
Morphological and molecular evidence for major re-circumscriptions in and eight new species of Melichrus R.Br. (Ericaceae subfam. Epacridoideae) in eastern Australia
Helen T. Kennedy A B * , Ian R. H. Telford A , Darren Crayn C D , Jeremy J. Bruhl A and Rose L. Andrew A
A B * , Ian R. H. Telford A , Darren Crayn C D , Jeremy J. Bruhl A and Rose L. Andrew A
A
B
C
D
Abstract
The genus Melichrus R.Br. has received very little taxonomic attention and treatments have largely disagreed on species delimitation. The eastern Australian clade of Melichrus was last revised in 1958. Over 60 years later we present new, in-depth evidence for species delimitation. A morphological dataset (90 individuals from 68 populations, scored for 26 characters) was analysed using NMDS ordination with Bayesian Inference cluster modelling (mclust) and UPGMA hierarchical clustering to detect morphological discontinuities in the genus that may indicate species boundaries. Discontinuities in the morphological analyses were compared with those apparent in clustering (principal component analysis, SplitsTree Neighbour-Net, STRUCTURE and conStruct) and statistical analyses (HE, HO, FIS and FST) of a DArTseq SNP dataset of 548 samples from 110 populations of Melichrus. These new lines of evidence form the basis of detailed recommendations for a revised species taxonomy of Melichrus including the description of eight new species, recircumscription of previously described species and the correction of a longstanding nomenclatural misapplication.
Keywords: DArTseq, Epacridoideae, Ericaceae, Melichrus, morphometrics, phenetics, reduced representation, species delimitation, taxonomy.
Introduction
Melichrus R.Br. (Ericaceae: Epacridoideae) is a genus of shrubs endemic to Australia. The taxonomy currently consists of six described species (Paterson 1958; Kennedy et al. 2020), all confined to eastern Australia. In addition, six phrase-named (Barker 2005) species are accepted by the Australian Plant Census (APC) (Council Heads of Australian Herbaria, CHAH, https://biodiversity.org.au/nsl/services/APC, accessed 4 May 2023), three in eastern Australia and three in Western Australia. The eastern taxa occur in Queensland (6 spp.), New South Wales (6 spp.) and Victoria (1 sp.), and are distributed from coastal to drier regions well inland (Fig. 1). Species of Melichrus from Western Australia and the eastern states form sister clades (Powell et al. 1997; Puente-Lelièvre et al. 2016), allowing this study to focus on the eastern Australian Melichrus. Herein, unless otherwise specified, ‘Melichrus’ refers to the eastern Australian clade.
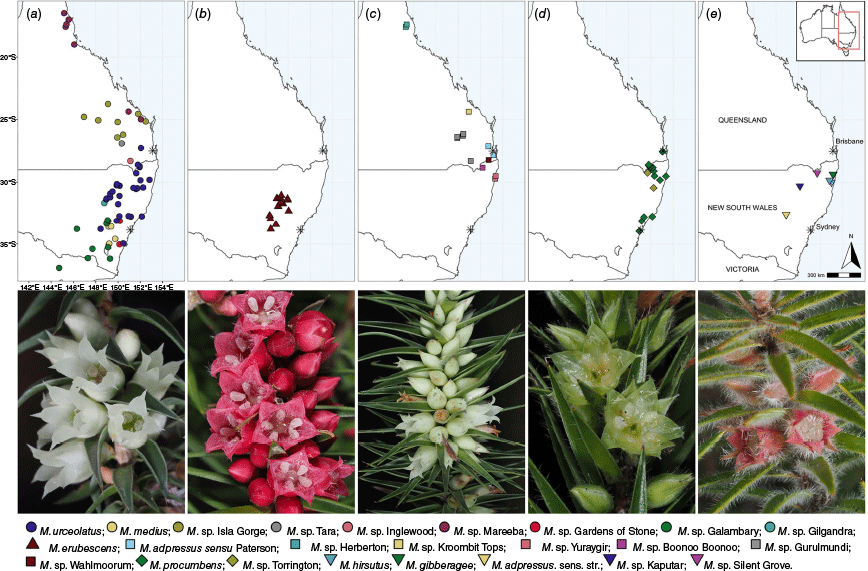
Geographic locations of all Melichrus samples included in the DArTseq analysis and images of a selection of species. Each panel contains samples from a single sensu Paterson group (see Table 1) and a photograph of one OTU from the respective group. Each point represents a population sampled. See Table 1 for a textual description of symbols. (a) M. urceolatus sensu Paterson: M. urceolatus from Gardens of Stone NP, NSW (NE 109885) by J.J. Bruhl. (b) M. erubescens sensu Paterson: M. erubescens from Goobang National Park, NSW (NE 111997) by H.T. Kennedy. (c) M. adpressus sensu Paterson: M. adpressus sensu Paterson from Plunkett CA, Qld (NE 110785) by H.T. Kennedy. (d) M. procumbens sensu Paterson: M. procumbens from Myall Lakes NP, NSW (NE 111989) by H.T. Kennedy. (e) OTUs that were not included in Paterson (1958): M. hirsutus from Flaggy Creek NR NSW (NE 109319) by J. J. Bruhl.
Melichrus has received very little taxonomic attention, with the treatments differing on species and generic delimitation (Brown 1810; De Candolle 1839; Bentham 1868; von Mueller 1868; Paterson 1958). Since the genus was last revised by Betsy R. Paterson (now Prof. Betsy R. Jackes AM) in 1958, several putative new species have been proposed. Melichrus requires revision using an updated approach to resolve problematic species boundaries, test putative new species and produce a biologically meaningful and usable species-level taxonomy.
A species name represents a scientific hypothesis of species boundaries that can be tested experimentally (Rouhan and Gaudeul 2021). This includes phrase names that are a means of recognising and labelling a putative new species for testing, prior to formal description – a well-established practice in Australia (e.g. https://florabase.dbca.wa.gov.au/help/names). As theoretical and technological advancements have been made, new sources of evidence have been used to test these hypotheses. The integrative taxonomic method combines different data sources and statistical analyses such as morphology, genetics and chemistry to test species hypotheses (Dayrat 2005; Padial et al. 2010; Carstens et al. 2013). Using several data sources concurrently has successfully addressed long-standing taxonomic questions, and can improve species discovery and provide a conservative evidence base for differentiating closely related species (see Denham et al. 2019; Hosegood et al. 2020; Collins et al. 2022).
The unified species concept equates species with separately evolving metapopulation lineages and importantly provides flexible criteria for delimiting species that can be met using multiple lines of evidence (de Queiroz 2007). Separately evolving lineages may be recognised through the identification of corresponding discontinuities between samples across separate data sources, in this case using morphological and genetic datasets. Phylogenetic information should be considered, with the aim of describing monophyletic or, with consideration of evolutionary processes, paraphyletic species (Hörandl and Stuessy 2010). The integrative taxonomic method with the adoption of the unified species concept provides a robust framework for species discovery and validation.
Despite the theoretical and technological advancements that have enabled the production of well-evidenced species-level taxonomies, challenges to identifying species boundaries remain. Speciation does not always follow a simple bifurcating tree, as sometimes imagined; reticulation, polyploidy, ongoing gene flow and population dynamics all create patterns that can increase the complexity of identifying separately evolving metapopulation lineages (Fujita et al. 2012). The process of speciation can produce discontinuities in some datasets (e.g. molecular data) that are not seen in or are contradicted by other datasets (e.g. morphological data); in some instances, these are referred to as cryptic species (Struck et al. 2018). Furthermore, different types of analysis carry assumptions and biases that can create challenges for taxonomic interpretation (e.g. PCA v. STRUCTURE). In such cases, looking for further evidence (e.g. anatomical or ploidy data) and new methods for analysing a dataset – as the integrative taxonomic method encourages before making taxonomic decisions – is wise. Pragmatic decisions, made on the balance of evidence and interpreted within a speciation theory framework can be required to produce a working taxonomy for use by stakeholders. In this scenario, being explicit about which evidence does or does not support the presented topological result and remembering that a species’ taxonomy, as any model, is a hypothesis, is important.
A taxonomy that captures both the evolutionary history and morphological diversity of a group of organisms provides an essential foundation for studies on the biology, and better management and conservation of the named species (Taxonomy Decadal Plan Working Group 2018; Rouhan and Gaudeul 2021). Species and subspecies are the units of diversity most commonly used for conservation listing, and therefore biodiversity management decisions and action (Wege et al. 2015). How well species are delimited has a profound effect on how effectively biodiversity is conserved (Mace 2004; Stanton et al. 2019; Ewart et al. 2020; Hosegood et al. 2020).
History of taxonomic study of Melichrus
Luis Née made the earliest known herbarium specimen collection of a Melichrus species in Port Jackson, New South Wales, in 1793 (Orchard 1999; JSTOR Global Plants, see http://plants.jstor.org/). The specimen, housed at Herbario Real Jardín Botánico (MA 476478), was later described by Antonio José Cavanilles, as Ventenatia procumbens Cav. (Cavanilles 1797, p. 28 & tab. 349). The first use of the name Melichrus was by Robert Brown in Prodr. Fl. Nov. Holland. (Brown 1810) in which two species were described; Melichrus urceolatus R.Br. and M. rotatus R.Br. Brown described Melichrus urceolatus from material collected by botanical illustrator Ferdinand Lukas Bauer in 1804, at ‘(J.) [Newcastle; New South Wales]’ (Brown 1810, p. 539; JSTOR Global Plants, see http://plants.jstor.org/; BM 000797778) and Melichrus rotatus from personal collections made at ‘(J.) [Sydney; New South Wales]’ and ‘(T.) [Fraser Island; Queensland]’ between 1802 and 1804 (Brown 1810, p. 539; Orchard 1999). Brown (1810) acknowledged that Melichrus rotatus was synonymous with Ventenatia procumbens but did not make the recombination using the epithet ‘procumbens’ within the new genus, as nomenclatural precedence was not yet a formal protocol at that time. When Brown (1810) transferred the only other congeneric Ventenatia humifusa to Astroloma R.Br., the name Ventenatia fell out of use. Later, nomenclatural precedence was acknowledged by English botanist George Claridge Druce, the name Melichrus procumbens (Cav.) Druce was formalised (Druce 1917) and M. rotatus R.Br. was synonymised as a superfluous name.
Brown (1810) also placed Melichrus in a hierarchical classification for the first time when erecting the family Epacridaceae and established two infrafamilial sections based on fruit type. Melichrus was included in section 1, characterised by indehiscent fruits with one ovule per locule. This section was subsequently formalised as the tribe Styphelieae (Bartling 1830) and remains currently recognised (Watson 1967; Kron et al. 2002; Puente-Lelièvre et al. 2016; Crayn et al. 2020).
Allan Cunningham collected and recognised three new species of Melichrus. The first was collected from the ‘Plains at Bathurst’ (Field 1825, p. 323) and called M. medius A.Cunn. due to appearing to be ‘intermediate between the two already described species [presumably referring to M. rotatus and M. urceolatus]’ (Field 1825, p. 323). Augustin Pyramis De Candolle described M. erubescens A.Cunn. ex DC. and M. adpressus A.Cunn. ex DC. from dried specimens collected by Cunningham in ‘plains … around Liverpool’ and ‘sterile land near Wellington Valley’ respectively (De Candolle 1839, p. 740).
In the late 1860s, both Ferdinand von Mueller (1868) and George Bentham (1868) revised Melichrus in respective treatises on the Australian flora. Bentham retained a great deal of the generic structure of Styphelieae established by Brown, whereas Mueller had a broader concept of the genus Styphelia and expanded this to incorporate many genera, including Melichrus. Generic delimitation within Styphelieae remained uncertain, with early authors on the family divided (see Bentham and Hooker 1876; Drude 1887; Watson 1967) until recent molecular work decisively delimited Melichrus as a monophyletic genus and Styphelia was recircumscribed (Puente-Lelièvre et al. 2016; Crayn et al. 2020).
Bentham and Mueller also differed regarding species delimitation within the group. Bentham, working entirely from dried specimens, could not detect notable morphological differences among most of the described species of Melichrus. Accordingly, M. adpressus, M. erubescens and M. medius were subsumed into M. urceolatus, and rotatus was retained (Bentham 1868). Mueller, like Bentham, synonymised M. erubescens and M. medius with M. urceolatus. Melichrus urceolatus was subsequently transferred to Styphelia as S. urceolata (R.Br.) F.Muell. and M. rotatus was retained as S. rotata F.Muell. A third species, Styphelia cunninghamii F.Muell. was named from two collections – Hermann Beckler: Hastings River (New South Wales) and Walter Hill from southern Queensland (von Mueller 1868, p. 39). Mueller chose to synonymise M. adpressus that was originally described from collections made in central western New South Wales with Styphelia cunninghamii presumably based on De Candolle’s description of M. adpressus as ‘a very distinctive species… [with] adpressedly imbricate leaves’ (De Candolle 1839, p. 740). Bentham also cited the Beckler and Hill specimens but did not find these to be morphologically distinct from M. urceolatus.
The species-level taxonomy of Melichrus was not studied again until 1958. On the basis of much wider sampling and more detailed morphological observations than earlier authors, Paterson (1958) recognised four species; M. urceolatus, M. erubescens, M. procumbens and M. adpressus.
Since Paterson’s revision, two narrowly endemic species of Melichrus were collected for the first time in northern New South Wales. These were recently described as M. hirsutus J.B.Williams ex H.T.Kenn. & I.Telford and M. gibberagee J.B.Williams ex H.T.Kenn. & J.J.Bruhl (Kennedy et al. 2020) based on strong morphological evidence, therefore six described species are currently recognised.
Taxonomic uncertainty, nomenclatural confusion and putative new species
In this study, Paterson’s (1958) revision (i.e. the status quo taxonomy) was used as a framework for explicitly testing species limits. Table 1 provides a full list of published and phrase-named species for Melichrus, accompanied by geographic distribution and a textual description of the symbol by which these are represented in figures.
| Operational taxonomic unit | Geographic distribution | Description of symbols used | |
|---|---|---|---|
| Melichrus sensu Paterson | |||
| M. urceolatus R.Br., Prodr. Fl. Nov. Holland: 539 (1810) | ‘Burnett River, Q’ld., to St Arnaud, Vict., with isolated specimens at Herberton and Jericho Q’ld.’ (Paterson 1958, p. 310) | dark blue circle | |
| M. erubescens A.Cunn. ex DC., Prodr. 7(2): 740 (1839) | ‘Central western slopes and plains of New South Wales’ (Paterson 1958, p. 309) | brown triangle | |
| M. adpressus A.Cunn. ex DC., Prodr. 7(2): 740 (1839) | ‘Isolated localities centred on Herberton, Inglewood and coastal area from Brisbane to Clarence River’ (Paterson 1958, p. 307) | light blue square | |
| M. procumbens (Cav.) Druce, Rep. Bot. Soc. Exch. Club Brit. Isles 4(Suppl. 2): 635 (1917) | ‘From Gympie through south-east Queensland and Northern Tablelands to Sydney’ Paterson 1958, p. 305) | dark green diamond | |
| Operational Taxonomic Units segregated from Melichrus urceolatus sensu Paterson | |||
| M. medius A.Cunn. Geogr. Mem. New South Wales [Field]: 341 (1825) | Near Bathurst NSW, as stated in the protologue | yellow circle | |
| M. sp. Isla Gorge (P.Sharpe + 601) Qld Herbarium A,B | QMA, QLE, QDD, QWB, QBN, QWA | olive-green circle | |
| M. sp. Tara (D.Halford Q2259) Qld Herbarium A | QDD, near Tara | grey circle | |
| M. sp. Inglewood (A.R.Bean 1652) Qld Herbarium A | QDD, near Inglewood | pink circle | |
| M. sp. Galambary (H.T. Kennedy 7) NE Herbarium | NCT, NST, NSP, VMI, VEH, CAN | dark green circle | |
| M. sp. Gardens of Stone (H.T. Kennedy 46) NE Herbarium | Gardens of Stone National Park & Morton National Park, NSW | red circle | |
| M. sp. Gilgandra (H.T. Kennedy 128) NE Herbarium | North east of Gilgandra, NSW | bluish-green circle | |
| M. sp. Mareeba (H.T. Kennedy 92) NE Herbarium | QPC, QBN, QKN, QCO | plum-coloured circle | |
| Operational Taxonomic Units segregated from Melichrus adpressus sensu Paterson | |||
| M. sp. Boonoo Boonoo (H.T. Kennedy 35) NE Herbarium | Boonoo Boonoo National Park, NSW | purple square | |
| M. sp. Gurulmundi (H.T. Kennedy 69) NE Herbarium | QMA, QLE, QDD, QBN | grey square | |
| M. sp. Herberton (H.T. Kennedy 91) NE Herbarium | Herberton, Qld | bluish-green square | |
| M. sp. Kroombit Tops (H.T. Kennedy 87) NE Herbarium | Kroombit Tops National Park, Qld | yellow square | |
| M. sp. Wahlmoorum (H.T. Kennedy 78) NE Herbarium | Mount Maroon summit in Barney National Park, Qld | brown square | |
| M. sp. Yuraygir (H.T. Kennedy 29) NE Herbarium | NNC, coastal QMO | pink square | |
| Operational Taxonomic Units segregated from Melichrus procumbens sensu Paterson | |||
| M. sp. Torrington (H.T. Kennedy 32) NE Herbarium | Torrington National Park, Cathedral Rock National Park and Glen Innes area, NSW | olive-green diamond | |
| Operational Taxonomic Units not included in Paterson (1958) | |||
| M. adpressus sens. str. | Wellington Valley, NSW, as stated in protologue. | yellow inverted triangle | |
| M. hirsutus J.B.Williams ex H.T.Kenn. & I.Telford, Telopea 23: 188 (2020) | From ~30 km north-west of Grafton south to Glenreagh and east to Wooli Road, NSW (14 Aug. 1993, J.B. Williams s.n., isotype: NE 85839) | blue inverted triangle | |
| M. gibberagee J.B.Williams ex H.T.Kenn. & J.J.Bruhl, Telopea 23: 191 (2020) | Single locality (<15 km2) in the Gibberagee area ~50 km south of Casino, NSW (25 July 2019, H.T. Kennedy 28, isotype: NE 109335) | green inverted triangle | |
| M. sp. Kaputar (H.T. Kennedy 25) NE Herbarium | The Governor, Mount Kaputar National Park NSW | dark blue inverted triangle | |
| M. sp. Silent Grove (H.T. Kennedy 33) NE Herbarium | Torrington State Conservation Area, NSW | purple inverted triangle | |
| M. sp. Colo Gorge (I.R. Telford 8659) NE Herbarium | Colo Gorge, NSW | NA | |
| M. sp. Salvator Rosa (M.B. Thomas 244) NE Herbarium | The western extent of the Carnarvon Range, Qld | NA | |
All operational taxonomic units (OTUs) used in this study are listed in column one. Column two provides the geographic distribution and column three the symbol by which these are represented in analyses. The status quo taxonomy as published in Paterson (1958) is listed first. Geographic distributions for these names are as appearing in Paterson (1958). Followed by all OTUs segregated from M. urceolatus sensu Paterson, M. adpressus sensu Paterson and M. procumbens sensu Paterson. The last group of names listed is not represented in Paterson (1958), including the strict application of M. adpressus that was excluded from the revision and several phrase names that represent morphologically distinctive populations or entities unknown in 1958. All OTUs segregated from M. urceolatus sensu Paterson are represented by circles; segregates of M. erubescens sensu Paterson by triangles; segregates of M. adpressus sensu Paterson by squares; segregates of M. procumbens sensu Paterson by diamonds; and all OTUs not included in Paterson (1958) by inverted triangles. Herbarium codes follow the Index Herbariorum. Codes used in the geographic distributions are abbreviations of botanical districts and follow the (Centre for Australian National Biodiversity Research 2018).
Melichrus urceolatus has been maintained as a species through various treatments, albeit with differing limits. Paterson circumscribed M. urceolatus to include a vast geographical area; from Herberton, northern Queensland to Saint Arnaud, Victoria, acknowledging that ‘within this range a large variety of form is shown…’ (Paterson 1958, p. 313). Paterson (1958) provided a brief description of five groups that differed morphologically from the ‘typical condition’ and the approximate geographic range but did not attempt formal subdivision (Paterson 1958, p. 313). A great deal of this variation was noted to occur in Queensland, where three phrase-named putative species have been accepted by the APC (CHAH, accessed 4 May 2023), namely M. sp. Inglewood (A.R.Bean 1652), M. sp. Isla Gorge (P.Sharpe + 601) and M. sp. Tara (D.Halford Q2259).
Several putative species have been segregated from Melichrus urceolatus and informally phrase-named at the N.C.W. Beadle Herbarium (NE) but are not yet listed on the APC, including M. sp. Mareeba (H.T. Kennedy 92) NE Herbarium, M. sp. Galambary (H.T. Kennedy 7) NE Herbarium, M. sp. Gilgandra (H.T. Kennedy 128) NE Herbarium, M. sp. Gardens of Stone (H.T. Kennedy 46) NE Herbarium and M. sp. Salvator Rosa (M.B. Thomas 244) NE Herbarium that were identified during exploratory herbarium and field observations made in the early stages of this study.
Paterson (1958) retained the synonymy of Melichrus medius under M. urceolatus established by Bentham, explaining that no difference was observed in the protologue descriptions or subsequent collections made from the type localities. Improved access to the type specimen and other early collections allows the original definition of M. medius to be considered anew in this study.
Paterson reinstated Melichrus erubescens to species status, explaining that ‘the taxonomic differences are such as to justify specific rank, although the two species [M. urceolatus and M. erubescens] are undoubtedly closely related’ (Paterson 1958, p. 310). As currently delimited, the boundary between M. urceolatus and M. erubescens is unclear. Melichrus sp. Gardens of Stone, M. sp. Gilgandra and M. sp. Galambary, though treated as M. urceolatus by Paterson, all share morphological affinities with M. erubescens, exemplifying the unworkable boundary between the species as currently described.
Several populations of Melichrus that are morphologically similar to both M. erubescens and M. urceolatus have been collected for the first time since Paterson’s revision. These gatherings cannot be confidently assigned to either species and have been given the phrase names M. sp. Silent Grove (H.T. Kennedy 33) NE Herbarium and M. sp. Kaputar (H.T. Kennedy 25) NE Herbarium (Table 1). A key aim of this study was to decisively clarify the boundary between M. erubescens, M. urceolatus and morphologically similar putative species M. sp. Inglewood, M. sp. Isla Gorge, M. sp. Tara, M. sp. Gardens of Stone, M. sp. Gilgandra, M. sp. Galambary, M. sp. Silent Grove and M. sp. Kaputar.
The nomenclatural history of Melichrus adpressus is convoluted. The name M. adpressus was first applied to a collection made by Cunningham near Wellington Valley, New South Wales (De Candolle 1839). Later, Mueller listed M. adpressus as synonymous with Styphelia cunninghamii (von Mueller 1868, p. 39), which was described from specimens collected in northern New South Wales and southern Queensland. In doing so, an illegitimate name was erected and applied to two morphologically and geographically disjunct entities. Paterson (1958, p. 308) stated that:
the type locality of Melichrus adpressus is Wellington Valley but specimens subsequently collected from this area do not agree with the original description, and unfortunately Cunningham’s specimens were unavailable for comparison.
There is uncertainty regarding which specimens Paterson was referring to as ‘subsequently collected’ because no specimens from the Wellington Valley were cited in the revision. Paterson chose to adopt Mueller’s concept of M. adpressus and excluded the strict application of the name in the revision. Easy access to digital images of herbarium specimens (e.g. from JSTOR Global Plants) now greatly facilitates comparison of specimens that were largely inaccessible to earlier workers. See, for example, a specimen of M. adpressus collected by A. Cunningham near Wellington Valley that was seen by De Candolle, compared with a specimen cited by Paterson (Fig. 2). The morphological disparity between these specimens, including differences in leaf size, shape, margin texture, indumentum and density on stems, suggests that the name M. adpressus is misapplied in Paterson (1958). Herein, the name Melichrus adpressus sensu Paterson (or abbreviated to ‘s. Paterson’ in figures) indicates the use in the sense of von Mueller (1868) and Paterson (1958). When referred to in the original, strict sense of De Candolle (1839) this is denoted M. adpressus sens. str. (see Table 1).
Comparison of the application of the name Melichrus adpressus. (a) H. Beckler’s M. adpressus sensu Paterson specimen (MEL 2185645) from the northern rivers region of NSW, likely seen by F. von Mueller and cited as M. adpressus in Paterson (1958). Image reproduced with permission from Royal Botanic Gardens Victoria. (b) A specimen of M. adpressus sens. str. collected by Cunningham near Wellington Valley NSW that was seen by De Candolle (1839) and is a candidate for lectotypification (G 00454459). Image © Conservatoire et Jardin botaniques de la Ville de Genève.

Paterson (1958) noted only minor morphological variability across the range of Melichrus adpressus sensu Paterson. However, observations of herbarium and field specimens made early in this study suggested that this taxonomic consideration may neglect substantial morphological variation. We represent this variation by several phrase names. The northernmost populations of M. adpressus sensu Paterson are restricted to the Herberton area in Queensland and highly geographically disjunct from the southern populations in the state, and were designated as M. sp. Herberton (H.T. Kennedy 91) NE Herbarium. Specimens from the North Coast of New South Wales were morphologically distinctive and phrase named M. sp. Yuraygir (H.T. Kennedy 29) NE. Melichrus sp. Gurulmundi (H.T.Kennedy 69) NE Herbarium was applied to populations of M. adpressus sensu Paterson, found in southern Queensland that also share some morphological affinities with M. urceolatus. Isolated populations of Melichrus resembling M. adpressus sensu Paterson are known from slightly below the rocky outcrop at the summit of Mount Maroon, Queensland (Wahlmoorum of the First Nations’ Yuggera language), the upper slopes of Kroombit Tops National Park, Queensland and the sandy flats of Boonoo Boonoo National Park, New South Wales. The earliest collections of these populations were made in the latter half of the 20th Century, after the publication of Paterson’s (1958) revision. Each population is morphologically distinctive and to each we assigned the respective phrase names M. sp. Wahlmoorum (H.T. Kennedy 78) NE Herbarium, M. sp. Kroombit Tops (H.T. Kennedy 87) NE Herbarium and M. sp. Boonoo Boonoo (H.T. Kennedy 35) NE Herbarium for this study.
Melichrus procumbens was the first species of Melichrus described (as M. rotatus) and the taxonomic status of this morphologically distinctive species has never been questioned (Bentham 1868; von Mueller 1868; Paterson 1958). This species has a geographically disjunct distribution and has been recorded from coastal sands at Hervey Bay south to Sydney, where the type was collected and on usually granitic soils in the New England Tableland bioregion from Stanthorpe to Wollomombi. Paterson (1958, p. 307) described the plants across the disjunct range as ‘morphologically similar’ with the exception of a population near Glen Innes that was ‘in all respects larger … than typical specimens’ and hypothesised to be an autopolyploid. This putative new species was phrase-named Melichrus sp. Torrington (H.T. Kennedy 32) NE Herbarium. Since Paterson’s (1958) revision, a morphologically allied population was collected in Colo Gorge, New South Wales as M. procumbens and is here given the phrase name M. sp. Colo Gorge (I.R. Telford 8659) NE Herbarium. Sampling across the geographic range of M. procumbens is needed to test the relationship between morphologically dissimilar and geographically disjunct populations.
Melichrus hirsutus and M. gibberagee were each collected separately for the first time several years after Paterson’s (1958) revision was published. Despite the geographic proximity in north-eastern NSW, the taxonomic status was strongly supported by distinct morphological characteristics and these were published as new species (Kennedy et al. 2020). These two species are included in this study to test their status as distinct species based on molecular evidence.
Aims and approach of the study
The species-level taxonomy for Melichrus has never been tested using either a numerical morphological approach or any form of genetic data. We use both in an integrative taxonomic approach to determine robust species boundaries in line with the unified species concept. We developed a new, comprehensive morphological dataset representing the eastern Australian Melichrus species and applied an analytical pipeline to thoroughly explore patterns of similarity in the dataset. A complementary DArTseq (Kilian et al. 2012) dataset for the genus was interrogated using various clustering, ancestry estimation and population genetics analyses to identify discontinuities in the dataset, examine population structure and therefore test boundaries of species in the study group. Within the broader aim of species delimitation we address several specific questions, namely: (1) are there six species of Melichrus in eastern Australia (M. urceolatus, M. erubescens, M. procumbens, M. adpressus, M. hirsutus and M. gibberagee) as currently described or are there up to 19 undescribed species as indicated by the phrase names in use at the Queensland Herbarium (BRI) and N.C.W. Beadle Herbarium (NE)? (2) Is Melichrus urceolatus a natural group or has this been used as a ‘catch-all’ taxon for specimens that could not be adequately identified under the status quo taxonomic classification? (3) Where does the boundary lie between the often-confused taxa, M. urceolatus and M. erubescens? We address these and other questions of species’ limits and provide detailed recommendations for a revised taxonomy of Melichrus.
Materials and methods
Name usage and sampling
To test species delimitation in Melichrus, ~150 populations were visited across eastern Australia and specimens collected by the authors and collaborators in 2019–2022. We identified extant populations for each described species that matched the type locality and morphology of the type specimen as closely as possible. These topotype populations represent each published name sensu stricto (e.g. M. urceolatus sens. str.) and are important for clarifying nomenclatural application. Collection sites were selected to represent the full geographic and environmental range of the genus. Collection site choice was informed by earlier collections recorded in The Australasian Virtual Herbarium (AVH, https://avh.chah.org.au, accessed 10 March 2019) and some sightings listed in The Atlas of Living Australia (ALA, https://www.ala.org.au/, accessed 10 March 2019).
Herbarium specimens from at least two plants per population were pressed in the field and dried at 16% relative humidity and 16°C before freezing for 1 week at −30°C for herbarium biosecurity. Flowers, fruits, stems and leaves were pickled in 70% ethanol. Where available, young leaves from at least five plants, spaced ~5 m apart (to avoid sampling direct siblings), were placed into breathable nylon bags and subsequently in sealed containers with silica gel desiccant (Si gel) for DNA extraction (Collins et al. 2022). Descriptions of habit, habitat and associated plants, topography, population size and soil were recorded at each collection site. Permission to collect plant material compliant with The Nagoya Protocol on Access and Benefit-Sharing (2010; see https://www.cbd.int/abs/resources/other.shtml) was acquired prior to each collection.
Early in the study, the most relevant collections of Melichrus and Epacridoideae ‘indets’ were examined including collections held at the Australian National Herbarium (CANB), BRI, NE and the National Herbarium of Victoria (MEL). The National Herbarium of New South Wales (NSW herbarium) was closed to visiting researchers for most of the duration of this study and specimen images were not yet available. The study of specimens facilitated the formation of hypotheses for species delimitation. In this study, such hypotheses are expressed as published names, APC-accepted phrase names (see https://biodiversity.org.au/) and informal phrase names (see https://ncw-beadleherbarium.une.edu.au/#!/explore), and are treated as operational taxonomic units (OTUs) in the analyses (Sokal 1963). Putative species segregated from the species recognised by Paterson or identified since the study follow standard APC format for phrase names (Barker 2005). We used Paterson’s (1958) taxonomy as a framework for testing species delimitation. This framework and name usage is summarised in Table 1. The species recognised in Paterson (1958) are listed first followed by OTUs segregated from each of these recognised species. Lastly, OTUs that were either intentionally excluded or not yet known at the time of Paterson (1958) are listed.
Each name is given a unique colour and symbol combination to label samples by OTU across analyses (see Table 1, Fig. 1). Although colour allocation is randomly assigned, the same symbol is given to all OTUs segregated from the same sensu Paterson species (e.g. all OTUs segregated from M. urceolatus sensu Paterson are indicated by a circle), allowing clustering patterns evident in the results to be easily compared with the sensu Paterson taxonomy.
Morphology
A total of 90 individuals from 68 populations was included in the final morphometric and phenetic analyses; 97% of the sampled populations were also included in the DArTseq sample set, to maximise the complementarity of the datasets. A list of specimens and associated metadata is provided in Supplementary Table S1. Specimens were chosen to capture the full geographic and morphological range of each OTU but availability of suitable material was a limiting factor in some cases. Two OTUs, Melichrus sp. Colo Gorge and M. sp. Salvator Rosa, could not be located in the field. These were excluded from the analyses due to inadequate sampling. Wherever possible, a minimum of four samples per OTU was included. Several OTUs represented by fewer samples in the analyses were either geographically narrowly defined, such as M. sp. Kaputar, M. sp. Wahlmoorum, M. sp. Inglewood and M. sp. Tara, or appeared to be naturally rare and narrowly distributed; e.g. M. sp. Torrington.
During an initial exploratory phase, 205 characters were examined across 85 specimens, to represent a cross-section of the genus. This pilot yielded a total of 21 quantitative and 5 (binary) qualitative characters optimal for analysis (Table 2). The characters selected for statistical analysis were those that: (1) varied between samples, (2) could be scored reasonably easily and accurately, and (3) were selected with the aim of including as many characters as possible while prioritising quantitative characters and minimising missing data.
| Character | Material observed | |
|---|---|---|
| Quantitative characters | ||
| Branchlet diameter (mm) | Dried voucher | |
| Bud length (mm) | EtOH | |
| Bud length/bud width (mm) | EtOH | |
| Sepal apex angle (mm) | EtOH | |
| Corolla lobe length (mm) | EtOH | |
| Corolla lobe length/corolla lobe width (mm) | EtOH | |
| Corolla subapical process length (mm) | EtOH | |
| Corolla subapical process hair length (mm) | EtOH | |
| Corolla tube length (mm) | EtOH | |
| Corolla tube width (mm) | EtOH | |
| Corolla tube length/corolla lobe length (mm) | EtOH | |
| Lamina abaxial surface vein count | Dried voucher | |
| Lamina margin hair maximum length (mm) | Dried voucher | |
| Lamina margin hair minimum length (mm) | Dried voucher | |
| Lamina margin width (mm) | Dried voucher | |
| Lamina tip length (mm) | Dried voucher | |
| Leaf density (leaf bases/2 cm) | Dried voucher | |
| Leaf length (mm) | Dried voucher | |
| Leaf width (mm) | Dried voucher | |
| Leaf length/width (mm) | Dried voucher | |
| Plant height (cm) | In field | |
| Binary characters | ||
| (1) Corolla lobes held erect before spreading (1Y) | Photograph or EtOH | |
| Corolla lobes spreading from base (1N) | ||
| (2) Corolla constricted below opening (2Y) | Photograph or EtOH | |
| Corolla not constricted below opening (2N) | ||
| (3) Leaf abaxial surface central nerve prominent (3Y) | Dried voucher | |
| Leaf abaxial surface central nerve not prominent (3N) | ||
| (4) Young leaves recurved (4Y) | Photograph, dried voucher, EtOH | |
| Young leaves straight or slightly incurved (4N) | ||
| (5) Plant habit dome shaped (5N) | In field | |
| Plant habit not dome shaped (5Y) | ||
Morphological characters were scored either in the field or from photographic, dried and 70% ethanol-fixed specimens (Table 2). Macroscopic characters were measured using digital callipers or a ruler. Micromorphological characters were examined using a Nikon SMZ25 stereo microscope with a Nikon SHR Plan Apo 0.5× or 1× lens and measurements were made using Nikon NIS-Elements software. Descriptive erminology follows Radford et al. (1974) or if absent there, the ‘Flora of Australia’ glossary (McCusker 1999).
Analyses were conducted on a genus-wide dataset and on numerous subsets (Table 3). R (ver. 4.2.0, R Foundation for Statistical Computing, Vienna, Austria, see https://www.r-project.org/) and RStudio (ver. 2.3.4, Posit Software, PBC, Boston, MA, USA, see https://posit.co/products/open-source/rstudio/) were used for all analyses. To assess the suitability of the dataset for phenetic analysis, R package DataExplorer (ver. 0.8.2, B. Cui; see https://CRAN.R-project.org/package=DataExplorer) was used to plot the distribution of scores for each character and calculate the percentage of missing data by character, sample and for the entire dataset. The Pearson’s coefficient of correlation between each pair of characters was calculated. Where characters had a very high degree of correlation (<−0.8 or >0.8) and there was a biological rationale indicating that these may not have been mutually independent, one of the pair of characters was removed. Four ratio characters were included in the dataset. Where one or both components of the ratio were strongly correlated to the ratio character, one of the component characters was removed.
| Dataset name | OTUs included | Percentage missing data; sample count; SNP count (molecular dataset only) | |
|---|---|---|---|
| Morphological dataset | |||
| All OTUs except M. sp. Colo Gorge and M. sp. Salvator Rosa | 3.3; 90 | ||
| Subsets | |||
| 1 | M. procumbens, M. sp. Torrington, M. hirsutus | 6.5; 9 | |
| 2 | M. erubescens, M. sp. Gardens of Stone, M. sp. Silent Grove | 10.1; 12 | |
| 3 | M. sp. Kaputar, M. adpressus sens. str., M. sp. Galambary | 1.0; 11 | |
| 4 | M. sp. Isla Gorge, M. sp. Tara, M. sp. Mareeba | 3.5; 10 | |
| 5 | M. sp. Yuraygir, M. sp. Boonoo Boonoo, M. sp. Herberton, M. sp. Kroombit Tops | 0; 10 | |
| 6 | M. sp. Gilgandra, M. sp. Inglewood | 0; 6 | |
| 7 | M. urceolatus, M. medius, M. sp. Gurulmundi | 2.8; 17 | |
| DArTseq dataset | |||
| Clustering and ancestry | All OTUs except M. sp. Colo Gorge and M. sp. Salvator Rosa | 2.6; 543; 2071 | |
| Population statistics | All OTUs except M. sp. Wahlmoorum, M. sp. Colo Gorge and M. sp. Salvator Rosa | 2.5; 524; 4936 | |
| Subsets | |||
| A (PCA clusters 1–3) | M. urceolatus, M. medius, M. sp. Silent Grove, M. sp. Inglewood, M. procumbens, M. sp. Torrington, M. sp. Yuraygir, M. sp. Boonoo Boonoo, M. sp. Wahlmoorum, M. sp. Gurulmundi, M. sp. Herberton, M. sp. Kroombit Tops, M. adpressus sensu Paterson | 2.3; 287; 2636 | |
| B (PCA clusters 7–10) | M. erubescens, M. sp. Kaputar, M. sp. Gardens of Stone, M. adpressus sens. str., M. sp. Galambary, M. sp. Gilgandra | 2.0; 147; 2710 | |
| C (PCA clusters 3–6) | M. hirsutus, M. gibberagee, M. sp. Isla Gorge, M. sp. Tara, M. sp. Mareeba; M. sp. Yuraygir, M. sp. Boonoo Boonoo, M. sp. Gurulmundi, M. sp. Herberton, M. sp. Kroombit Tops, M. adpressus sensu Paterson | 2.1; 187; 2915 | |
| D (PCA cluster 1) | M. urceolatus, M. medius, M. sp. Silent Grove, M. sp. Inglewood | 2.0; 148; 3024 | |
| E (PCA cluster 2) | M. procumbens, M. sp. Torrington | 1.7; 63; 2725 | |
| F (PCA cluster 3) | M. sp. Yuraygir, M. sp. Boonoo Boonoo, M. sp. Wahlmoorum, M. sp. Gurulmundi, M. sp. Herberton, M. sp. Kroombit Tops | 1.6; 76; 2717 | |
| G (PCA cluster 5) | M. sp. Isla Gorge, M. sp. Tara | 1.5; 60; 2581 | |
| H (PCA cluster 6) | M. hirsutus, M. gibberagee | 0; 16; 1568 | |
| I (PCA cluster 7) | M. adpressus sens. str., M. sp. Galambary, M. sp. Gilgandra | 1.2; 56; 2064 | |
| J (Fig. 8c ) | Northern populations of M. urceolatus (nNNT, QDD), M. sp. Silent Grove, M. sp. Inglewood | 1.6; 43; 2648 | |
Dataset name: number (morphological data) or letter (DArTseq data) chosen to represent each subset. OTUs included: list of OTUs included in each dataset. For subsets of both the morphological and DArTseq datasets the percentage of missing data and number of samples after filtering are provided, additionally for the DArTseq dataset the number of SNPs in each subset is given. Subsets of the morphological dataset are based on the clusters observed in the genus-wide mclust analysis (Fig. 3). Most subsets of the DArTseq dataset are based on clusters arising in the genus-wide PCA (Fig. 7), except subset J, that was based on the ‘northern cluster’ apparent among M. urceolatus samples in the PCA of subset D (i.e. cluster labelled ‘nNNT, QDD’ in Fig. 8c). Codes used to indicate geographic distributions are abbreviations of botanical districts and follow (Centre for Australian National Biodiversity Research 2018), ‘nNNT’ is modified to indicate the northern extent of the Northern Tablelands NSW.
The character matrix was converted to a dissimilarity matrix using Gower’s distance coefficient (package cluster, function ‘daisy’, ver. 2.1.3, M. Maechler, P. Rousseeuw, A. Struyf, M.Hubert and K. Hornik, see https://cran.r-project.org/package=cluster). Gower’s distance coefficient is the most appropriate coefficient for use with datasets comprising a mixture of quantitative and qualitative characters, and a small amount of missing data that are weighted as zero in analyses (Gower 1971). Phenetic methods in which qualitative characters have been used have received criticism, as these can potentially have a disproportionate influence on the result. To better understand the influence of each qualitative character on the results, analyses were run numerous times, using quantitative characters only initially and subsequently adding the qualitative characters in one by one and interrogating the influence on sample distributions (Supplementary Fig. S1).
Sample similarity was analysed and visualised in two complementary ways. A hierarchical clustering phenogram was generated (R package stats, function ‘hclust’, ver. 4.4.2, see https://stat.ethz.ch/R-manual/R-devel/library/stats/html/00Index.html) using the unweighted pair group method with arithmetic mean (UPGMA). Non-metric multidimensional scaling (NMDS) was performed in vegan (function ‘metaMDS’, ver. 2.6-2, J. Oksanen, G. L. Simpson, F. G. Blanchet, et al., see https://CRAN.R-project.org/package=vegan) to visualise clustering patterns in the data using 2–4 dimensions (k). UPGMA and a non-metric ordination algorithm are appropriate metrics for this dataset, as most characters were not normally distributed (Supplementary Fig. S2). The NMDS was run with 30 random starts for k = 2–5. This protocol was determined experimentally to be sufficient to reach a convergent solution for all values of k. To better understand how many dimensions are needed to best visualise the dataset, the ordination distance was plotted against the observed dissimilarity (function ‘stressplot’). A stress value is considered ‘fair’ if <0.2 and ‘good’ if <0.1 (Kruskal 1964). In addition to considering the stress inherent in each ordination, ordinations were visually inspected and where adding an additional dimension did not change the pattern observed, the lower value of k was preferred.
Ordination was performed for subsets 1–7 (Table 3). The subsets were based on the clustering pattern observed in the genus-wide ordination and phenogram, and designed to examine fine-scale morphological patterns in the data.
The phenogram and ordinations were visually inspected for clustering patterns that were potentially representative of taxonomic groupings. Additionally, the mclust program (function ‘mclust’, ver. 5.4.9, see https://CRAN.R-project.org/package=mclust; Scrucca et al. 2016, 2023) was used to investigate clustering patterns that best represent the data under a series of models (Fraley and Raftery 2002; Meudt et al. 2020). mclust chooses among 14 different models describing the shape, volume and orientation of clusters. For a given number of clusters, G, ranging from 1 to 9, mclust will choose the best-fitting model based on the Bayesian information criterion (BIC). mclust also allocates individual observations to each cluster under the optimal model and assesses the uncertainty of the classification for each specimen. Only results from the best-fitting model are shown.
To be potentially recognised as a cluster in mclust analyses, an OTU must have at least one more specimen sampled than the number of dimensions used (Hausdorf and Hennig 2010; Edwards and Knowles 2014). In our dataset the OTUs that did not fulfil this requirement at k = 2 and k = 3 were M. sp. Torrington (n = 2), M. sp. Inglewood (n = 2), M. sp. Kaputar (n = 2), M. sp. Tara (n = 2), M. sp. Wahlmoorum (n = 1) and additionally at k = 3, M. adpressus sens. str. (n = 3).
The influence of each character on the distribution of points in the ordination was examined to help identify diagnostic characters for putative species. The ‘envfit’ function in vegan was used to fit the quantitative characters to the ordination and those with a significant influence (P < 0.05) were visualised in a biplot with arrow length proportional to the correlation between the data for that character and the ordination configuration. The direction of each arrow indicates the axis of the ordination space across which that character changes most rapidly. Examining the magnitude and orientation of each arrow provides insight into potentially diagnostic characters for OTUs. For qualitative characters, the average value for each state was plotted on the ordination to examine the influence of the character on the distribution of samples.
Ordination analysis was replicated for subsets 1–7 (Table 3) to search for potentially diagnostic characters between morphologically similar OTUs that were not detectable at the coarser, genus-wide scale. Subsets were based on the clusters apparent in the genus-wide phenogram and mclust analysis of the NMDS ordination (clusters 1–7 in Fig. 3; subsets summarised in Table 3). These subsets were not subjected to mclust analysis as sample numbers were too low for this analysis to be effective, therefore the NMDS ordinations were visually inspected for clustering patterns. Cluster 8, as defined in the k = 3 genus-wide ordination (Fig. 3c), was not considered suitable for ordination due to inadequate representation of M. sp. Wahlmoorum in the analysis. Cluster 6 also had a low number of samples that should be considered when interpreting NMDS results for this subset.
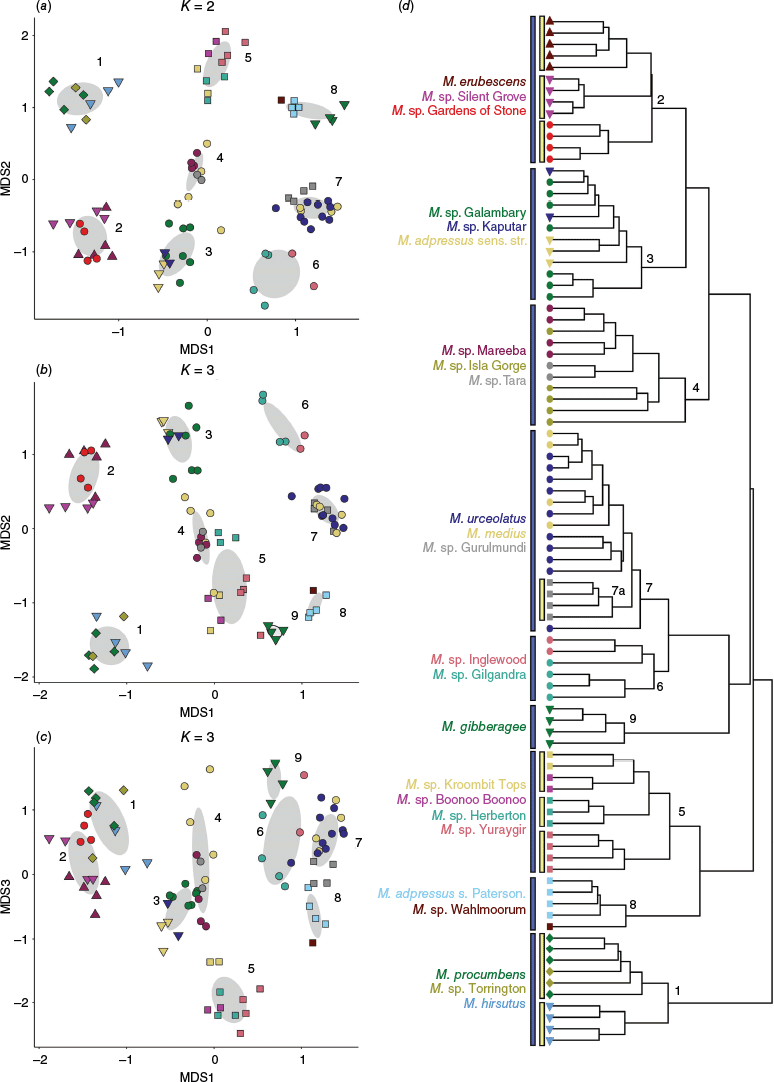
Morphometric analysis of 90 specimens of Melichrus scored for 26 characters showing NMDS ordination and mclust (grey ellipses) results. (a) k = 2, model ‘EII’, best number of clusters = 8. (b, c) k = 3, model ‘EEI’, best number of clusters = 9, axes 1 & 2 and axes 1 & 3. (d) A hierarchical clustering phenogram. Blue bars indicate clusters that correspond between the ordinations and phenogram, yellow bars indicate further clusters that are apparent only in the phenogram. Points represent individual plants. See Fig. 1 for a visual key and Table 1 for a textual description of symbols.
A table of differences was constructed to highlight diagnostic characters (Table 4a–c), using characters that varied between OTUs but that were not captured in the phenetic analyses. These characters were scored for the same specimens used in the phenetic analyses.
| (a) OTUs from clusters 1–3 | Branch habit | Lamina margin hair type | Lamina margin hairs longest near | Leaf apex pungent | Sepal apex angle at fruiting | Corolla colour | Corolla lobe hair distribution | Dry fruit texture | Calyx L. w.r.t. fruit apex | |
|---|---|---|---|---|---|---|---|---|---|---|
| M. procumbens | Decumbent | Scabrous–hispid + pilose | Apex | No | Acute–acuminate | Cream, with or without purple lobes | Pilose all over or only at margins | Wrinkly | > | |
| M. sp. Torrington | Decumbent | Scabrous to hispid + pilose | Apex | No | Unknown | Cream tube, purple lobes | Pilose all over or only at margins | Not seen | Not seen | |
| M. hirsutus | Ascending | Pilose | Base or entire length | Yes | Acuminate | Pink | Pilose at throat | Smooth–wrinkly | > | |
| M. erubescens | Ascending | Minutely scabrous | Base | Yes | Obtuse–subacute | Pink or white | Pilose all over | Smooth–wrinkly | < | |
| M. sp. Silent Grove | Ascending–erect | Minutely scabrous | Base | Yes | Acuminate | White | Pilose at throat | Smooth at apex | > | |
| M. sp. Gardens of Stone | Ascending | Minutely scabrous | Base | Yes | Acuminate | Pink | Pilose all over | Smooth–wrinkly | > | |
| M. sp. Galambary | Decumbent–ascending | Minutely scabrous | Base | Yes | Obtuse–subacute | White | Pilose at throat | Smooth–wrinkly | < or = | |
| M. adpressus sens. str. | Ascending | Minutely scabrous | Base | Yes | Obtuse–subacute | White | Pilose at throat | Smooth–wrinkly | < | |
| M. sp. Kaputar | Ascending | Minutely scabrous | Base | Yes | Obtuse | White | Pilose at throat | Smooth–wrinkly | < |
| (b) OTUs from clusters 4–5 | Branch habit | Lamina margin hair type | Furrows between veins | Sepals pilose | Calyx L. w.r.t. fruit apex | |
|---|---|---|---|---|---|---|
| M. sp. Isla Gorge | Decumbent–ascending | Scabrous–hispid + pilose | Wide | Present or absent | < or = | |
| M. sp. Tara | Ascending | Scabrous–hispid + pilose | Wide | Present | Unknown | |
| M. sp. Mareeba | Decumbent | Scabrous–hispid | Intermediate | Absent | < | |
| M. sp. Herberton | Laxly erect | Scabrous–hispid | Narrow | Absent | < | |
| M. sp. Kroombit Tops | Laxly erect | Scabrous–hispid | Narrow | Present | = or > | |
| M. sp. Boonoo Boonoo | Erect | Scabrous–hispid | Narrow | Present | = or > | |
| M. sp. Yuraygir | Erect | Sparsely scabrous–hispid | Narrow | Present | = or > |
| (c) OTUs from clusters 6–9 | Branch habit | Number of stems at base | Upper lamina surface colour | Furrows between veins | Sepal hairs | Dry fruit texture | Calyx L. w.r.t. fruit apex | |
|---|---|---|---|---|---|---|---|---|
| M. sp. Gilgandra | Ascending | 1(–3) | Glaucous + blue | Narrow | ±Glabrous | Smooth–wrinkly | < | |
| M. sp. Inglewood | Ascending | Many | Greyish-green | Narrow | ±Glabrous | Smooth at apex | > | |
| M. medius | Ascending | 1–5 | Green | Narrow | Pilose | Smooth at apex | > | |
| M. urceolatus | Decumbent– erect | 1–many | Greyish-green–green | Narrow | ±Glabrous or pilose or puberulent | Smooth at apex | > | |
| M. sp. Gurulmundi | Erect | 1(–3) | Green | Narrow | Pilose | Smooth at apex | > | |
| M. adpressus sensu Paterson | Ascending–erect | 2–6 | Shiny + green | Narrow | Pilose | Smooth at apex | > | |
| M. sp. Wahlmoorum | Ascending | 1–5 | Shiny + green | Narrow | ±Glabrous | Not seen | Not seen | |
| M. gibberagee | Erect | 1(–3) | Green | Wide | ±Glabrous | Wrinkly | > |
Abbreviations: <, shorter than; =, equal to; >, longer than; w.r.t., with reference to.
Molecular
A total of 572 dried leaf samples were submitted to Diversity Arrays Technology Pty Ltd (DArT) for extraction of total genomic DNA (Supplementary Table S1). In addition to the 548 samples from 110 populations of eastern Australian Melichrus, this included 12 samples representing the three western Australian phrase-named entities, ten outgroup samples from other genera of Styphelieae (for use in phylogenetic analyses reported on in a future publication) and two technical duplicates (that were found to be surplus to the requirements of the analyses). A single sample, obtained from the only collection of Melichrus sp. Bruce Rock housed at the Western Australian Herbarium (PERTH), failed to produce sequence data.
Single nucleotide polymorphism (SNP) genotypes were determined from sequence reads by DArT using proprietary DArTseq pipelines (Kilian et al. 2012). These pipelines filter and remove poor quality sequence reads before de novo assembly of loci and calling of genotypes. Technical replicates of multiple samples are included and consistency of genotype calls between the replicates scored to provide a ‘reproducibility’ score for each locus. SNP genotypes were called on the whole sample set and subsequently called on a separate partition of samples that included only eastern Melichrus and a reduced selection of outgroup samples. Exploration of the two datasets revealed very little difference in the quality of SNPs between datasets and no difference in the results of analyses. We report only on the original, pre-partitioning sample set, as this returned a larger number of SNPs (95,319 SNPs) than the reduced partition (82,287 SNPs).
The statistical software R (ver. 4.2.0) and RStudio (ver. 2.3.492) were used for all statistical analyses. The package dartR (ver. 1.9.9.1, see https://CRAN.R-project.org/package=dartR; Gruber et al. 2018; Mijangos et al. 2022) was used to manipulate, filter and analyse the data. Only the eastern Australian Melichrus samples were required for this study, therefore the outgroup taxa and two technical duplicates were removed. Any resulting monomorphic loci were removed and the dataset metrics recalculated. The resultant dataset consisted of 548 individual samples and 78,474 SNPs, 76% of which had a reproducibility of 100% (93% of SNPs had a reproducibility of ≥98%). Approximately 24% of SNPs (18,940 SNPs) had <10% missing data and the average call rate for samples was 76%.
Initial exploratory filtering of DArTseq data at different levels of stringency confirmed only minor effects on the clustering of samples in PCA or SplitsTree4 (ver. 4.1, see https://github.com/husonlab/splitstree4; Huson and Bryant 2006). Refined sets of filtered SNPs were subsequently used in all analyses for improved calculation efficiency (Rutherford et al. 2018, Collins et al. 2022). For the entire dataset, all SNPs with a reproducibility of less than 98% were removed (gl.filter.RepAvg) and all remaining loci with more than 5% missing SNP calls were subsequently removed (gl.filter.callrate). To prevent linkage disequilibrium from influencing analyses, one SNP was randomly selected to retain from each of the loci that contained more than one SNP (gl.filter.secondaries). Any individual sample with more than 10% missing data was removed (gl.filter.callrate), except that for data subset F (Table 3), the filter was relaxed to allow up to 11% missing data to retain the single specimen of M. sp. Wahlmoorum in the analysis. SNPs where the frequency of the minor allele (MAF) was less than 0.05 were removed (gl.filter.maf). Finally, any monomorphic loci (gl.filter.monomorphs) were removed and the data metrics recalculated (gl.recalc.metrics). This filtration protocol was used on all sets of data for all analyses except for the population statistics. The filtered dataset used for population statistics was not filtered for minor allele frequency as this may affect estimates of observed heterozygosity (Linck and Battey 2019).
To improve resolution among OTUs and the associated populations in clustering analyses, several subsets of the dataset were made (summarised in Table 3). The dataset was divided into subsets based on the clusters observable in the results of a genus-wide PCA and the major clades in a Neighbour-Net analysis. Subsets were derived from the unfiltered in-group only dataset and subsequently filtered following the procedure outlined above.
PCA (dartR function ‘gl.pcoa’) was used to explore genetic structure between putative species and populations. PCA was performed on the entire dataset and all subsets of the data outlined in Table 3.
SplitsTree4 was used to conduct a Neighbour-Net analysis (Bryant and Moulton 2004) producing a distance-based network of relatedness for samples. The network can represent evolutionary histories with substantial reticulation or discordance, caused by incomplete lineage sorting and introgression (Huson and Bryant 2006). A relationship network of the total dataset was computed using the default software settings.
A Bayesian-model based procedure implemented in STRUCTURE (ver. 2.3.4, Pritchard et al. 2000) was used to further assess population structure and individual ancestry. STRUCTURE assigns individuals or a proportion of an individual’s ancestry, when using an admixture model, to K genetic groups based on allele frequencies for multi-locus genotypes without any prior assumptions of population, geographic or ancestral groups.
An admixture model with correlated allele frequencies and no location priors was used. Estimation of ancestry was performed on all the molecular subsets listed in Table 3. The ANCESTDIST option was used to return 90% probability intervals around each ancestry estimate. Each run included 400,000 MCMC steps and a burn-in period of 100,000 steps. Ten independent replicate runs were conducted for each value of K, for at least K = 1–10 and up to K = 13 for larger subsets.
Determining the best-supported number of genetically distinct groups (K) can be challenging. Several biases in a sample set, such as population divergence caused by isolation-by-distance, subtle subdivisions within diverged groups and recent genetic bottlenecks can lead to misinterpretation of results (Lawson et al. 2018). We took a cautious approach to identifying the best supported values of K, following the recommendations of Janes et al. (2017), Lawson et al. (2018) and Funk et al. (2020). This involved a two-pronged approach. We closely examined all bar plots produced by each STRUCTURE analysis at different values of K (from multiple runs and iterations), seeking consistency among results. We also plotted the natural logarithm of the likelihood (ln Pr(X/K)) for all values of K tested, using STRUCTURE HARVESTER (ver. 0.6.94, see http://taylor0.biology.ucla.edu/structureHarvester, accessed 30 September 2023). When interpreting the STRUCTURE results, we described the ancestral assignment for several values of K in relation to known natural history data (Funk et al. 2020). Admixture plots were generated using CLUMPAK (beta version, see http://clumpak.tau.ac.il/; Kopelman et al. 2015); all admixture plots are included in the Supplementary material (Supplementary Fig. S6–S9). CLUMPAK identifies distinct clustering modes from multiple independent runs at each value of K in the space of possible solutions for that value of K, and the runs are sorted into the predominant or ‘major’ clustering mode and any alternative or ‘minor’ clustering modes that occur less frequently. CLUMPAK also aligns inferred clusters across runs with different values of K for ease of plotting.
The R package conStruct (ver. 1.05, see https://cran.r-project.org/package=conStruct; Bradburd et al. 2018) was used to investigate the role of isolation by distance in producing some of the observed clustering patterns in the STRUCTURE and PCA results. conStruct is a model-based clustering method similar to STRUCTURE but with several important differences. When assigning ancestry, conStruct models ancestral populations as spatial layers incorporating isolation by distance to explain observed genetic variation, only ascribing discrete population structure to the dataset when the genetic differentiation significantly deviates from that explained by geographic distance. conStruct also provides a non-spatial model similar to STRUCTURE for comparison with the spatial model. When combined with STRUCTURE analyses conStruct provides a robust method to test whether sampled variation is discrete or continuous across a landscape. conStruct uses population-based allele frequencies, rather than individual genotypes as used by STRUCTURE and incorporates geographic information. Only populations with reliable latitude and longitude information available, and population level sampling of three or more individuals were used in this analysis, resulting in 16 populations being excluded on this basis. Only one OTU was entirely lost from the analysis through filtration, M. sp. Wahlmoorum, otherwise all OTUs retained adequate representation.
Basic runs (function ‘construct’) were performed on the specified data subsets. Initially the number of iterations, replicates and value of K, were explored and 20,000 iterations for 10 replicates of each value of K from 1 to 10 were found to produce results that converged largely on one solution and for which the most trace plots showed little autocorrelation and had settled into a solution space (see vignette ‘model-comparison’; Bradburd et al. 2018).
To help determine the best value of K for each subset analysed, cross validation (function ‘x.validation’) analysis was performed using the same run parameters as for basic runs. We plotted the mean predictive accuracy for each value of K to find the value(s) for which the data best fitted the model, and to compare the spatial and non-spatial models. To avoid over-parameterisation, we also calculated layer contributions (function ‘layer.contribution’) for each value of K. This analysis provides insight into how much each layer contributes to the total covariance; if the contribution of a layer is small to negligible it indicates that fewer layers are likely to be equally descriptive. Similar to STRUCTURE analysis, considering multiple values of K and placing the results within a biological framework when interpreting the implications of the results are important.
Genetic diversity parameters were estimated for each population, with three or more individuals sampled, to provide insight into gene flow within and between populations, and to look for evidence of hybridisation, polyploidy and apomixis. The expected heterozygosity (HE), observed heterozygosity (HO) and degree of inbreeding (FIS) were estimated using the hierfstat package (function ‘basic stats’, ver. 0.5-11, J. Goudet and T. Jombart, see https://cran.r-project.org/package=hierfstat).
To investigate genetic differentiation between all populations, the pairwise FST was estimated using STAMPP (function ‘stamppFst’, ver. 1.6.3, see https://cran.r-project.org/package=StAMPP; Pembleton et al. 2013). Where pairwise FST values between taxa are high relative to intraspecific pairwise values within congenerics, while maintaining high levels of outcrossing (low FIS), these can be considered genetically differentiated (Rutherford et al. 2018).
Results
Morphology
Two characters, ‘bud width’ and ‘corolla lobe width’ were removed from the analysis as these were highly correlated with several other characters and there was a biological rationale to suggest a lack of independence (Supplementary Table S2). An analysis of only the quantitative characters showed the basic structure of an emerging pattern in the data. However, several clusters in the phenogram and ordination were not clearly resolved, including OTUs from the M. erubescens sensu Paterson and M. urceolatus sensu Paterson groups. Qualitative characters were included one at a time and analyses rerun (Supplementary Fig. S1). The underlying pattern in the data did not change with the addition of each character but the distinction between clusters became clearer. After filtration, a dataset comprising 90 samples (Supplementary Table S1), 21 quantitative variables and 5 binary characters (Table 2) was deemed suitable for the final analysis.
The total amount of missing data in the genus-wide final dataset was 3.3%. Corolla characters had the most data missing (up to 8.9% per character; Supplementary Table S3a) due to the practical difficulty of aligning fieldwork with the period of flowering. All the missing data were contributed by 10 individual samples, from six putative taxa, for which incomplete fertile material was available (Supplementary Table S3b). The analyses were run both with and without these samples and the sample inclusion did not influence the general patterns observed. These were retained in the final analysis to boost representation for otherwise under-represented taxa.
Most quantitative characters were not normally distributed. Some characters were bimodal and many showed a skewed distribution (Supplementary Fig. S2). This supported the justification for use of a non-metric ordination approach and the UPGMA hierarchical clustering method.
The pattern of clustering in the phenogram (Fig. 3d) was consistent with that visualised in the NMDS ordinations (Fig. 3a–c). In Fig. 3d, clusters that correspond between the phenogram and NMDS ordinations are indicated by the blue bars, whereas further clusters apparent only in the phenogram are shown with yellow bars. The clustering pattern reflected many of the species boundaries hypothesised early in this study, expressed as phrase-named OTUs (Table 1). The NMDS ordination for both k = 2 and k = 3 returned fair stress values of 0.1920 and 0.1062 respectively, whereas k = 4 returned a good stress value of 0.0799. The clustering pattern produced by each analysis was similar and iterative, with the addition of more dimensions improving the resolution of clusters. Results for k = 2 and k = 3 NMDS ordinations are shown (Fig. 3a–c). The mclust model selected as the best model to represent the genus-wide data for k = 2 was ‘EII’ (spherical, equal volume) and the best number of clusters was eight. For k = 3, ‘EEI’ (diagonal, equal volume and shape), the optimal number of clusters was nine. For k = 3, eight of the nine clusters identified were also recovered in the k = 2 analysis (Fig. 3a–c).
The influence of each character on the clustering patterns in the ordination was examined to better understand what was driving the clustering pattern and identify potentially diagnostic characters for putative species. Results for the k = 3 ordination (Supplementary Fig. S3) were concordant with the k = 2 results (Fig. 4). All 26 characters had a statistically significant impact on the ordination configuration for k = 3, providing some further insight into diagnostic characters, especially for Melichrus gibberagee.
Character importance analysis of an NMDS ordination (k = 2) for a morphometric dataset comprising 26 characters scored for 90 specimens of Melichrus. Points represent individual plants. See Fig. 1 for a visual key and Table 1 for a textual description of symbols. Each vector represents the magnitude and direction of the influence of a quantitative character that had a statistically significant influence on the ordination space. The average value for each state (Y, affirmative; N, negative) for each binary character is plotted, the corresponding character for numbers 1–5 are listed in Table 2. Abbreviations: C., corolla; L., length; W., width.
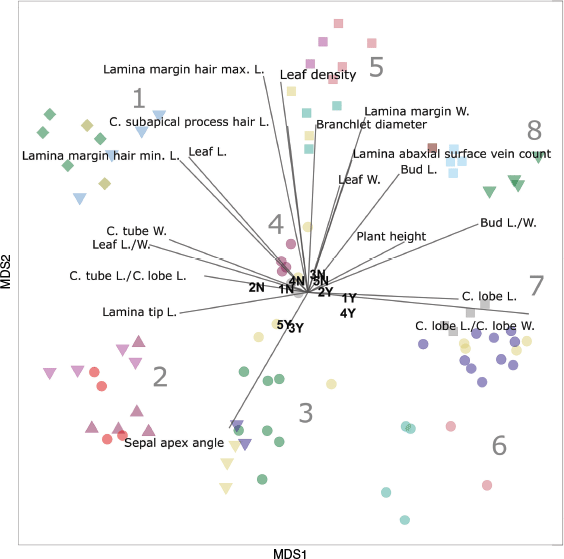
The distribution of samples across axis 1 primarily reflected the fact that taxa in clusters 1 and 2 have a cup-shaped or rotate corolla (C. tube L./C. lobe L.; C. tube W.; 2N), with corolla lobes that are spreading from the base (1N; e.g. see Fig. 5), relatively narrow leaves (Leaf L./W.) and young leaves that are straight or slightly incurved (4N; e.g. see Fig. 5a, b). By contrast, taxa in clusters 6 and 7 have an urceolate corolla, with lobes that are held erect before spreading, leaves are generally ovate and young leaves are recurved (Fig. 4).
Selected morphological characters in Melichrus. (a) Young leaves recurved (M. urceolatus, H.T. Kennedy 130). (b) Young leaves straight to slightly incurved (M. sp. Galambary, H.T. Kennedy 136). (c) Fruit elliptical, sepal apex acuminate and exceeding fruit (M. gibberagee, H.T. Kennedy 28). (d) Fruit ovoid, sepal apex obtuse and not exceeding fruit (M. adpressus sens. str., H.T. Kennedy 140). (e) Fruit spheroid, sepal apex acute and exceeding fruit (M. urceolatus, H.T. Kennedy 61). (f) Corolla urceolate (M. urceolatus H.T. Kennedy 130). (g) Corolla cup-shaped (M. sp. Galambary, H.T. Kennedy 137). (h) Corolla rotate (M. procumbens, H.T. Kennedy 123). Images by J. J. Bruhl (c & e) and H. T. Kennedy (a, b, d, f, g & h).

Cluster 3 occupies a similar area of the ordination space to cluster 2, and these share several characters, including a broad sepal apex (Sepal apex angle), a domed habit (5Y) and a prominent central nerve on the abaxial leaf surface (3Y). OTUs in cluster 3 differ from those in cluster 2 by the broader leaves and some samples have an urceolate corolla shape (2Y).
Several characters contributed to the pronounced isolation of cluster 5 in the ordination space (Fig. 4). These characters included dense, imbricate leaves (Leaf density), robust branchlets (Branchlet diameter) and a wide lamina margin (Lamina margin W.; e.g. see Fig. 6a–c). The maximum length of hairs on the lamina margin (Lamina margin hair max. L.) strongly distinguished clusters 5, 8 and 1 across axis 2 from taxa with much shorter lamina hairs in clusters 2, 3, 6 and 7 (e.g. see Fig. 6a–c).
Selected micromorphological characters in Melichrus. (a) Upper leaf surface showing narrow coloured margin, minutely scabrous (M. adpressus sens. str., H.T. Kennedy 11). (b) Upper leaf surface showing wide, white, thin margin, scabrous to hispid (M. adpressus sensu Paterson, H.T. Kennedy 82). (c) Upper leaf surface margin not distinct from leaf surface, hairs pilose (M. hirsutus, H.T. Kennedy 112). (d) Lower leaf surface showing many veins with narrow furrows between veins that have minute barbs on the margins (M. sp. Herberton from QCO, H.T. Kennedy 91). (e) Lower leaf surface showing medium number of veins with wide furrows between veins that have short papillae on the margins (M. sp. Mareeba, H.T. Kennedy 94). (f) Lower leaf surface showing few veins with very wide furrows between veins that have long papillae on the margins (M. sp. Isla Gorge, H.T. Kennedy 84). Scale bars: 1 mm. Images: H. T. Kennedy.

Clusters 6 and 7 shared a similar area of the ordination space reflecting shared floral characters and that the young leaf growth is often recurved (4Y). Taxa in cluster 6 are distinguished from cluster 7 in the analyses by the especially small, sparse leaves (Fig. 4).
Cluster 4 is the most broadly spread cluster identified in the analysis, suggesting that this has the most intracluster differences (Fig. 4). This cluster also occupies the middle of the ordination, indicating a lack of unique characters captured in the analysis for taxa in this group. Table 4b highlights some additional characters that distinguish OTUs in this cluster from the proximal, cluster 5. Particularly, OTUs in cluster 4 all share a character in which the furrows between the veins on the undersides of leaves are particularly wide and have long hairs on the margins of the furrows (e.g. see Fig. 6d–f). Melichrus hirsutus (cluster 1) and M. gibberagee (cluster 9) are the only other taxa that share this character.
The k = 3 ordination split cluster 8 (from the k = 2 ordination) into two groups separated by significant morphological differences (Supplementary Fig. S3). Taxa in both clusters 8 and 9 have long, urceolate flowers, long buds and sepals with a narrow apex. However Melichrus gibberagee has far narrower leaves (Leaf L./W.) and flowers (Corolla lobe L. & Corolla lobe L./corolla lobe W.), far fewer veins on the abaxial leaf surface (Lamina veins count) and longer lamina margin hairs (Lamina margin hair max. L.) than in M. sp. Wahlmoorum and M. adpressus sensu Paterson. Further differences between the three OTUs are included in the table of differences (Table 4c).
NMDS ordination of the subsets of the data (see Table 3 for a summary of data subsets analysed) recovered the same additional clusters that were observable in the genus-wide phenogram, indicated in Fig. 3d by yellow bars. Supplementary Fig. S5 includes the most informative NMDS ordinations and character importance analyses for each data subset, the results of which are summarised here.
The NMDS ordination of a subset of the data including only OTUs from cluster 1 (subset 1), distinguished strongly between Melichrus hirsutus and M. procumbens plus M. sp. Torrington (Supplementary Fig. S4a). Several characters had a significant influence on the ordination pattern, particularly the lack of a white lamina margin, taller stature, cup-shaped corolla, more slender buds, acuminate sepal apex and wider leaves in M. hirsutus. Table 4a highlights several more characters that morphologically differentiate M. hirsutus from other OTUs in cluster 1, including an ascending habit, lamina margin hairs are all a similar length or longer at the base of the lamina and a pink corolla.
Ordination of subset 2 showed Melichrus erubescens, M. sp. Silent Grove and M. sp. Gardens of Stone samples clustering in three discrete groups corresponding with OTU designation (Supplementary Fig. S4b, c). Samples of M. sp. Gardens of Stone were distinguished by a relatively long, narrow, cup-shaped corolla with long corolla lobes. Melichrus sp. Silent Grove was distinguished in the ordination by a tall, erect stature and narrow corolla tube with relatively short corolla lobes. Melichrus sp. Gardens of Stone and M. sp. Silent Grove are further distinguishable from M. erubescens by acuminate sepals at fruiting that are longer than the fruit. Melichrus sp. Silent Grove also has a different fruit texture, similar to that seen in M. urceolatus where there is a distinctly smooth, slightly raised distal section (Table 4a).
Morphological analyses did not return any reliable diagnostic characters differentiating the three morphologically similar OTUs Melichrus sp. Kaputar, M. adpressus sens. str. and M. sp. Galambary (Supplementary Fig. S4d, e). The results did suggest that some of the southern populations of M. sp. Galambary (Gal3 & 4) were morphologically different from the more northern populations. In particular, southern populations had a longer corolla tube that was subtly urceolate with lobes that were held erect before spreading, the leaves were generally narrower and the dome-shaped habit was lacking.
Melichrus sp. Mareeba specimens in the ordination of subset 4 were differentiated primarily by the distinctive leaves that were larger and had a broader white margin, similar to the OTUs in cluster 5 (Supplementary Fig. S4f, g). Melichrus sp. Isla Gorge specimens clustered loosely in the ordination, demonstrating the variability for the chosen characters. The sample Isl5 of M. sp. Isla Gorge is nested within the M. sp. Mareeba samples in the genus-wide phenogram and M. sp. Isla Gorge and M. sp. Mareeba samples were not well separated in the ordination. Melichrus sp. Mareeba can be distinguished from M. sp. Isla Gorge and M. sp. Tara by several characters not included in phenetic analyses, such as a decumbent plant habit, the absence of pilose hairs on leaves and glabrous sepals (Table 4b). The two M. sp. Tara specimens included in the analysis were distinguished in the ordination by a broad sepal apex angle and short buds and flowers.
Samples in subset 5 separated into three distinct groups across the ordination space (Supplementary Fig. S4h). Samples of Melichrus sp. Herberton clustered together, defined by broader leaves and sepal apex angle and as the only OTU in this cluster with glabrous sepals (Table 4c). Melichrus sp. Yuraygir samples showed a high degree of variability in some corolla and indumentum characters but clustered discreetly based on the wide white margin, high density of short, wide leaves and a short pungent tip at the leaf apex. Samples of M. sp. Boonoo Boonoo and M. sp. Kroombit Tops occupied a similar part of the ordination, though these were not tightly clustered. This part of the ordination space was defined by longer, slightly narrower leaves and generally longer hairs across various plant parts.
Several vegetative and fertile characters separated Melichrus sp. Gilgandra and M. sp. Inglewood in the ordination of subset 6 (Supplementary Fig. S4i). Melichrus sp. Gilgandra samples generally had a wider lamina with a longer pungent tip, and flowers with a broad sepal apex and relatively short corolla lobes. Additionally, M. sp. Gilgandra leaves are strongly glaucous, the texture of the fruit is variably wrinkly all over and the calyx does not exceed the apex of the fruit (Table 4c).
All samples of Melichrus sp. Gurulmundi shared a similar area of the ordination for subset 7 (Supplementary Fig. S4j, k). This area was defined by a high density of leaves on flowering branches, a broad branchlet diameter, broad sepal apex angle and more veins on the abaxial leaf surface. Some samples of M. urceolatus, particularly Urc4 and Urc6 also fell within this area of the ordination, in evidence of the similarity that M. sp. Gurulmundi has with some of the more robust forms of M. urceolatus. Although M. sp. Gurulmundi is morphologically similar to M. urceolatus, these can be distinguished by a combination of characters. Melichrus sp. Gurulmundi rarely has more than one stem at the base, is robust, erect, with a candelabra-like branching pattern, has densely imbricate leaves, and sepals are pilose and have a broad apex angle (Supplementary Fig. S4j, k and Table 4c). Samples of M. medius were not distinguished from M. urceolatus in the phenetic analyses. The table of differences highlights the particularly densely pilose sepals but some other populations of M. urceolatus also exhibit this character, particularly populations on the Northern Tablelands of New South Wales (Table 4c).
Molecular
Filtration of the dataset for PCA and Neighbour-Net analysis produced a set of 2071 high-quality SNPs and retained 543 individuals. No entire populations or OTUs were lost from the analysis through filtration.
The first three axes of the ‘genus-wide’ PCA (Fig. 7a, b) explained 20.6, 18.2, and 11.4% of the total variation respectively. OTUs segregated from Melichrus urceolatus sensu Paterson (represented by circles) separated into at least five distinct groups occupying the extremities of all axes (Fig. 7 clusters 1, 4, 5, 7 & 9). Cluster 1 included M. urceolatus, M. medius and M. sp. Inglewood, and M. sp. Silent Grove. In further analyses this is referred to as the ‘M. urceolatus cluster’. All OTUs attributed to M. procumbens sensu Paterson (1958) (represented by diamonds) clustered closely together (cluster 2; ‘M. procumbens cluster’) adjacent to the M. urceolatus cluster (cluster 1) and cluster 3 that contained all M. adpressus sensu Paterson (‘M. adpressus sensu Paterson cluster’) segregates (represented by squares). Note that the distinctions between clusters 1–3 are more easily observed in the network analysis and PCA of subset A (see below). Cluster 4 included all six M. sp. Mareeba populations, and cluster 5 included all eight M. sp. Isla Gorge and the single M. sp. Tara populations (‘M. sp. Isla Gorge cluster’). Melichrus hirsutus and M. gibberagee clustered closely together (cluster 6; ‘M. hirsutus cluster’). Axis 3 defined clusters corresponding to OTUs or groups thereof: cluster 7, consisting of M. sp. Galambary, M. sp. Gilgandra and M. adpressus sens. str. (‘M. sp. Galambary cluster’); cluster 8, comprising M. sp. Kaputar (except one individual that is displaced from the group); cluster 9, with M. sp. Gardens of Stone; and cluster 10, consisting only of M. erubescens samples (represented by triangles).
Melichrus genus-wide PCA and Neighbour-Net analysis of a DArTseq dataset of 2071 SNPs. (a) PCA axes 1 & 2. (b) PCA axes 1 & 3. (c) SplitsTree Neighbour-Net. In the ordinations (a, b) individuals are represented by single points, in the SplitsTree network (c) each terminus represents a single population. Point shape indicates membership to a sensu Paterson group. Numbered clusters on the SplitsTree network correspond with number clusters on the PCA ordinations. See Fig. 1 for a visual key and Table 1 for a textual description of symbols.
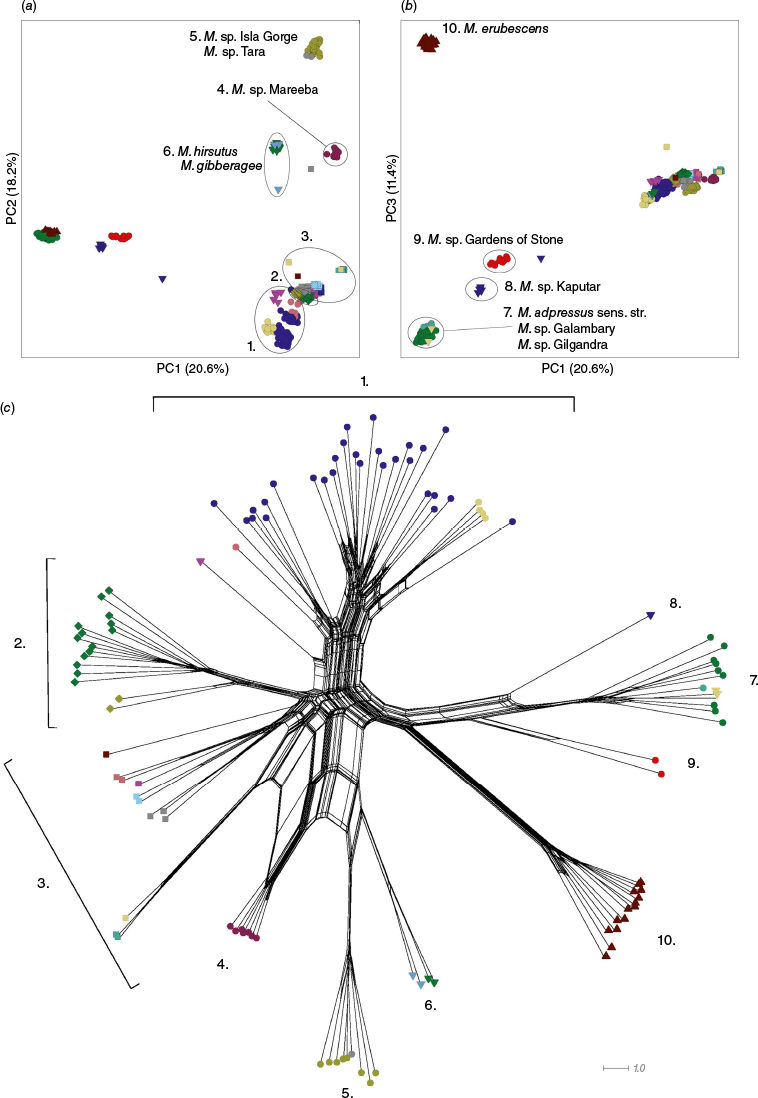
The groups observed in the SplitsTree Neighbour-Net analysis (Fig. 7c) mirrored the PCA, with some additional insights provided by the distance-based relatedness network. In a relationship network, the parallel lines reflect splits in the data, with the longer lines indicating more support for that particular split (Huson and Bryant 2006). The split separating the Melichrus erubescens cluster from the remainder of the genus is especially strong in this analysis, as are the splits defining the M. procumbens (cluster 2), M. sp. Isla Gorge (5) and the M. sp. Galambary (7) clusters. The network confirms that which is not necessarily distinct in the PCA, that the M. urceolatus (1), M. procumbens (2) and M. adpressus sensu Paterson (3) clusters are distinct groups. The higher number of parallel edges in the sections corresponding with the M. urceolatus (1) and M. adpressus sensu Paterson (3) clusters respectively indicate a larger degree of discordance in these sections of the dataset. The M. sp. Mareeba cluster (4) shares multiple parallel lines with both M. sp. Isla Gorge (5) and M. sp. Herberton + M. sp. Kroombit Tops (3) that may indicate a hybrid ancestry for M. sp. Mareeba. Parallel edges between M. sp. Silent Grove and M. procumbens may indicate some introgression, as do the parallel lines between M. sp. Kaputar and M. sp. Galambary.
PCA of subset A, that includes only the samples from the genus-wide PCA clusters 1–3 (see Table 3 for a summary of subsets used in analyses; Fig. 8a), exhibited three broad clusters distributed across axes 1 (28.4%) and 2 (14.1%). Each group matched with the Melichrus urceolatus (1), M. procumbens (2) and M. adpressus sensu Paterson (3) clusters as seen in the Neighbour-Net analysis.
The most informative principal component axes (PCs) plotted for select subsets of a DArTseq dataset of 2071 SNPs. (a) Subset A; PC1, PC2. (b) Subset E; PC1, PC2. (c) Subset D; PC1, PC2. (d) Subset J; PC1, PC2. (e) Subset F; PC1, PC2. (f) Subset F; PC1, PC3. (g) Subset I; PC1, PC2. (h) Subset I; PC1, PC3. (i) Subset H, PC1, PC2. (j) Subset G, PC1, PC2. Individuals are represented by single points. See Fig. 1 for a visual key and Table 1 for a textual description of symbols. The OTUs included in each subset are listed in Table 3. Codes used to indicate geographic distributions are abbreviations of botanical districts and follow (Centre for Australian National Biodiversity Research 2018), except ‘nNNT’ and ‘sNNT’ that are modified to indicate the northern and southern extents of the Northern Tablelands NSW (see Supplementary Table S1 for further geographic information).
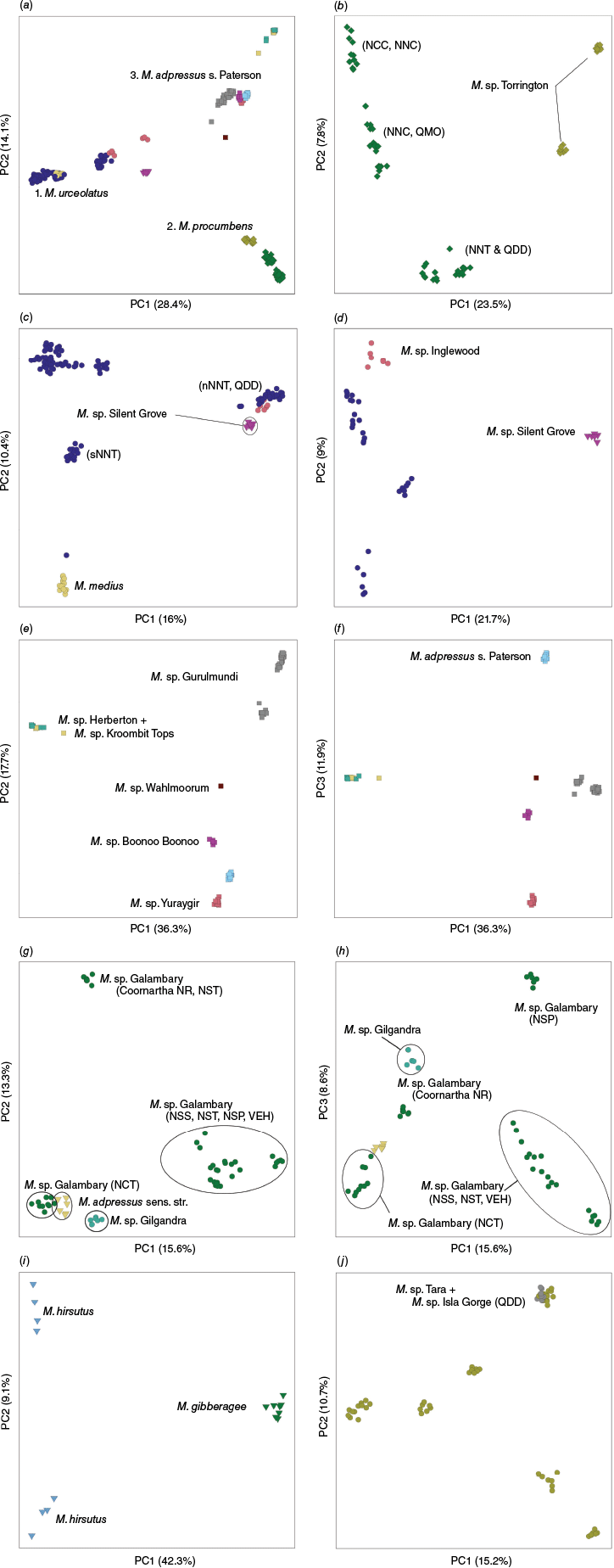
In a PCA of the Melichrus procumbens cluster (subset E), the only axis that explained more than 10% of the variation in the dataset was PC1 (23.5%). Along this axis, the two populations of M. sp. Torrington were distinct from M. procumbens samples. Melichrus procumbens showed some geographic grouping corresponding with coastal and tableland populations (Fig. 8b).
PC1 (16%) from the PCA of subset D, defined a ‘northern cluster’ (Fig. 8c) containing Melichrus urceolatus specimens collected north of Henry River, New South Wales in the Northern Tablelands (nNNT) and Queensland Darling Downs (QDD), and the single populations of M. sp. Silent Grove and M. sp. Inglewood respectively. Along PC2 (10.4%) three additional clusters were apparent: the first containing all M. medius (Southern Tablelands, NST) populations; the second, populations collected on the Northern Tablelands south of Henry River (sNNT), the third contained M. urceolatus samples from the North Western Slopes (NNS), Central Tablelands (NCT), Central Coast (NCC) and South Coast (NSC) botanical districts of New South Wales. The single M. urceolatus plant found in Conimbla National Park (NCT) clustered most closely to the M. medius samples. In a PCA of only the ‘northern cluster’ (subset J), M. sp. Silent Grove was distinct (PC1, 21.7%) from M. sp. Inglewood and the northern M. urceolatus specimens (Fig. 8d).
Subset F returned several distinct clusters corresponding with hypothesised species limits (Fig. 8e, f: PC1 = 36.3%; PC2 = 17.7%; PC3 = 11.9%). Melichrus sp. Herberton samples clustered with M. sp. Kroombit Tops, despite a vast geographic disjunction and all M. sp. Gurulmundi samples formed a distinct cluster. The remaining samples in this subset formed four discrete clusters corresponding with the OTU designations, M. adpressus sensu Paterson, M. sp. Boonoo Boonoo, M. sp. Yuraygir and M. sp. Wahlmoorum.
Melichrus adpressus sens. str. clustered with the populations of M. sp. Galambary to which this is geographically closest (NCT) across the first three axes of a PCA of subset I (Fig. 8g, h: PC1 = 15.6%; PC2 = 13.3%; PC3 = 8.6%). Other populations of M. sp. Galambary were spread across the three axes in clusters apparently corresponding with the geographical proximity or environmental continuity. The single M. sp. Gilgandra population included in the analysis clustered near to but always separate from the Central Tablelands M. sp. Galambary and M. adpressus sens. str. clusters.
In subset H, Melichrus gibberagee populations were strongly separated from the M. hirsutus populations across PC1 (42.3%: Fig. 8i). Melichrus sp. Tara samples clustered with the two M. sp. Isla Gorge populations to which this is geographically closest across all major PCA axes (subset G; Fig. 8j). All other Melichrus sp. Isla Gorge samples clustered by population, according to geographic proximity to each other.
For subset A, an ancestry model with eight populations (K = 8) had the highest mean log-likelihood (Supplementary Fig. S5a, b) and the individual log-likelihood values for all 10 runs at K = 8 were similar. The major (5/10) and minor (3/10 and 2/10) modes showed similar patterns of admixture (Supplementary Fig. S6). For K = 9, two runs with outlier values of log-likelihood were removed (Supplementary Fig. S5b), producing a slightly higher mean log-likelihood than that of K = 8. This indicated that an admixture model with eight or nine ancestral populations likely best represents this subset of the data (Fig. 9). Log-likelihood values for runs at a higher value of K (K = 10–13) did not converge. Ancestry models with K = 8 and K = 9 were best supported but across all values of K, consistent patterns of ancestry and admixture emerged (Supplementary Fig. S6).
STRUCTURE bar plots for Subset A, a dataset of 2636 DArTseq SNPs. The major clustering modes for the best supported number of ancestral populations, K = 8 and K = 9 are shown. White lines separate individuals. Black lines separate populations. Each OTU is labelled and the botanical district of each population is given (Centre for Australian National Biodiversity Research 2018). Within each OTU, populations are arranged by latitude. Ancestral populations are coloured by the OTU (see Table 1 and Fig. 1) that these primarily but not necessarily exclusively represent. The asterisk (*) indicates M. sp. Wahlmoorum.

Corroborating results observed in the PCA, all OTUs from the Melichrus procumbens cluster shared a common ancestry distinct from other OTUs in the STRUCTURE analysis of subset A (Fig. 9: green). Samples of M. procumbens from the southern extent of the species range drew exclusively from this ancestral population, with populations of M. procumbens further north showing admixture with an additional ancestral population. Melichrus sp. Torrington and M. sp. Silent Grove that co-occur in Torrington National Park (NNT) also drew substantially from this population (Fig. 9: purple).
All samples of Melichrus medius shared a single ancestral population in the STRUCTURE analysis of subset A (Fig. 9: yellow). Melichrus urceolatus from the southern extent of the Northern Tablelands (sNNT) partially shared ancestry with M. medius and some with M. urceolatus (Fig. 9: dark blue). Mirroring results observed in the PCA (Fig. 8c), M. urceolatus samples showed a major divide in genetic clustering corresponding to a north-south geographic division. Melichrus urceolatus from most of New South Wales, ranging from the South Coast through to the North Western Slopes shared most of the ancestry (Fig. 9: dark blue). Melichrus urceolatus from north of this divide – occurring in the northernmost extent of the Northern Tablelands and the North Coast in New South Wales and the Darling Downs in Queensland – derived most ancestry from a separate population that the species shares with M. sp. Inglewood (Fig. 9: light blue). There is a small but notable amount of admixture between these two ancestral populations in samples from the North Western Slopes. The possibility that this pattern of differentiation is a product of isolation by distance was explored through conStruct analysis (see below).
The STRUCTURE analysis of subset A provided strong support for Melichrus sp. Herberton and M. sp. Kroombit Tops as conspecific, distinct from the other M. adpressus sensu Paterson segregates (Fig. 9: bluish-green). Melichrus sp. Gurulmundi was also supported as a distinct entity (Fig. 9: grey), though this shared some ancestry with samples of M. adpressus sensu Paterson from the Moreton district (Fig. 9: pink). At K = 8, M. sp. Boonoo Boonoo and M. sp. Yuraygir also shared this cluster but at K = 9, an additional ancestral population, primarily corresponding with M. sp. Yuraygir but also with M. sp. Boonoo Boonoo is introduced (Fig. 9: brown). As in other samples from the Northern Tablelands, M. sp. Boonoo Boonoo also shared the ancestral population that defines M. sp. Silent Grove (Fig. 9: purple). As expected, the program did not return a distinct grouping for the single, isolated specimen of M. sp. Wahlmoorum.
For a STRUCTURE analysis of subset B, an ancestry model with four ancestral populations (K = 4) had the highest mean log-likelihood (Supplementary Fig. S5c, d) and the individual log-likelihood values for all 10 runs at K = 4 were similar. The major (6/10) and minor (4/10) modes showed similar patterns of admixture in the plots, with one important difference (Fig. 10). The ancestry of the only known population of M. sp. Kaputar was derived from the population’s own exclusive cluster in the major mode, whereas most of the ancestry in the minor mode was shared with M. sp. Galambary. Two runs for K = 5 were removed as these had very different log-likelihood values from the remaining eight runs (Supplementary Fig. S5d). Once the outliers were removed, K = 5 had a slightly higher mean log-likelihood than K = 4, indicating that an admixture model with four or five admixing populations likely best represents this subset of the SNP data.
STRUCTURE bar plots for Subset B, a dataset of 2710 DArTseq SNPs. The major and minor clustering modes for the best supported number of ancestral populations, K = 4 (major and minor clustering modes) and K = 5 (major mode: 6/10) are shown. White lines separate individuals. Black lines separate populations. Each OTU is labelled and the botanical district of each population is given (Centre for Australian National Biodiversity Research 2018). Within each OTU, populations are arranged by latitude. Ancestral populations are coloured by the OTU (see Table 1 and Fig. 1) that these primarily but not necessarily exclusively represent.
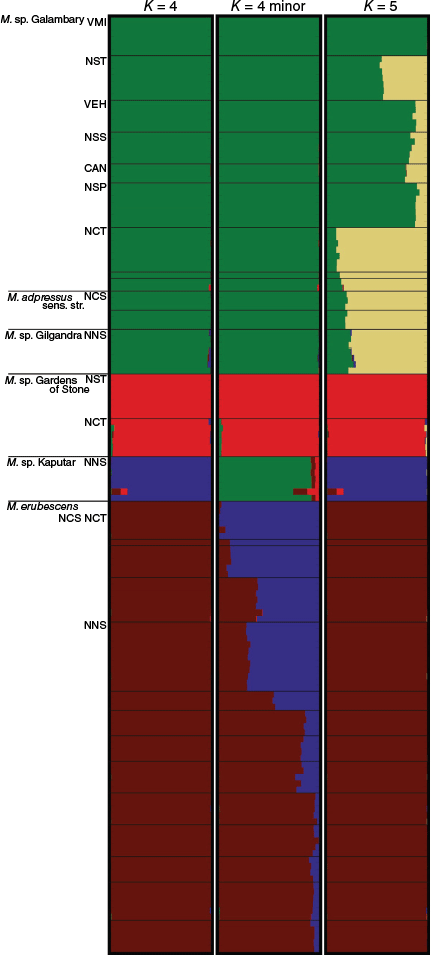
Melichrus sp. Gardens of Stone was represented primarily by a single exclusive ancestral population in the STRUCTURE analysis of subset B (Fig. 10: red). All samples of M. erubescens shared either one (Fig. 10: brown: K = 4 major mode, K = 5 all modes) or two ancestral populations (Fig. 10: brown and dark blue: K = 4 minor mode). At K = 4, M. sp. Galambary, M. sp. Gilgandra and M. adpressus sens. str. shared a single population (Fig. 10: dark green). At K = 5 an additional population was introduced, from which northern populations generally derived more ancestry (Fig. 10: yellow). The possibility that the pattern of differentiation observed in this subset was a product of isolation by distance was explored through conStruct analysis (see below). STRUCTURE plots for all values of K are included in Supplementary Fig. S7.
For a STRUCTURE analysis of subset C, an ancestry model with four populations (K = 4) had the highest mean log-likelihood (Supplementary Fig. S5e, f) and the individual log-likelihood values for all 10 runs at K = 4 were similar. A model with four admixing ancestral populations likely best represents this subset of the SNP data. For the major mode (9/10), all populations of M. sp. Mareeba showed approximately equal admixture from ancestral populations corresponding to M. sp. Isla Gorge and M. sp. Herberton + M. sp. Kroombit Tops respectively (Fig. 11: olive-green and bluish-green). This pattern, characteristic of hybridisation, was evident across most values of K (Supplementary Fig. S8). At all levels of parametrisation in this analysis, populations of M. hirsutus and M. gibberagee shared a single ancestral population (Fig. 11: blue). A STRUCTURE run consisting of only these two OTUs (subset H) returned a result where K = 2 was decisively the best supported number of populations (Supplementary Fig. S5g) and the two ancestral populations corresponded with each species, with negligible admixture detected (Fig. 11: blue and dark green; all plots included in Supplementary Fig. S9).
STRUCTURE bar plots for subset C and subset H, datasets of 2915 and 1568 DArTseq SNPs respectively. Left: subset C, best supported number of ancestral populations, K = 4 major clustering mode. Right: subset H, best supported number of ancestral populations, K = 2, major clustering mode. White lines separate individuals. Black lines separate populations. Each OTU is labelled and the botanical district of each population is given (Centre for Australian National Biodiversity Research 2018). Within each OTU, populations are arranged by latitude. Ancestral populations are coloured by the OTU (see Table 1 and Fig. 1) that these primarily but not necessarily exclusively represent. The asterisk (*) indicates M. sp. Wahlmoorum.

To test how isolation by distance may have contributed to the formation of the northern Melichrus urceolatus genetic group observable in the PCA and STRUCTURE results, subset D (see Table 3) was analysed with conStruct. The spatial model had a higher predictive accuracy overall than the non-spatial model (Supplementary Fig. S10) and the spatial model with six layers (K = 6) had the highest mean predictive accuracy of any model. For the non-spatial model, K = 8 had the highest mean predictive accuracy. The layer contribution analysis showed that for the spatial model only the first five layers contributed significantly to the total explained variation (Supplementary Fig. S11). As models with four or five layers had similar mean predictive accuracy, these were approximately equally suitable models for the dataset (K = 5, spatial (a) and non-spatial (b) are shown in Fig. 12).
conStruct analysis results for subsets D and B of the DArTseq SNP dataset (see Table 3). (a) Subset D; K = 5 spatial model. (b) Subset D; K = 5 non-spatial model. (c) Subset B; K = 4 spatial model. (d) Subset B; K = 5 spatial model. Each pie chart represents a single population. For figures (a) and (b) all unlabelled populations are M. urceolatus. For figures (c) and (d) the populations that are not labelled are either M. sp. Galambary and M. adpressus sens. str. (primarily drawn from the dark green layer) or M. erubescens (primarily drawn from the brown layer). The scale represents geographic distance. Layers are coloured by the OTU (see Table 1 and Fig. 1) that these primarily but not necessarily exclusively represent.
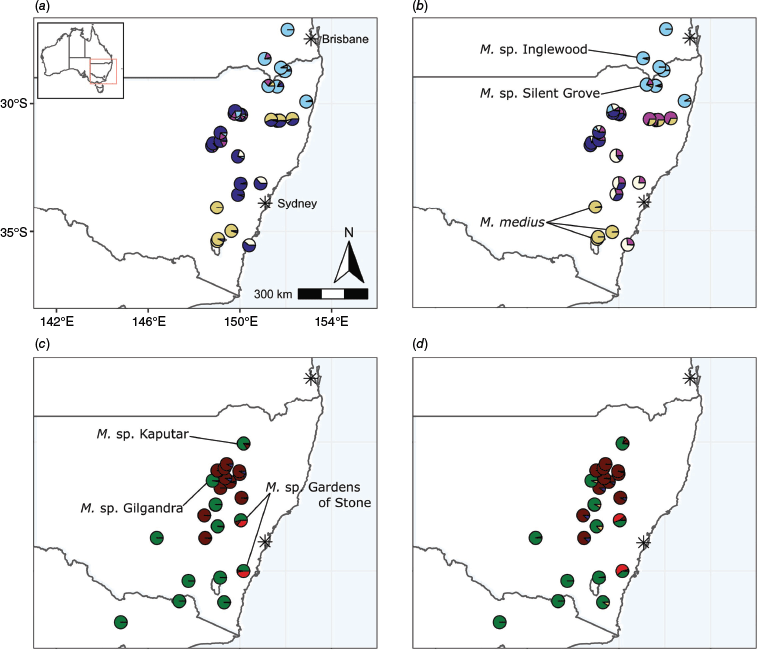
Across several values of K, including the best supported (K = 5, Fig. 12a), the spatial model delivered a result with a clear distinction between the northern and southern M. urceolatus groups. This pattern was also evident in the non-spatial model but was less clear in the noise of several other layers describing differentiation across the geographic range. This indicates that the ancestry pattern observed in the STRUCTURE and PC analyses is likely substantially independent of the influence of isolation by distance. Both the spatial and non-spatial models found that all M. medius populations drew nearly exclusively from a single layer, across several levels of parameterisation (including K = 5). The spatial model showed a stronger relationship between the M. medius and M. urceolatus populations from the southern extent of the Northern Tablelands than was found by the non-spatial model.
Subset B was subjected to a conStruct analysis to test if the distinction between Melichrus sp. Kaputar, M. sp. Galambary and M. sp. Gardens of Stone observed in the PC and STRUCTURE analyses could be attributed to isolation by distance. The spatial model had a higher predictive accuracy overall than the non-spatial model (Supplementary Fig. S12) and the spatial model with six layers (K = 6) had the highest mean predictive accuracy of any model. For the non-spatial model, K = 9 had the highest mean predictive accuracy. The layer contribution analysis showed that for the spatial model only the first four or five layers contributed significantly to the total explained variation (Supplementary Fig. S13). The model with five layers had a higher mean predictive accuracy than K = 4 but contributed less in the layer contribution analysis, therefore on balance spatial models with K = 4 or 5 are likely the most suitable models for the dataset (Fig. 12c, d).
Across several values of K, including the best supported, the layer composition of Melichrus sp. Kaputar differed only slightly from that of M. sp. Galambary populations across both the spatial and non-spatial analyses (Fig. 12c, d). Melichrus sp. Gardens of Stone, however, while showing some amount of admixture with M. sp. Galambary, drew substantially from an independent layer (Fig. 12c, d, shown in red).
Filtration of the dataset for population statistics produced a set of 4936 high-quality SNPs and retained 524 individuals across 95 populations. Observed heterozygosity (HO) was generally slightly lower than expected heterozygosity (HE) and FIS was moderately positive or negative for most populations (Fig. 13). The notable exception was Melichrus sp. Mareeba, for which all populations had HO values approximately twice the magnitude of the HE values and strongly negative FIS values (Supplementary Table S4).
Observed heterozygosity (HO), expected heterozygosity (HE) and inbreeding coefficient (FIS) for all populations of Melichrus with three or more individuals for a DArTseq dataset of 4936 SNPs. Each tick represents one population. Populations are labelled by OTU (Table 1).
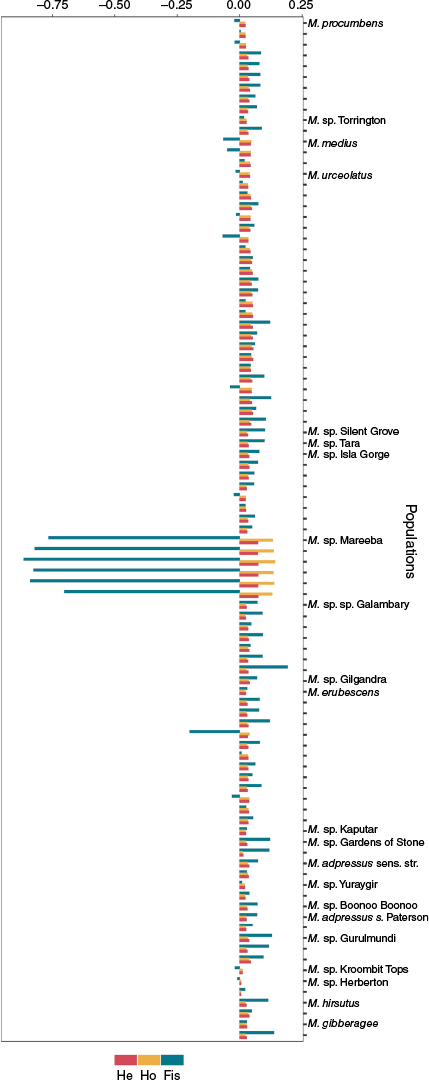
Pairwise FST values estimated for all pairs of populations in the study ranged from 0.01–0.93 (Supplementary Table S5). Intraspecific FST values for Melichrus sp. Mareeba populations (0.02–0.06) were an order of magnitude lower than interspecific FST values between M. sp. Mareeba and all other populations (0.43–0.75). Comparison of FST values between populations was consistent with the pattern of diversity observed in the PCA and STRUCTURE results.
Discussion
This study is the first to apply molecular and quantitative morphological analyses to attempt to resolve species boundaries in Melichrus. Using an integrative taxonomic approach, we combined multiple data sources to test hypothesised species boundaries, expressed as Operational Taxonomic Units (OTUs). Results were considered through the lens of the unified species concept to provide recommendations for changes to the species-level taxonomy of Melichrus. Morphological analyses resolved numerous distinct clusters corresponding with hypothesised species boundaries and in many cases these clusters were subsequently supported by analyses of the DArTseq molecular data. Although reconciling the patterns observed in the morphological and molecular analyses was challenging in some instances (see Table 5 for a summary of evidence and recommended taxonomic changes), the new evidence produced by this study supports considerable clarification of and improvement in the species-level taxonomy of Melichrus.
| OTU | Morphology | Molecular | Corresponding discontinuities | Taxonomic recommendation | |
|---|---|---|---|---|---|
| M. adpressus sensu Paterson | Y (similar to M. sp. Wahlmoorum). | Y (relationship to other M. adpressus sensu Paterson segregates uncertain). | Y, but with some uncertainty. | Redefine the M. adpressus sensu Paterson taxonomic boundary. Describe as new species with new name, to rectify misapplication. | |
| M. adpressus sens. str. | Y | Y | Y | Reinstate sens. str. taxonomic concept to rectify misapplication of name. Include M. sp. Galambary and M. sp. Kaputar as synonyms. | |
| M. sp. Boonoo Boonoo | N = M. sp. Herberton | Y, groups closest to, but distinct from M. adpressus sensu Paterson and M. sp. Yuraygir. | N, evidence types in conflict. | Exclude from taxonomic revision, further research needed. | |
| M. sp. Colo Gorge | – | – | – | Exclude from taxonomic revision, could not locate any living specimens. | |
| M. erubescens | Y | Y | Y | Retain status quo taxonomic concept. Provide updated morphological and geographic descriptions. | |
| M. sp. Galambary | N = M. adpressus sens. str. | N = M. adpressus sens. str. | N = M. adpressus sens. str. | Recognise as synonymous with M. adpressus sens. str. Investigate further the north-south disjunction in both molecular and morphological data for this OTU. | |
| M. sp. Gardens of Stone | Y | Y | Y | Describe as a new species. | |
| M. gibberagee | Y | Y | Y | Retain status quo taxonomic concept. | |
| M. sp. Gilgandra | Y | N = M. adpressus sens. str. | N | Describe as a new subspecies of M. adpressus sens. str. | |
| M. sp. Gurulmundi | Y | Y | Y | Describe as a new species. | |
| M. sp. Herberton | Y | Y | Y | Describe as a new species. | |
| Include M. sp. Kroombit Tops as synonym. Relationship to M. sp. Boonoo Boonoo and M. sp. Wahlmoorum remains unresolved. | |||||
| M. hirsutus | Y | Y | Y | Retain status quo taxonomic concept. | |
| M. sp. Inglewood | Weakly Y | N = M. urceolatus | N = M. urceolatus | Recognise as synonymous with M. urceolatus. | |
| M. sp. Isla Gorge | Y | Y | Y | Describe as a new species. | |
| Include M. sp. Tara as synonym. | |||||
| M. sp. Kaputar | N = M. adpressus sens. str. | Y (related to but diverged from from M. adpressus sens. str. lineage). | N = M. adpressus sens. str. | Recognise as synonymous with M. adpressus sens. str. | |
| Note: Highly restricted distribution may have driven divergent genetics. | |||||
| M. sp. Kroombit Tops | N = M. sp. Herberton | N = M. sp. Herberton | N = M. sp. Herberton | Describe as a new species synonymous with M. sp. Herberton. | |
| M. sp. Mareeba | Y | Y | Y | Describe as a new species. | |
| M. medius | N (some distinctive characters, but inadequate for diagnosis). | N (evidence of some history of divergence from M. urceolatus). | Somewhat | Retain synonymy under M. urceolatus. | |
| M. procumbens | Y | Y | Y | Retain status quo taxonomic concept. Provide updated morphological and geographic descriptions. | |
| M. sp. Salvator Rosa | – | – | – | Exclude from taxonomic revision, could not locate any living specimens. | |
| M. sp. Silent Grove | Y | Y | Y | Describe as a new species. | |
| M. sp. Tara | N = M. sp. Isla Gorge | N = M. sp. Isla Gorge | N = M. sp. Isla Gorge | Describe as a new species synonymous with M. sp. Isla Gorge. | |
| M. sp. Torrington | N = M. procumbens | Y, some evidence of divergence from M. procumbens lineage. | N = M. procumbens | Recognise as synonymous with M. procumbens. | |
| M. urceolatus | Y | Y | Y | Retain name with significantly revised boundaries. | |
| Include M. sp. Inglewood and M. medius as synonyms. | |||||
| Further investigate the north-south disjunction in molecular data for this OTU. | |||||
| M. sp. Wahlmoorum | Y (similar to M. adpressus sensu Paterson) but sampling inadequate. | Unknown-insufficient sampling. | N | Exclude from taxonomic revision, further sampling and analysis needed. | |
| M. sp. Yuraygir | Y | Y, but relationship to other M. adpressus sensu Paterson segregates uncertain. | Y | Describe as a new species. | |
| Relationship to M. sp. Boonoo Boonoo and M. sp. Wahlmoorum remains unresolved. |
Morphological: Y, distinctive and diagnosable. Molecular: Y, evidence that the entity is a separately evolving lineage. Corresponding discontinuities: Y, discontinuities seen in morphological analyses agree with those seen in the molecular analyses.
Melichrus urceolatus and M. erubescens
In the genus-wide phenetic analysis, the Melichrus urceolatus segregates (see Table 1 for summary of OTUs) clustered in five separate groups, spread across the ordination space and phenogram (Fig. 3). Strikingly, a very similar pattern of variation was returned in a genus-wide PCA and SplitsTree analysis of the DArTseq dataset (Fig. 7), where the M. urceolatus segregates also clustered in five highly distinct clusters but with some variation in which OTUs grouped together. These results demonstrated that M. urceolatus has been used as a ‘catch all’ species requiring significant taxonomic restructuring.
Both the morphological and molecular analyses demonstrated that Melichrus erubescens and M. urceolatus are highly distinct from each other, despite decades of confusion regarding the boundary. The analyses also showed that some OTUs segregated from M. urceolatus, such as M. sp. Galambary and M. sp. Gardens of Stone, had more in common with M. erubescens genetically, morphologically or in some cases both. This explains why the status quo taxonomic boundary between M. urceolatus and M. erubescens has been so difficult to apply in practice.
Across all analyses, Melichrus adpressus sens. str. was supported as distinct from M. urceolatus, with which this was synonymised by Bentham; distinct from M. adpressus sensu Paterson, to which the name was misapplied by Mueller and Paterson; and distinct from M. erubescens sensu Paterson, with which many of the populations have been confused in herbarium collections. The results for M. adpressus sens. str. corroborate the recognition as a unique species, as originally proposed by Cunningham and formalised by De Candolle in 1839. As such, the name M. adpressus should be restored to the original application, sensu De Candolle (1839).
On the balance of evidence, we recommend that the OTUs Melichrus sp. Galambary and M. sp. Kaputar be synonymised with M. adpressus sens. str. Morphological and molecular evidence indicated a close relationship between M. sp. Galambary and M. adpressus sens. str. across all analyses. There was some indication of an approximately north-south disjunction in morphology and genetics among the populations examined of M. sp. Galambary. This observation requires targeted sampling and further testing before accepting this phrase name as a separate species. Melichrus sp. Kaputar, known only from a single, isolated outcrop on Mount Kaputar, New South Wales is morphologically consistent with M. adpressus sens. str. Molecular analyses on the other hand, suggested that the OTU is related to a lineage containing M. adpressus sens. str. but is genetically divergent. This was particularly evident in the PCA and SplitsTree analysis. However, the results of the STRUCTURE analysis were more equivocal and the conStruct analysis suggested that this was only minimally differentiated from M. adpressus sens. str. Mount Kaputar is a known area of endemism (New South Wales Department of Planning, Industry and Environment 2021) the uniqueness of M. sp. Kaputar contributes to the conservation value of the area. However, in the absence of diagnostic characters to distinguish M. sp. Kaputar reliably from M. adpressus sens. str., we recommend that this be synonymised.
Melichrus sp. Gilgandra is strongly morphologically distinct from M. adpressus sens. str. but only weakly genetically diverged. This putative species with urceolate flowers and small, broad, often recurved leaves had more in common in the phenetic analyses with M. sp. Inglewood and M. urceolatus than M. adpressus sens. str. (Fig. 4). However M. sp. Gilgandra does share some morphological characters, such as fruit texture and calyx length, with M. adpressus sens. str. (Table 4c). The combination of strongly glaucous, blue-coloured leaves, pink petiole margins, urceolate flowers and wrinkly fruit epidermis make this putative species highly recognisable. The strong morphological but weak genetic discontinuity between M. sp. Gilgandra and M. adpressus sens. str. could indicate that M. sp. Gilgandra has only relatively recently diverged from a lineage containing M. adpressus sens. str. and is yet to accumulate many differences in allele frequency through genetic drift. The area around Gilgandra in New South Wales where this putative entity occurs is at the more arid end of the environmental envelope in which M. adpressus sens. str. occurs, therefore some of the morphological differences seen in M. sp. Gilgandra, such as smaller, glaucous leaves could be evidence of local selective adaptation (Solbrig 1994). These may also represent a plastic response to the environmental conditions (Carvajal et al. 2017). Ideally, this would be directly tested through greenhouse experiments (see Collins et al. 2022) but as species of Melichrus are challenging to cultivate from seed and cuttings, this has not been feasible.
As explained in the Introduction, to satisfy requirements for species status under the unified species concept, evidence should indicate that M. sp. Gilgandra is an independently evolving metapopulation lineage (de Queiroz 2007), identifiable by corresponding discontinuities in a variety of data sources. The evidence from morphological analyses supports M. sp. Gilgandra as a distinct species but the molecular data suggests that this is a morphologically distinct population of M. adpressus sens. str. Collections of new populations and data or greenhouse experiments may tell us more about the evolutionary trajectory of M. sp. Gilgandra. Until further evidence becomes available, we recommend that this be described as a subspecies of M. adpressus sens. str. Recognition as a subspecies will allow formal description of the unique morphology of this OTU. This is important for identification purposes and communicating the diversity value to conservationists.
A specimen of Melichrus sp. Gardens of Stone collected by Erwin Gauba near ‘Pigeon House Mountains’ (CANB 003865) was listed in Paterson (1958) under M. urceolatus but in both the molecular and morphological analyses all samples of M. sp. Gardens of Stone clustered closer to M. erubescens. More detailed analyses showed this OTU to be similar to, but diagnosable both morphologically and genetically as distinct from M. erubescens. We therefore recommend that this be described as a new species.
Melichrus sp. Silent Grove clustered near to, but distinct from M. erubescens in the morphological analyses. However, PCA and SplitsTree genetic analysis demonstrated that M. sp. Silent Grove is more genetically similar to, yet distinct from, M. urceolatus. Melichrus sp. Silent Grove does share some morphological traits with M. urceolatus, particularly the surface texture of the dried fruits but is easily distinguished by several characters, especially the adpressed and straight young leaves (v. young leaves erect to strongly spreading, often recurved) and the absence of a white margin on leaves (v. leaves with a white margin ~1 mm wide). Given the distinctiveness of M. sp. Silent Grove across analyses, we recommend recognising this narrow range endemic as a new species. The results of the STRUCTURE analysis support this recommendation but raise some questions regarding the evolutionary history of this new species. In the STRUCTURE analysis, M. sp. Silent Grove was represented almost entirely by a single ancestral population. This ancestral population was shared in part by all other geographically proximal Melichrus species, including M. sp. Torrington, M. urceolatus and M. sp. Boonoo Boonoo. There are plausible mechanistic explanations for such a pattern of admixture including that northern New South Wales is an evolutionary centre of diversity for this genus and that this layer represents an old, widely shared ancestral population. This pattern could also be an artefact of the analysis, as M. sp. Silent Grove was only represented in analyses by the single known population and thus warrants further testing.
Melichrus sp. Inglewood grouped with M. urceolatus in all genetic analyses. In morphological analyses, this clustered close to M. urceolatus but was separated by the particularly small leaves. No further characters were found to reliably segregate this putative species from all M. urceolatus populations. We recommend this be treated as a synonym of M. urceolatus.
A northern cluster was detected among the Melichrus urceolatus samples across all analyses. conStruct analysis indicated that the discontinuity observed in the STRUCTURE results was likely substantially independent of the influence of isolation by distance. Despite the strong genetic discontinuity between the northern cluster and remaining M. urceolatus samples, there was no morphological evidence to support species delimitation. Similarly, STRUCTURE results indicated substantial divergence and interesting patterns of admixture between populations of M. medius and some M. urceolatus. This genetic divergence was not matched by a corresponding discontinuity in morphological characteristics sufficient to recommend description as a separate species. The continued synonymy of M. medius with M. urceolatus is recommended.
Molecular data decisively placed Melichrus sp. Tara as conspecific with M. sp. Isla Gorge, and together, both were highly disjunct from M. urceolatus, despite being previously included under a broader concept of this species by Paterson (1958). Morphological analyses also strongly supported M. sp. Isla Gorge plus M. sp. Tara as distinct from M. urceolatus and we recommend that the two be described as a single new species.
Molecular and morphological results supported Melichrus sp. Mareeba as a species distinct from M. urceolatus. Furthermore, molecular results indicated that M. sp. Mareeba may have evolved from a hybrid or allopolyploid ancestor(s), where the closest living relatives of the ancestral parental lineages are M. sp. Isla Gorge plus M. sp. Tara and M. sp. Herberton plus M. sp. Kroombit Tops. This result is most strikingly apparent in the admixture pattern observed in the SplitsTree and STRUCTURE analyses (Figs. 7, 11). The elevated HE and HO of these populations, and the extremely negative FIS are more consistent with an allopolyploid or possibly a clonal F1 diploid hybrid (Fig. 13). A sexual diploid hybrid can likely be ruled out, as a single generation of outcrossing would be expected to bring FIS much closer to zero, though chromosome counts are needed to understand the possible involvement of polyploidy. Morphological phenetic analyses weakly separated M. sp. Mareeba from M. sp. Isla Gorge and M. sp. Tara samples clustered close to M. sp. Mareeba. Melichrus sp. Mareeba shares some morphological characters with M. sp. Herberton, most notably the presence of a wide white leaf margin. Several characters for the decisive diagnosis of M. sp. Mareeba as distinct from M. sp. Isla Gorge plus M. sp. Tara and M. sp. Herberton were identified (Table 4b). On the basis of this evidence, we recommend that M. sp. Mareeba be described as a new species. Results of unpublished phylogenetic analyses (Kennedy et al. unpublished manuscript) that interrogate the putative hybrid ancestry of M. sp. Mareeba shed more light on this taxon and bolster evidence supporting the delimitation (see Nauheimer et al. 2021).
The final segregate from Melichrus urceolatus was M. sp. Salvator Rosa but this was unable to be located in the field and thus excluded from the analyses, therefore the status of this segregate remains unresolved.
Melichrus adpressus sensu Paterson
In the genus-wide morphological analyses, segregates of M. adpressus sensu Paterson were spread across three distinct clusters. Melichrus sp. Gurulmundi clustered with M. urceolatus, due to similarities in both vegetative and floral morphology. Melichrus sp. Gurulmundi was found to be reliably diagnosable through a combination of several characters. Molecular evidence decisively demonstrated the genetic distinctiveness and as such we consider that this should be described as a new species.
Samples of M. adpressus sensu Paterson clustered with the single M. sp. Wahlmoorum sample in morphological analyses based on the shared floral morphology but in molecular analyses M. sp. Wahlmoorum did not group with any M. adpressus sensu Paterson segregates. This may indicate some divergence but is more likely an artefact due to inadequate sampling of M. sp. Wahlmoorum. We therefore recommend not recognising the entity as a distinct species until further information is available and we exclude this from the updated Melichrus taxonomy.
The third group recovered in the morphological analyses contained Melichrus sp. Herberton, M. sp. Kroombit Tops, M. sp. Boonoo Boonoo and M. sp. Yuraygir. Analysis of a subset of the morphological data containing only samples from this cluster demonstrated M. sp. Yuraygir to be morphologically distinct. Discontinuities in the results of the molecular analyses also supported M. sp. Yuraygir and M. adpressus sensu Paterson as distinct species. Melichrus sp. Herberton and M. sp. Kroombit Tops clustered tightly in all molecular analyses, despite the geographic disparity. Melichrus sp. Boonoo Boonoo clustered closely with M. sp. Kroombit Tops and near to M. sp. Herberton in morphological analyses but not in the DArTseq PCA and SplitsTree analyses. In the STRUCTURE analysis, the single population of M. sp. Boonoo Boonoo drew from two ancestral populations shared by M. sp. Yuraygir and M. adpressus sensu Paterson respectively. The discontinuities observed in the morphological analyses could not be readily reconciled with those observed in the molecular analyses. We recommend that M. sp. Boonoo Boonoo be excluded from taxonomic revision until further information is available. Denser sampling, particularly in northern New South Wales and southern Queensland may reveal a more detailed picture of species boundaries in this group.
Melichrus procumbens
We found that most evidence supported no change to the status quo taxonomy for Melichrus procumbens. The proportions of M. sp. Torrington plants were generally large, as noted by Paterson (1958) but this variation fell within the observed range for M. procumbens and so is not consistent with Paterson’s hypothesis that M. sp. Torrington is an autopolyploid. However, the two populations of M. sp. Torrington included in the molecular analyses did cluster together, separate from nearby M. procumbens samples. This pattern is consistent with M. sp. Torrington as an autopolyploid or a seperately evolving metapopulation but a chromosomal study is required for confirmation. As the discontinuity in the genetic dataset is not accompanied by a corresponding morphological discontinuity, we recommend that M. sp. Torrington be considered a synonym of M. procumbens.
Since Paterson’s revision, a morphologically distinctive taxon similar to Melichrus procumbens was collected in Colo Gorge, New South Wales and phrase-named M. sp. Colo Gorge (I.R.Telford 8659) NE Herbarium. This OTU could not be located in the field as efforts were impeded in part by bushfires and drought in the area. As such, this is excluded from the taxonomic revision pending new collections.
Melichrus hirsutus and Melichrus gibberagee
Melichrus hirsutus and M. gibberagee were collected for the first time several years after Paterson’s revision was published. The species were validly published in 2020 on the basis of qualitative morphology. The species status of both entities was strongly supported by both the morphological and genetic analyses. Similarities in the molecular analyses suggest the two species may be more closely related than would be expected based on strong morphological dissimilarity. Phylogenetic analyses should interrogate the relationship further.
Conclusion
Multiple lines of evidence, including genotypic differences, common ancestry and distinct morphological characteristics support the recognition of eight new species and one new subspecies of Melichrus, most of which are segregated from M. urceolatus demonstrating that this has long been used as a ‘catch-all’ name in the absence of a working taxonomy. Melichrus adpressus sens. str. should be restored to species status since having been subsumed into Melichrus urceolatus by Bentham in 1868. Long overdue recognition of the sensu stricto concept of M. adpressus has proved to be the ‘missing link’ in clarifying the long troubling and confused boundary between M. urceolatus and M. erubescens. Phylogenetic inference of relationships of the species demonstrated in this study is a natural next step towards understanding speciation in this group. That study and a formal taxonomic revision of the genus form two further manuscripts for imminent submission to Australian Systematic Botany.
Data availability
Raw morphological data and the DArTseq SNP dataset will be made available 12 months after the publication date, as forthcoming manuscripts are based on these datasets. These will be fully accessible through the University of New England data repository: https://rune.une.edu.au/web/index.jsp.
Conflicts of interest
Darren Crayn is Editor in Chief and Jeremy J. Bruhl is an Associate Editor for Australian Systematic Botany but were not involved in the peer review or decision-making process for this paper. Australian Systematic Botany encourages its editors to publish in the journal and they are kept totally separate from the decision-making process for their manuscripts. The authors have no further conflicts of interest to declare.
Declaration of funding
This research was supported by an Australian Government Research Training Program (RTP) Scholarship and a grant from the Australian Biological Resources Study National Taxonomy Research Grant Program. Further funding was gratefully received from the Friends of Grasslands and the University of New England School of Environmental and Rural Science.
Acknowledgements
We thank the directors, curators and staff of the following institutes for facilitating the use of the collections, through loans and visitation: BRI, CANB, CNS, MEL, NSW, PERTH, SEV and MA. Thank you to G and MEL for the provision of specimen images. We gratefully acknowledge permission from the following authorities to collect material in management areas: New South Wales Office of Environment and Heritage, Queensland Department of Environment and Heritage Protection, Victoria Department of Environment Land, Water and Planning, Australian Capital Territory Government and the many private land holders who kindly granted us access to their properties. We extend heartfelt thanks to all the staff, volunteers and students at the N.C.W. Beadle Herbarium for significant contributions to this project. We thank Peter Bostock and Karen Wilson for assistance translating Latin protologues. We thank Josh Zimmerman for assistance with troubleshooting DNA extractions, and Shelley Rowntree, Tareg Shaldoom, Anne-Cecile Colin and Nicolas Campione for assistance with R. We are grateful to Sangay Dema, Tim Collins, Oliver Marshall, Shelley Rowntree, Ruth Palsson, Dick Medd, John Edwards, Josh Brown, Fanie Venter, Eris Kennedy and Teresa Kennedy for assistance with specimen collection, and Betsy Jackes for her generous advice and continuing interest in Melichrus.
References
Barker WR (2005) Standardising informal names in Australian publications. Australian Systematic Botany Society Newsletter 122, 11-12.
| Google Scholar |
Bradburd GS, Coop GM, Ralph PL (2018) Inferring continuous and discrete population genetic structure across space. Genetics 210, 33-52.
| Crossref | Google Scholar | PubMed |
Bryant D, Moulton V (2004) Neighbor-Net: an agglomerative method for the construction of phylogenetic networks. Molecular Biology and Evolution 21, 255-265.
| Crossref | Google Scholar | PubMed |
Centre for Australian National Biodiversity Research (2018) Botanical Districts of Australia. (Australian National Herbarium) Available at https://www.anbg.gov.au/cpbr/anhsir/anhsir-manual/botanical-districts.html [Verified 31 March 2025]
Carstens BC, Pelletier TA, Reid NM, Satler JD (2013) How to fail at species delimitation. Molecular Ecology 22, 4369-4383.
| Crossref | Google Scholar | PubMed |
Carvajal DE, Loayza AP, Rios RS, Gianoli E, Squeo FA (2017) Population variation in drought-resistance strategies in a desert shrub along an aridity gradient: Interplay between phenotypic plasticity and ecotypic differentiation. Perspectives in Plant Ecology, Evolution and Systematics 29, 12-19.
| Crossref | Google Scholar |
Collins TL, Schmidt-Lebuhn AN, Andrew RL, Telford IRH, Bruhl JJ (2022) There’s gold in them thar hills! Morphology and molecules delimit species in Xerochrysum (Asteraceae; Gnaphalieae) and reveal many new taxa. Australian Systematic Botany 35, 120-185.
| Crossref | Google Scholar |
Crayn DM, Hislop M, Puente-Lelièvre C (2020) A phylogenetic recircumscription of Styphelia (Ericaceae, Epacridoideae, Styphelieae). Australian Systematic Botany 33, 137-168.
| Crossref | Google Scholar |
Dayrat B (2005) Towards integrative taxonomy. Biological Journal of the Linnean Society 85, 407-415.
| Crossref | Google Scholar |
Denham SS, Brignone NF, Johnson LA, Pozner RE (2019) Using integrative taxonomy and multispecies coalescent models for phylogeny reconstruction and species delimitation within the “Nastanthus–Gamocarpha” clade (Calyceraceae). Molecular Phylogenetics and Evolution 130, 211-226.
| Crossref | Google Scholar | PubMed |
de Queiroz K (2007) Species concepts and species delimitation. Systematic Biology 56, 879-86.
| Crossref | Google Scholar | PubMed |
Druce GC (1917) Nomenclatural Notes: chiefly African and Australian. Botanical Society and Exchange Club of the British Isles Report for 1916 4, 635.
| Google Scholar |
Edwards DL, Knowles LL (2014) Species detection and individual assignment in species delimitation: can integrative data increase efficacy? Proceedings of the Royal Society of London – B. Biological Sciences 281(1777), 20132765.
| Crossref | Google Scholar | PubMed |
Ewart K, Lo N, Ogden R, Joseph L, Ho S, Frankham G, Eldridge M, Schodde R, Johnson R (2020) Phylogeography of the iconic Australian red-tailed black-cockatoo (Calyptorhynchus banksii) and implications for its conservation. Heredity 125, 167.
| Crossref | Google Scholar |
Field B (Ed.) (1825) ‘Geographical Memoirs on New South Wales; by Various Hands.’ (John Murray: London, UK) Available at http://gutenberg.net.au/ebooks13/1304421h.html
Fraley C, Raftery AE (2002) Model-based clustering, discriminant analysis, and density estimation. Journal of the American Statistical Association 97, 611-631.
| Crossref | Google Scholar |
Fujita MK, Leaché AD, Burbrink FT, McGuire JA, Moritz C (2012) Coalescent-based species delimitation in an integrative taxonomy. Trends in Ecology & Evolution 27, 480-488.
| Crossref | Google Scholar | PubMed |
Funk SM, Guedaoura S, Juras R, Raziq A, Landolsi F, Luís C, Martínez AM, Musa Mayaki A, Mujica F, Oom MdM, Ouragh L, Stranger Y-M, Vega-Pla JL, Cothran EG (2020) Major inconsistencies of inferred population genetic structure estimated in a large set of domestic horse breeds using microsatellites. Ecology and Evolution 10, 4261-4279.
| Crossref | Google Scholar | PubMed |
Gower JC (1971) A general coefficient of similarity and some of its properties. Biometrics 27, 857-871.
| Crossref | Google Scholar |
Gruber B, Unmack PJ, Berry OF, Georges A (2018) dartR: an R package to facilitate analysis of SNP data generated from reduced representation genome sequencing. Molecular Ecology Resources 18, 691-699.
| Crossref | Google Scholar | PubMed |
Hausdorf B, Hennig C (2010) Species delimitation using dominant and codominant multilocus markers. Systematic Biology 59(5), 491-503.
| Crossref | Google Scholar | PubMed |
Hörandl E, Stuessy TF (2010) Paraphyletic groups as natural units of biological classification. Taxon 59, 1641-1653.
| Crossref | Google Scholar |
Hosegood J, Humble E, Ogden R, de Bruyn M, Creer S, Stevens GMW, Abudaya M, Bassos-Hull K, Bonfil R, Fernando D, Foote AD, Hipperson H, Jabado RW, Kaden J, Moazzam M, Peel LR, Pollett S, Ponzo A, Poortvliet M, Salah J, Senn H, Stewart JD, Wintner S, Carvalho G (2020) Phylogenomics and species delimitation for effective conservation of manta and devil rays. Molecular Ecology 29, 4783-4796.
| Crossref | Google Scholar | PubMed |
Huson DH, Bryant D (2006) Application of phylogenetic networks in evolutionary studies. Molecular Biology and Evolution 23, 254-267.
| Crossref | Google Scholar | PubMed |
Janes JK, Miller JM, Dupuis JR, Malenfant RM, Gorrell JC, Cullingham CI, Andrew RL (2017) The K=2 conundrum. Molecular Ecology 26, 3594-3602.
| Crossref | Google Scholar | PubMed |
Kennedy HT, Telford IRH, Crayn DM, Bruhl JJ (2020) Validation of two informally named species of Melichrus (Ericaceae: Epacridoideae) from north-eastern New South Wales. Telopea 23, 187-196.
| Crossref | Google Scholar |
Kilian A, Wenzl P, Huttner E, Carling J, Xia L, Blois H, Caig V, Heller-Uszynska K, Jaccoud D, Hopper C, Aschenbrenner-Kilian M, Evers M, Peng K, Cayla C, Hok P, Uszynski G (2012) Diversity Arrays Technology: a generic genome profiling technology on open platforms. In ‘Data Production and Analysis in Population Genomics: Methods and Protocols’. (Eds F Pompanon, A Bonin) pp. 67–89. (Humana Press: Totowa, NJ, USA)
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Molecular Ecology Resources 15, 1179-1191.
| Crossref | Google Scholar | PubMed |
Kron KA, Judd WS, Stevens PF, Crayn DM, Anderberg AA, Gadek PA, Quinn CJ, Luteyn JL (2002) Phylogenetic classification of Ericaceae: molecular and morphological evidence. Botanical Review 68, 335-423.
| Crossref | Google Scholar |
Kruskal JB (1964) Multidimensional scaling by optimizing goodness of fit to a nonmetric hypothesis. Psychometrika 29, 1-27.
| Crossref | Google Scholar |
Lawson DJ, van Dorp L, Falush D (2018) A tutorial on how not to over-interpret STRUCTURE and ADMIXTURE bar plots. Nature Communications 9, 3258.
| Crossref | Google Scholar | PubMed |
Linck E, Battey CJ (2019) Minor allele frequency thresholds strongly affect population structure inference with genomic data sets. Molecular Ecology Resources 19, 639-647.
| Crossref | Google Scholar | PubMed |
Mace GM (2004) The role of taxonomy in species conservation. Philosophical Transactions of the Royal Society of London – B. Biological Sciences 359, 711-719.
| Crossref | Google Scholar | PubMed |
Meudt HM, Thorsen MJ, Prebble JM (2020) Taxonomic revision of the Myosotis australis group (Boraginaceae) native to Australia, New Zealand and New Guinea. Australian Systematic Botany 33, 477-524.
| Crossref | Google Scholar |
Mijangos JL, Gruber B, Berry O, Pacioni C, Georges A (2022) dartR v2: an accessible genetic analysis platform for conservation, ecology and agriculture. Methods in Ecology and Evolution 13, 2150-2158.
| Crossref | Google Scholar |
Nauheimer L, Weigner N, Joyce E, Crayn D, Clarke C, Nargar K (2021) HybPhaser: a workflow for the detection and phasing of hybrids in target capture datasets. Applications in Plant Sciences 9, e11441.
| Crossref | Google Scholar | PubMed |
Padial JM, Miralles A, De la Riva I, Vences M (2010) The integrative future of taxonomy. Frontiers in Zoology 7, 16.
| Crossref | Google Scholar | PubMed |
Paterson BR (1958) Revision of the genus Melichrus R.Br. (Epacridaceae). Proceedings of the Linnean Society of New South Wales 82, 303-313.
| Google Scholar |
Pembleton LW, Cogan NO, Forster JW (2013) StAMPP: an R package for calculation of genetic differentiation and structure of mixed-ploidy level populations. Molecular Ecology Resources 13, 946-952.
| Crossref | Google Scholar | PubMed |
Powell JM, Morrison DA, Gadek PA, Crayn DM, Quinn CJ (1997) Relationships and generic concepts within Styphelieae (Epacridaceae). Australian Systematic Botany 10, 15-29.
| Crossref | Google Scholar |
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155, 945-959.
| Crossref | Google Scholar | PubMed |
Puente-Lelièvre C, Hislop M, Harrington M, Brown EA, Kuzmina M, Crayn DM (2016) A five-marker molecular phylogeny of the Styphelieae (Epacridoideae, Ericaceae) supports a broad concept of Styphelia. Australian Systematic Botany 28, 368-387.
| Crossref | Google Scholar |
Rouhan G, Gaudeul M (2021) Plant taxonomy: a historical perspective, current challenges, and perspectives. Methods in Molecular Biology 2222, 1-38.
| Crossref | Google Scholar | PubMed |
Rutherford S, Rossetto M, Bragg JG, McPherson H, Benson D, Bonser SP, Wilson PG (2018) Speciation in the presence of gene flow: population genomics of closely related and diverging Eucalyptus species. Heredity 121, 126-141.
| Crossref | Google Scholar | PubMed |
Scrucca L, Fop M, Murphy TB, Raftery AE (2016) mclust 5: clustering, classification and density estimation using Gaussian finite mixture models. The R Journal 8, 289-317.
| Google Scholar | PubMed |
Scrucca L, Fraley C, Murphy TB, Raftery AE (2023) ‘Model-based clustering, classification, and density estimation using mclust in R.’ (Chapman and Hall: New York, NY, USA) doi:10.1201/9781003277965
Sokal RR (1963) The principles and practice of numerical taxonomy. Taxon 12, 190-199.
| Crossref | Google Scholar |
Stanton DWG, Frandsen P, Waples RK, Heller R, Russo I-RM, Orozco-terWengel PA, Pedersen C-ET, Siegismund HR, Bruford MW (2019) More grist for the mill? Species delimitation in the genomic era and its implications for conservation. Conservation Genetics 20, 101-113.
| Crossref | Google Scholar |
Struck TH, Feder JL, Bendiksby M, Birkeland S, Cerca J, Gusarov VI, Kistenich S, Larsson KH, Liow LH, Nowak MD, Stedje B, Bachmann L, Dimitrov D (2018) Finding evolutionary processes hidden in cryptic species. Trends in Ecology and Evolution 33, 153-163.
| Crossref | Google Scholar | PubMed |
von Mueller F (1868) ‘Fragmenta Phytographiae Australiae vol. VI.’ (Government Printer: Melbourne, Colony of Victoria) [In Latin] doi:10.5962/bhl.title.287
Watson L (1967) Taxonomic implications of a comparative anatomical study of Epacridaceae. New Phytologist 66, 495-504.
| Crossref | Google Scholar |
Wege JA, Thiele KR, Shepherd KA, Butcher R, Macfarlane TD, Coates DJ (2015) Strategic taxonomy in a biodiverse landscape: a novel approach to maximizing conservation outcomes for rare and poorly known flora. Biodiversity and Conservation 24, 17-32.
| Crossref | Google Scholar |