibetan Plateau. BMC Plant Biology 22, 195.
| Crossref | Google Scholar |
Caron H, Molino J-F, Sabatier D, Léger P, Chaumeil P, Scotti-Saintagne C, Frigério J-M, Scotti I, Franc A, Petit RJ (2019) Chloroplast DNA variation in a hyperdiverse tropical tree community. Ecology and Evolution 9, 4897-4905.
| Crossref | Google Scholar |
Charboneau JLM, Cronn RC, Liston A, Wojciechowski MF, Sanderson MJ (2021) Plastome structural evolution and homoplastic inversions in Neo-Astragalus (Fabaceae). Genome Biology and Evolution 13, evab215.
| Crossref | Google Scholar |
Chase MW, Fay MF (2009) Barcoding of plants and fungi. Science 325, 682-683.
| Crossref | Google Scholar |
Chaturvedi V, Goyal S, Mukim M, Meghani M, Patwekar F, Patwekar M, Khan SK, Sharma GN (2022) A comprehensive review on Catharanthus roseus L.(G.) Don: clinical pharmacology, ethnopharmacology and phytochemistry. Journal of Pharmacological Research and Developments 4, 17-36.
| Crossref | Google Scholar |
Chen Y, Zhou B, Li J, Tang H, Tang J, Yang Z (2018) Formation and change of chloroplast-located plant metabolites in response to light conditions. International Journal of Molecular Sciences 19, 654.
| Crossref | Google Scholar |
Cingolani P, Platts A, Wang Le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80-92.
| Crossref | Google Scholar |
Collard BCY, Mackill DJ (2008) Marker-assisted selection: an approach for precision plant breeding in the twenty-first century. Philosophical Transactions of the Royal Society B: Biological Sciences 363, 557-572.
| Crossref | Google Scholar |
de Groot GA, During HJ, Maas JW, Schneider H, Vogel JC, Erkens RHJ (2011) Use of rbcL and trnL–F as a two-locus DNA barcode for identification of NW-European ferns: an ecological perspective. PLoS ONE 6, e16371.
| Crossref | Google Scholar |
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, Del Angel G, Rivas MA, Hanna M, Mckenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature Genetics 43, 491-498.
| Crossref | Google Scholar |
Dierckxsens N, Mardulyn P, Smits G (2017) NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Research 45, e18.
| Crossref | Google Scholar |
Dierckxsens N, Mardulyn P, Smits G (2020) Unraveling heteroplasmy patterns with NOVOPlasty. NAR Genomics and Bioinformatics 2, lqz011.
| Crossref | Google Scholar |
Dong W, Xu C, Wen J, Zhou S (2020) Evolutionary directions of single nucleotide substitutions and structural mutations in the chloroplast genomes of the family Calycanthaceae. BMC Evolutionary Biology 20, 96.
| Crossref | Google Scholar |
Drouin G, Daoud H, Xia J (2008) Relative rates of synonymous substitutions in the mitochondrial, chloroplast and nuclear genomes of seed plants. Molecular Phylogenetics and Evolution 49, 827-831.
| Crossref | Google Scholar |
Duret L (2002) Evolution of synonymous codon usage in metazoans. Current Opinion in Genetics & Development 12, 640-649.
| Crossref | Google Scholar |
El-Domyati F, Ramadan A, Gadalla N, Edris S, Shokry A, Hassan S, Hassanien S, Baeshen MN, Hajrah N, Al-Kordy M (2012) Identification of molecular markers for flower characteristics in Catharanthus roseus producing anticancer compounds. Life Sci Journal 9, 5949-5960.
| Google Scholar |
Endress ME, Bruyns PV (2000) A revised classification of the Apocynaceae s.l. The Botanical Review 66, 1-56.
| Crossref | Google Scholar |
Fenster CB, Armbruster WS, Wilson P, Dudash MR, Thomson JD (2004) Pollination syndromes and floral specialization. Annual Review of Ecology, Evolution, and Systematics 35, 375-403.
| Crossref | Google Scholar |
Forsythe ES, Williams AM, Sloan DB (2021) Genome-wide signatures of plastid-nuclear coevolution point to repeated perturbations of plastid proteostasis systems across angiosperms. The Plant Cell 33, 980-997.
| Crossref | Google Scholar |
Frazão A, Thode VA, Lohmann LG (2023) Comparative chloroplast genomics and insights into the molecular evolution of Tanaecium (Bignonieae, Bignoniaceae). Scientific Reports 13, 12469.
| Crossref | Google Scholar |
Gao W, Chen X, He J, Sha A, Luo Y, Xiao W, Xiong Z, Li Q (2024) Intraspecific and interspecific variations in the synonymous codon usage in mitochondrial genomes of 8 pleurotus strains. BMC Genomics 25, 456.
| Crossref | Google Scholar |
Goswami S, Ali A, Prasad ME, Singh P (2024) Pharmacological significance of Catharanthus roseus in cancer management: a review. Pharmacological Research – Modern Chinese Medicine 11, 100444.
| Crossref | Google Scholar |
Gu J, Li M, He S, Li Z, Wen F, Tan K, Bai X, Hu G (2024) Comparative chloroplast genomes analysis of nine Primulina (Gesneriaceae) rare species, from karst region of southwest China. Scientific Reports 14, 30256.
| Crossref | Google Scholar |
Guimarães G, Cardoso L, Oliveira H, Santos C, Duarte P, Sottomayor M (2012) Cytogenetic characterization and genome size of the medicinal plant Catharanthus roseus (L.) G. Don. AoB Plants 2012, pls002.
| Crossref | Google Scholar |
Hao DC, Huang BL, Chen SL, Mu J (2009) Evolution of the chloroplast trnLtrnF region in the gymnosperm lineages Taxaceae and Cephalotaxaceae. Biochemical Genetics 47, 351-369.
| Crossref | Google Scholar |
Hosni HA, Shamso EM (2022) Contribution to the flora of Egypt: taxonomic and nomenclature changes. Taeckholmia 42, 12-26.
| Crossref | Google Scholar |
Hu X, Li Y, Meng F, Duan Y, Sun M, Yang S, Liu H (2024) Analysis of chloroplast genome characteristics and codon usage bias in 14 species of Annonaceae. Functional & Integrative Genomics 24, 109.
| Crossref | Google Scholar |
Jamal QMS, Ahmad V (2024) Identification of metabolites from Catharanthus roseus leaves and stem extract, and in vitro and in silico antibacterial activity against food pathogens. Pharmaceuticals 17, 450.
| Crossref | Google Scholar |
Jiang D, Cai X, Gong M, Xia M, Xing H, Dong S, Tian S, Li J, Lin J, Liu Y, Li H-L (2023) Complete chloroplast genomes provide insights into evolution and phylogeny of Zingiber (Zingiberaceae). BMC Genomics 24, 30.
| Crossref | Google Scholar |
Kalwij JM (2012) Review of ‘The Plant List, a working list of all plant species’. Journal of Vegetation Science 23, 998-1002.
| Crossref | Google Scholar |
Kan J, Nie L, Wang M, Tiwari R, Tembrock LR, Wang J (2024) The Mendelian pea pan-plastome: insights into genomic structure, evolutionary history, and genetic diversity of an essential food crop. Genomics Communications 1, e004.
| Crossref | Google Scholar |
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution 30, 772-780.
| Crossref | Google Scholar |
Krawczyk K, Paukszto Ł, Maździarz M, Sawicki J (2023) The low level of plastome differentiation observed in some lineages of Poales hinders molecular species identification. Frontiers in Plant Science 14, 1275377.
| Crossref | Google Scholar |
Kress WJ, Erickson DL (2007) A two-locus global DNA barcode for land plants: the coding rbcL gene complements the non-coding trnH–psbA spacer region. PLoS ONE 2, e508.
| Crossref | Google Scholar |
Kulagina N, Méteignier L-V, Papon N, O’connor SE, Courdavault V (2022) More than a Catharanthus plant: a multicellular and pluri-organelle alkaloid-producing factory. Current Opinion in Plant Biology 67, 102200.
| Crossref | Google Scholar |
Kumar A, Singhal KC, Sharma RA, Vyas GK, Kumar V (2014) Molecular characterisation of Catharanthus roseus cultivars from various regions of rajasthan based on rapd marker. International Journal of Pharmaceutical Sciences and Research 5, 3936-3941.
| Crossref | Google Scholar |
Leypold NA, Speicher MR (2021) Evolutionary conservation in noncoding genomic regions. Trends in Genetics 37, 903-918.
| Crossref | Google Scholar |
Li H, Xiao W, Tong T, Li Y, Zhang M, Lin X, Zou X, Wu Q, Guo X (2021) The specific DNA barcodes based on chloroplast genes for species identification of Orchidaceae plants. Scientific Reports 11, 1424.
| Crossref | Google Scholar |
Li C, Wood JC, Vu AH, Hamilton JP, Rodriguez Lopez CE, Payne RME, Serna Guerrero DA, Gase K, Yamamoto K, Vaillancourt B, Caputi L, O’Connor SE, Robin Buell C (2023) Single-cell multi-omics in the medicinal plant Catharanthus roseus. Nature Chemical Biology 19, 1031-1041.
| Crossref | Google Scholar | PubMed |
Liu L, Lin N, Liu X, Yang S, Wang W, Wan X (2020) From chloroplast biogenesis to chlorophyll accumulation: the interplay of light and hormones on gene expression in Camellia sinensis cv. Shuchazao leaves. Frontiers in Plant Science 11, 256.
| Crossref | Google Scholar |
Liu H, Ye H, Zhang N, Ma J, Wang J, Hu G, Li M, Zhao P (2022) Comparative analyses of chloroplast genomes provide comprehensive insights into the adaptive evolution of Paphiopedilum (Orchidaceae). Horticulturae 8, 391.
| Crossref | Google Scholar |
Lubna , Asaf S, Jan R, Asif S, Bilal S, Khan AL, Kim K-M, Lee I-J, AL-Harrasi A (2024) Plastome diversity and evolution in mosses: insights from structural characterization, comparative genomics, and phylogenetic analysis. International Journal of Biological Macromolecules 257, 128608.
| Crossref | Google Scholar |
Mallott EK, Garber PA, Malhi RS (2018) trnL outperforms rbcL as a DNA metabarcoding marker when compared with the observed plant component of the diet of wild white-faced capuchins (Cebus capucinus, Primates). PLoS ONE 13, e0199556.
| Crossref | Google Scholar |
Matiz-Ceron L, Reyes A, Anzola J (2022) Taxonomical evaluation of plant chloroplastic markers by bayesian classifier. Frontiers in Plant Science 12, 782663.
| Crossref | Google Scholar |
Meena RK, Negi N, Uniyal N, Shamoon A, Bhandari MS, Pandey S, Negi RK, Sharma R, Ginwal HS (2020) Chloroplast-based DNA barcode analysis indicates high discriminatory potential of matK locus in Himalayan temperate bamboos. 3 Biotech 10, 534.
| Crossref | Google Scholar |
Mokhtar MM, Alsamman AM, El Allali A (2023) MegaSSR: a web server for large scale microsatellite identification, classification, and marker development. Frontiers in Plant Science 14, 1219055.
| Crossref | Google Scholar |
Müller KF, Borsch T, Hilu KW (2006) Phylogenetic utility of rapidly evolving DNA at high taxonomical levels: contrasting matK, trnT–F, and rbcL in basal angiosperms. Molecular Phylogenetics and Evolution 41, 99-117.
| Crossref | Google Scholar |
Nazar N, Goyder DJ, Clarkson JJ, Mahmood T, Chase MW (2013) The taxonomy and systematics of Apocynaceae: where we stand in 2012. Botanical Journal of the Linnean Society 171, 482-490.
| Crossref | Google Scholar |
Nazar N, Clarkson JJ, Goyder D, Kaky E, Mahmood T, Chase MW (2019) Phylogenetic relationships in Apocynaceae based on nuclear PHYA and plastid trnL–F sequences, with a focus on tribal relationships. Caryologia 72, 55-81.
| Crossref | Google Scholar |
Pham HNT, Sakoff JA, Vuong QV, Bowyer MC, Scarlett CJ (2019) Phytochemical, antioxidant, anti-proliferative and antimicrobial properties of Catharanthus roseus root extract, saponin-enriched and aqueous fractions. Molecular Biology Reports 46, 3265-3273.
| Crossref | Google Scholar |
Ravi V, Khurana JP, Tyagi AK, Khurana P (2008) An update on chloroplast genomes. Plant Systematics and Evolution 271, 101-122.
| Crossref | Google Scholar |
Ravikumar B, Dhatt KK (2024) Genetic analysis of flower colour variation in periwinkle (Catharanthus roseus L.) inbred lines. Genetic Resources and Crop Evolution 71, 2247-2253.
| Crossref | Google Scholar |
Robbins EHJ, Kelly S (2023) The evolutionary constraints on angiosperm chloroplast adaptation. Genome Biology and Evolution 15, evad101.
| Crossref | Google Scholar |
Salama I, Ragab E, Mohamed MH (2020) Impact of environmental diversity in Egypt on Catharanthus roseus cultivars genome and assessment that by different DNA markers. Egyptian Journal of Radiation Sciences and Applications 33, 33-44.
| Crossref | Google Scholar |
Samiyarsih S, Naipospos N, Palupi D (2019) Variability of Catharanthus roseus based on morphological and anatomical characters, and chlorophyll contents. Biodiversitas Journal of Biological Diversity 20, 2986-2993.
| Crossref | Google Scholar |
Schneider H, Ranker TA, Russell SJ, Cranfill R, Geiger JMO, Aguraiuja R, Wood KR, Grundmann M, Kloberdanz K, Vogel JC (2005) Origin of the endemic fern genus Diellia coincides with the renewal of Hawaiian terrestrial life in the Miocene. Proceedings of the Royal Society B: Biological Sciences 272, 455-460.
| Crossref | Google Scholar |
Sebastin R, Kim J, Jo I-H, Yu J-K, Jang W, Han S, Park H-S, Algarawi AM, Hatamleh AA, So Y-S, Shim D, Chung J-W (2024) Comparative chloroplast genome analyses of cultivated and wild capsicum species shed light on evolution and phylogeny. BMC Plant Biology 24, 797.
| Crossref | Google Scholar |
Shamso EM, Hosni HA, Hosny AI, Rabei SH, Elgamal IAEr (2023) Contribution to the flora of Egypt: a critical inventory of newly recorded vascular taxa of Egypt. Scientific Journal for Damietta Faculty of Science 13, 111-149.
| Crossref | Google Scholar |
Shang C, Li E, Yu Z, Lian M, Chen Z, Liu K, Xu L, Tong Z, Wang M, Dong W (2022) Chloroplast genomic resources and genetic divergence of endangered species Bretschneidera sinensis (Bretschneideraceae). Frontiers in Ecology and Evolution 10, 873100.
| Crossref | Google Scholar |
Sharma MK, Kumar M, Renu (2021) Biosynthesis and modulation of terpenoid indole alkaloids in Catharanthus roseus: a review of targeting genes and secondary metabolites. Journal of Pure and Applied Microbiology 15, 1745-1758.
| Crossref | Google Scholar |
Shaw J, Shafer HL, Leonard OR, Kovach MJ, Schorr M, Morris AB (2014) Chloroplast DNA sequence utility for the lowest phylogenetic and phylogeographic inferences in angiosperms: the tortoise and the hare IV. American Journal of Botany 101, 1987-2004.
| Crossref | Google Scholar |
She J, Yan H, Yang J, Xu W, Su Z (2019) croFGD: Catharanthus roseus Functional Genomics Database. Frontiers in Genetics 10, 238.
| Crossref | Google Scholar |
Shi N, Yuan Y, Huang R, Wen G (2024) Analysis of codon usage patterns in complete plastomes of four medicinal Polygonatum species (Asparagaceae). Frontiers in Genetics 15, 1401013.
| Crossref | Google Scholar |
Stelkens R, Seehausen O (2009) Genetic distance between species predicts novel trait expression in their hybrids. Evolution 63, 884-897.
| Crossref | Google Scholar |
Stevens PF, Davis H (2001) Angiosperm phylogeny website. Missouri Botanical Garden, St Louis, MO, USA. Available at http://www.mobot.org/MOBOT/research/APweb/
Stevens PF, Davis HM (2005) The angiosperm phylogeny Website – a tool for reference and teaching in a time of change. Proceedings of the American Society for Information Science and Technology 42,.
| Crossref | Google Scholar |
Stothard P (2000) The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. BioTechniques 28, 1102-1104.
| Crossref | Google Scholar |
Sun J, Wang Y, Liu Y, Xu C, Yuan Q, Guo L, Huang L (2020) Evolutionary and phylogenetic aspects of the chloroplast genome of Chaenomeles species. Scientific Reports 10, 11466.
| Crossref | Google Scholar |
Susanto AH, Dwiati M (2024) Genetic comparison among some cultivars of Catharanthus roseus (L.) G.Don. using three intergenic spacers of the chloroplast genome. Biodiversitas Journal of Biological Diversity 25, 2999-3007.
| Crossref | Google Scholar |
Suzuki H, Morton BR (2016) Codon adaptation of plastid genes. PLoS ONE 11, e0154306.
| Crossref | Google Scholar |
The Angiosperm Phylogeny Group, Chase MW, Christenhusz MJM, Fay MF, Byng JW, Judd WS, Soltis DE, Mabberley DJ, Sennikov AN, Soltis PS, Stevens PF (2016) An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Botanical Journal of the Linnean Society 181, 1-20.
| Crossref | Google Scholar |
Tiernan N, Nakamura K, Burns C, Jestrow B, Oviedo Prieto R, Francisco-Ortega J (2023) Phylogenetic relationships of Cuban and Caribbean Plumeria (Apocynaceae) based on the plastid genome. Biological Journal of the Linnean Society 140, 397-412.
| Crossref | Google Scholar |
Tillich M, Lehwark P, Pellizzer T, Ulbricht-Jones ES, Fischer A, Bock R, Greiner S (2017) GeSeq – versatile and accurate annotation of organelle genomes. Nucleic Acids Research 45, W6-W11.
| Crossref | Google Scholar |
Torka P, Przespolewski E, Evens AM (2022) Treatment strategies for advanced classical Hodgkin lymphoma in the times of dacarbazine shortage. JCO Oncology Practice 18, 491-497.
| Crossref | Google Scholar | PubMed |
Triest L (2008) Molecular ecology and biogeography of mangrove trees towards conceptual insights on gene flow and barriers: a review. Aquatic Botany 89, 138-154.
| Crossref | Google Scholar |
Turudić A, Liber Z, Grdiša M, Jakše J, Varga F, Šatović Z (2021) Towards the well-tempered chloroplast DNA sequences. Plants 10, 1360.
| Crossref | Google Scholar |
Van Der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, Banks E, Garimella KV, Altshuler D, Gabriel S, Depristo MA (2013) From FastQ data to high-confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Current Protocols in Bioinformatics 43, 11.10.1-11.10.33.
| Crossref | Google Scholar |
Wang L, Xing H, Yuan Y, Wang X, Saeed M, Tao J, Feng W, Zhang G, Song X, Sun X (2018) Genome-wide analysis of codon usage bias in four sequenced cotton species. PLoS ONE 13, e0194372.
| Crossref | Google Scholar |
Wang J, Yan Z, Zhong P, Shen Z, Yang G, Ma L (2022a) Screening of universal DNA barcodes for identifying grass species of Gramineae. Frontiers in Plant Science 13, 998863.
| Crossref | Google Scholar |
Wang S, Gao J, Chao H, Li Z, Pu W, Wang Y, Chen M (2022b) Comparative chloroplast genomes of Nicotiana Species (Solanaceae): insights into the genetic variation, phylogenetic relationship, and polyploid speciation. Frontiers in Plant Science 13, 899252.
| Crossref | Google Scholar |
Wang Y, Zhang C-F, Odago WO, Jiang H, Yang J-X, Hu G-W, Wang Q-F (2023a) Evolution of 101 Apocynaceae plastomes and phylogenetic implications. Molecular Phylogenetics and Evolution 180, 107688.
| Crossref | Google Scholar |
Wang W, Wang X, Shi Y, Yin Q, Gao R, Wang M, Xiang L, Wu L (2023b) Identification of Laportea bulbifera using the complete chloroplast genome as a potentially effective super-barcode. Journal of Applied Genetics 64, 231-245.
| Crossref | Google Scholar |
Wang J, Kan J, Wang J, Yan X, Li Y, Soe T, Tembrock LR, Xing G, Li S, Wu Z, Jia M (2024a) The pan-plastome of Prunus mume: insights into Prunus diversity, phylogeny, and domestication history. Frontiers in Plant Science 15, 1404071.
| Crossref | Google Scholar |
Wang X, Guo L, Ding L, Medina L, Wang R, Li P (2024b) Comparative plastome analyses and evolutionary relationships of 25 East Asian species within the medicinal plant genus Scrophularia (Scrophulariaceae). Frontiers in Plant Science 15, 1439206.
| Crossref | Google Scholar |
Wu H, Li D-Z, Ma P-F (2024) Unprecedented variation pattern of plastid genomes and the potential role in adaptive evolution in Poales. BMC Biology 22, 97.
| Crossref | Google Scholar |
Xiang Y-N, Wang X-Q, Ding L-L, Bai X-Y, Feng Y-Q, Qi Z-C, Sun Y-T, Yan X-L (2024) Deciphering the plastomic code of chinese hog-peanut (Amphicarpaea edgeworthii Benth., Leguminosae): comparative genomics and evolutionary insights within the phaseoleae tribe. Genes 15, 88.
| Crossref | Google Scholar |
Xu Z, Wang G, Wang Q, Li X, Zhang G, Qurban A, Zhang C, Zhou Y, Si H, Hu L, Wang F, Wang Y, Tian Z, Chen W, Jin S, Ding F (2023) A near-complete genome assembly of Catharanthus roseus and insights into its vinblastine biosynthesis and high susceptibility to the Huanglongbing pathogen. Plant Communications 4, 100661.
| Crossref | Google Scholar |
Yang Z (1994) Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. Journal of Molecular Evolution 39, 306-314.
| Crossref | Google Scholar |
Yang J, Vázquez L, Chen X, Li H, Zhang H, Liu Z, Zhao G (2017) Development of chloroplast and nuclear DNA markers for Chinese oaks (Quercus subgenus Quercus) and assessment of their utility as DNA barcodes. Frontiers in Plant Science 8, 816.
| Crossref | Google Scholar |
Yang L, Zhang S, Wu C, Jiang X, Deng M (2024) Plastome characterization and its phylogenetic implications on Lithocarpus (Fagaceae). BMC Plant Biology 24, 1277.
| Crossref | Google Scholar |
Zeb U, Wang X, AzizUllah A, Fiaz S, Khan H, Ullah S, Ali H, Shahzad K (2022) Comparative genome sequence and phylogenetic analysis of chloroplast for evolutionary relationship among Pinus species. Saudi Journal of Biological Sciences 29, 1618-1627.
| Crossref | Google Scholar |
Zhang Y, Tian L, Lu C (2023) Chloroplast gene expression: recent advances and perspectives. Plant Communications 4, 100611.
| Crossref | Google Scholar |
Zhang E, Ma X, Guo T, Wu Y, Zhang L (2024) Comparative analysis and phylogeny of the complete chloroplast genomes of nine Cynanchum (Apocynaceae) species. Genes 15, 884.
| Crossref | Google Scholar |
Zhou Z, Dang Y, Zhou M, Li L, Yu C-H, Fu J, Chen S, Liu Y (2016) Codon usage is an important determinant of gene expression levels largely through its effects on transcription. Proceedings of the National Academy of Sciences 113, E6117-E6125.
| Crossref | Google Scholar |
Zhou C, Tao F, Long R, Yang X, Wu X, Xiang L, Zhou X, Girdthai T (2024) The complete chloroplast genome of Mussaenda pubescens and phylogenetic analysis. Scientific Reports 14, 9131.
| Crossref | Google Scholar |
Zoschke R, Bock R (2018) Chloroplast translation: structural and functional organization, operational control, and regulation. The Plant Cell 30, 745-770.
| Crossref | Google Scholar |
Zupok A, Kozul D, Schöttler MA, Niehörster J, Garbsch F, Liere K, Fischer A, Zoschke R, Malinova I, Bock R, Greiner S (2021) A photosynthesis operon in the chloroplast genome drives speciation in evening primroses. The Plant Cell 33, 2583-2601.
| Crossref | Google Scholar |