

Enriched enzymes and crosstalking KEGG pathways in the rhizospheric soil fungiome of the wild plant Moringa oleifera
Rewaa S. Jalal A * , Abeer S. Aloufi B , Abeer Al-Andal C , Nahaa M. Alotaibi B , Haneen W. Abuauf D , Fatimah M. Alshehrei E , Mohammed Y. Refai A , Sahar A. Alshareef F , Alaa A. Alnahari A , Fatmah O. Sefrji G , Abeer M. Almutrafy G and Aala A. Abulfaraj H *
A * , Abeer S. Aloufi B , Abeer Al-Andal C , Nahaa M. Alotaibi B , Haneen W. Abuauf D , Fatimah M. Alshehrei E , Mohammed Y. Refai A , Sahar A. Alshareef F , Alaa A. Alnahari A , Fatmah O. Sefrji G , Abeer M. Almutrafy G and Aala A. Abulfaraj H *
A
B
C
D
E
F
G
H
Abstract
We aimed to identify the genes encoding predominant KEGG enzymes within the rhizospheric soil fungiome of Moringa oleifera. We also aimed to uncover how the rhizospheric fungiome drives intricate biochemical networks that bolster soil health, plant vitality, nutrient cycling, metabolic efficiency and resilience to environmental stress. These findings offer valuable insights that could enhance the efficacy of innovative agricultural practices. Previous research has focused on the role of soil microbiomes, including both bacteriomes and fungiomes, in the ecological dynamics of native and cultivated plants. The rhizospheric fungiome plays a critical role in plant health by suppressing pathogens, decomposing plant residues and facilitating nutrient assimilation in various environmental conditions. Fungal taxa from the phylum Mucoromycota, including Rhizophagus, Mucor ambiguus, Phycomyces blakesleeanus, Mortierella elongata, Absidia glauca, Mucor circinelloides and the taxon Basidiobolus meristosporus from Zoopagomycota, were identified as primary hosts of Kyoto Encyclopedia of Genes and Genomes (KEGG)-enriched enzymes in the rhizospheric soil of M. oleifera. These enzymes participate in crosstalk pathways within KEGG categories such as ‘Metabolism’, ‘Genetic Information Processing’, and ‘Environmental Information Processing’. These fungal enzymes contribute to the biosynthesis of critical metabolites, including carbamoyl-P, lipoyllysine, acetyl-CoA, isoleucine, valine and nucleotides (dADP, dGDP, dCDP, dUDP) that are essential for cellular functions such as DNA repair, replication and transcription. The symbiotic relationship between these enzymes and plant roots regulates nitrogen levels in the rhizosphere and supports mitochondrial stability. Metabolites also aid in cellular development, membrane metabolism, plant signal transduction and energy metabolism, including fueling the citric acid cycle. Our findings highlight the potential of crosstalking pathways in the rhizospheric fungiome of M. oleifera to enhance energy metabolism and maintain plant cell integrity. We propose that this research can serve as a foundation for advancing sustainable agricultural practices.
Keywords: Acetyl-CoA, BCAA, carbamoyl-phosphate, DNA replication and repair, energy metabolism, fatty acids, Mucor ambiguus, Rhizophagus, symbiotic relationship.
Introduction
Moringa oleifera, a member of the Moringaceae family, is native to several regions across Africa and Asia, including Saudi Arabia (Gupta and Ahmed 2020). Commonly referred to as drumstick, horseradish tree or malunggay, this remarkable plant boasts a wide range of applications in nutrition, pharmaceuticals, industry and agriculture (Kalibbala et al. 2009; Serafico et al. 2015; Gopalakrishnan et al. 2016; Palada 2019). Notably, M. oleifera is rich in vital nutrients, including essential vitamins C and A, minerals such as calcium, potassium and iron, and antioxidants such as flavonoids, polyphenols and ascorbic acid (Rockwood et al. 2013; Milla et al. 2021). The pharmaceutical relevance is underscored by the production of key compounds such as glycosides, β-sitosterol and N-α-rhamnophyranosyl vincosamide that contribute to regulating blood pressure and managing cholesterol levels (Panda et al. 2013). Additionally, M. oleifera is recognised for skin health benefits, effectiveness in treating chronic illnesses, and protective properties against liver oxidation and toxicity (Kumar et al. 2016; Zouboulis et al. 2023). Beyond these health-related applications, M. oleifera shows promise in diverse areas such as water purification, cosmetics, livestock feed, soil fertilisation and biofuel production (Rockwood et al. 2013; Gómez and Angulo 2014).
Recent investigations have increasingly focused on the contributions of soil rhizospheric microbiomes – including both bacteriomes and fungiomes – to the health and growth of both wild and cultivated plant species (Borrel et al. 2020; Ashy et al. 2023; Tashkandi and Baz 2023). The symbiotic relationship between M. oleifera and the associated rhizospheric soil microbiome has been empirically supported in multiple studies (Shami et al. 2022; Tashkandi et al. 2022; Ashy et al. 2023; Tashkandi and Baz 2023). The rhizospheric fungiome is particularly influential in enhancing plant growth and overall vitality, contributing to pathogen resistance, the decomposition of plant residues and nutrient availability for plant uptake (Wang et al. 2017). The tripartite symbiotic interactions among plants, bacteria and fungi, facilitated by fungal arbuscules and mycelium, promote nutrient exchange and absorption across these three kingdoms, thereby increasing plant resilience to challenging environmental conditions (Lidoy et al. 2023).
Soil fungi possess a remarkable ability to produce a diverse array of extracellular enzymes, significantly enhancing plant vitality through essential mechanisms such as nutrient cycling and bolstering resilience against biotic and abiotic stressors (Frąc et al. 2018). These fungi excel in the degradation of soil cellulose while simultaneously acting as biosorbents for harmful heavy metals, including cadmium, mercury and lead (Baldrian 2003). Moreover, the fungi are integral to improving soil structure by creating water-stable aggregates and facilitating pore space development that enhance both water retention and drainage capabilities (Frąc et al. 2018). Among these, mycorrhizal fungi stand out for the crucial role in promoting plant health and complex interactions with surrounding microenvironments (Barea et al. 2005; Buscot and Varma 2005). The mycelium of mycorrhizae serves as a vital channel for the transport of phosphorus in a bioavailable form, readily taken up by neighboring plants (Barea et al. 2005).
We aimed to explore the enriched enzymes and associated KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways within the rhizospheric soil fungi associated with Moringa oleifera, focusing specifically on the functional category of ‘Metabolism’. Additionally, we explored potential symbiotic interactions between plants and fungi, further elucidating the complexities of the relationships in the plant rhizospheric region. This study also aims to provide insights into how fungiome within the rhizosphere of M. oleifera contribute to complex biochemical networks that support plant health, energy metabolism and environmental adaptability.
Materials and methods
DNA extraction and whole genome shotgun sequencing
Rhizospheric soil samples were collected in triplicate from naturally growing Moringa oleifera in the Mecca district, Saudi Arabia (21°12′17.8″N 39°31′26.4″E) (Al-Eisawi and Al-Ruzayza 2015). Corresponding bulk soil samples (~1 m away from each of the three plants) served as controls. These samples were obtained under similar environmental conditions to maintain consistency. Both soil types were rapidly frozen in liquid nitrogen and stored at −20°C. The freezing process was carried out immediately after collection to preserve the microbial diversity present in the samples. DNA extraction followed the CTAB method, as previously described (Hurt et al. 2001). Subsequent treatment included RNase A (10 μm) at 37°C to eliminate RNA contaminants, followed by agarose gel electrophoresis (1%) for validation of DNA integrity. The electrophoresis was used to confirm the absence of degraded DNA and assess the size distribution. DNA concentration adjustment to 10 ng/μL utilised the dsDNA Assay kit using a Nanodrop spectrophotometer. Six samples (1 μg DNA each) were sent to Novogene Co. Ltd for next generation sequencing and biocomputational analysis.
DNA sequencing and bioinformatics analysis
Prior to sequencing, Covaris Sonicator physically fractionated DNA samples to achieve a 350 bp-band size. The use of sonication ensured uniform DNA fragmentation that is critical for generating libraries with optimal fragment size for sequencing. Subsequent processes included end-polishing, A-tailing and ligation with full-length adaptors for PCR using i7/i5 dual index primer set 1 (E7600). These processes facilitate the generation of high-quality sequencing libraries with accurate index tagging for multiplexing. The Ultra DNA Library Prep kit for Illumina (NEB, USA) facilitated the generation of sequencing libraries, following the manufacturer’s instructions. Illumina HiSeq 2500 platform was employed for sequencing. Quality assurance measures involved the recovery of filtered clean data, removal of low-quality bases (Q-value ≤ 38) exceeding a 40-bp threshold, and elimination of reads with N nucleotides >10-bp threshold and overlapped reads with adapters’ >15-bp threshold. These processes facilitate the generation of high-quality sequencing libraries with accurate index tagging for multiplexing. The resulting sequencing data were deposited in the European Nucleotide Archive (https://www.ebi.ac.uk/ena/browser/) under bioproject no. PRJEB55112 with run accession nos. ERR12764799, ERR10100771 and ERR10100772 for rhizosphere soil biosamples and ERR12764800, ERR10100774 and ERR10100781 for bulk soil biosamples. De novo assembly utilised Metagenome Assembly for Large-Scale High-Throughput Sequencing (MEGAHIT) (K-mer = 55), with removal of chimeras as described (Karlsson et al. 2012; Mende et al. 2012; Oh et al. 2014). The K-mer size was optimised to ensure maximal sensitivity for recovering short genomic sequences typical in metagenomic datasets. Subsequently, unassembled reads of all samples were de novo assembled to generate NOVO_MIX scaffolds as described (Mende et al. 2012; Nielsen et al. 2014). Mapping employed Soap 2.21, and effective individual and NOVO_MIX scaftigs were utilised. Prediction of gene was done using MetaGeneMark (Nielsen et al. 2014) and the resulting genes underwent dereplication using Cluster Database at High Identity with Tolerance (CD-HIT) (Li and Godzik 2006; Fu et al. 2012). Annotation used the binning reference-based classification method Metagenome Analyser (MEGAN) (Huson et al. 2011, 2016). Gene redundancy removal involved a greedy pairwise comparison (Li et al. 2014), leading to the construction of non-redundant gene catalogues (nrGC) used for subsequent analysis. Taxonomic ranks and functional levels were assigned to non-redundant genes using a Novogene Co.-provided pipeline. Accordingly, novel gene families were identified and functional categorisation was performed using pathway enrichment analysis tools as described (Reimand et al. 2019). Furthermore, taxonomic classification was corroborated through the use of both marker gene-based and whole-genome alignment approaches to ensure robustness and accuracy.
To identify specific microbial taxa or functional pathways that are differentially abundant between rhizosphere and bulk soils, the EdgeR algorithm (https://bioinformaticshome.com/db/tool/edgeR) was applied. These methods are widely used for count-based data, such as microbial sequence abundance and apply negative binomial models to identify features that exhibit statistically significant differences in abundance between conditions (Robinson et al. 2010).
Construction of functional KEGG hierarchy
Metagenomic gene-coding sequences were functionally annotated based on the similarity to sequences in the KEGG database (https://www.genome.jp/kegg/pathway.html), including KEGG PATHWAY and KEGG ENZYME (EC) (Kanehisa et al. 2006, 2014, 2016a, 2016b; Segata et al. 2011). The KO database (https://www.genome.jp/kegg/ko.html) was employed for molecular function detection, while the KEGG PATHWAY database mediated category, sub-category and pathway mapping, and KEGG ENZYME for functional annotation (Karlsson et al. 2012, 2013; Li et al. 2014). Function abundance data were subjected to principle coordinate analysis (PCoA) for validation of microbiome signatures. This method transforms the multidimensional dataset into a set of orthogonal axes, where the distances between samples are reflective of the dissimilarity. Bray–Curtis dissimilarity was typically employed as the distance measure for microbial community composition. PCoA results were visualised as scatter plots, allowing for the identification of groupings or clusters that corresponded to specific soil types, thereby facilitating an intuitive understanding of the microbiome’s diversity (Legendre and Anderson 1999; Anderson 2001).
Moreover, to assess the biological relevance of the microbiome data, the analysis incorporated pathway-based clustering and co-occurrence network modeling to identify key microbial interactions and metabolic processes, thus providing a comprehensive overview of microbial functionality as described (Layeghifard et al. 2017).
Results
Data validation
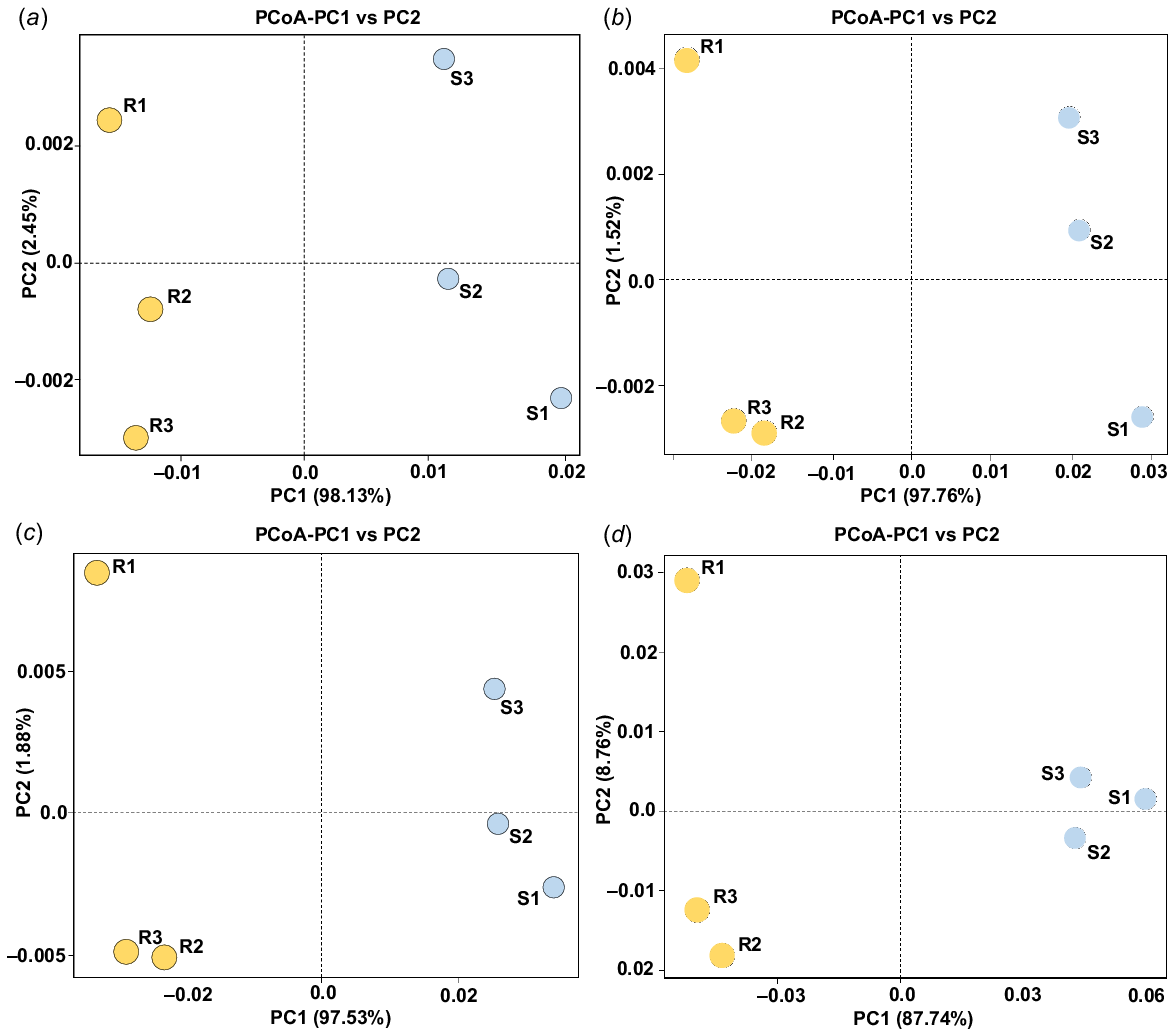
The metagenomic events of rhizospheric and bulk soils of Moringa oleifera were assessed across four functional levels, namely category, sub-category, pathway and orthologous group (OK). PCoA was employed for validation, as depicted in Fig. 1. The analysis revealed genetic variation between the microbiome samples of the two soil types. Notably, at the four functional levels, rhizospheric soil microbiome samples predominantly clustered on the negative side of the PC1 dimension, while bulk soil microbiome samples were situated on the other side (Fig. 1), signifying distinct compositional differences. PC2 dimension results indicated no clear trend, suggesting a high diversity level at both taxonomic and functional levels among samples of each soil type.
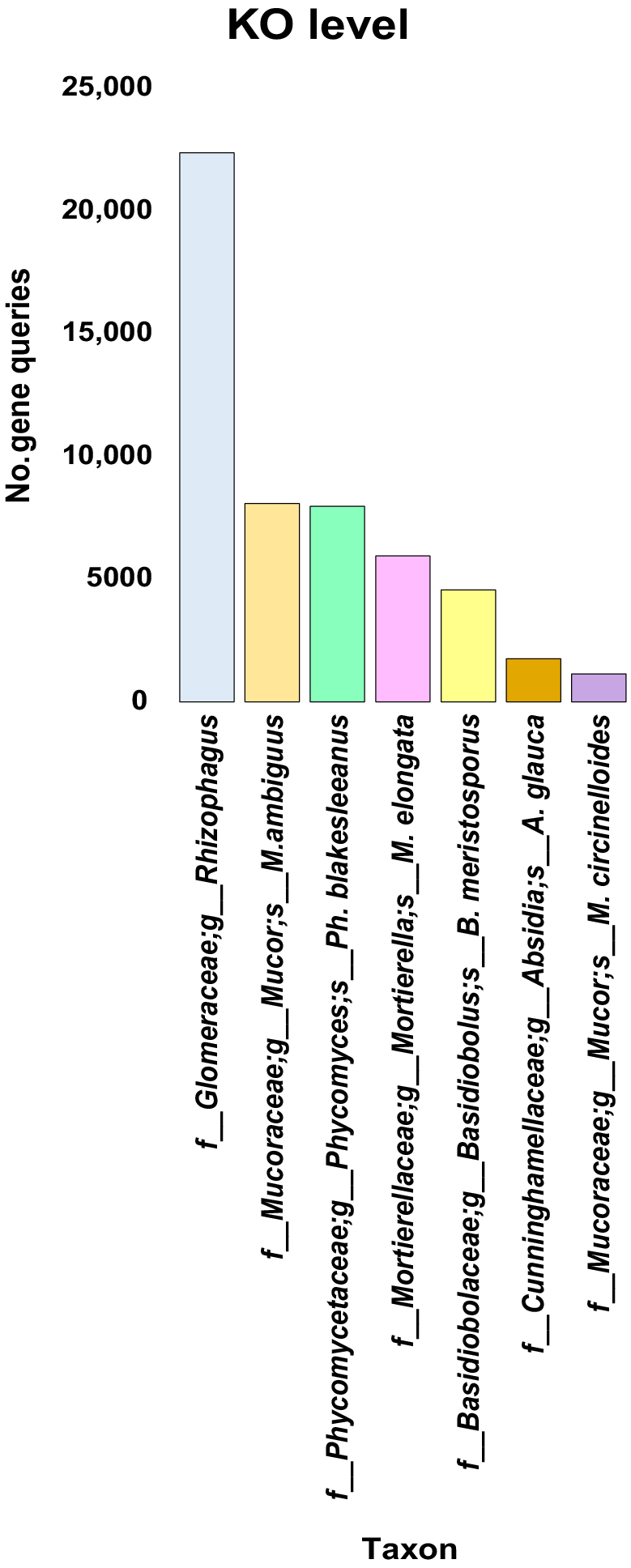
Major players of the rhizospheric soil fungiome of Moringa oleifera
In this study, we initially identified the enriched KEGG enzymes across the four microorganism kingdoms archaeome, bacteriome, fungiome and virome, and the two types of M. oleifera soil (Supplementary Table S1). We subsequently identified the top prominent fungal taxa (7) based on the abundance of KEGG-related gene queries (≥1000). Six of these taxa, e.g. genus Rhizophagus and the five species Mucor ambiguus, Phycomyces blakesleeanus, Mortierella elongata, Absidia glauca and Mucor circinelloides, belong to the phylum Mucoromycota (Fig. 2, Table S2). The seventh is the species Basidiobolus meristosporus that belongs to the phylum Zoopagomycota. Among these seven taxa, the genus Rhizophagus showed the highest number of gene queries (22,361) followed by the fungal species M. ambiguous (8078), Ph. blakesleeanus (7959) and M. elongata (5931) (Fig. 2).
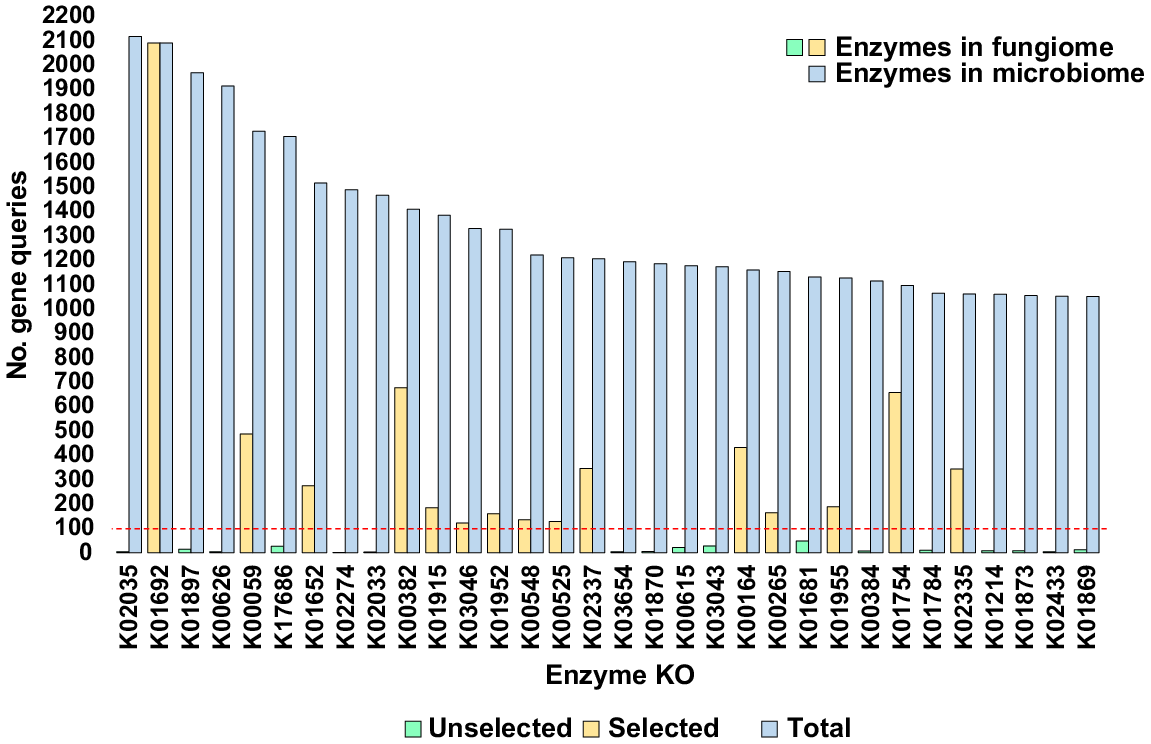
Enriched KEGG enzymes in soil rhizosphere of Moringa oleifera
We explored the top KEGG enzymes (32) that were shared among the four kingdoms with a total of ≥1000 gene queries across soil microbiomes (Table S3). Among these enzymes, we selected 15, with ≥100 gene queries in the fungiome of M. oleifera soil, for further analysis (Fig. 3, Table S2).
The top KEGG enzymes identified by the number of gene queries (≥1000) in the rhizospheric and bulk soil microbiomes of Moringa oleifera are highlighted. KEGG enzymes from the fungiome with at least 100 gene queries were chosen for further analysis. Additional information can be found in Tables S2 and S3.
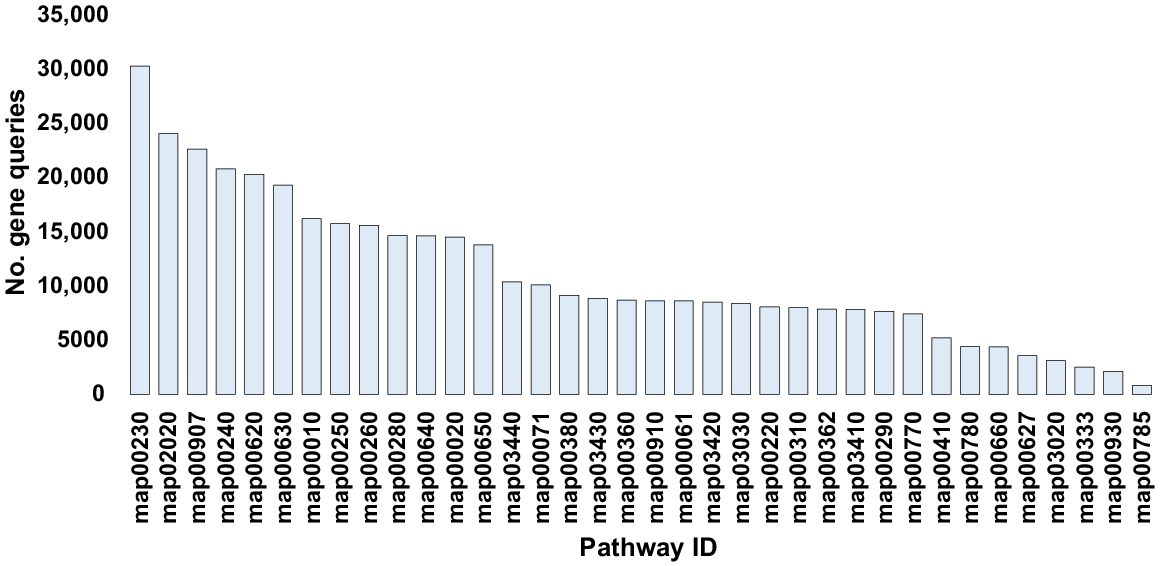
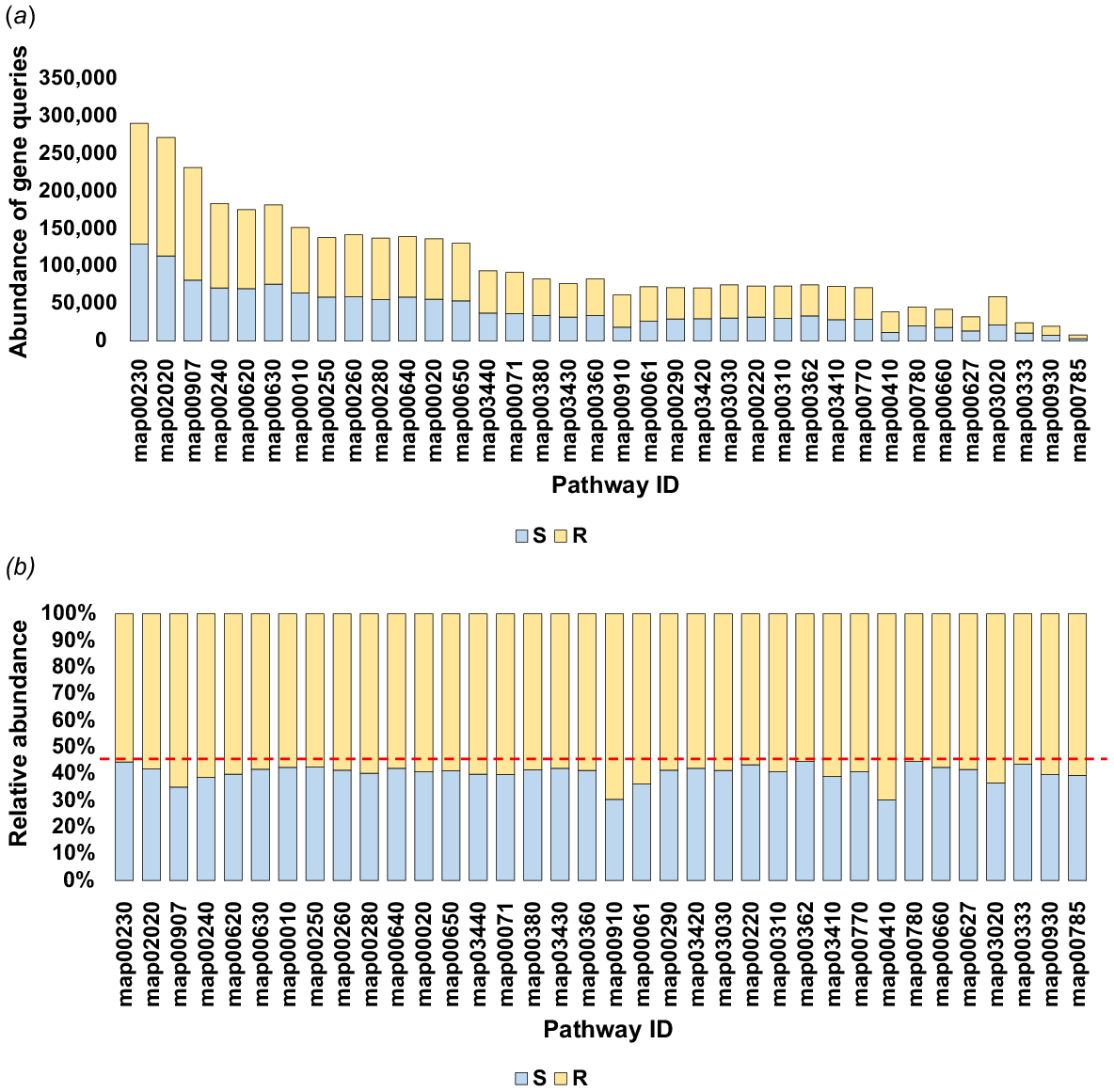
The enriched enzymes were associated with 43 KEGG pathways across 14 subcategories within the categories of ‘Metabolism’, ‘Genetic Information Processing’ and ‘Environmental Information Processing’ (Table S4). Average gene query numbers for these three functional levels across soil type were 77,899–323,697 at the category level, and 3623–101,777 and 831–30,295 at subcategory and KEGG pathway levels respectively (Figs S1a, S1b and 4, Tables S5, S6 and S7, respectively). Notably, the three functional levels exhibited ranges of gene query abundance in rhizospheric soil of ~392,912–1,877,367 at the category level, ~55,410–664,988 at the subcategory level, and ~5002–161,331 at the KEGG pathway level (Figs S2a, S2b and 5a, Tables S8, S9 and S10, respectively). The abundance ranges in bulk soil at the three functional levels were ~306,374–1,416,764, ~28,960–385,904 and ~3016–128,403 respectively. Consequently, relative abundance of gene queries in rhizospheric soil was >50% across the three functional levels compared to that of bulk soil of M. oleifera (Figs S3a, S3b and 5b, Tables S8, S9 and S10 respectively).
Number of gene queries at the KEGG pathway level pertaining to the most abundant KEGG enzymes found in the rhizosphere and bulk soil fungiomes of M. oleifera. More information is available in Table S7.
The abundance (a) and relative abundance (b) of gene queries at the KEGG pathway level in the rhizosphere (R) and bulk (S) soil microbiomes of Moringa oleifera, highlighting the most abundant KEGG enzymes in both soil fungiomes. The red dotted line indicates a 45% relative abundance at the KEGG pathway level, emphasising the greater abundance in the rhizosphere compared to the bulk soil. Additional details can be found in Table S10.
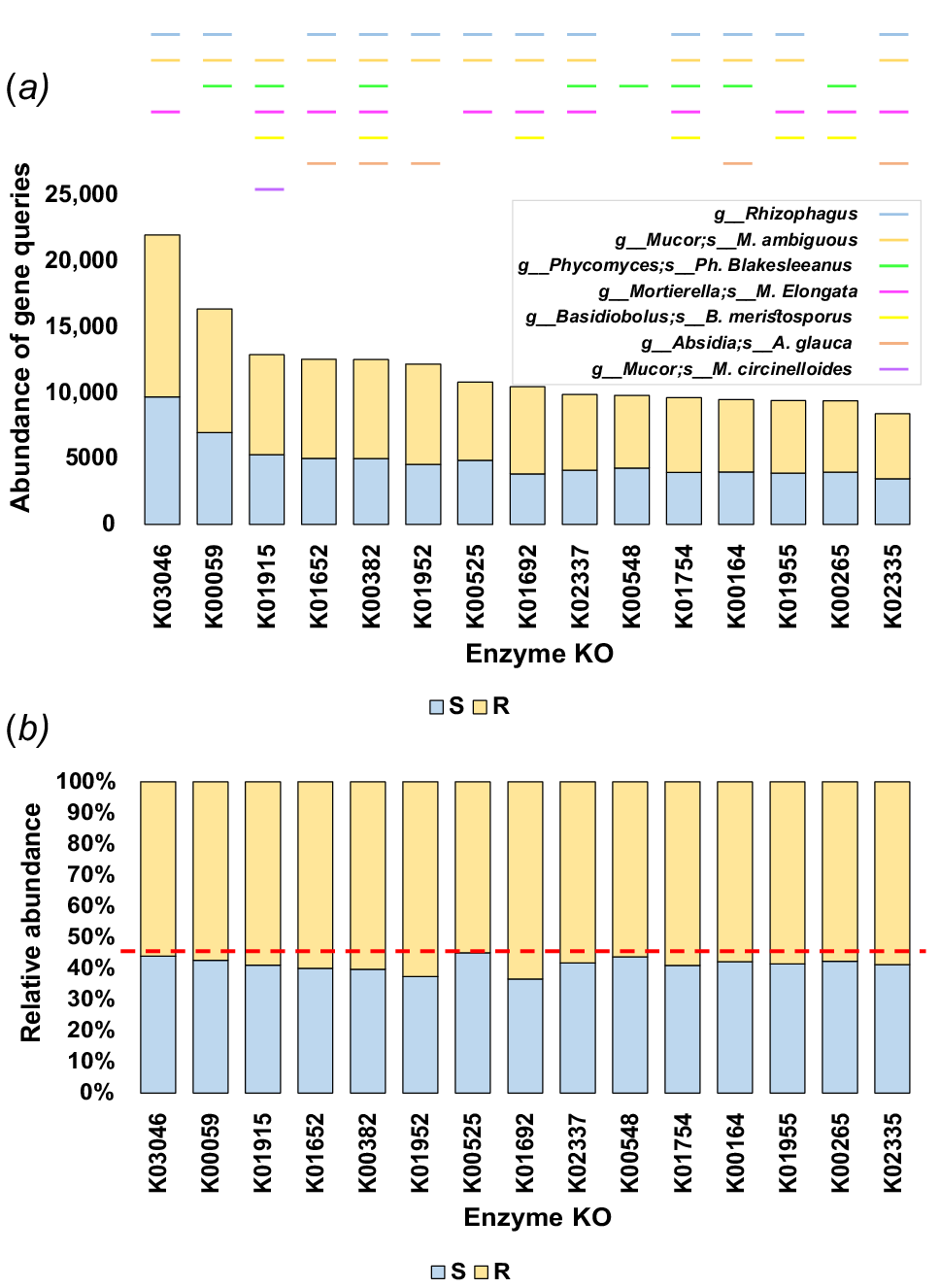
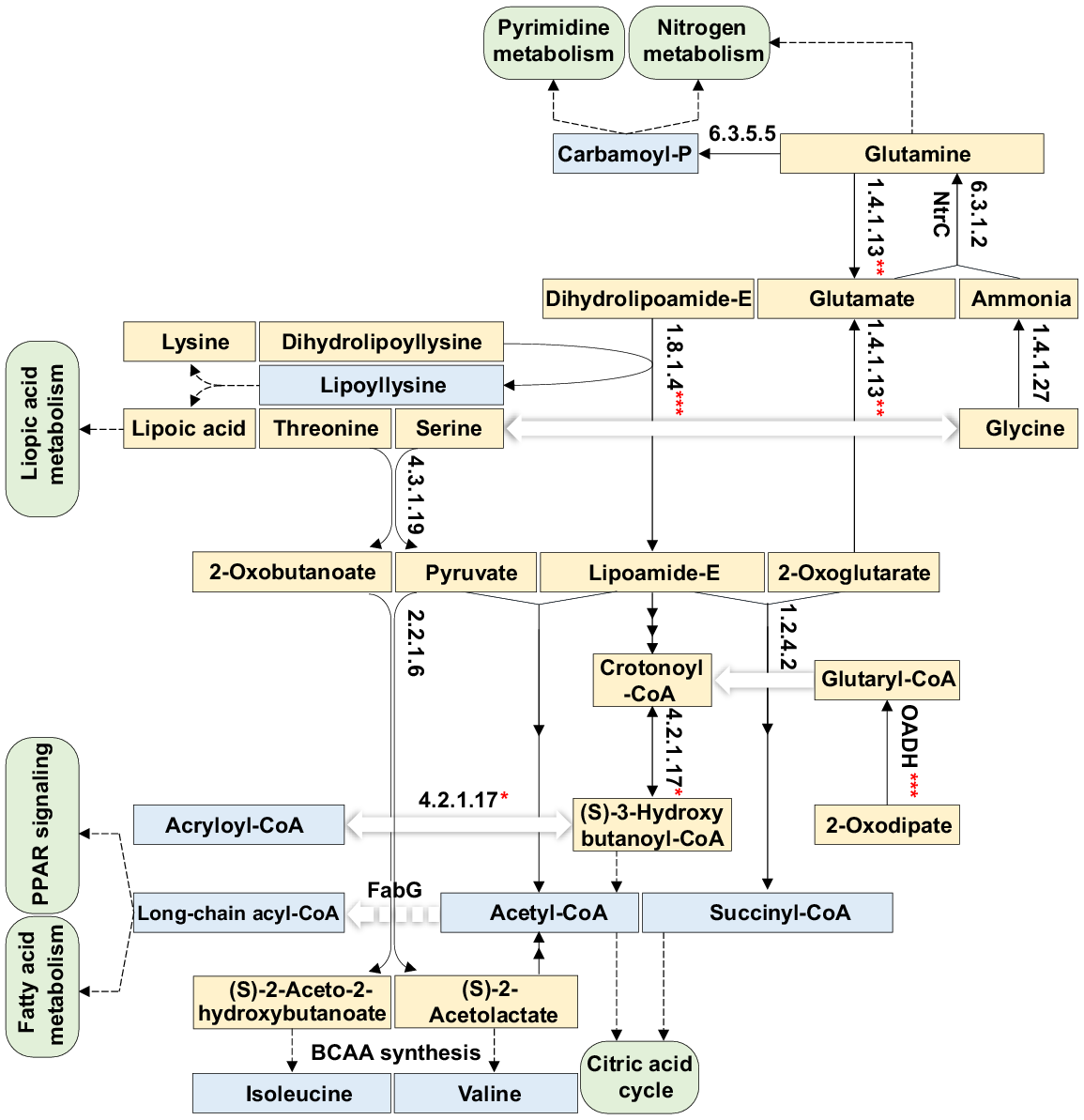
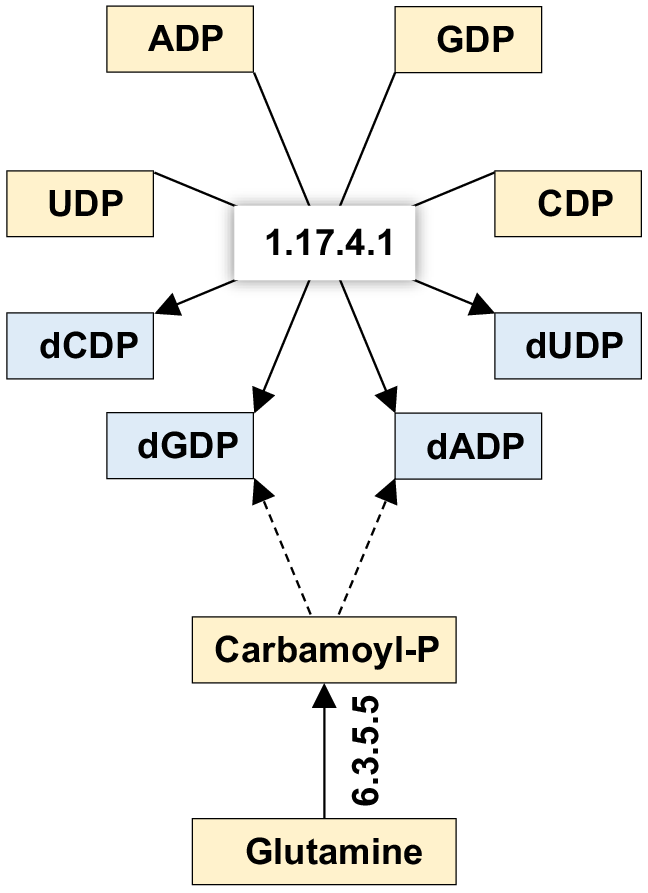
To streamline the analysis, pathways belonging to the subcategory ‘Global and Overview Maps’ were excluded due to the high complexity (seven pathways). Additionally, the enzyme 5-methyltetrahydrofolate--homocysteine methyltransferase (K00548, EC:2.1.1.13) was exclusively present in the pathway ‘Biosynthesis of amino acids’ within the subcategory ‘Global and Overview Maps’ and was not further analysed. Consequently, 14 enzymes existing in 36 pathways were subjected to in-depth analysis (Figs S4–S39). The results for the abundance (Fig. 6a, Table S11) and relative abundance (Fig. 6b, Table S11) of gene queries at the enzyme KO level were also higher in the rhizospheric soil fungiome of M. oleifera than in the bulk soil fungiome. Genes encoding most of the 14 enzymes were highly abundant in the fungal taxa Rhizophagus, Mucor ambiguus and Mortierella elongata, while that in Mucor circinelloides showed high abundance of one gene only encoding glutamine synthetase (K01915, EC:6.3.1.2) (Fig. 6a). A total of 10 out of these 14 enzymes mediate crosstalking of 27 pathways towards the production of several crucial metabolites, including carbamoyl-P, lipoyllysine, propanoyl-CoA, crotonoyl-CoA, acryloyl-CoA, long-chain acyl-CoA, acetyl-CoA, succinyl-CoA, isoleucine and valine (Figs 7, S4, S6, S8–S16, S18–S32 and S39). Other enriched enzymes in the rhizospheric soil fungiome of M. oleifera include ribonucleoside-diphosphate reductase alpha chain (EC:1.17.4.1, K00525) that exists in the two pathways ‘Purine metabolism’ (map00230) and ‘Pyrimidine metabolism’ (map00240) and was proven to convert the nucleotides UDP, CDP, GDP and ADP to dUDP, dCDP, dGDP and dADP, respectively (Figs 8, S7 and S8).
Abundance (a) and relative abundance (b) of gene queries at the KEGG enzyme level in rhizosphere (R) and bulk (S) soil microbiomes of M. oleifera referring to the most abundant KEGG enzymes across the two types of soil fungiomes. Color boxes in top of different enzyme IDs refer to the most abundant fungus/fungi encoding these enzymes. Enzyme with KO of K00548 exists only in pathway ‘Biosynthesis of amino acids’ of subcategory ‘Global and overview maps’ that is difficult to analyse and was therefore removed from further analysis and discussion. The red dotted line refers to 45% relative abundance at the KEGG enzyme level to ensure higher abundance in the rhizosphere than that in the bulk soil. More information is available in Tables S2 and S11.
Avenues extracted from 27 crosstalking KEGG pathways referring to 10 highly enriched enzymes in the rhizospheric fungiome of M. oleifera. Enzyme EC with one asterisk, with double asterisks or with three asterisks exists twice. Metabolites in orange refer to substrates or intermediate steps of a given avenue, while those in blue refer to vital outputs of these interacting avenues. Green boxes refer to some selected downstream pathways. EC:2.2.1.6 = K01652 = acetolactate synthase I/II/III large subunit, OADH = EC:1.8.1.4 = K00382 = dihydrolipoamide dehydrogenase, EC:1.2.4.2 = K00164 = 2-oxoglutarate dehydrogenase E1 component, EC:6.3.5.5 = K01955 = carbamoyl-phosphate synthase large subunit, EC:6.3.1.2 = K01915 = GlnA = glutamine synthetase, EC:1.4.1.13 = K00265 = glutamate synthase (NADPH/NADH) large chain, EC:4.3.1.19 = K01754 = threonine dehydratase, EC:4.2.1.17 = K01692 = enoyl-CoA hydratase, EC:1.4.1.27 = K00382 = glycine cleavage system, FabG = EC:1.1.1.100 = K00059 = 3-oxoacyl-[acyl-carrier protein] reductase, NtrC = the DNA binding transcriptional regulator of the NtrBC two-component system that induces transcription of GlnALG operon as a response to nitrogen deprivation. BCAA = branched-chain amino acid More information is available in Table S4 and in Figs S4, S6, S8–S16, S18–S32 and S39.
Function of ribonucleoside-diphosphate reductase alpha chain (EC:1.17.4.1 = K00525) as a core enzyme to reduce (or dephosphorylate) ribonucleoside diphosphates and carbamoyl-phosphate synthase large subunit (EC:6.3.5.5 = K01955) to biosynthesise pyrimidines in the rhizosphere fungiome of M. oleifera. Metabolites in orange refer to substrates of a given avenue, while those in blue refer to vital outputs of these avenues. Further details can be found in Table S4, Figs S7 and S8.
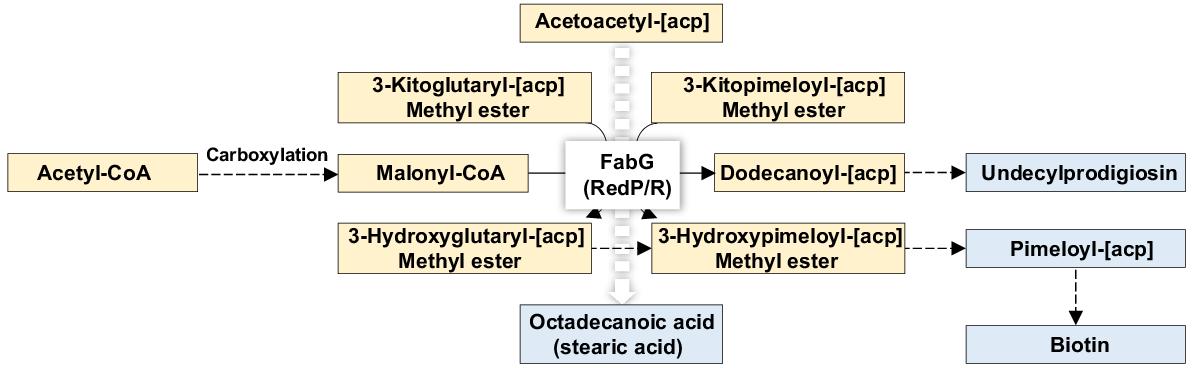
Moreover, the enzyme 3-oxoacyl-[acyl-carrier protein] reductase (FabG, RedP/R, EC:1.1.1.100, K00059) shown in Fig. 9 also participates in a number of metabolic processes in the three pathways ‘Biotin metabolism’, ‘Prodigiosin biosynthesis’ and ‘Fatty acid biosynthesis’ (Figs S5, S17 and S26, respectively). Note that an extra passage for this enzyme exists in Fig. 7. Important metabolites downstream the avenues that involves the latter enzyme include long-chain Acyl-CoA, undecylprodigiosin, pimeloyl-[acp] and octadecanoic acid (Figs 7 and 9). Three other enzymes of which two have the same EC number, e.g. EC:2.7.7.7 (K02335 and K02337), while one has a separate EC number, e.g. EC:2.7.7.6, were enriched in rhizospheric fungiome of M. oleifera (Figs S33–S38, Table S4). These three enzymes are DNA polymerase III subunit alpha of pathways ‘DNA replication’, ‘Mismatch repair’ and ‘Homologous recombination’, DNA polymerase I of pathways ‘DNA replication’, ‘Base excision repair’, ‘Nucleotide excision repair’ and ‘Homologous recombination’ and DNA-directed RNA polymerase subunit beta of pathway ‘RNA polymerase’ (Table S4).
Function of FabG (RedP/R) referring to 3-oxoacyl-[acyl-carrier protein] reductase (EC:1.1.1.100 = K00059) as a core enzyme to orchestrate several vital metabolic processes in the rhizospheric fungiome of M. oleifera. Metabolites in orange refer to substrates or intermediate steps of a given avenue, while those in blue refer to vital outputs of these interacting avenues. One more function of this enzyme is shown in Fig. 7. More information is available in Table S4 and in Figs S5, S17 and S26.
Discussion
Symbiotic associations of the rhizospheric soil fungiome of Moringa oleifera
In this section, we explore the potential symbiotic relationships among the diverse fungal community present in the rhizosphere of M. oleifera. Our KEGG enzyme analysis has identified several dominant fungal genera, notably Rhizophagus, Mucor, Phycomyces, Mortierella, Absidia and Basidiobolus (Fig. 2). Among these, Rhizophagus, Mucor, and Mortierella exhibit a high abundance of genes associated with various KEGG enzymes (Fig. 6a), indicating the pivotal roles in nutrient cycling and plant growth. The extension of fungal hyphae as root-like structures allows plants to efficiently access soil moisture and essential nutrients (Floudas et al. 2020). This interaction not only facilitates a reprogramming of metabolic pathways within both plant and fungiome but also establishes a crucial plant-fungus biological circuit that enhances the acquisition of nutrients and metabolites from the rhizospheric fungiome (Kaur and Suseela 2020; Rayko et al. 2021). Moreover, these fungal genera, particularly Rhizophagus, Mortierella, and Absidia (Ozimek and Hanaka 2021; Zhao et al. 2022), provide multiple ecological benefits, including carbon and nitrogen cycling, and phytohormone production (Floudas et al. 2020).
Recent studies emphasise the significance of mycorrhiza helper bacteria (MHB) in facilitating the growth and colonisation of mycorrhizal fungi (Xing et al. 2018). In this tripartite interaction, arbuscular mycorrhizal fungi (AMF) promote root exudation, thereby enhancing bacterial proliferation and activity (Hodge and Storer 2015). Additionally, phosphate-solubilising bacteria in the soil contribute to mineral solubilisation, while mycorrhizal mycelia transport solubilised phosphate back to the plant roots (Gahan and Schmalenberger 2014; Hodge and Storer 2015). The symbiotic relationship between plants and AMF, particularly those in the genus Rhizophagus from the phylum Glomeromycota, is mediated by phytochemicals such as flavonoids and strigolactones that stimulate fungal spore germination and subsequent colonisation (Santoyo et al. 2021; Lidoy et al. 2023). AMF act as obligatory symbionts, forming associations with a wide array of plant species, providing essential nutrients, including nitrogen and phosphorus, in exchange for carbon and lipids (Chen et al. 2018). Moreover, AMF enhance the capacity of plants to biosynthesise primary and secondary metabolites, including amino acids and fatty acids that are crucial for pathogen resistance and environmental stress tolerance (Cartabia et al. 2021).
The symbiotic relationships between plants and soil endophytic fungi, particularly within the genus Mucor, are primarily mediated by strigolactone that promotes fungal colonisation and stimulates hyphal growth (Rozpądek et al. 2018a, 2018b). In exchange, Mucor enhances plant growth, metal tolerance and nutrient uptake while relying on reduced carbon from the host plant (Baron and Rigobelo 2022). Similarly, the saprotrophic genus Mortierella plays a critical role in degrading hemicelluloses, thus generating sugars that are accessible to plants in the soil (Boddy and Hiscox 2016). In addition, Mortierella provides polyunsaturated fatty acids that are vital for the composition of membrane phospholipids, facilitating signal transduction in plants, and organic acids with antifungal, nematocidal and antibacterial properties (Shemshura et al. 2018; DiLegge et al. 2019). Emerging evidence suggests that the saprotrophic genus Absidia also has beneficial interactions with plants, supplying essential compounds such as chitin and chitosan (Kaczmarek et al. 2019; Zhao et al. 2022). Despite this, there is limited understanding of the substantial enzyme enrichment attributed to Phycomyces blakesleeanus and Basidiobolus meristosporus within the rhizospheric soil of M. oleifera. Prior studies indicate that P. blakesleeanus exhibits phototropic and gravitropic growth and is responsive to various chemicals (Cerdá-Olmedo 2001). This suggests that the enzyme abundance may be influenced by specific root exudates from M. oleifera. Moreover, certain Basidiobolus species are known to thrive on decayed plant matter in the soil (Yang 1962) that may explain the enzyme prevalence linked to organic decay. Currently, the literature lacks substantial evidence regarding the potential symbiotic relationships between these fungi and plants or bacteria, highlighting the need for further investigation into the ecological roles.
Contribution of fungal KEGG enzymes to the growth of Moringa oleifera
We reveal that 10 out of 14 enzymes involved in 27 metabolic pathways are significantly enriched within the rhizospheric fungiome of M. oleifera (Fig. 7). These enzymes play crucial roles in the production of various intermediate metabolites such as carbamoyl-P, lipoyllysine, propanoyl-CoA, crotonoyl-CoA, acryloyl-CoA, long-chain acyl-CoA, acetyl-CoA, succinyl-CoA, isoleucine and valine (Fig. 7). For example, the enzyme carbamoyl-phosphate synthase large subunit (EC:6.3.5.5) facilitates the production of carbamoyl-P that is integral in pyrimidine synthesis and nitrogen metabolism (Strauss et al. 2020) (Figs 7, 8, S8 and S10). The upstream metabolic processes involve the glycine cleavage system (EC:1.4.1.27) that generates ammonia from glycine (Fig. S21) and glutamate synthase (NADPH/NADH) large chain (EC:1.4.1.13), converting 2-Oxoglutarate (Fig. S10) and glutamine (Fig. S25) to glutamate (Fig. 7). Glutamate synthetase (EC:6.3.1.2) subsequently catalyses the condensation of glutamate and ammonia to produce glutamine (Figs 7 and S10). The regulation of this metabolic pathway is mediated by the DNA-binding transcriptional regulator/activator (NtrC) of the NtrBC two-component system (Figs 7 and S39) (Yang et al. 2021) that triggers the transcription of the GlnALG operon to maintain nitrogen homeostasis, especially during nitrogen depletion (De Carlo et al. 2006).
The enzyme dihydrolipoamide dehydrogenase (EC:1.8.1.4) plays a critical role in linking mitochondrial protein lysine residues with lipoic acid to form lipoyllysine, and mediating the oxidation of dihydrolipoyllysine to produce lipoamide-E (Figs 7 and S11). Lipoyllysine functions as a storage compound and is involved in the metabolism of lipoic acid, that is essential for cellular growth and energy coordination (Solmonson and DeBerardinis 2018). Additionally, the enrichment of 2-oxoglutarate dehydrogenase E1 component (EC:1.2.4.2) suggests a vital step in the biosynthesis of succinyl-CoA, initially initiated by dihydrolipoamide dehydrogenase (EC:1.8.1.4). This enzyme catalyses the decarboxylation of oxoglutarate to yield succinyl-CoA (Figs 7 and S6), an important intermediate in the citric acid cycle and a high-energy thiol ester molecule (Fig. 7) (Cole and Eastoe 2014). Dihydrolipoamide dehydrogenase (EC:1.8.1.4) is also involved in the oxidative decarboxylation of pyruvate into acetyl-CoA (Figs 7 and S6) (Xiao et al. 2018). Previous research has indicated the significance of this enzyme in eukaryotic energy metabolism (Babady et al. 2007), emphasising the necessity for both fungi and plants. The enzyme known as 2-oxoadipate dehydrogenase is involved in the oxidative decarboxylation of 2-oxoadipate (OA), leading to the production of glutaryl-CoA, crucial for mitochondrial metabolism (Figs 7 and S6) (Nemeria et al. 2018).
The enzyme responsible for crotonoyl-CoA biosynthesis that serves as a substrate for acetyl-CoA synthesis, utilises glutaryl-CoA (Figs 7 and S14). The enriched enzyme enoyl-CoA hydratase (EC:4.2.1.17) produces (S)−3-Hydroxybutanoyl-CoA by hydrating the double bond between the second and third carbons on 2-trans/cis-enoyl-CoA (or Crotonoyl-CoA) (Figs 7 and S14). This enzyme plays an essential role in fatty acid metabolism for ATP and acetyl-CoA production (Figs 7 and S27), and facilitates the dehydration of 3-hydroxypropionyl-CoA to yield acryloyl-CoA that has nutritional and membrane-stabilising properties (Figs 7 and S28) (Teufel et al. 2009). Acetyl-CoA is a key player in energy and carbon synthesis through various metabolic pathways, including lipid and carbohydrate metabolism, and is regarded as a fundamental component of life (Ljungdahl 2009; Martin 2020). The fungiome of M. oleifera demonstrates an enriched downstream pathway featuring the enzyme 3-oxoacyl-[acyl-carrier protein] reductase (FabG, EC:1.1.1.100), indicating involvement in long-chain acyl-CoA biosynthesis, where acetyl-CoA acts as the primary substrate (Figs 7 and S26). Long-chain acyl-CoA participates in the signaling cascade of peroxisome proliferator-activated receptors (PPAR), influencing fatty acid metabolism and overall cellular development (Ezzeddini et al. 2021). PPAR is a class of receptor proteins that function as transcription factors controlling gene expression for cellular development, differentiation and metabolism (carbohydrate, lipid, etc.) (Dunning et al. 2014).
The pathway for the interconversion of amino acids, particularly between serine and glycine, is prominent in the ‘Glyoxylate and dicarboxylate metabolism’ (Figs 7 and S21). In the context of M. oleifera’s fungiome, the pathway appears to favour the conversion of glycine to serine that subsequently feeds into the production of glutamine and carbamoyl-phosphate via several key enzymes, including the glycine cleavage system (EC:1.4.1.27), glutamine synthetase (EC:6.3.1.2) and carbamoyl-phosphate synthase large subunit (EC:6.3.5.5) (Figs 7, S8 and S10). The reverse reaction involves threonine dehydratase (EC:4.3.1.19) and acetolactate synthase I/II/III large subunit (EC:2.2.1.6), crucial for branched-chain amino acid (BCAA) synthesis, specifically isoleucine and valine (Figs 7, S11 and S13) (Neinast et al. 2019). The bi-functional threonine dehydratase catalyses the conversion of L-threonine to 2-oxobutanoate and ammonia while also converting serine to pyruvate (Figs 7, S11 and S13) (Fuller and Petzke 2017). The resulting 2-oxobutanoate can be transformed into L-isoleucine through intermediate metabolites generated by acetolactate synthase I/II/III (EC:2.2.1.6). BCAAs, such as valine and isoleucine, are essential for various plant functions, enhancing signaling pathways, glucose metabolism and protein synthesis (Neinast et al. 2019).
The enzyme carbamoyl-phosphate synthase large subunit (EC:6.3.5.5), alongside ribonucleoside-diphosphate reductase alpha chain (EC:1.17.4.1), serves as a crucial component for pathways associated with ‘Replication and repair.’ The activities of DNA polymerase III subunit alpha and DNA polymerase I (both EC:2.7.7.7) further illustrate the role of these enzymes in DNA replication (Figs 8 and S33–S37, Table S4). Additionally, the enzyme DNA-directed RNA polymerase subunit beta’ (EC:2.7.7.6), enriched in the rhizospheric fungiome of M. oleifera, is vital for RNA synthesis (Figs 8 and S38, Table S4). Notably, ribonucleoside-diphosphate reductase generates dUDP, a nucleotide not commonly used in standard DNA replication or transcription, highlighting the potential utility within eukaryotic organisms, including both plants and fungi. This enzyme is capable of deoxidising three other nucleotides – CTP, ADP and GTP – raising the possibility of dUDP serving a unique role in nucleotide metabolism. The enriched fungiome of M. oleifera may provide essential nucleotides for replication and repair processes, including base excision and mismatch repair (Figs S33–S38).
The enzyme 3-oxoacyl-[acyl-carrier protein] reductase (FabG, RedP/R, EC:1.1.1.100) is integral to several critical metabolic pathways within the rhizospheric fungiome of M. oleifera (Figs 9, S5, S17 and S26). Main metabolites generated through these pathways include undecylprodigiosin (Figs 9 and S17), pimeloyl-acyl carrier protein [acp] (Figs 9 and S5), octadecanoic acid (Figs 9 and S26) and biotin (Figs 9 and S5). FabG is essential in fatty acid and polyunsaturated fatty acid biosynthesis that constitutes key components of plant and fungal membrane phospholipids, thus promoting plant signal transduction (Shimakata and Stumpf 1982). The biosynthesis of undecylprodigiosin requires Malonyl-CoA, formed through the carboxylation of acetyl-CoA (Figs 9 and S26), serving as a primary substrate that plays a crucial role in chain elongation during fatty acid biosynthesis (Nelson et al. 2008). Furthermore, undecylprodigiosin is noted for the potential pharmaceutical applications, including anticancer properties, although enrichment thereof in the rhizospheric soil of M. oleifera remains unexplained. Pimeloyl-[acp] is a key intermediate in biotin biosynthesis (Fig. 9) (Zeng et al. 2020), while biotin (vitamin H/B7) is an essential micronutrient necessary for various cellular functions, acting as a cofactor for carboxylation and decarboxylation reactions (Beckett 2007). Additionally, octadecanoic acid (or stearic acid) is a vital saturated fatty acid with numerous biological roles (Soliman and Zahran 2022), further underscoring the nutritional contributions of the rhizospheric fungiome to M. oleifera.
The findings from the rhizospheric fungiome of M. oleifera present a compelling landscape of intricate biochemical and metabolic interactions, crucial for plant vitality and environmental adaptation. When comparing these insights with data from other studies, certain thematic overlaps and notable divergences emerge, reflecting the unique ecological roles of rhizospheric microorganisms across different plant species. In the case of M. oleifera, the dominance of fungi from the phylum Mucoromycota, particularly Rhizophagus, Mucor ambiguus and Mortierella elongata, underscores the importance of these fungal taxa in facilitating key metabolic processes within the rhizosphere. The synthesis of vital metabolites such as acetyl-CoA, succinyl-CoA and isoleucine is critical for cellular development, energy metabolism and membrane integrity. These metabolites contribute to DNA repair mechanisms, transcription and signaling pathways – critical for plant health under varying environmental conditions. Such enzymatic activity appears to align with the general trend seen in other studies, where microbial communities, particularly in the rhizosphere, contribute significantly to plant growth and stress tolerance. For example, the research on Bacopa monnieri (Kushwaha et al. 2023) highlights the beneficial role of metal-tolerant plant growth-promoting rhizobacteria (PGPR) such as Cupriavidus basilensis and Ralstonia syzygii, that enhance plant biomass and photosynthetic capacity under copper and iron stress. This parallels the role of rhizospheric fungi in M. oleifera, where fungal enzymes not only regulate nitrogen but also contribute to stress tolerance by facilitating energy metabolism and stabilising cellular complexes. Both fungal and bacterial symbioses mitigate stress responses through metabolite production, indicating a shared mechanism of promoting plant resilience in adverse environmental conditions.
Furthermore, the studies on Aspalathus linearis (Muofhe and Dakora 2000) and Cicer arietinum (Wouterlood et al. 2005) provide additional context for understanding how root-associated microorganisms modify rhizosphere conditions to enhance nutrient uptake. While the former focuses on pH modulation by legumes to counteract soil acidity, the latter reveals the constitutive exudation of carboxylates in response to phosphorus deficiency. These mechanisms, that involve exudation of organic acids to solubilise nutrients, can be seen as complementary to the metabolic activity of fungal taxa in Moringa rhizospheres. Both Moringa and Aspalathus demonstrate adaptive traits for improving nutrient acquisition – via microbial symbiosis or direct biochemical manipulation of the rhizosphere – thereby reinforcing plant growth and stress tolerance under challenging soil conditions.
In contrast, the investigation of Canola (Rumberger and Marschner 2004) and Chickpea (Wouterlood et al. 2005) reveals the dynamic relationship between plant growth and the composition of rhizospheric microbial communities. Specifically, Canola cultivars demonstrated variation in the concentration of 2-phenylethylisothiocyanate (PEITC) and the influence thereof on bacterial community dynamics, suggesting that plant-derived metabolites directly shape rhizosphere microbiomes. This observation complements findings from M. oleifera, where fungal enzymes produce metabolites that drive not only the plant’s metabolic pathways but also the microbial interactions. In both cases, metabolites serve as signals, guiding microbial activity and contributing to a robust, dynamic symbiosis. Lastly, the research on olive trees (Bonetto et al. 2024) highlights the influence of arbuscular mycorrhizal fungi (AMF) in mitigating drought stress through enhanced water and nutrient uptake. This reinforces the idea that fungal associations in Moringa oleifera may similarly enhance plant adaptability to abiotic stresses. Both studies emphasise the role of fungi in improving plant resilience – whether through water regulation, nutrient acquisition or metabolite biosynthesis – by fostering symbiotic relationships that optimise resource utilisation.
Thus, while each plant species exhibits unique microbial interactions and metabolic pathways tailored to the associated environmental conditions, a common thread emerges: the rhizospheric microbiomes – whether fungal or bacterial – serve as critical players in regulating plant growth, nutrient dynamics and stress responses. The insights drawn from Moringa oleifera not only align with established findings across different plant species but also offer a distinctive perspective on how fungiome can orchestrate complex biochemical networks that underlie plant health, energy metabolism and environmental adaptability.
Conclusion
In conclusion, this study underscores the pivotal role of KEGG enzyme-encoding genes in the soil rhizospheric fungiome of M. oleifera, highlighting the symbiotic plant-fungi interactions that enhance plant vitality. Our findings corroborate previous research on fungi’s role in boosting plant health through nutrient modulation and stress resilience. Fungi exhibit exceptional abilities, including cellulose degradation, heavy metal biosorption and soil structure enhancement, improving both moisture retention and drainage. Nevertheless, further exploration is necessary to elucidate the ecological impact of these enzymes. Metabolic engineering and genetic transformation may offer pathways to optimise enzyme activity, enhancing plant resilience across diverse environments.
Data availability
The datasets generated and analysed during the current study are available in the European Nucleotide Archive (ENA) (https://www.ebi.ac.uk/ena/browser/) repository under bioproject no. PRJEB55112 with run accession nos. ERR12764799, ERR10100771 and ERR10100772 for rhizosphere soil biosamples and ERR12764800, ERR10100774 and ERR10100781 for bulk soil biosamples.
Declaration of funding
This research received funding from Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2025R357), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
Author contributions
Conceptualisation, ASA, AAl-Andal, NMA, HWA, FMA, MYR, SAA, AAAlnahari, FOS, AMA, RSJ and AAAbulfaraj; methodology, ASA, AAl-Andal, NMA, HWA and FMA; software, MYR, SAA, AAAlnahari, FOS, AMA, RSJ and AAAbulfaraj; validation, ASA, AAl-Andal, NMA, HWA, FMA and MYR; formal analysis, ASA, AAl-Andal, NMA, HWA, FMA, MYR and SAA; investigation, AAAlnahari, FOS, AMA, RSJ and AAAbulfaraj; resources, SAA, AAAlnahari, RSJ and AAAbulfaraj; data curation, ASA, AAl-Andal, NMA, HWA, FMA, MYR, SAA, AAAlnahari, FOS, AMA, RSJ and AAAbulfaraj; writing – original draft preparation, ASA, AAl-Andal, NMA, HWA, FMA, MYR, SAA, AAAlnahari, FOS, AMA, RSJ and AAAbulfaraj; writing – review and editing, RSJ and AAAbulfaraj; visualisation, ASA, AAl-Andal, NMA, HWA, FMA, MYR, SAA, AAAlnahari, FOS, AMA, RSJ and AAAbulfaraj; supervision, ASA, AAl-Andal, NMA, HWA, FMA, MYR, SAA, AAAlnahari, FOS, AMA, RSJ and AAAbulfaraj; project administration, RSJ, AAAbulfaraj and ASA; funding acquisition, ASA. All authors have read and agreed to the published version of the manuscript.
Acknowledgements
The authors acknowledge with thanks Princess Nourah bint Abdulrahman University Researcher Supporting Project number (PNURSP2024R357), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
References
Al-Eisawi DM, Al-Ruzayza S (2015) The flora of holy Mecca district, Saudi Arabia. International Journal of Biodiversity and Conservation 7(3), 173-189.
| Crossref | Google Scholar |
Anderson MJ (2001) A new method for non-parametric multivariate analysis of variance. Austral Ecology 26(1), 32-46.
| Crossref | Google Scholar |
Ashy RA, Jalal RS, Sonbol HS, Alqahtani MD, Sefrji FO, Alshareef SA, et al. (2023) Functional annotation of rhizospheric phageome of the wild plant species Moringa oleifera. Frontiers in Microbiology 14, 1166148.
| Crossref | Google Scholar |
Babady NE, Pang Y-P, Elpeleg O, Isaya G (2007) Cryptic proteolytic activity of dihydrolipoamide dehydrogenase. Proceedings of the National Academy of Sciences of the United States of America 104(15), 6158-6163.
| Crossref | Google Scholar |
Baldrian P (2003) Interactions of heavy metals with white-rot fungi. Enzyme and Microbial Technology 32(1), 78-91.
| Crossref | Google Scholar |
Baron NC, Rigobelo EC (2022) Endophytic fungi: a tool for plant growth promotion and sustainable agriculture. Mycology 13(1), 39-55.
| Crossref | Google Scholar |
Beckett D (2007) Biotin sensing: universal influence of biotin status on transcription. Annual Review of Genetics 41, 443-464.
| Crossref | Google Scholar | PubMed |
Boddy L, Hiscox J (2016) Fungal ecology: principles and mechanisms of colonization and competition by saprotrophic fungi. Microbiology Spectrum 4(6),.
| Crossref | Google Scholar |
Bonetto M, Cofré N, Calvo F, Silvente S (2024) Effects of arbuscular mycorrhizal fungi in the rhizosphere of two olive (Olea europaea) varieties Arbequina and Barnea under water deficit conditions. Functional Plant Biology 51(7), FP24108.
| Crossref | Google Scholar |
Borrel G, Brugere J-F, Gribaldo S, Schmitz RA, Moissl-Eichinger C (2020) The host-associated archaeome. Nature Reviews Microbiology 18(11), 622-636.
| Crossref | Google Scholar |
Cartabia A, Tsiokanos E, Tsafantakis N, Lalaymia I, Termentzi A, Miguel M, et al. (2021) The arbuscular mycorrhizal fungus Rhizophagus irregularis MUCL 41833 modulates metabolites production of Anchusa officinalis L. under semi-hydroponic cultivation. Frontiers in Plant Science 12, 724352.
| Crossref | Google Scholar |
Cerdá-Olmedo E (2001) Phycomyces and the biology of light and color. FEMS Microbiology Reviews 25(5), 503-512.
| Crossref | Google Scholar |
Chen M, Arato M, Borghi L, Nouri E, Reinhardt D (2018) Beneficial services of arbuscular mycorrhizal fungi–from ecology to application. Frontiers in Plant Science 9, 1270.
| Crossref | Google Scholar |
De Carlo S, Chen B, Hoover TR, Kondrashkina E, Nogales E, Nixon BT (2006) The structural basis for regulated assembly and function of the transcriptional activator NtrC. Genes & Development 20(11), 1485-1495.
| Crossref | Google Scholar |
DiLegge MJ, Manter DK, Vivanco JM (2019) A novel approach to determine generalist nematophagous microbes reveals Mortierella globalpina as a new biocontrol agent against Meloidogyne spp. nematodes. Scientific Reports 9(1), 7521.
| Crossref | Google Scholar |
Dunning KR, Anastasi MR, Zhang VJ, Russell DL, Robker RL (2014) Regulation of fatty acid oxidation in mouse cumulus-oocyte complexes during maturation and modulation by PPAR agonists. PLoS ONE 9(2), e87327.
| Crossref | Google Scholar |
Ezzeddini R, Taghikhani M, Salek Farrokhi A, Somi MH, Samadi N, Esfahani A, Rasaee MJ (2021) Downregulation of fatty acid oxidation by involvement of HIF-1α and PPARγ in human gastric adenocarcinoma and related clinical significance. Journal of Physiology and Biochemistry 77, 249-260.
| Crossref | Google Scholar | PubMed |
Floudas D, Bentzer J, Ahren D, Johansson T, Persson P, Tunlid A (2020) Uncovering the hidden diversity of litter-decomposition mechanisms in mushroom-forming fungi. The ISME Journal 14(8), 2046-2059.
| Crossref | Google Scholar |
Frąc M, Hannula SE, Bełka M, Jędryczka M (2018) Fungal biodiversity and their role in soil health. Frontiers in Microbiology 9, 707.
| Crossref | Google Scholar |
Fu L, Niu B, Zhu Z, Wu S, Li W (2012) CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28(23), 3150-3152.
| Crossref | Google Scholar |
Fuller BT, Petzke KJ (2017) The dietary protein paradox and threonine 15N-depletion: pyridoxal-5’-phosphate enzyme activity as a mechanism for the δ15N trophic level effect. Rapid Communications in Mass Spectrometry 31(8), 705-718.
| Crossref | Google Scholar | PubMed |
Gahan J, Schmalenberger A (2014) The role of bacteria and mycorrhiza in plant sulfur supply. Frontiers in Plant Science 5, 723.
| Crossref | Google Scholar |
Gómez AV, Angulo KJO (2014) Revisión de las características y usosde la planta Moringa Oleífera. Investigación & Desarrollo 22(2), 309-330.
| Google Scholar |
Gopalakrishnan L, Doriya K, Kumar DS (2016) Moringa oleifera: a review on nutritive importance and its medicinal application. Food Science and Human Wellness 5(2), 49-56.
| Crossref | Google Scholar |
Gupta B, Ahmed K (2020) Moringa oleifera: a bibliometric analysis of international publications during 1935–2019. Pharmacognosy Reviews 14(28), 82-90.
| Crossref | Google Scholar |
Hodge A, Storer K (2015) Arbuscular mycorrhiza and nitrogen: implications for individual plants through to ecosystems. Plant and Soil 386, 1-19.
| Crossref | Google Scholar |
Hurt RA, Qiu X, Wu L, Roh Y, Palumbo AV, Tiedje JM, Zhou J (2001) Simultaneous recovery of RNA and DNA from soils and sediments. Applied and Environmental Microbiology 67(10), 4495-4503.
| Crossref | Google Scholar |
Huson DH, Mitra S, Ruscheweyh H-J, Weber N, Schuster SC (2011) Integrative analysis of environmental sequences using MEGAN4. Genome Research 21(9), 1552-1560.
| Crossref | Google Scholar | PubMed |
Huson DH, Beier S, Flade I, Górska A, El-Hadidi M, Mitra S, et al. (2016) MEGAN community edition-interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Computational Biology 12(6), e1004957.
| Crossref | Google Scholar |
Kaczmarek MB, Struszczyk-Swita K, Li X, Szczesna-Antczak M, Daroch M (2019) Enzymatic modifications of chitin, chitosan, and chitooligosaccharides. Frontiers in Bioengineering and Biotechnology 7, 243.
| Crossref | Google Scholar |
Kalibbala HM, Wahlberg O, Hawumba TJ (2009) The impact of Moringa oleifera as a coagulant aid on the removal of trihalomethane (THM) precursors and iron from drinking water. Water Supply 9(6), 707-714.
| Crossref | Google Scholar |
Kanehisa M, Goto S, Hattori M, Aoki-Kinoshita KF, Itoh M, Kawashima S, et al. (2006) From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Research 34(90001), D354-D357.
| Crossref | Google Scholar |
Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M (2014) Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Research 42(D1), D199-D205.
| Crossref | Google Scholar |
Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M (2016a) KEGG as a reference resource for gene and protein annotation. Nucleic Acids Research 44(D1), D457-D462.
| Crossref | Google Scholar |
Kanehisa M, Sato Y, Morishima K (2016b) BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. Journal of Molecular Biology 428(4), 726-731.
| Crossref | Google Scholar |
Karlsson FH, Fåk F, Nookaew I, Tremaroli V, Fagerberg B, Petranovic D, et al. (2012) Symptomatic atherosclerosis is associated with an altered gut metagenome. Nature Communications 3(1), 1245.
| Crossref | Google Scholar |
Karlsson FH, Tremaroli V, Nookaew I, Bergström G, Behre CJ, Fagerberg B, et al. (2013) Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 498(7452), 99-103.
| Crossref | Google Scholar |
Kaur S, Suseela V (2020) Unraveling arbuscular mycorrhiza-induced changes in plant primary and secondary metabolome. Metabolites 10(8), 335.
| Crossref | Google Scholar |
Kumar A, Naaz F, Kushwaha A, Chaudhary P, Srivastav P (2016) Present review on phytochemistry, neutraceutical, antimicrobial, antidiabetic, biotechnological and pharmacological characteristics of Moringa oleifera Linn. BMR Phytomedicine 2(1), 1-17.
| Google Scholar |
Kushwaha RK, Joshi SM, Bajaj R, Mastan A, Kumar V, Patel H, et al. (2023) Copper and iron metal resistant rhizospheric bacteria boost the plant growth and bacoside A content in Bacopa monnieri under stress conditions. Functional Plant Biology 50(6), 482-496.
| Crossref | Google Scholar | PubMed |
Layeghifard M, Hwang DM, Guttman DS (2017) Disentangling interactions in the microbiome: a network perspective. Trends in Microbiology 25(3), 217-228.
| Crossref | Google Scholar | PubMed |
Legendre P, Anderson MJ (1999) Distance-based redundancy analysis: testing multispecies responses in multifactorial ecological experiments. Ecological Monographs 69(1), 1-24.
| Crossref | Google Scholar |
Li W, Godzik A (2006) Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22(13), 1658-1659.
| Crossref | Google Scholar |
Li J, Jia H, Cai X, Zhong H, Feng Q, Sunagawa S, et al. (2014) An integrated catalog of reference genes in the human gut microbiome. Nature Biotechnology 32(8), 834-841.
| Crossref | Google Scholar |
Lidoy J, Berrio E, García M, España-Luque L, Pozo MJ, López-Ráez JA (2023) Flavonoids promote Rhizophagus irregularis spore germination and tomato root colonization: a target for sustainable agriculture. Frontiers in Plant Science 13, 1094194.
| Crossref | Google Scholar |
Ljungdahl LG (2009) A life with acetogens, thermophiles, and cellulolytic anaerobes. Annual Review of Microbiology 63, 1-25.
| Crossref | Google Scholar | PubMed |
Martin WF (2020) Older than genes: the acetyl CoA pathway and origins. Frontiers in Microbiology 11, 817.
| Crossref | Google Scholar |
Mende DR, Waller AS, Sunagawa S, Järvelin AI, Chan MM, Arumugam M, et al. (2012) Assessment of metagenomic assembly using simulated next generation sequencing data. PLoS ONE 7(2), e31386.
| Crossref | Google Scholar |
Milla PG, Penalver R, Nieto G (2021) Health benefits of uses and applications of Moringa oleifera in bakery products. Plants 10(2), 318.
| Crossref | Google Scholar |
Muofhe ML, Dakora FD (2000) Modification of rhizosphere pH by the symbiotic legume Aspalathus linearis growing in a sandy acidic soil. Functional Plant Biology 27(12), 1169-1173.
| Crossref | Google Scholar |
Neinast M, Murashige D, Arany Z (2019) Branched chain amino acids. Annual Review of Physiology 81, 139-164.
| Crossref | Google Scholar | PubMed |
Nemeria NS, Gerfen G, Nareddy PR, Yang L, Zhang X, Szostak M, Jordan F (2018) The mitochondrial 2-oxoadipate and 2-oxoglutarate dehydrogenase complexes share their E2 and E3 components for their function and both generate reactive oxygen species. Free Radical Biology and Medicine 115, 136-145.
| Crossref | Google Scholar | PubMed |
Nielsen HB, Almeida M, Juncker AS, Rasmussen S, Li J, Sunagawa S, et al. (2014) Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nature Biotechnology 32(8), 822-828.
| Crossref | Google Scholar | PubMed |
Oh J, Byrd AL, Deming C, Conlan S, NISC Comparative Sequencing Program, Kong HH, Segre JA (2014) Biogeography and individuality shape function in the human skin metagenome. Nature 514(7520), 59-64.
| Crossref | Google Scholar |
Ozimek E, Hanaka A (2021) Mortierella species as the plant growth-promoting fungi present in the agricultural soils. Agriculture 11(1), 7.
| Crossref | Google Scholar |
Panda S, Kar A, Sharma P, Sharma A (2013) Cardioprotective potential of N,α-L-rhamnopyranosyl vincosamide, an indole alkaloid, isolated from the leaves of Moringa oleifera in isoproterenol induced cardiotoxic rats: in vivo and in vitro studies. Bioorganic & Medicinal Chemistry Letters 23(4), 959-962.
| Crossref | Google Scholar |
Rayko M, Sokornova S, Lapidus A (2021) Fungal metagenome of chernevaya taiga soils: taxonomic composition, differential abundance and factors related to plant gigantism. Journal of Fungi 7(11), 908.
| Crossref | Google Scholar |
Reimand J, Isserlin R, Voisin V, Kucera M, Tannus-Lopes C, Rostamianfar A, et al. (2019) Pathway enrichment analysis and visualization of omics data using g: profiler, GSEA, Cytoscape and EnrichmentMap. Nature Protocols 14(2), 482-517.
| Crossref | Google Scholar | PubMed |
Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26(1), 139-140.
| Crossref | Google Scholar |
Rockwood JL, Anderson BG, Casamatta DA (2013) Potential uses of Moringa oleifera and an examination of antibiotic efficacy conferred by M. oleifera seed and leaf extracts using crude extraction techniques available to underserved indigenous populations. International Journal of Phytotherapy Research 3(2), 61-71.
| Google Scholar |
Rozpądek P, Domka A, Ważny R, Nosek M, Jędrzejczyk R, Tokarz K, Turnau K (2018a) How does the endophytic fungus Mucor sp. improve Arabidopsis arenosa vegetation in the degraded environment of a mine dump? Environmental and Experimental Botany 147, 31-42.
| Crossref | Google Scholar |
Rozpądek P, Domka AM, Nosek M, Ważny R, Jędrzejczyk RJ, Wiciarz M, Turnau K (2018b) The role of strigolactone in the cross-talk between Arabidopsis thaliana and the endophytic fungus Mucor sp. Frontiers in Microbiology 9, 441.
| Crossref | Google Scholar |
Rumberger A, Marschner P (2004) 2-Phenylethylisothiocyanate concentration and bacterial community composition in the rhizosphere of field-grown canola. Functional Plant Biology 31(6), 623-631.
| Crossref | Google Scholar | PubMed |
Santoyo G, Gamalero E, Glick BR (2021) Mycorrhizal-bacterial amelioration of plant abiotic and biotic stress. Frontiers in Sustainable Food Systems 5, 672881.
| Crossref | Google Scholar |
Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C (2011) Metagenomic biomarker discovery and explanation. Genome Biology 12(6), R60.
| Crossref | Google Scholar |
Shami AY, Abulfaraj AA, Refai MY, Barqawi AA, Binothman N, Tashkandi MA, et al. (2022) Abundant antibiotic resistance genes in rhizobiome of the human edible Moringa oleifera medicinal plant. Frontiers in Microbiology 13, 990169.
| Crossref | Google Scholar |
Shemshura ON, Shemsheyeva ZN, Sadanov AK, Lozovicka B, Kamzolova SV, Morgunov IG (2018) Antifungal potential of organic acids produced by Mortierella alpina. International Journal of Engineering and Technology 7(4.38 Special Issue 38), 1218-1221.
| Crossref | Google Scholar |
Shimakata T, Stumpf PK (1982) Purification and characterizations of β-Ketoacyl-[acyl-carrier-protein] reductase, β-hydroxyacyl-[acyl-carrier-protein] dehydrase, and enoyl-[acyl-carrier-protein] reductase from Spinacia oleracea leaves. Archives of Biochemistry and Biophysics 218(1), 77-91.
| Crossref | Google Scholar |
Soliman HM, Zahran HA (2022) Synthesis of a new hydrophobic coating film from stearic acid of buffalo fat. Scientific Reports 12(1), 18465.
| Crossref | Google Scholar |
Solmonson A, DeBerardinis RJ (2018) Lipoic acid metabolism and mitochondrial redox regulation. Journal of Biological Chemistry 293(20), 7522-7530.
| Crossref | Google Scholar |
Tashkandi M, Baz L (2023) Function of CAZymes encoded by highly abundant genes in rhizosphere microbiome of Moringa oleifera. Saudi Journal of Biological Sciences 30(3), 103578.
| Crossref | Google Scholar |
Tashkandi MA, Jalal RS, Baz L, Refai MY, Shami A, Ashy RA, et al. (2022) Functional interpretation of cross-talking pathways with emphasis on amino acid metabolism in rhizosphere microbiome of the wild plant Moringa oleifera. Agriculture 12(11), 1814.
| Crossref | Google Scholar |
Teufel R, Kung JW, Kockelkorn D, Alber BE, Fuchs G (2009) 3-hydroxypropionyl-coenzyme A dehydratase and acryloyl-coenzyme A reductase, enzymes of the autotrophic 3-hydroxypropionate/4-hydroxybutyrate cycle in the Sulfolobales. Journal of Bacteriology 191(14), 4572-4581.
| Crossref | Google Scholar |
Wang Z, Li T, Wen X, Liu Y, Han J, Liao Y, DeBruyn JM (2017) Fungal communities in rhizosphere soil under conservation tillage shift in response to plant growth. Frontiers in Microbiology 8, 1301.
| Crossref | Google Scholar |
Wouterlood M, Lambers H, Veneklaas EJ (2005) Plant phosphorus status has a limited influence on the concentration of phosphorus-mobilising carboxylates in the rhizosphere of chickpea. Functional Plant Biology 32(2), 153-159.
| Crossref | Google Scholar | PubMed |
Xiao R, Wang X, Jiang L, Tang H (2018) Research and application of lipoic acid in plants. IOP Conference Series: Earth and Environmental Science 108, 042100.
| Crossref | Google Scholar |
Xing R, Yan H-Y, Gao Q-B, Zhang F-Q, Wang J-L, Chen S-L (2018) Microbial communities inhabiting the fairy ring of Floccularia luteovirens and isolation of potential mycorrhiza helper bacteria. Journal of Basic Microbiology 58(6), 554-563.
| Crossref | Google Scholar |
Yang B-Y (1962) Basidiobolus meristosporus of Taiwan. Taiwania 8, 17-27.
| Google Scholar |
Yang Z, Li Q, Yan Y, Ke X, Han Y, Wu S, et al. (2021) Master regulator NtrC controls the utilization of alternative nitrogen sources in Pseudomonas stutzeri A1501. World Journal of Microbiology and Biotechnology 37(10), 177.
| Crossref | Google Scholar |
Zeng Q, Yang Q, Jia J, Bi H (2020) A Moraxella virulence factor catalyzes an essential esterase reaction of biotin biosynthesis. Frontiers in Microbiology 11, 148.
| Crossref | Google Scholar |
Zhao H, Nie Y, Zong T-K, Wang Y-J, Wang M, Dai Y-C, Liu X-Y (2022) Species diversity and ecological habitat of Absidia (Cunninghamellaceae, Mucorales) with emphasis on five new species from forest and grassland soil in China. Journal of Fungi 8(5), 471.
| Crossref | Google Scholar |
Zouboulis CC, Hossini AM, Hou X, Wang C, Weylandt KH, Pietzner A (2023) Effects of Moringa oleifera seed oil on cultured human sebocytes in vitro and comparison with other oil types. International Journal of Molecular Sciences 24(12), 10332.
| Crossref | Google Scholar |