

Arsenic binding to organic and inorganic sulfur species during microbial sulfate reduction: a sediment flow-through reactor experiment
Raoul-Marie Couture A B C F , Dirk Wallschläger D , Jérôme Rose E and Philippe Van Cappellen A BA Georgia Institute of Technology, 311 Ferst Drive, Atlanta, GA 30332, USA.
B University of Waterloo, 200 University Avenue West, Waterloo, ON, N2L 3G1, Canada.
C Norwegian Institute for Water Research, Gaustadalléen 21, N-0349 Oslo, Norway.
D Trent University, 1600 West Bank Drive, Peterborough, ON, K9J 7B8, Canada.
E CNRS-Aix Marseille University UMR 7330 CEREGE, Europôle de l’Arbois, 13545 Aix-en-Provence, France.
F Corresponding author. Email: rmc@niva.no
Environmental Chemistry 10(4) 285-294 https://doi.org/10.1071/EN13010
Submitted: 18 January 2013 Accepted: 26 May 2013 Published: 5 August 2013
Environmental context. The use of water contaminated with arsenic for drinking and irrigation is linked to water and food borne diseases throughout the world. Although reducing conditions in soils and sediments are generally viewed as enhancing arsenic mobility in subsurface environments, we show they can actually promote As sequestration in the presence of reduced sulfur species and labile organic matter. We propose that sulfurisation of organic matter and subsequent binding of As to thiol groups may offer an innovative pathway for As remediation.
Abstract. Flow-through reactors (FTRs) were used to assess the mobility of arsenic under sulfate reducing conditions in natural, undisturbed lake sediments. The sediment slices in the FTRs were supplied continuously with inflow solutions containing sulfate and soluble AsIII or AsV and, after 3 weeks, also lactate. The experiment ran for a total of 8 weeks. The dissolved iron concentration, pH, redox potential (Eh), as well as aqueous As and sulfur speciation were monitored in the outflow solutions. In FTRs containing surface sediment enriched in labile organic matter (OM), microbial sulfate reduction led to an accumulation of organically bound S, as evidenced by X-ray absorption spectroscopy. For these FTRs, the inflowing dissolved As concentration of 20 μM was lowered by two orders of magnitude, producing outflow concentrations of 0.2 μM monothioarsenate and 0.1 μM arsenite. In FTRs containing sediment collected at greater depth, sulfide and zero-valent S precipitated as pyrite and elemental S, while steady-state outflow arsenite concentrations remained near 5 μM. The observations thus suggest that As sequestration is enhanced when sediment OM buffers the free sulfide and zero-valent S concentrations. An updated conceptual model for the fate of As in the anoxic As–C–S–Fe system is presented based on the results of this study.
Introduction
Arsenic, a highly toxic metalloid, is a worldwide public health concern as As remobilisation from soil and sediment can contaminate water used for drinking and irrigation.[1–3] The high affinity of As for metal oxyhydroxides, in particular those of ferric iron, often leads to very low As concentrations in the aqueous phase at comparatively high solid phase concentrations. The latter nonetheless pose a threat as flooding, a rise in watertable or an increased supply of electron donors may create reducing conditions, which in turn may translocate As to the aqueous phase through a variety of processes, for instance microbial FeIII oxyhydroxide respiration coupled to the degradation of organic matter (OM).[3] Because under reducing conditions As can be sequestered by co-precipitation with,[4] or adsorption onto[5–7] Fe sulfides, such as pyrite (FeS2(s)) and mackinawite (FeSm(s)), production of sulfide as a result of microbial sulfate (SO42–) reduction has been suggested as a possible As remediation pathway for subsurface environments.[8] The caveat, however, is that the redox boundary separating FeIII oxyhydroxides from FeII and As sulfides is potentially a zone of high As remobilisation.
The environmental fate and transport of As is thus closely related to the biogeochemical cycling of iron, sulfur and carbon, as well as their relative abundances.[9–12] In reducing soils and sediments, in the presence of complex mineral assemblages,[13–16] As sorption onto Fe sulfides is often of minor importance except at high As loading and slightly acidic pH. Under the latter conditions, As uptake by Fe monosulfide (FeSm(s)) occurs through the formation of a sorption complex akin to realgar (AsS(s)), as shown by Gallegos et al.[17] In environments where all the reactive Fe is transformed into Fe sulfides, the remaining free sulfide (S–II) becomes available to complex As, allowing for the precipitation of As sulfide minerals, such as AsS(s) and orpiment (As2S3(s)), and the formation of thiolated As species.[18] At the present time, the mechanisms by which aqueous S, including sulfide (S(aq)–II), zero-valent sulfur (S(aq)0) and polysulfides (Sn0S(aq)–II), all ubiquitous in the aquatic environment,[19] promote thioarsenite and thioarsenate formation are not completely understood.[20]
The key influence of Fe and S on the geochemical behaviour of As has led to the classification of subsurface environments as exhibiting either Fe- or S-controlled As sequestration.[11] Because this classification does not explicitly consider the role of organic C – other than as an energy source for microbial metabolism[13,14] – it may overlook the full spectrum of reactions involving As and natural OM. For instance, it was recently shown that thiols (C–SH) present in OM-rich wetlands and peaty soils can efficiently bind arsenite.[21,22] Such geochemical conditions are likely not restricted to wetlands and may occur in other soils and sediments where thiols form as a result of assimilatory SO42– reduction and OM sulfurisation.[23] Nevertheless, although there is compelling evidence for the abundance of thiols, or reduced organic S (Sorg), in marine[24] and lake sediments,[25,26] thiol formation has so far been a neglected process in laboratory studies focussing on As mobility under sulfidic conditions. These studies typically only consider sulfide minerals as the dominant sink for reduced S.
Here, we used flow through reactors[27] (FTRs) containing undisturbed lake sediments to study As and S biogeochemistry under controlled, near steady state conditions. The FTR approach causes minimal disturbance of the physical and microbiological structure of the natural sediment and, hence, allows one to obtain information under environmentally relevant conditions. Our main objectives were to: (i) quantify the extent of thiol formation during microbial SO42– reduction in aquatic sediments containing contrasting initial OM and Fe contents and (ii) identify the dominant aqueous and solid-phase As species formed during the microbially mediated formation of S(aq)0, Sn0S(aq)–II and S(aq)–II, with a particular emphasis on organic and inorganic thiolated As.
The experimental design is illustrated in Fig. 1. The microbial community naturally present in the lake sediment samples were stimulated by adding lactate and sulfate to induce microbial sulfidogenesis in four FTRs, two of them filled with the upper two centimetres of sediment from duplicate cores, and two with sediment from a deeper depth interval (4–6 cm). Sediment from the 0–2-cm depth interval was enriched in recently deposited (labile) OM, relative to the deeper sediment. The FTRs were supplied with solutions containing sulfate, and either dissolved AsIII or AsV. After 3 weeks, the inflow solutions were further amended with a labile organic electron donor (lactate). The outflow solutions of the FTRs were analysed for dissolved constituents using a variety of analytical techniques, whereas solid-phase speciation of As and S were analysed by X-ray absorption spectroscopy (XAS).
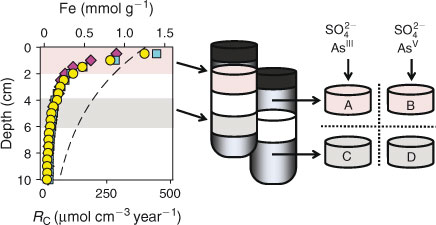
|
Methodology
Field site and sediment sampling
Lake Tantaré (47°4′4″N, 71°32′59″W) is a slightly acidic (pH 5.8), perennially oxygenated (>3.75 mg O2 L–1), oligotrophic headwater lake located in the Province of Québec, Canada. Three cores were collected by divers in September 2010 in the deepest, central part of the lake. Cores #1 and #2 supplied the sediment slices for the FTR, whereas core #3 was used to characterise the initial condition of the sediment. Sampling was carried out with shuttle corers[27] consisting of a continuous stacking of Plexiglas reactor cells. Once on shore, undisturbed sediment slices of 2-cm thickness and 4.7-cm diameter were recovered in an anaerobic glove bag (PE glove-bag, Alfa-Aesar, Ward Hill, MA, USA) purged with a continuous N2 flow. The FTRs were assembled within 30 min by placing nitrite (NBR) O-rings on the reactor cells, covering the sediment with 0.2-µm pore size membranes (poly-ether sulfone, Pall Corporation, Port Washington, NY, USA), backing filters (fibreglass, Pall Corporation) and sealing the cells with high density polyethylene (HDPE) covers with input and output channels. The sealed FTRs were kept at 4 °C during shipment by plane from Québec City to the Georgia Institute of Technology in Atlanta, where the experiments were performed.
Sediments from the deepest part of Lake Tantaré have previously been shown to be laterally homogenous. The topmost sediment layer is enriched in diagenetic Fe oxyhydroxides (~1 mmol Fe g–1, see Fig. 1),[26] consisting mainly of poorly crystalline ferrihydrite, lepidocrocite and goethite.[28] The sediments are organic-rich (~20 mmol Corg g–1), with OM representing on average approximately half of the sediment’s dry weight. The lability of sediment OM decreases exponentially with depth, as evidenced by the rates of Corg degradation (RC, µmol C cm–3 year–1) measured independently in batch incubations.[26] The concurrent decrease of the concentration of Fe oxyhydroxides and of the lability of OM (Fig. 1) is attributed to OM degradation coupled to dissimilatory Fe reduction in the sediments,[26] a process that has been shown to induce profound changes in the reactivity of both OM and Fe oxyhydroxides over time.[29] Based on these expected differences in OM and Fe reactivity as a function of depth in the sediments, and on the previously determined depth profiles of solid-phase Fe and RC, depth intervals 0–2 and 4–6 cm were selected for the FTR experiment. The uppermost sediments (0–2 cm), enriched in labile OM and in diagenetic Fe oxyhydroxides, are labelled FTRs A and B, whereas the deeper sediments (4–6 cm), depleted in labile OM and Fe oxyhydroxides (relative to the depth interval 0–2 cm) are labelled FTRs C and D.
FTR experiments and aqueous phase characterisation
General methods in the laboratory
Glass- and plastic-ware were cleaned by soaking in 10 % (v/v) HNO3 (VWR, ACS grade, Atlanta, GA, USA) for at least 24 h, followed by 24 h in 1 % (v/v) HNO3 and repeated rinsing with Milli-Q water (resistivity of 18.2 MΩ cm–1). Acids used for reagent solutions were of ultra-pure grade (VWR, Aristar grade), and all solutions were prepared using Milli-Q water, which was brought to boil and cooled while bubbling with high purity N2. As a precautionary measure, laboratory ware, (sealed) input solutions and coring equipment were sterilised by autoclaving or washing with an anti-bacterial agent.
Experimental conditions
The experiment was conducted over 8 weeks (56 days) in an O2-free (2 % H2 in N2 with O2(g) <1 ppm) anaerobic chamber (Type A Vinyl Anaerobic Chamber with Pd catalyst, Coy Laboratories, Grass Lake, MI, USA). FTRs A and C were supplied with inflow solutions containing 20 µM of dissolved arsenate (NaHAsVO4·7H2O(s); Sigma–Aldrich), whereas FTRs B and D were supplied with inflow solutions containing 20 µM of dissolved arsenite (Na2AsIIIO2(s); Sigma–Aldrich). All inflow solutions were made using 0.2-µm filtered deoxygenated Milli-Q water (see above) and contained 200 µM Na2SO4(s) (Alfa–Aesar). After 21 days, the inflow solutions to all FTRs were amended with 200 µM Na–lactate (Sigma–Aldrich) as an electron donor. Throughout the experiment, input solutions were adjusted to the lake’s natural pH (5.8) and ionic strength (I = 0.5 mM) respectively using HCl–NaOH and NaCl (all Suprapur grade, EMD Chemicals). The solutions were supplied at an imposed constant flow rate of 1.0 ± 0.1 mL h–1, or ~1 pore volume day–1, using a peristaltic pump (ICP-N, Ismatec, Wertheim, Germany). The FTR outflows were collected in the anaerobic chamber using a computer-controlled fraction collector in vials containing, if needed, appropriate reagents for sample preservation (see below) and, unless otherwise noted, kept under N2 until analysis. The following parameters were measured every two days in the FTR outflows: redox potential (Eh), pH, [SO42–], total concentration of aqueous sulfides (ΣS(aq)–II), total concentration of aqueous zero-valent S (ΣS(aq)0 = S(aq)0 + Sn0S(aq)–II), total concentration of dissolved Fe and total dissolved As (ΣAs). Detailed aqueous As speciation was performed six times throughout the experiment, that is, every 7 days with the exception of day 35 where sample preservation could not be performed because of a liquid N2 shortage. At the end of the FTR experiment the solid-phase was pressure-filtered under N2, homogenised and preserved in aliquots for analysis.
Sample preservation
Samples for SO42– measurements were delivered to polypropylene tubes (Eppendorf) and those for major cations were delivered to 4-mL HDPE vials (VWR) containing 45 µL of 1.5 M HNO3 (Aristar VWR); the tubes were kept at 4 °C until analysis. Samples for ΣS–II measurements were delivered to 4-mL PTFE-lined septa amber vials (VWR) containing 40 µL of 2.7 mM N,N′-dimethyl-p-phenylenediamine sulfate (Sigma–Aldrich) and 5.5 mM FeCl3 (Alfa-Aesar). Samples for ΣS(aq)0 measurement were delivered to 4-mL PTFE-lined septa amber vials (VWR) containing 2 mL of double-distilled ethanol, 40 µL of tetrahydrofuran (THF, Sigma–Aldrich, HPLC grade), 0.4 mL of 1 M NaNO3 (Merck, Suprapur grade), and 10 µL of 1 M HNO3. Samples for total As measurements were delivered to 3.5-mL HDPE vials (Camenco Cryovials), immediately flash-frozen in liquid N2 and placed in a –80 °C freezer. The subset of samples selected for detailed aqueous As speciation was shipped overnight on dry-ice to Trent University (Canada), where the As speciation measurements were performed, and stored in a –80 °C freezer until analysis.
Analytical techniques
The pH measurements were carried out in the anaerobic chamber using a gel-filled, epoxy-sealed electrode (VWR), and the redox potentials were measured using a 3.5 M KCl-filled Pt electrode (VWR) and converted into potential values relative to a standard hydrogen electrode. Dissolved concentrations of SO42– were analysed by ion chromatography (Dionex DX-300 system with AMMS-2 conductivity suppressor, IONPAC AG-14 column and AS-14 pre-column, Dionex Thermo-Fisher Scientific, Sunnyvale, CA, USA). Dissolved Fe concentrations were determined by inductively coupled plasma–optical emission spectroscopy (ICP-OES Ultima 2C, HORIBA Instruments, Chicago, IL, USA) and ΣS(aq)–II by UV–Vis spectroscopy (DU Series 700, Beckman-Coulter, Atlanta, GA, USA). ΣS(aq)0 was analysed by square-wave cathodic stripping voltammetry (SW-CSV) in hanging Hg drop mode (Metrohm 940 VA Computrace, Metrohm USA, Riverview, FL, USA) according to Wang et al.[30] Total aqueous As concentrations were determined by atomic fluorescence spectroscopy coupled to continuous flow hydride generation (HG-AFS, PSA 10.055 Millenium Excalibur, PS Analitical, Orpington, UK). To eliminate the possibility of As sulfide precipitation within the flow-injection coil,[31] the pH of the sample was raised to pH >10 with 1 N NaOH and then oxidised with 30 % H2O2 (VWR), thus oxidising As species to AsV and S species to SVI.[32] The solid-bound Corg and total sulfur (Stot) concentrations were determined on a CHNS analyzer (EA 1108, Carlo-Erba, Milan, Italy).
Aqueous As speciation was analysed by anion exchange chromatography (AEC, Dionex Thermo-Fisher Scientific) coupled to inductively coupled plasma–mass spectrometry (ICP-MS, Elan DRC II, Perkin-Elmer, Woodbridge, ON, Canada) according to Wallschläger and London.[33] Briefly, the separation was conducted at alkaline pH on a column with low hydrophobicity to allow intact and rapid elution of the acid-labile As–S compounds, and eluant elimination with a membrane suppressor was not used to prevent losses of certain methylated As species in the suppressor. Both As and S were measured as their oxides in dynamic reaction cell mode, using oxygen as the reaction gas. The following As species, shown in their completely deprotonated form (along with the abbreviation used for each throughout the manuscript), were separated: arsenite (AsO33–, AsIII), arsenate (AsO43–, AsV), monothioarsenate (AsOS3–, MTAsV), dithioarsenate (AsO2S23–, DTAsV), trithioarsenate (AsOS33–, TTAsV), tetrathioarsenate (AsS43–, T4TAsV), monomethylarsenite ((CH3)AsO22–; MMAsIII), monomethylarsenate ((CH3)AsO32–, MMAsV), monomethylmonothioarsenate ((CH3)AsSO22–, MMMTAsV), monomethyldithioarsenate ((CH3)AsS2O2–, MMDTAsV), mono-methyltrithioarsenate ((CH3)AsS32–, MMTTAsV), dimethylarsenite ((CH3)2AsO, DMAsIII), dimethylarsenate ((CH3)2AsO2–, DMAsV), dimethylmonothioarsenate ((CH3)2AsOS–, DMMTAsV) and dimethyldithioarsenate ((CH3)2AsS2–, DMDTAsV). In addition, the total aqueous As concentration was determined independently by ICP-MS in the dynamic reaction cell mode, using oxygen as the reaction gas. The speciation mass balance (= Σ(detected As species)/(total As concentration)) averaged 107 ± 18 % (1σ) over all measured samples, confirming that all quantitatively significant As species were detected in the speciation analyses.
Solid-phase characterisation
Solid-phase aliquots were collected from the sediment core #3 (initial conditions) as well as from each of the four reactors at the end of the experiment. Samples were either flash-frozen in liquid N2 and kept at –80 °C until XAS analysis or frozen at –20 °C until sequential extraction of S phases (see below).
Arsenic speciation in the sediment samples was examined by extended X-ray absorption fine structure spectroscopy (EXAFS) and by X-ray absorption near edge structure spectroscopy (XANES) at the As K-edge (11 867 eV) at the bending magnet beamline BM30b of the European Synchrotron Radiation Facility (ESRF, Grenoble, France). Sulfur speciation in the sediment samples was examined by XANES at the S K-edge (2472 eV) at the wriggler beamline 4–3 of the Stanford Synchrotron Radiation Laboratory (SSRL, Stanford, CA, USA). The X-ray energy resolution was achieved by a Si(111) monochromator calibrated relative to the L3-edge energy of an elemental gold (Au(s)) standard (Sigma–Aldrich) at 11 919 eV (As) or to the white line of a Na2S2O3(s) standard (Sigma–Aldrich) at 2479.3 eV (S). Spectra were collected in transmission mode using an ionisation chamber or in fluorescence mode using a 30-element array Canberra germanium solid-state detector (for As) or a 4-element solid-state Si detector (for S). Analyses for As were carried out at t < 15 K in a helium (He) cryostat to limit As photooxidation or reduction, whereas those for S were carried with the sample compartment and detector under slight He over-pressure. For S, higher-order harmonics were reduced by detuning the second monochromator to 20 % of its maximum intensity. Reference compounds were prepared as described in the Supplementary material.
For all edge-normalised, background corrected spectra, the relative contributions of the different As and S reference compounds were calculated by linear combination fitting (LCF) using the ATHENA software package.[34] The LCF procedure first aimed at reproducing quantitatively all the features of the spectra, using the smallest number of components, and removing those contributing to less than 5 % of the sum. This step yielded fits with 1–3 components. Visually similar fits were further evaluated by selecting those with the lowest R-factor values, and permutations were tested by selecting the combinations that lowered the reduced Chi-square (χred2) values by at least 20 %, which was deemed a significant improvement. The suite of reference materials included was as follows: (i) the sodium salts of AsIII and AsV (Sigma–Aldrich), MTAsV and TTAsV prepared according to Wallschläger and Stadey,[35] (ii) the minerals AsS(s) (Sigma–Aldrich), As2S3(s) (Alfa-Aesar), nano-crystalline As2S3(s) (digitised from Helz et al.[36]), scorodite and arsenopyrite (D. Testemale, pers. comm. and James-Smith et al,[37]), (iii) the sorption complexes of AsIII, AsV, MTAsV and TTAsV sorbed onto two-lines ferryhydrite, goethite, FeSm(s) and FeS2(s) (Couture et al.[38]) and (iv) the thiol-bound AsIII species glutamyl-cysteinyl-glycinyl-thioarsenite (AsIII-glu; C. Mikutta, pers. comm and Langner et al.[21]). For S K-edge XANES, the suite of reference material included was as follows: FeS2(s) (Strem Chemicals), rhombic elemental sulfur (S(α)8(s); Alpha-Aesar), cysteine (Scyst; Sigma–Aldrich), methionine sulfoxide (Sigma–Aldrich), cysteic acid (Sigma–Aldrich) and chondroitin sulfate (Sigma–Aldrich). To determine changes in solid-phase S composition over the course of the experiment, initial lake sediment samples from the two depth intervals were included with the reference compounds.
Despite precautions taken during sample preparation, S K-edge XANES can display non-linear additive behaviour depending on the S content and particle size distribution of the sample,[39] thus introducing uncertainty in the LCF estimations. To complement the XANES results, three inorganic solid-phase S pools were therefore operationally defined based on the classic sequential extraction scheme for acid-volatile sulfide (AVS), chromium-reducible sulfur (CRS) and elemental sulfur (ES), and nominally ascribed to the species FeSm(s), FeS2(s) and S(α)8(s). The extraction scheme chosen, detailed in the Supplementary material, was optimised for room temperature and is safer than protocols involving heated acidic oxidising solutions.[40,41] Sulfur not released by the sequential extraction scheme was assumed to represent organic S (Sorg = Stot – [AVS + CRS + ES]). Note that we elected not to apply sequential extractions for As and Fe because the outcomes of published extraction schemes in the presence of reduced S species have been criticised.[10]
Thermodynamic calculations
Thermodynamic calculations were performed with the public domain computer code PHREEQC, Version 2.17.5.[42] To be used with PHREEQC, Eh values were converted into pe (negative logarithm of the hypothetical electron activity) using the Nernst Equation (Eh = 0.059 × pe). The following calculations were performed: (i) the theoretical pe based on the ratios of the measured redox couples and (ii) the saturation state of the FTR outflows with respect to selected sulfur minerals (AsS(s), As2S3(s), AsFeS(s), FeS(s), FeSm(s), FeSt(s), FeS2(s) and αS8(s)). Details on the thermodynamic calculations and databases used are given in the Supplementary material.
Results
Solid phase transformations during FTR experiments
The quantity and speciation of S sequestered in the FTRs over the course of the experiment differed strikingly between the 0–2- and 4–6-cm depth intervals. FTRs A and B (0–2 cm, OM respiration rates ~600 µmol C g–1 year–1) sequestered 80 and 87 µmol S g–1, whereas FTRs C and D (4–6 cm, OM respiration rates ~65 µmol C g–1 year–1) sequestered 35 and 53 µmol S g–1. Both the sequential extractions and LCF of XANES spectra at the S K-edge (Table 1 and Fig. 1) implied that the majority of S accumulated in FTRs A and B was in the reduced Sorg fraction (estimated by LCF using cysteine as a reference compound), whereas most S accumulated in the FeS2(s) and S(α)8(s) fractions in FTRs C and D. Negative values of AVS (Table 1) indicate that they represented a smaller proportion of total S after the incubation than in the initial sediment.

|
Of the 26 reference compounds considered, six were necessary for the LCF procedure to reproduce the As K-edge XAS spectra of the entire set of samples (Table 2 and Fig. 2b–c). The positions of the As K-edge XANES peaks indicate that As accumulated predominantly as AsIII in the FTRs (Table 2). In FTRs A and B, 45 % of solid-phase As was attributed to AsS(s), 35–45 % to thiol-bound As (AsIII-glu) and the remaining to As sorbed onto goethite initially present in the 0–2-cm depth interval sediment. The high proportion of AsS(s) in FTRs A and B (Table 2) is compatible with the observation that almost all S is under the organic form (Table 1), because mass-balance calculations indicate that AsS(s) contributes, at most, to 3 % of the total S, a proportion lower than the precision of ±5 % typically afforded by LCF procedures. For the deeper sediments (FTRs C and D), the dominant As species was identified as As2S3(s). In FTR C, LCF suggests that an AsV–S compound (using TTAsV as a reference material) contributed on the order of 35 % of the total As. FeAsS(s), as well as AsIII and AsV sorbed onto FeSm(s) or FeS2(s), which were previously shown to produce distinctly different EXAFS spectra than either AsIII–glu, AsS(s) or As2S3(s),[4,21] were included in the LCF procedure, but were not found to contribute to the samples’ fluorescence.

|

|
Aqueous chemistry of the FTR outflows
Concentration time-series of the outflows of the FTRs are shown in Fig. 3. The first 21 days are referred to as the ‘acclimation phase’ and the period from day 36 till the end of the experiment (day 56), during which the concentrations of SO42– in the outflows of all FTRs reached near constant values of ~2 µM, is referred to as the ‘steady state phase’. During the acclimation phase, FTRs A and B consumed SO42– faster than FTRs C and D. After ~15 days, outflow SO42– concentrations levelled off for FTRs C and D, but they started to increase for FTRs A and B, indicating that SO42–-reducing microorganisms were depleting the readily available OM pool. We then added lactate, a substrate known to stimulate sulfate-reducing microorganisms,[15,43] to the inflowing solutions on day 21. Outflow ΣS(aq)0 concentrations were systematically lower in FTRs A and B, compared with FTRs C and D. Upon the addition of lactate, ΣS(aq)0 in FTRs A and B peaked briefly to 3.5 µM and then returned to 1–2 µM, whereas in FTRs C and D ΣS(aq)0 increased to 7–17 µM for 12 days before dropping to values in the range of 2–5 µM.
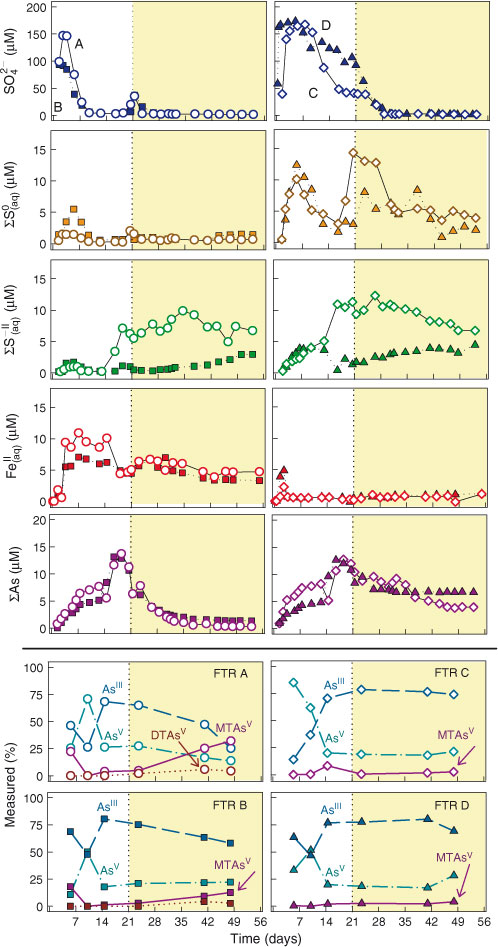
|
Outflow ΣS–II was the only aqueous species that showed systematic differences between the AsIII and the AsV-fed FTRs, being higher for AsV (FTRs B and D) than for AsIII (FTRs A and C) by ~5 µM. Outflow ΣS(aq)–II concentrations in FTRs B and D remained comparatively low, between 0.5 and 5 µM, throughout the experiment, whereas in FTRs A and C they increased steadily during the acclimation phase, plateaued at 10–12 µM after the addition of lactate, and decreased slightly towards the end of the experiment. The outflow Fe concentrations were higher for the 0–2-cm depth interval sediment FTRs (A and B), reaching values up to 10 µM during the acclimation phase and remaining at ~5 µM during the steady state phase. In comparison, Fe concentrations remained <1 µM for the 4–6 depth interval sediment FTRs (C and D) throughout the experiment.
Outflow ΣAs concentrations increased progressively at the beginning of the experiments, peaking at ~15 µM on day 20 (Fig. 3). They decreased abruptly for all FTRs upon lactate addition. Towards the end of the experiment, ΣAs was an order of magnitude lower in the outflows of FTRs A and B than in those of FTRs C and D, with average values of ~0.5 and ~5.0 µM. Aqueous As speciation analyses (Fig. 3) revealed measurable quantities of methylated-As species (limits of detection (LODs) of ~2 nmol L–1 for As and 50 nmol L–1 for S), with MMAsV being the most abundant. Only AsIII, AsV, MTAsV and DTAsV represented >2 % of the total outflowing dissolved As concentration at any given time. Irrespective of the oxidation state of As in the input solutions, the outflows contained between 75 and 25 % AsV during the acclimation phase and ~25 % AsV during the steady state phase (Fig. 3 and Table S1). Thiolated-As species were more abundant in the outflows of FTRs A and B, with MTAsV representing ~25 % of the total dissolved As during the first week of the experiment, decreasing to 1–2 % towards the end of the acclimation phase, and increasing again during the steady state phase. At the end of the experiment, MTAsV concentrations reached 12.4 µM in FTR A and 13.1 µM in FTR B, becoming the dominant aqueous As species in the 0–2-cm depth interval sediment FTR supplied with AsIII (FTR A) representing 32 % of the total dissolved As leaving the reactor.
Discussion
Sinks for reduced sulfur during microbial sulfate reduction
The sediments in FTRs C and D sequestered S as FeS2(s) and S(α)8(s) (Table 1), in line with recent studies on microbial sulfidogenesis in soils, sediments and laboratory columns reporting that these S species are the primary end products of SO42– reduction.[13,14,24,44] A possible mechanism for the accumulation of elemental S in the sediments is its precipitation from S(aq)0 produced during the oxidation of S(aq)–II by Fe oxyhydroxides.[45] Thermodynamic calculations indicate that S(α)8(s) was oversaturated by 0.5–2 orders of magnitude (saturation index = log IAP/Ksp where IAP is the ion activity product and Ksp is the solubility product) in the outflow of all FTRs throughout the experiment.
In the surface sediments (0–2 cm, FTRs A and B), the results suggest that the dominant sink for S(aq)–II and S(aq)0 produced as a results of microbial sulfate reduction is thiol formation (Table 1). Sulfurisation of OM as a sink for reduced S species has been observed in natural environments, and has been proposed to be a key early diagenetic process in sediments overlain by sulfidic waters.[23] The dominance of thiol formation in the upper 2 cm of sediment is likely related to the presence of labile OM, which is needed both as a substrate to support bacterial SO42– reduction and as a reactant for S(aq)–II and S(aq)0 to form thiols.[46] The observation that outflow Fe concentrations in FTRs A and B are an order of magnitude higher than in the outflow of FTRs C and D (Fig. 3) suggests that Fe is outcompeted by OM as a sink for reduced S. In addition, lower outflow S(aq)–II concentrations in FTRs B and D than in FTRs A and C are consistent with the observation that the former FTRs accumulated more solid-phase S than the latter FTRs during the incubations (see the Solid phase transformations during FTR experiments section). This difference in the S(aq)–II dynamic between AsIII and AsV-fed FTRs may be due to a greater affinity of S(aq)–II for AsV than for AsIII, consistent with the current best estimates of the free energy values for the sulfidation of these As species.[47]
Sulfurisation of OM has mostly been studied in marine sediments, where it is typically slower than the formation of Fe sulfides and occurs over time scales of 102–104 years.[46] In contrast, in this study with freshwater lake sediments, thiols form on time scales of days to weeks, most likely as a result of a combination of factors, such as the high rates of sulfate reduction and the nature of the labile OM deposited from the water column. One hypothesis worth further investigation is that soil-derived OM in lake sediments may be promoting fast thiol formation because of its high content of oxidised S (up to 98 % total Sorg[48]), mainly in the form of easily hydrolysable ester-SO4[25,49] or sulfonate.[50]
Controls on As mobility
XAS results show that As sorbed onto Fe oxyhydroxides is present at the end of the experiment (Table 2), suggesting incomplete sulfidisation of the Fe oxyhydroxides. Consistent with previous reports,[13,14,16] we see no evidence for extensive As adsorption onto Fe sulfides in the FTRs. LCF does suggest, however, the formation of AsS(s), despite the fact that the outflow of all FTRs is undersaturated by >2 orders of magnitudes with respect to AsS(s). This apparent contradiction has been previously reported at circum-neutral pH[15] and can be ascribed to a surface reaction of As onto Fe sulfides which lead to a local As coordination that, using XAS, is indistinguishable from an AsS(s)-like solid phase.[6,7,12,17,51]
In the FTRs A and B, where thiol formation was the dominant sink for reduced S, LCF suggests that thiol-bound AsIII accounts for a third of the total As. The sequestration of AsIII by organic S moieties of natural OM through the formation of thiol-bound As has been reported only very recently in peat samples.[21,22] To our knowledge, this process has never before been demonstrated in controlled experiments with sediments. For peat, Langner et al.[21] hypothesised that AsIII complexation by freshly formed thiol, or the reaction of thiolated-As directly with OM, is a dominant sink for As during sulfate reduction. According to these authors, LCF of As K-edge EXAFS spectra is able to discriminate between As2S3(s), both crystalline and nano-crystalline, and AsIII coordinated to thiol. They used Morlet wavelet analysis to show that a spectroscopic feature of the As2S3(s) spectra, characteristic of As backscattering, is absent from the spectra of As–thiol compounds. Although evidence based solely on LCF is not definitive, the results presented here provide further grounds for more detailed investigations of thiol-formation as an As sequestration pathway.
In FTR C, LCF suggests the presence of an AsV–S bond, based on TTAsV as a reference compound (Fig. 1). The presence of AsV–S is supported by the analysis of the EXAFS data using Fourier transform (see Fig. S1), revealing that the average As–S bond distances (uncorrected for backscattered phase shift) in this sample is equal to the previously reported value for AsV–S,[18] which is ~0.10 Å shorter than for AsIII–S. However, based on our current understanding of the geochemistry of TTAsV, it is doubtful that it is formed in FTR C, because TTAsV is unstable at pH <10.[52] Indeed, TTAsV was not detected in the outflow of any the FTRs. Instead, other species such as di- or tri-thioarsenate are potentially present, both being stable at circumneutral pH and likely to form in FTR C which produced the highest outflow ΣS–II and ΣS(aq)0 concentrations (Fig. 2). The presence of these S species are thought to promote AsIII oxidation and sulfidation[35,47,53] through an electrophonic addition of an S0 atom to the free electron pair of AsIII,[35] yielding an AsV–S complex.
In all FTRs, the outflow ΣAs concentrations peaked during the transition period separating maximum aqueous Fe and maximum ΣS(aq)–II concentrations (Fig. 3). High As mobility at this redox transition has been previously ascribed to the time lag between the onset of reductive dissolution of Fe oxyhydroxides, to which As is likely sorbed, and that of free sulfide accumulation in the pore water, which ultimately lead to the precipitation of As sulfide phases.[11] During this transition, the aqueous As in the FTR outflows progresses from equal proportions of AsV and AsIII towards the predominance of AsIII, irrespective of what As species is supplied with the inflow (Fig. 3). Arsenic speciation evolves towards higher proportions of MTAsV in all FTRs, becoming the dominant dissolved As species in the outflow of FTR A by the end of the experiment. Aqueous As speciation thus appears to be closely controlled by the biogeochemical conditions established in the FTRs, driven by the microbial community utilising S, and likely also Fe and As as energy sources. Calculation of the theoretical pe (Fig. S1) using the SO4–HS–, S(aq)0–HS–, AsIII–AsV and Fe(aq)II–FeIII(OH)3(s) redox couples reveal that only the pe values calculated using the S(aq)0–HS– redox couple remained within 1 pe unit of the measured values, suggesting, as previously hypothesised,[47,53] that this couple may be the effective redox buffer controlling As speciation (Fig. S2).
Conceptual model for As sequestration in sulfidic sediment
In the reactors containing the topmost sediment (FTRs A and B), the formation of thiols appears to be the primary sink for sulfides and other reduced S species produced by microbial SO42– reduction. This leads to enhanced As sequestration through the formation of AsS(s) (or of an analogue surface precipitate) and thiol-bound AsIII, suggesting that thiol formation, resulting from OM sulfurisation, could be used as a remediation pathway for metalloids such as As. In the FTRs with the deeper sediment (FTRs C and D), which presumably contained less labile OM,[26] the dominant sink for sulfides is the formation of Fe-minerals and elemental S. The remaining sulfides are available to complex As and form As sulfide minerals. We also observed the formation of thiolated AsV species, both in the aqueous and in the solid phase, despite the reducing conditions. In the aqueous phase, all FTRs produced MTAsV, a species not observed before in pore waters of sulfidic sediments. The biogeochemistry of MTAsV is currently under intense scrutiny because of its mobility,[54] kinetic stability[55] and possible innocuity.[56,57] Thus, the sequestration of As and the concomitant mobilisation of small quantities of MTAsV represent a scenario where both sequestration and potential detoxification of As contribute to reducing the risk of As contamination.
Based on these results, we propose an updated conceptual model for the fate of As in OM-rich sediments, which includes the role of OM as a sink for S and its control on As sequestration through the formation of thiol-bound AsIII (Fig. 4). This model complements the generally accepted idea that Fe is the effective buffer for S–II produced during SO42– reduction, and the current classification of As sequestration in sediments as being either Fe- or S-controlled.[11] The implications of the prevalence of thioarsenates and thiol-bound As species for the mobility and remediation of As, and thus for the quality of the water used worldwide for drinking and irrigation, are significant The stimulation of thiol formation through microbial sulfidogenesis has yet to be considered as a pathway of remediation for metalloids in the subsurface, which could be relevant for aquifers subjected to naturally high levels of sediment-bound As, such as those of South-east Asia.[2]
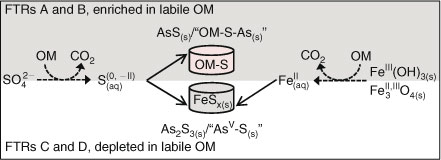
|
Acknowledgements
The authors thank D. Campisi, J. Evans, N. Kumar, K. Mitchell and M. Trail for research assistance in the field and in the laboratory. They acknowledge the ESRF for provision of synchrotron facilities and thank D. Testemale for assistance at the FAME beamline BM30b. A portion of this research was performed at SSRL, a user facility supported by the US Department of Energy, Office of Sciences; the authors thank E. Nelson for assistance at beamline 4–3. They are indebted to C. Mikutta (ETH, Switzerland) for providing the reference spectrum of AsIII-glu and to D. Testemale (ESRF) and J. Brugger (University of Adelaide) for the reference spectrum of scorodite and arsenopyrite. Logistical support from C. Gobeil and R. Rodrigue (INRS-ETE) and permission to work in the Tantaré Ecological Reserve granted by the Québec MEDDEP is gratefully acknowledged. K. Mueller kindly accepted to edit the manuscript. R.-M. Couture was financially supported by a postdoctoral fellowship from the Fonds de recherche du Québec – Nature et technologies. P. Van Cappellen acknowledges funding from the Georgia Research Alliance and the Canada Excellence Research Chair (CERC) program.
References
[1] D. Polya, L. Charlet, Environmental science: rising arsenic risk? Nat. Geosci. 2009, 2, 383.| Environmental science: rising arsenic risk?Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXms1Ortbc%3D&md5=10103852831db6e3cd0d7786f2dafb8bCAS |
[2] M. L. Polizzotto, B. D. Kocar, S. G. Benner, M. Sampson, S. Fendorf, Near-surface wetland sediments as a source of arsenic release to ground water in Asia. Nature 2008, 454, 505.
| Near-surface wetland sediments as a source of arsenic release to ground water in Asia.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXovV2mtLs%3D&md5=1c8d2a7c780e18daac4364b85a296ec0CAS | 18650922PubMed |
[3] C. F. Harvey, C. H. Swartz, A. B. M. Badruzzaman, N. Keon-Blute, W. Yu, M. A. Ali, J. Jay, R. Beckie, V. Niedan, D. Brabander, P. M. Oates, K. N. Ashfaque, S. Islam, H. F. Hemond, M. F. Ahmed, Arsenic mobility and groundwater extraction in Bangladesh. Science 2002, 298, 1602.
| Arsenic mobility and groundwater extraction in Bangladesh.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38Xosl2ktb0%3D&md5=55f23a5e3f8700c429b43c7c5d6a028fCAS | 12446905PubMed |
[4] K. S. Savage, T. N. Tingle, P. A. O’Day, G. A. Waychunas, D. K. Bird, Arsenic speciation in pyrite and secondary weathering phases, mother lode gold district, tuolumne county, california. Appl. Geochem. 2000, 15, 1219.
| Arsenic speciation in pyrite and secondary weathering phases, mother lode gold district, tuolumne county, california.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXisFaksrs%3D&md5=4648221cfa722372af30305c4520c178CAS |
[5] M. Wolthers, L. Charlet, C. H. van Der Weijden, P. R. van der Linde, D. Rickard, Arsenic mobility in the ambient sulfidic environment: Sorption of arsenic(V) and arsenic(III) onto disordered mackinawite. Geochim. Cosmochim. Acta 2005, 69, 3483.
| Arsenic mobility in the ambient sulfidic environment: Sorption of arsenic(V) and arsenic(III) onto disordered mackinawite.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXmtlKltbw%3D&md5=3542dbe8b308dd650bac989224d2c483CAS |
[6] B. C. Bostick, S. Fendorf, Arsenite sorption on troilite (FeS) and pyrite (FeS2). Geochim. Cosmochim. Acta 2003, 67, 909.
| Arsenite sorption on troilite (FeS) and pyrite (FeS2).Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXht1WgtLw%3D&md5=e86bb8a437785f288fd9158773bb48fdCAS |
[7] T. J. Gallegos, S. P. Hyun, K. F. Hayes, Spectroscopic investigation of the uptake of arsenite from solution by synthetic mackinawite. Environ. Sci. Technol. 2007, 41, 7781.
| Spectroscopic investigation of the uptake of arsenite from solution by synthetic mackinawite.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhtFOrs7zJ&md5=696b1ea8c382826d1c7481f1db2fabaaCAS | 18075088PubMed |
[8] J. A. Saunders, M. K. Lee, M. Shamsudduha, P. Dhakal, A. Uddin, M. T. Chowdury, K. M. Ahmed, Geochemistry and mineralogy of arsenic in (natural) anaerobic groundwaters. Appl. Geochem. 2008, 23, 3205.
| Geochemistry and mineralogy of arsenic in (natural) anaerobic groundwaters.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtlWkurrN&md5=b5209398b3a77e9a1a023d0807e27a8aCAS |
[9] C. F. Harvey, K. N. Ashfaque, W. Yu, A. B. M. Badruzzaman, M. A. Ali, P. M. Oates, H. A. Michael, R. B. Neumann, R. Beckie, S. Islam, M. F. Ahmed, Groundwater dynamics and arsenic contamination in bangladesh. Chem. Geol. 2006, 228, 112.
| Groundwater dynamics and arsenic contamination in bangladesh.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XjsFehs7o%3D&md5=6d3e210de096c871f093e674bd8c0dcdCAS |
[10] R. T. Wilkin, R. G. Ford, Arsenic solid-phase partitioning in reducing sediments of a contaminated wetland. Chem. Geol. 2006, 228, 156.
| Arsenic solid-phase partitioning in reducing sediments of a contaminated wetland.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XjsFehs7g%3D&md5=fc0f81c8199f356e1c59111799269e52CAS |
[11] P. A. O’Day, D. Vlassopoulos, R. Root, N. Rivera, The influence of sulfur and iron on dissolved arsenic concentrations in the shallow subsurface under changing redox conditions. Proc. Natl. Acad. Sci. USA 2004, 101, 13703.
| The influence of sulfur and iron on dissolved arsenic concentrations in the shallow subsurface under changing redox conditions.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXotVygtrk%3D&md5=7e1010b7a4d91da4126ee9a3033ca595CAS | 15356340PubMed |
[12] B. C. Bostick, C. Chen, S. Fendorf, Arsenite retention mechanisms within estuarine sediments of Pescadero, CA. Environ. Sci. Technol. 2004, 38, 3299.
| Arsenite retention mechanisms within estuarine sediments of Pescadero, CA.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXjs1Kht7c%3D&md5=69749f03b6f9dfc5af566b77359031b6CAS | 15260327PubMed |
[13] M. F. Kirk, E. E. Roden, L. J. Crossey, A. J. Brealey, M. N. Spilde, Experimental analysis of arsenic precipitation during microbial sulfate and iron reduction in model aquifer sediment reactors. Geochim. Cosmochim. Acta 2010, 74, 2538.
| Experimental analysis of arsenic precipitation during microbial sulfate and iron reduction in model aquifer sediment reactors.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXjvFSnsbs%3D&md5=6f1f2a7fb46238063f5f21d7b12a13feCAS |
[14] B. D. Kocar, T. Borch, S. Fendorf, Arsenic repartitioning during biogenic sulfidization and transformation of ferrihydrite. Geochim. Cosmochim. Acta 2010, 74, 980.
| Arsenic repartitioning during biogenic sulfidization and transformation of ferrihydrite.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhs1SlsbrN&md5=340b591e48216db213c0248a74b6d15dCAS |
[15] E. D. Burton, S. G. Johnston, R. T. Bush, Microbial sulfidogenesis in ferrihydrite-rich environments: effects on iron mineralogy and arsenic mobility. Geochim. Cosmochim. Acta 2011, 75, 3072.
| Microbial sulfidogenesis in ferrihydrite-rich environments: effects on iron mineralogy and arsenic mobility.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXltlyksbk%3D&md5=caf6b42cafe9ec5d1bd925bd7ac9c6d2CAS |
[16] S. L. Saalfield, B. C. Bostick, Changes in iron, sulfur, and arsenic speciation associated with bacterial sulfate reduction in ferrihydrite-rich systems. Environ. Sci. Technol. 2009, 43, 8787.
| Changes in iron, sulfur, and arsenic speciation associated with bacterial sulfate reduction in ferrihydrite-rich systems.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtlaltbjK&md5=98b2e3ee1a69fc5efe6bd60d783d86a8CAS | 19943647PubMed |
[17] T. J. Gallegos, Y.-S. Han, K. F. Hayes, Model predictions of realgar precipitation by reaction of AsIII with synthetic mackinawite under anoxic conditions. Environ. Sci. Technol. 2008, 42, 9338.
| Model predictions of realgar precipitation by reaction of AsIII with synthetic mackinawite under anoxic conditions.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtlKlsrvM&md5=3d837bdf5a75ab12ddba947b7a5041acCAS | 19174913PubMed |
[18] E. Suess, A. C. Scheinost, B. C. Bostick, B. J. Merkel, D. Wallschläger, B. Planer-Friedrich, Discrimination of thioarsenites and thioarsenates by X-ray absorption spectroscopy. Anal. Chem. 2009, 81, 8318.
| Discrimination of thioarsenites and thioarsenates by X-ray absorption spectroscopy.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtFGls7nF&md5=6561f450cad2cabd3162a92fb4b70ac6CAS | 19764741PubMed |
[19] F. Wang, A. Tessier, Polysulfide and metal speciation in sediment porewaters of freshwater lakes. Environ. Sci. Technol. 2009, 43, 7252.
| Polysulfide and metal speciation in sediment porewaters of freshwater lakes.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXjtFyrtrc%3D&md5=e32c3cbe38c2e34f851156fe80f529bdCAS | 19848130PubMed |
[20] R. M. Couture, C. Gobeil, A. Tessier, Arsenic, iron and sulfur co-diagenesis in lake sediments. Geochim. Cosmochim. Acta 2010, 74, 1238.
| Arsenic, iron and sulfur co-diagenesis in lake sediments.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXjtVCjtg%3D%3D&md5=368fbb75654d99ddcb78aed836594a80CAS |
[21] P. Langner, C. Mikutta, R. Kretzschmar, Arsenic sequestration by organic sulphur in peat. Nat. Geosci. 2012, 5, 66.
| Arsenic sequestration by organic sulphur in peat.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsFeit7%2FK&md5=f2819aacab8d1ab22895cac04c915435CAS |
[22] M. Hoffmann, C. Mikutta, R. Kretzschmar, Bisulfide reaction with natural organic matter enhances arsenite sorption: insights from x-ray absorption spectroscopy. Environ. Sci. Technol. 2012, 46, 11788.
| Bisulfide reaction with natural organic matter enhances arsenite sorption: insights from x-ray absorption spectroscopy.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhsVWhur7J&md5=a1c01ec591da015ca2c321dd4a1feef8CAS | 23075303PubMed |
[23] J. P. Werne, T. W. Lyons, D. J. Hollander, S. Schouten, E. C. Hopmans, J. S. Sinninghe Damsté, Investigating pathways of diagenetic organic matter sulfurization using compound-specific sulfur isotope analysis. Geochim. Cosmochim. Acta 2008, 72, 3489.
| Investigating pathways of diagenetic organic matter sulfurization using compound-specific sulfur isotope analysis.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXotFarsb8%3D&md5=78f751f53af9315375b7f6c0472e9bdfCAS |
[24] M. Yücel, S. K. Konovalov, T. S. Moore, C. P. Janzen, G. W. Luther, Sulfur speciation in the upper black sea sediments. Chem. Geol. 2010, 269, 364.
| Sulfur speciation in the upper black sea sediments.Crossref | GoogleScholarGoogle Scholar |
[25] N. R. Urban, K. Ernst, S. Bernasconi, Addition of sulfur to organic matter during early diagenesis of lake sediments. Geochim. Cosmochim. Acta 1999, 63, 837.
| Addition of sulfur to organic matter during early diagenesis of lake sediments.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXkt1Sns78%3D&md5=23c00a0fb22c4516e54938d5c22f4fffCAS |
[26] R. M. Couture, B. Shafei, P. Van Cappellen, A. Tessier, C. Gobeil, Non-steady state modeling of arsenic diagenesis in lake sediments. Environ. Sci. Technol. 2010, 44, 197.
| Non-steady state modeling of arsenic diagenesis in lake sediments.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsFSrtrfL&md5=cefd82fef8b62a62a12dba889f09f1ccCAS | 19957997PubMed |
[27] C. Pallud, C. Meile, A. M. Laverman, J. Abell, P. Van Cappellen, The use of flow-through sediment reactors in biogeochemical kinetics: methodology and examples of applications. Mar. Chem. 2007, 106, 256.
| The use of flow-through sediment reactors in biogeochemical kinetics: methodology and examples of applications.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXovFSmurk%3D&md5=07cd7535213cf8d931e1b3a708b465ddCAS |
[28] D. Fortin, G. G. Leppard, A. Tessier, Caracteristics of lacustrine diagenetic iron oxyhydroxide. Geochim. Cosmochim. Acta 1993, 57, 4391.
| Caracteristics of lacustrine diagenetic iron oxyhydroxide.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2cXlvV2ktA%3D%3D&md5=7cd7f9335869e66b65e96f429d36c4e7CAS |
[29] S. G. Benner, C. M. Hansel, B. W. Wielinga, T. M. Barber, S. Fendorf, Reductive dissolution and biomineralization of iron hydroxide under dynamic flow conditions. Environ. Sci. Technol. 2002, 36, 1705.
| Reductive dissolution and biomineralization of iron hydroxide under dynamic flow conditions.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XhvFymsbk%3D&md5=ea57554880f3791e56b668d043451b54CAS | 11993867PubMed |
[30] F. Wang, A. Tessier, J. Buffle, Voltammetric determination of elemental sulfur in pore waters. Limnol. Oceanogr. 1998, 43, 1353.
| Voltammetric determination of elemental sulfur in pore waters.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXntlOnu7g%3D&md5=f29fd23b6915187881a49ef45d25f880CAS |
[31] B. Planer-Friedrich, D. Wallschläger, A critical investigation of hydride generation-based arsenic speciation in sulfidic waters. Environ. Sci. Technol. 2009, 43, 5007.
| A critical investigation of hydride generation-based arsenic speciation in sulfidic waters.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXmtlelsbg%3D&md5=082d56d4231117db3374fc11330d12ffCAS | 19673299PubMed |
[32] D. G. Beak, R. T. Wilkin, R. G. Ford, S. D. Kelly, Examination of arsenic speciation in sulfidic solutions using X-ray absorption spectroscopy. Environ. Sci. Technol. 2008, 42, 1643.
| Examination of arsenic speciation in sulfidic solutions using X-ray absorption spectroscopy.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXht1CjtLk%3D&md5=e0c8104498519db6bcbaf2aaa21e0262CAS | 18441815PubMed |
[33] D. Wallschläger, J. London, Determination of methylated arsenic-sulfur compounds in groundwater. Environ. Sci. Technol. 2008, 42, 228.
| Determination of methylated arsenic-sulfur compounds in groundwater.Crossref | GoogleScholarGoogle Scholar | 18350901PubMed |
[34] B. Ravel, M. Newville, Athena, artemis, hephaestus: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 2005, 12, 537.
| Athena, artemis, hephaestus: data analysis for X-ray absorption spectroscopy using IFEFFIT.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXltlCntLo%3D&md5=3352866b98f8433d863db2225c798ea1CAS | 15968136PubMed |
[35] D. Wallschläger, C. J. Stadey, Determination of (oxy)thioarsenates in sulfidic waters. Anal. Chem. 2007, 79, 3873.
| Determination of (oxy)thioarsenates in sulfidic waters.Crossref | GoogleScholarGoogle Scholar | 17437336PubMed |
[36] G. R. Helz, J. A. Tossell, J. M. Charnock, R. A. D. Pattrick, D. J. Vaughan, D. Garner, Oligomerization in AsIII sulfide solutions: theoretical constraints and spectroscopic evidence. Geochim. Cosmochim. Acta 1995, 59, 4591.
| Oligomerization in AsIII sulfide solutions: theoretical constraints and spectroscopic evidence.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXps1ylsr8%3D&md5=8232b4ae22c627acb46ce629a289ba42CAS |
[37] J. James-Smith, J. Cauzid, D. Testemale, W. H. Liu, J. L. Hazemann, O. Proux, B. Etschmann, P. Philippot, D. Banks, P. Williams, J. Brugger, Arsenic speciation in fluid inclusions using micro-beam X-ray absorption spectroscopy. Am. Mineral. 2010, 95, 921.
| Arsenic speciation in fluid inclusions using micro-beam X-ray absorption spectroscopy.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXpt1ymt7Y%3D&md5=7ddf9108461c61f8e5db2382c581e5deCAS |
[38] R.-M. Couture, J. C. Rose, N. Kumar, K. Mitchell, D. Wallschläger, P. Van Cappellen, Sorption of arsenite, arsenate and thioarsenates to iron oxides and iron sulfides: a kinetic and spectroscopic investigation. Environ. Sci. Technol. 2013, 47, 5652.
| Sorption of arsenite, arsenate and thioarsenates to iron oxides and iron sulfides: a kinetic and spectroscopic investigation.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXmt1ymu7s%3D&md5=70c491409988f77d332f5efe30501f8bCAS | 23607702PubMed |
[39] A. Manceau, K. L. Nagy, Quantitative analysis of sulfur functional groups in natural organic matter by xanes spectroscopy. Geochim. Cosmochim. Acta 2012, 99, 206.
| Quantitative analysis of sulfur functional groups in natural organic matter by xanes spectroscopy.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xhs12ksrzP&md5=91028b8e6f1a56930fd295e6f043027fCAS |
[40] Y. P. Hsieh, Y. N. Shieh, Analysis of reduced inorganic sulfur by diffusion methods: Improved apparatus and evaluation for sulfur isotopic studies. Chem. Geol. 1997, 137, 255.
| Analysis of reduced inorganic sulfur by diffusion methods: Improved apparatus and evaluation for sulfur isotopic studies.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXjtlGqsr8%3D&md5=f7dd067c9410f1d4dbe07ada38401691CAS |
[41] E. D. Burton, L. A. Sullivan, R. T. Bush, S. G. Johnston, A. F. Keene, A simple and inexpensive chromium-reducible sulfur method for acid-sulfate soils. Appl. Geochem. 2008, 23, 2759.
| A simple and inexpensive chromium-reducible sulfur method for acid-sulfate soils.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtVGnsLjK&md5=24d8a0a2f98b046bb8fad3cf9cc39db4CAS |
[42] D. L. Parkhurst, C. A. J. Apello, User’s guide to PHREEQC (Version 2) : a computer program for speciation, batch-reaction, one-dimensional transport, and inverse geochemical calculations. Water-Resources Investigations Report 99-4259 1999 (US Geological Survey: Denver, CO). Available at http://pubs.er.usgs.gov/publication/wri994259 [Verified 28 June 2013].
[43] D. Canfield, E. Kristensen, B. Thamdrup, Advances in Marine Biology: Aquatic Geomicrobiology, vol. 48 (Eds AJ Southward, PA Tyler, CM Young, LA Fuiman) 2005 (Elsevier Academic Press: San Diego, CA).
[44] E. D. Burton, R. T. Bush, S. G. Johnston, L. A. Sullivan, A. F. Keene, Sulfur biogeochemical cycling and novel Fe–S mineralization pathways in a tidally re-flooded wetland. Geochim. Cosmochim. Acta 2011, 75, 3434.
| Sulfur biogeochemical cycling and novel Fe–S mineralization pathways in a tidally re-flooded wetland.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXmtVWnt78%3D&md5=2c2e41f285ef36f6eac2da6be3e19d13CAS |
[45] S. W. Poulton, M. D. Krom, R. Raiswell, A revised scheme for the reactivity of iron (oxyhydr)oxide minerals towards dissolved sulfide. Geochim. Cosmochim. Acta 2004, 68, 3703.
| A revised scheme for the reactivity of iron (oxyhydr)oxide minerals towards dissolved sulfide.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXnt1Cns70%3D&md5=a5b95d94d8af7f886bc8773d9b862891CAS |
[46] J. P. Werne, D. J. Hollander, T. W. Lyons, J. S. Sinninghe Damsté, Organic sulfur biogeochemistry: recent advances and future research directions. Spec. Pap. Geol. Soc. Am. 2004, 379, 135.
[47] G. R. Helz, J. A. Tossell, Thermodynamic model for arsenic speciation in sulfidic waters: a novel use of ab initio computations. Geochim. Cosmochim. Acta 2008, 72, 4457.
| Thermodynamic model for arsenic speciation in sulfidic waters: a novel use of ab initio computations.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtVGjtrfL&md5=97501cf0aef922b5a8b9599ecc86550aCAS |
[48] D. Solomon, J. Lehmann, M. Tekalign, F. Fritzsche, W. Zech, Sulfur fractions in particle-size separates of the sub-humid ethiopian highlands as influenced by land use changes. Geoderma 2001, 102, 41.
| Sulfur fractions in particle-size separates of the sub-humid ethiopian highlands as influenced by land use changes.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXjsFWmtb0%3D&md5=7a1d6d0d6be2c998772d6a503d9557d8CAS |
[49] J. r. Prietzel, A. Botzaki, N. Tyufekchieva, M. Brettholle, J. r. Thieme, W. Klysubun, Sulfur speciation in soil by s k-edge xanes spectroscopy: comparison of spectral deconvolution and linear combination fitting. Environ. Sci. Technol. 2011, 45, 2878.
| Sulfur speciation in soil by s k-edge xanes spectroscopy: comparison of spectral deconvolution and linear combination fitting.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjtFKqtro%3D&md5=5bae1da47bde46ff8c244b9664493284CAS |
[50] B. Morgan, E. D. Burton, A. W. Rate, Iron monosulfide enrichment and the presence of organosulfur in eutrophic estuarine sediments. Chem. Geol. 2012, 296–297, 119.
| Iron monosulfide enrichment and the presence of organosulfur in eutrophic estuarine sediments.Crossref | GoogleScholarGoogle Scholar |
[51] M. L. Farquhar, J. M. Charnock, F. R. Livens, D. J. Vaughan, Mechanisms of arsenic uptake from aqueous solution by interaction with goethite, lepidocrocite, mackinawite, and pyrite: an X-ray absorption spectroscopy study. Environ. Sci. Technol. 2002, 36, 1757.
| Mechanisms of arsenic uptake from aqueous solution by interaction with goethite, lepidocrocite, mackinawite, and pyrite: an X-ray absorption spectroscopy study.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XhvVeqtL8%3D&md5=805ea76aaf0962c3036a7fef61b30ae7CAS | 11993874PubMed |
[52] B. Planer-Friedrich, E. Suess, A. C. Scheinost, D. Wallschlger, Arsenic speciation in sulfidic waters: reconciling contradictory spectroscopic and chromatographic evidence. Anal. Chem. 2010, 82, 10 228.
| Arsenic speciation in sulfidic waters: reconciling contradictory spectroscopic and chromatographic evidence.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsVyitLbL&md5=6fe5f147d49ad776b6d334afb0273478CAS |
[53] R.-M. Couture, P. Van Cappellen, Reassessing the role of sulfur geochemistry on arsenic speciation in reducing environments. J. Hazard. Mater. 2011, 189, 647.
| Reassessing the role of sulfur geochemistry on arsenic speciation in reducing environments.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXmtlems7o%3D&md5=226c6f05a31a4cf998267c74b1054ab9CAS | 21382662PubMed |
[54] E. Suess, B. Planer-Friedrich, Thioarsenate formation upon dissolution of orpiment and arsenopyrite. Chemosphere 2012, 89, 1390.
| Thioarsenate formation upon dissolution of orpiment and arsenopyrite.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XpvFGmtrg%3D&md5=9c7184d6375ad2249ce7613bff4f0647CAS | 22771176PubMed |
[55] E. Suess, D. Wallschläger, B. Planer-Friedrich, Stabilization of thioarsenates in iron-rich waters. Chemosphere 2011, 83, 1524.
| Stabilization of thioarsenates in iron-rich waters.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXmtFylt7c%3D&md5=553d677d9fe8ec4bde7c8791e68d55f8CAS | 21324509PubMed |
[56] B. Planer-Friedrich, D. Franke, B. Merkel, D. Wallschläger, Acute toxicity of thioarsenates to Vibrio fischeri. Environ. Toxicol. Chem. 2008, 27, 2027.
| Acute toxicity of thioarsenates to Vibrio fischeri.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXht1Sgur%2FN&md5=419e407f98c7e6a96d381a5d934e028dCAS | 18422398PubMed |
[57] R.-M. Couture, A. Sekowska, G. Fang, A. Danchin, Linking selenium biogeochemistry to the sulfur-dependent biological detoxification of arsenic. Environ. Microbiol. 2012, 14, 1612.
| Linking selenium biogeochemistry to the sulfur-dependent biological detoxification of arsenic.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xhtleht7%2FO&md5=d9c848fbbaf9baf21b6b4230b0b2af9fCAS | 22515279PubMed |