

Secondary organic aerosol formation from the oxidation of a series of sesquiterpenes: α-cedrene, β-caryophyllene, α-humulene and α-farnesene with O3, OH and NO3 radicals
Mohammed Jaoui A C , Tadeusz E. Kleindienst B , Kenneth S. Docherty A , Michael Lewandowski B and John H. Offenberg BA Alion Science and Technology, PO Box 12313, Research Triangle Park, NC 27709, USA.
B US Environmental Protection Agency, Office of Research and Development, National Exposure Research Laboratory, Research Triangle Park, NC 27711, USA.
C Corresponding author. Email: jaoui.mohammed@epa.gov
Environmental Chemistry 10(3) 178-193 https://doi.org/10.1071/EN13025
Submitted: 1 February 2012 Accepted: 28 May 2013 Published: 28 June 2013
Journal Compilation © CSIRO Publishing 2013 Open Access CC BY-NC-ND
Environmental context. Sesquiterpenes, chemicals emitted by terrestrial vegetation, are oxidised in the ambient atmosphere leading to the formation of secondary organic aerosol. Although secondary organic aerosol can have significant effects on air quality from local to global scales, considerable gaps remain in our understanding of their various sources and formation mechanisms. We report studies on the oxidation of sesquiterpenes aimed at improving aerosol parameterisation for these reactions for incorporation into future air quality models.
Abstract. A series of sesquiterpenes (SQT) were individually oxidised under a range of conditions, including irradiation in the presence of NOx, reactions with O3 or reactions with NO3 radicals. Experiments were conducted in either static mode to observe temporal evolution of reactants and products or in dynamic mode to ensure adequate collection of aerosol at reasonably low reactant concentrations. Although some measurements of gas-phase products have been made, the focus of this work has been particle phase analysis. To identify individual products, filter samples were extracted, derivatised and analysed using gas chromatography mass spectrometry techniques. The results indicate that secondary organic aerosol (SOA) is readily formed from SQT oxidation. The high reactivity of these systems and generally high conversion into SOA products gives rise to high SOA levels. SOA yields (ratio of SOA formed to hydrocarbon reacted) averaged 0.53 for ozonolysis, 0.55 for photooxidation and 1.19 for NO3 reactions. In select experiments, SOA was also analysed for the organic matter/organic carbon (OM/OC) ratio, and the effective enthalpy of vaporisation (ΔHvapeff). The OM/OC ranged from 1.8 for ozonolysis and photooxidation reactions to 1.6 for NO3 reactions, similar to that from SOA generated in monoterpene systems. ΔHvapeff was measured for β-caryophyllene–NOx, β-caryophyllene–O3, β-caryophyllene–NO3, α-humulene–NOx and α-farnesene–NOx systems and found to be 43.9, 41.1, 44.9, 48.2 and 27.7 kJ mol–1. Aerosol yields and products identified in this study are generally in good agreement with results from several studies. A detailed examination of the chamber aerosol for the presence of chemical tracer compounds was undertaken. Only β-caryophyllinic acid, observed mainly under β-caryophyllene photooxidation and ozonolysis experiments, was detected in ambient aerosol. Chemical analysis yielded compounds having oxygen and nitrogen moieties present, which indicates continued evolution of the particles over time and presents high dependence on the SQT–oxidant system studied. This study suggests that SOA from laboratory ozonolysis experiments may adequately represent ambient aerosol in areas with SQT emissions.
Additional keywords: biogenic SOA, fine particulate matter, night-time chemistry, ozonolysis, PM2.5, SOA.
Introduction
Organic compounds of biogenic origin (BOCs) including isoprene (C5H8), monoterpenes (C10H16), sesquiterpenes (C15H24) and oxygenated hydrocarbons are considered an important class of organic species emitted into the troposphere from vegetation.[1,2] Since Went reported that the oxidation of volatile organic compounds (VOCs) emitted by terrestrial vegetation could lead to the formation of organic aerosol,[3] considerable efforts have been devoted to understanding secondary organic aerosol (SOA) formation from these reactions.[4,5] SOA often constitutes a sizable fraction of fine particulate matter (PM2.5) and can have significant effects on both the physical and chemical characteristics of ambient aerosol. PM2.5 negatively affects the air quality on a local, regional and global scale through visibility degradation[6] and climate change by its role in radiative forcing.[7] Exposure to PM2.5 has also been associated with increases in human mortality and morbidity levels, and decreased particulate matter (PM) levels have been associated with increased life expectancy.[8] As our understanding of the toxicology associated with these particles develops, more accurate compositional data likely will be required.
Despite recent progress to explain the organic mass associated with gas and aerosol phases in the Earth’s atmosphere, degradation of known emitted VOCs cannot account for the total organic mass, leaving a substantial fraction unresolved.[9–11] Over the past decade, numerous studies have shown evidence of a substantial amount of unmeasured highly reactive compounds in forest ecosystems.[10–14] Hence, understanding the gas-phase chemistry of these unmeasured species is key to assessing their involvement in both air quality and climate issues.
Sesquiterpenes (SQTs) have become target analytes in biogenic emission analyses because their molecular mass and reactivity make them more efficient SOA precursors than other BOCs. Recent flux studies within the forest canopy in the Amazon and in an orange grove in Spain have shown that SQTs undergo oxidation reactions (e.g. ozonolysis) near the ground and are undetected above the canopy level.[15,16] Based on these observations, it has been proposed that SQTs may serve as endogenous antioxidants in plants by reducing oxidative damage during the stress-induced accumulation of reactive oxygen species.[17] For example, Jardine et al. established that SQTs act as an effective ozone sink within the plant canopy and that their emissions might significantly reduce harmful ozone uptake and its associated oxidative damage to plants within the canopy.[15] Jardine et al.[15] also found that the mean daytime ozone loss due to gas-phase reactions with SQTs represents a significant fraction (7–28 %) of calculated ozone fluxes within the central Amazon canopy during the dry season. Although uncertainties remain regarding the role of environmental factors in these emissions, SQTs emissions have been found to be sensitive to light, temperature and ecological factors.[18] SQTs on a reacted mass basis have much higher aerosol formation potential than monoterpenes.[19] In addition, the presence of one, two, three or four double bonds in SQTs and their fast reactions with oxidants, mainly with ozone (O3),[20–22] clearly suggest the importance of SQT chemistry in the atmosphere.
Although numerous SQTs of interest to atmospheric scientists have been detected in ambient atmosphere (e.g. α-cedrene, β-cedrene, longifolene, α-humulene, α-bergamotene and α-farnesene), their role in ambient SOA formation is still not fully understood because of the lack of a direct link to ambient organic aerosol (e.g. lack of chemical tracer identification in PM2.5) and further investigation is needed. One exception is β-caryophyllene, which has been studied extensively in chamber experiments[19,23–35] and used to estimate the contribution of SQTs to ambient PM2.5.[33,36–38] Although gas- and particle-phase products from a series of SQTs have been reported,[23,29,30,35–42] only β-caryophyllinic acid, an oxidation product from β-caryophyllene, has been detected in ambient organic aerosol as a unique tracer.[33] Detection of specific SQT reaction products in chamber and ambient field measurements is further evidence of the possible influence of SQTs in regions with high SQT emissions. Although SQT emissions are not fully understood and cover a wide range of values,[2] the rapid oxidation of these hydrocarbons leads to contributions to the chemistry of hydrogen oxide radicals (HOx), producing O3, formaldehyde, acetone and other carbonyl compounds. In addition, dicarbonyls possibly formed through secondary reactions can lead to increased SOA production in an aqueous or non-aqueous aerosol phase.[43–46] Finally, several theoretical studies have reported on the degradation of β-caryophyllene and other SQTs (see Jenkin et al.[38] and references therein).
Given the uncertainties in emissions, sensitivity to environmental factors and the contribution from the atmospheric oxidation of a variety of SQTs to ambient organic aerosol, additional studies are needed to provide a more complete understanding of SOA formation from these compounds. The present study examined gas and aerosol products formed from the smog chamber oxidation of α-cedrene (αCed), β-caryophyllene, (βCar), α-humulene (αHum) and α-farnesene (αFarn) respectively containing one, two, three and four double bonds with the hydroxyl (OH) and NO3 radicals as well as O3 (Table 1). Aerosol yields and other aerosol properties were measured. Moreover, a detailed examination of organic compounds comprising the aerosol was undertaken with the goal of identifying possible unique tracer compounds of SQTs oxidation. Most literature studies of SOA formation have examined reactions occurring in the presence of ozone. However, SOA formation can also occur at night when the NO3 radicals can react with unsaturated hydrocarbons, typically those emitted by trees and other vegetation. This study also examines the chemical characteristics and physical properties of organic aerosol formed under these conditions including under potentially important night-time conditions.

|
Experimental methods
Apparatus and reactants
All chemicals, including derivatisation reagents (O-(2,3,4,5,6-pentafluorobenzyl)hydroxylamine hydrochloride (PFBHA), BF3–methanol and N,O-bis(trimethylsilyl)trifluoroacetamide (BSTFA) (1 % trimethylchlorosilane), were purchased from Aldrich Chemical Co. (Milwaukee, WI) at the highest purity available and were used without further purification.
The experiments were conducted in a 14.5-m3 stainless-steel, fixed-volume chamber with 40-μm polytetrafluoroethylene (PTFE) Teflon-coated walls. The chamber incorporates a combination of fluorescent bulbs that provide radiation distributed over the actinic portion of the spectrum similar to solar radiation, from 300 to 400 nm. Experiments were conducted in either the presence of NOx, O3 or NO3 radicals. The chamber was illuminated only when NOx was present in the chamber. The chamber was operated either in static mode (as a conventional batch reactor) or in dynamic mode (as a flow reactor) to produce a steady-state concentration of gas- and particle-phase reaction products over an extended period of time. The ozonolysis experiments, with the exception of ER533, were conducted in a 9-m3 chamber similar to the 14.5-m3 chamber where ozone, hydrocarbon and organic aerosol concentrations could be measured as a function of time. Ozone was added to the chamber using an electrical discharge generator and allowed to stabilise before the start of the reaction. SQT was added to the chamber rapidly by injecting the neat liquid into a heated inlet. A fan was used to mix the reactants in the chamber. However, mixing was performed cautiously and not extensively due to reported particle losses attributed to the use of a mixing fan during SOA formation.[47]
Night-time experiments involved dark oxidations (reactions conducted in the dark to avoid the rapid photolysis of NO3) in the smog chamber of a series of SQTs (Table 1) in the presence of N2O5. NO3 radicals were produced within the chamber by the in situ thermal decomposition of nitric acid anhydride (N2O5, dinitrogen pentoxide). Because NO3 radicals do not propagate a radical chain reaction, the level of hydrocarbon reactant removal is dictated largely by the ratio of the initial reactant concentrations. N2O5 was generated by a synthetic approach developed in our laboratory that involves reaction of ozone with nitrogen dioxide (see ‘NO3 radical synthesis’ section). Hydrocarbon and oxidant reactants were then admitted into the chamber in a flow mode to allow for measurement of SOA formation parameters and chemical properties. Two dark experiments were also conducted in static mode.
Temperature and relative humidity within both chambers were determined with an Omega Engineering, Inc. (Stamford, CT) digital thermo-hygrometer (Model RH411). Light intensity (where applicable) was continuously monitored with an integrating radiometer (Eppley Laboratory, Inc., Newport, RI). The reactant generation system provided the chamber’s constant sources of zero air, gas-phase reactants, external radical sources, water vapour and seed aerosol. The seed aerosol, used only in the 14.5-m3 chamber, was generated by nebulising a 10 mg L–1 aqueous solution of ammonium sulfate. Nitric oxide (NO) was obtained from a high-concentration cylinder. For the 14.5-m3 chamber, SQTs, which are liquids at room temperature, were vaporised by injecting the liquid into a heated bulb at 85 °C. Additional details of the chamber and its operation have been described by Kleindienst et al.[48] Reactants were added to the chamber to the target initial concentrations. In dynamic mode, reactants were added to a manifold in a continuous fashion using mass flow controllers while the effluent used for analysis was withdrawn from the chamber. The average residence time for these experiments was ~4 h, and the reaction mixture reached a steady-state before sampling. For the static experiments, reactant flows were turned off after the initial concentrations were reached, leaving on only the dilution flow, necessary due to the chamber’s fixed volume.
NO3 radical synthesis
Dinitrogen pentoxide (N2O5) can be produced from the dehydration of nitric acid or from the reaction of NO3 with NO2, with NO3 being generated from the reaction of NO2 with O3. The chemical reactions involved in this synthesis are as follows:

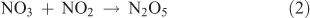
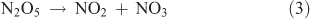
where Reactions 2 and 3 are reversible reactions and form an equilibrium.
In this particular method, the means for generating NO2 from the reaction of O3 with NO shown by Reaction 4 represents the crucial step in the synthesis:
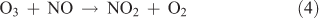
The advantage of using this method for generating NO2 lies in the physical properties of NO and NO2. NO is available in considerably higher purity than NO2, as either a pure compound or a high-concentration gas mixture (e.g. 1 % in N2). The fact that the boiling point of NO (–152 °C) is considerably lower than that of either NO2 or N2O5 is an important consideration in trapping N2O5 in the presence of the remaining reactants and other nitrogen oxide impurities.
The key to the synthesis has been to use sufficiently high reactant concentrations, which allow Reaction 4 followed by Reactions 1–3 to be completed in a glass bulb at room temperature before the product stream reaches the collection stage of the manifold. Given the rate constants for Reactions 1–4, trial concentrations of 1500 ppm for ozone and 750 ppm for NO were found to be required. Under these conditions, all NO in the system was converted into NO2 in 8 ms. The reaction of ozone with NO2 (~500 times slower than the O3 + NO reaction) occurred with a lifetime of 0.65 s–1. Once NO3 starts to form, the equilibrium with NO2 occurs and NO3 can be immediately siphoned from the system. As long as ozone and NO2 are present, the equilibrium is forced towards the generation of N2O5, which is immediately subject to condensation in a cold trap. The reaction continues until virtually all of the NO2 has reacted. Under these conditions, NO does not react with N2O5 because it continues to be removed from the system.
Under the designated reactant concentrations, 375 ppm N2O5 is formed with 0.66 ppm NO2 remaining in the product stream. Assuming a reactant flow rate of 2 L min–1, 3.32 mg of N2O5 should be formed for each minute of reaction. For these flow conditions and a manifold volume of 250 mL, which sets the residence time, the conditions favour the formation of almost pure N2O5. At the dry ice–acetone trapping temperature of –77 °C, the entire N2O5 product can be collected with a minimum of NO2 being trapped. With the fully titrated, the high ozone concentration remaining in the system passes through the trap given the low O3 boiling point (–112 °C) and high trap temperature. Trap-to-trap distillation of the N2O5 product is generally not required under these synthesising conditions. Operation of the manifold in a flow mode is thought to generate 300–500 mg of N2O5 during a typical 2-h synthesis period. This mass of N2O5 has generally been found to be sufficient for a multi-day flow-mode reactor experiment.
The measurement of N2O5 is based on gas-phase titration of NO3 by NO. Measurements of this type are based on the thermal decomposition of N2O5 at elevated temperature and have been described in the literature.[49–52] When the temperature is raised, the N2O5 equilibrium is shifted to favour the NO2 + NO3 decomposition products. By adding an excess amount of NO to the heated analyte stream, NO3 can be removed from the system according to the following reaction:
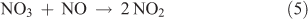
The room temperature rate constant for Reaction 5 is equal to 2.6 × 10–11 cm3 molecules–1 s–1. At a concentration of 0.1 ppm for NO, the lifetime for this reaction is 15.6 ms, much faster than any other process likely to occur under these conditions. Thus, measuring a decrease in added NO or an increase in NO2 can serve as a metric for the amount of N2O5 present in the system. Fried et al. have already discussed the difficulty of reliably measuring the increase in NO2 because of the possibility of additional NO2 or NOy sources,[50] and thus NO is typically measured by this technique. In these experiments, a heated zone in the sampling manifold at 125 °C was used to decompose the N2O5. NO at a known concentration was admitted to the analyte stream immediately before entry into the heated zone. On a time scale based on the analyte flows, the reaction was complete before the mixture leaves the heated zone. The analyte stream was then allowed to equilibrate to room temperature before NO was measured with a conventional NOx analyser. The concentration of N2O5 was determined from the difference between the initial and final NO concentrations.
Gas-phase measurements
NO and NOx were monitored with a TECO (Franklin, MA) oxides of nitrogen analyser (Model 42C) with an upstream, in-line nylon filter used to prevent nitric acid from entering the analyser. Ozone was measured with a Bendix (Lewisburg, WV) ozone monitor (Model 8002). SQT concentrations in the inlet and within the chamber were measured in a semi-continuous fashion by gas chromatography with flame ionisation detection (GC-FID). NO3 radicals were measured using the techniques reported above. SQTs and selected carbonyls were also measured using a Hewlett–Packard (HP) (Palo Alto, CA) GC-FID instrument (Model 5890) and a HP gas chromatography mass spectrometry (GC-MS) instrument (Model 6890 GC, Model 5972 MS). A fused column (60 m × 0.32-mm inner diameter) with a 1-μm DB-1 coating (J&W Scientific, Folsom, CA) was used for both GC systems. Liquid nitrogen was used to preconcentrate the analyte compounds in a sampling loop. The condensed organics were then injected onto the GC column using gas-sampling valves. The GC-FID and GC-MS operating parameters have been described previously by Blunden et al.[53]
Gas-phase hydrocarbon reactants were measured by GC-FID incorporating a double-focussing cryogenic system. Samples were initially collected in a sampling loop held at –180 °C. The loop was then flash heated to inject the analytes onto the GC column initially held at –60 °C. This system provided a reasonably fast, accurate determination for a broad range of organic reactants in the system. A semicontinuous GC system with an HP5890 GC-FID operated in an isothermal mode was also used. The system uses a 5-mL gas-sampling loop and a duty cycle of 20 min to periodically measure the hydrocarbon reactant in the chamber.
Aerosol-phase measurements
Data analysis included the measurement of aerosol yield, OM/OC ratio, ΔHvapeff and the individual products associated with organic aerosol. Organic compounds were polar, highly oxygenated and, in several cases, nitrogenated. To identify individual products, filter samples were extracted, derivatised and analysed using GC-MS techniques.
Organic carbon (OC) concentrations were measured using a semi-continuous elemental carbon–organic carbon (EC-OC) instrument (Sunset Laboratories, Tigard, OR). The instrument operates with a quartz filter positioned within the oven housing used for the analysis. The pumping system chamber drew effluent through the filter at a rate of 8 L min–1. A carbon-strip denuder was placed in-line before the quartz filter to remove gas-phase organic compounds in the effluent that might interfere with the OC measurement. With a sample collection time of 0.5 h and an analysis time of 0.25 h, the integrated sample resolution for the measurement of OC was 0.75 h. For experiments conducted early in this study, SOA from the chamber was collected on quartz filters (QFs) and then analysed for OC content using a similar offline EC-OC analyser.[54]
Aerosol size distribution, volume and total number density were measured with a scanning mobility particle sizer (SMPS) (Model 3071A, TSI, Inc., Shoreview, MN) and a condensation particle counter (CPC) (Model 3010, TSI, Inc., Shoreview, MN). The operating conditions for the SMPS were as follows: sample flow 0.2 L min–1, sheath flow 2 L min–1 and size scan from 19 to 982 nm. The SMPS was also used to determine effective enthalpies of vaporisation of SQT aerosol by adding a heated inlet, which allows the aerosol to be subjected to a range of temperatures.[55]
The composition of chamber-generated SOA was also monitored in real time using either a unit-mass resolution quadrupole aerosol mass spectrometer (Q-AMS, ER533 and ER563) or a high-resolution time-of-flight aerosol mass spectrometer (HR-AMS, ER581 and ER632). Details of Q-AMS sampling and data analysis have been discussed previously by Docherty et al.[56] ToF-AMS and Q-AMS sampling were similar and ToF-AMS data were analysed using the standard ToF-AMS Squirrel data analysis package (v1.51H).[57] To account for instrument-specific fragmentation patterns and contributions of gas-phase species that are dependent on the sampling environment, the unit-resolution mass spectrometric fragmentation table for each analysis was modified using spectra obtained during periods both before and following each experiment when high-efficiency particulate air (HEPA)-filtered chamber air was sampled into the AMS.
The aerosol formed in the chamber was also collected with 47-mm glass-fibre (GF) filters (Pall Gelman Laboratory, Ann Arbor, MI) for off-line analysis. Samples were collected at a flow rate of 15 L min–1 for dynamic experiments and 10 L min–1 for static experiments. These samples were then extracted by sonication with methanol, and the filter extracts were derivatised with BSTFA, PFBHA, PFBHA–BF3 and PFBHA–BSTFA double derivatisation.[58] All extracts were analysed by GC-MS on a ThermoQuest (Austin, TX) gas chromatograph coupled with an ion trap mass spectrometer. The injector, heated to 270 °C, was operated in splitless mode. Compounds were separated on a 60-m, 0.25-mm internal diameter, RTx-5MS column (Restek, Inc., Bellefonte, PA) that had a 0.25-µm film thickness. The oven temperature program started isothermally at 84 °C for 1 min followed by a temperature ramp of 8 °C min–1 to 200 °C, followed by a 2-min hold, and then a second ramp of 10 °C min–1 to 300 °C. The ion source, ion trap and interface temperatures were 200, 200 and 300 °C.
Results and discussion
Initial conditions and aerosol parameters
Table 2 shows the initial conditions for experiments conducted in this study. SQT initial concentrations ranged from 0.12 to 4.95 ppm C. In experiments involving photooxidation, NO concentrations ranged from 87 to 497 ppb, and for the dark ozonolysis reactions, O3 concentrations ranged from 135 to 720 ppb. For the dark reactions conducted with NO3 radicals, concentrations of 25 to 252 ppb were used. For experiments conducted in the 14.5-m3 chamber, ammonium sulfate at ~1 μg m–3 was added to the reaction mixture to aid in secondary product condensation onto chamber aerosols. No seed aerosol was used in the 9-m3 chamber. Experiments involving farnesene oxidation were performed with a mixture of α- and β-farnesene isomers (ER575 and ER569) and α-farnesene at greater than 90 % purity in ER632. The purity of farnesene was evaluated in our laboratory using a 30-L bag filled with 1 µL of the SQT to be analysed and 25 L of zero hydrocarbon air. A sample from the bag was then analysed by GC-FID and GC-MS. Chamber operating temperatures ranged between 21 and 25 °C, with the irradiated NOx experiments operating at the upper end of this range. All of the NO3 reactions (along with a few select static runs) were conducted dry (relative humidity <3 %), due primarily to the sensitivity of N2O5 to the presence of water. Typical static experiments started with an initial relative humidity of 30 %, which decreased over time due to dilution, whereas dynamic experiments were maintained continuously at 30 % relative humidity.

|
All photooxidation experiments conducted in the 14.5-m3 chamber were static except for ER393, ER632, ER648 and ER649 (Table 2). β-Caryophyllene, O3, NO, NOx–NO and integrated SMPS volume concentrations measured during ER282 are shown as a function of irradiation time in Fig. 1 (top). Time profiles of NO, NOx–NO and O3 show characteristics of a typical VOC–NOx static irradiation. This trend was observed for all photooxidation experiments conducted in this study. As can be seen from the integrated SMPS volume concentration time series in Fig. 1, particle formation from the photooxidation of β-caryophyllene occurred almost immediately, before ozone started to build up in the chamber and despite the presence of NO in the system. The concentration of particles continues to increase as the NO is removed from the system due to reaction and reaches a maximum when O3 starts building up in the chamber. This early SOA formation is different from particle volume concentration profiles observed from the photooxidation of monoterpenes and 2-methyl-3-buten-2-ol (MBO),[59,60] where negligible SOA formed at the beginning of the experiment before the depletion of NO. For example, Fig. 1 (bottom) shows similar time profiles for an additional experiment involving α-pinene photooxidation in the presence of NOx. The experimental conditions for the α-pinene experiment were similar to those described in the text for static runs. The initial conditions for α-pinene are shown in Fig. 1 (bottom). An early particle formation trend was observed for all photooxidation experiments conducted in this study reflecting the rapid photooxidation of SQTs typically within the first 20 min of the reaction, which is slower than observed for ozonolysis reactions. This early formation of particles is likely due to the high rate constant of the oxidation of SQTs with O3 as well as the presence of more than one double bond in the SQTs (Table 1). This is consistent with the proposal of Jardine et al.[15] that SQTs may serve as O3 sinks in highly vegetative ambient environments. Edney et al.[61] suggested that SOA from irradiated α-pinene–NOx–air mixtures better represented ambient PM2.5 in Research Triangle Park, NC than that generated by ozonolysis of α-pinene. Due to the apparent dominance of ozonolysis reactions in the SQT–NOx photochemical systems, it appears that simple ozonolysis dark reactions may be far more representative of ambient conditions for SQTs than for monoterpenes or other SOA precursors.
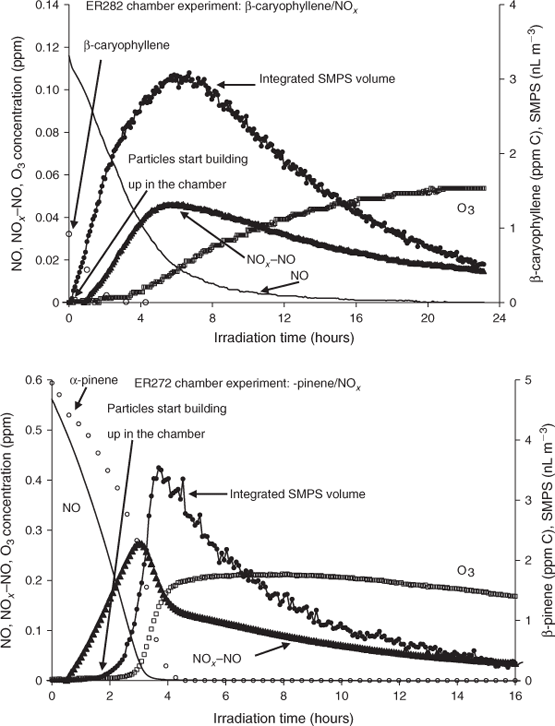
|
Table 3 shows the reacted SQT concentrations for seven experiments conducted in dynamic mode: one O3 experiment, four photooxidation experiments in the presence of NOx and two experiments in the presence of NO3 radicals. The reacted SQT concentration, determined from the difference between the initial and steady-state SQT concentrations, ranged from 0.12 to 0.63 ppm C (66 to 347 µg m–3). The uncertainty in the reacted SQT results arising from the reproducibility of both the initial and steady-state hydrocarbon values by GC is estimated to range between 30 and 50 % due to the relatively low volatility of these compounds compared to monoterpenes. Note that substantial challenges exist for adding SQTs into the chamber in a controlled fashion at target concentrations. In static mode, for example, it is difficult to determine SQT time profiles due to their low volatility and high reactivity towards oxidants. Therefore, aerosol yields were measured only from dynamic experiments.

|
The major aerosol parameters measured, including SOA and secondary organic carbon (SOC), are given in Table 3 for experiments conducted in dynamic mode. Uncertainties in the SOC values are taken from the reproducibility of the semicontinuous measurement and are typically better than 10 % for a single run, although this does not include possible systematic error. For OM, the uncertainties are determined from the reproducibility of side-by-side filter measurements, which are typically better than 5 %. An estimate of the systematic errors due to minor changes in reactant concentrations, minor variations in chamber temperature and other similar factors bring the total uncertainty to between 15 and 25 % for these parameters.
The SOA yield (YSOA) and secondary organic carbon yield (YSOC) are generally defined using the following relationships:
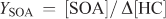
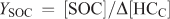
where [SOC] is the background corrected organic carbon concentration (μg C m–3), Δ[HC] is the reacted hydrocarbon mass concentration (μg m–3) and Δ[HCC] is the reacted carbon mass concentration of the hydrocarbon (μg C m–3). Using the relationship above, β-caryophyllene YSOA values of 0.53, 0.30 and 1.19 (average of two data points) were calculated for the measured aerosol mass for reactions involving ozonolysis, photooxidation and NO3 reaction. Uncertainties in the calculated yields arise from the experimental uncertainties in determining SOA mass and the uncertainties associated with determining reacted SQT concentrations as discussed above. YSOC values are also reported in Table 3 and for β-caryophyllene ranged from 0.28 for ozonolysis, 0.19 for photooxidation to 0.83 (average of two data points) for NO3 reactions. These yields are in reasonable agreement with literature data (Chen et al.[35] and references therein).
The effective enthalpy of vaporisation was determined for the bulk aerosol in select dynamic experiments. The values were calculated from the slope of a plot of ln V v. T–1 (V represents the integrated aerosol volume at each temperature (T) of the heated tube) which when multiplied by the gas constant (R) gives a ΔHvapeff. The effective enthalpies of vaporisation for aerosol generated from β-caryophyllene were found to be ~43.9 kJ mol–1 for photooxidation, 41.1 kJ mol–1 for ozonolysis and 44.9 kJ mol–1 for the β-caryophyllene–NO3 system. A value of 27.7 kJ mol–1 was found for the α-farnesene–NOx system and 48.2 kJ mol–1 for the α-humulene–NOx system. These values are generally consistent with the ranges of values measured for SOA generated in the photooxidation of other terpenes.[55,62] For example, Offenberg et al. reported an effective enthalpy of vaporisation of 38 kJ mol–1 for α-pinene and 18 kJ mol–1 for isoprene.[55]
Particle-phase products analysis
For the systems studied, sufficient aerosol masses were collected on GF filters to be solvent extracted, derivatised and analysed by GC-MS. Aerosol extracts were derivatised using various combinations of BSTFA, PFBHA, PFBHA–BF3 or PFBHA–BSTFA derivatisations. These techniques provide a sensitive method for identifying and quantifying low concentrations of lightly to highly oxidised organic compounds. The BSTFA single derivatisation technique provides good quantitative analysis due to both its simplicity and efficiency.[58] Although they are not quantitatively rigorous, the double derivatisations provide additional structural information which aids in the identification of SOA constituents, including carbonyls that are not detected by BSTFA derivatisation alone.
Many compounds identified in SQT SOA do not have an authentic standard, so their identification was based on interpretation of the mass spectra of the derivatised compound. For compounds that have standards, identification was based on comparisons between mass spectra of the derivatised samples and the authentic spectra in chemical ionisation (CI) or electron impact (EI) mode, as well as on retention times. For compounds with no standard, initial identification was made based on fragment and adduct peaks in CI mode that permit determination of the number and identity of functional groups and the molecular weight (MW) of the derivative. Additional analyses were made using double derivatisations to confirm the identity of potentially ambiguous functional groups. Analysis of CI mass spectra of the derivatives involved recognition of characteristic ions associated with a particular derivatisation scheme. A detailed description of this method can be found in Jaoui et al.[58]
In the present study, GC-MS mass spectra for more than 150 compounds have been recorded. The approach used for their detection is as follows: peaks detected in blank and background samples were eliminated first; a peak was associated with a reaction product only if its corresponding mass spectrum was consistent with the fragmentation pattern of the derivatisation reagent used. Given the large number of peaks generated from these systems, an extensive evaluation was made by comparing products identified in this study with those from our previous work or other laboratories to determine the degree of consistency and to provide information on which chemical systems might be subject to the greatest uncertainties.[23,29,30,33,38–40,63]
The majority of organic products structurally identified in this study from α-cedrene, α-humulene and β-caryophyllene (Table 4) have been reported in the literature[23,29,30,33,39,40] and therefore only a few examples are presented here. The reader should consult the original papers for more information. For experiments involving farnesene, for which only very limited data are available for gas-phase compounds, a detailed chemical analysis is reported elsewhere (M. Jaoui, K. S. Docherty, M. Lewandowski, T. E. Kleindienst, J. H. Offenberg, unpubl. data) and only limited data are provided here. We have included in this paper representative examples of only products observed either for the first time or observed previously in ozonolysis (or photooxidation) experiments and detected in this study in photooxidation (or ozonolysis) experiments. Additional compounds (e.g. high-molecular-weight organics and organonitrates) might have been present in the SOA but could not be detected based on the analytical techniques used in this study. In this section, our main purpose is to compare and highlight the chemical characteristics of SOA generated from the different systems studied (e.g. photooxidation v. ozonolysis v. NO3 oxidation).

|
Typical total ion chromatograms from the BSTFA derivatisation of SOA extracts from α-humulene–NO3, α-humulene–NOx and α-humulene–O3 systems are shown in Fig. 2. The chromatograms related to photooxidation (middle) and ozonolysis (bottom) experiments are similar. A detailed analysis of spectra associated with each peak in these chromatograms reveals that more than 80 % of peaks eluted in both chromatograms were identical, suggesting similar chemistry involved in both ozonolysis and photooxidation in the presence of NOx. Identified peaks are in good agreement with our previous chemical analysis of SOA formed from the same systems showing the presence of similar products.[39] Literature data exists only for α-humulene photooxidation and β-caryophyllene ozonolysis.

|
Analysis of mass spectra associated with peaks detected in Fig. 2 from the α-humulene–NO3 system (top) compared with those for α-humulene–O3 and α-humulene–NOx systems shows that the majority of peaks detected in the NO3 system were not present in the two other systems. Also, there are fewer peaks observed in chromatograms of aerosol generated from reaction with NO3, and most peaks are eluted earlier in the chromatogram than those detected in the photooxidation and ozonolysis reactions. A similar analysis undertaken for the β-caryophyllene–NO3 and α-farnesene–NO3 systems shows similar trends to the α-humulene–NO3 system. This suggests that the composition produced by night-time chemistry of SQTs is considerably different than that involved during the daytime. These results are similar to those obtained from monoterpene experiments conducted in our laboratory (results not shown here), in which organic compounds detected in the photooxidation system were absent in the NO3 reaction and only a small number of peaks were observed in all the SQT systems.
In general, the mass spectrometric signals associated with the majority of chromatographic peaks detected from NO3 chamber oxidations of β-caryophyllene, α-humulene and α-farnesene using BSTFA derivatisation had predominant peaks that showed odd values. Note that BSTFA and other derivatisations of organic nitrates are not well understood and the analysis of SOA from NO3 reaction is still associated with high uncertainties, mainly for the detection of organic nitrates. For example, some reaction products may contain more than one nitrogen atom moiety (due either to the presence of more than one double bond in the parent SQT or due to chemistry), leading to a more challenging and complicated interpretation of mass spectra. In addition, the high GC inlet temperatures used create the potential for thermal decomposition of organic nitrates. For example, a HR-AMS mass spectrum of β-caryophyllene–NO3 aerosol is shown in Fig. 3c. A Q-AMS spectrum of isoprene–NO3 aerosol and an HR-AMS spectrum of α-pinene–NO3 aerosol are provided in Fig. 3a and 3b for comparison. Each of these spectra were first normalised by the total spectrometric signal and the fragment intensity was subsequently adjusted to foster comparison. Overall, the spectrum of β-caryophyllene–NO3 aerosol has characteristic differences to the other mass spectra, particularly in the presence and intensity of fragments in the medium (e.g. 51–200 amu) and high (201–500 amu) mass-to-charge ratio (m/z) range. As Fig. 3 shows, the intensity of fragments in the high mass range proceeds in the order isoprene > α-pinene > β-caryophyllene, whereas the intensity of fragments in the low mass range (e.g. 10–49 amu) follows the opposite trend. Fig. 3d summarises the relative contribution of mass spectrometric fragments (within 50 amu segments) to the total unadjusted mass spectrum and shows these differences more clearly. Overall, fragments in the low mass region dominate the isoprene–NO3 aerosool spectrum, whereas contributions in the intermediate and high mass ranges are small or negligible. Low mass fragments appear to have similar high contributions to the α-pinene–NO3 SOA mass spectrum. However, the contributions of fragments in the intermediate mass range are higher than those in the isoprene spectrum, whereas those in the high mass range are relatively similar. The relative contributions of fragments in the high mass range of β-caryophyllene–NO3 SOA are highest among this set of spectra with distinct structures observed up to m/z 375. These results suggest that high MW compounds are present in β-caryophyllene–NO3 SOA and further suggests that the detection of fewer smaller compounds by GC-MS could be due to their thermal decomposition in the heated inlet or the column.
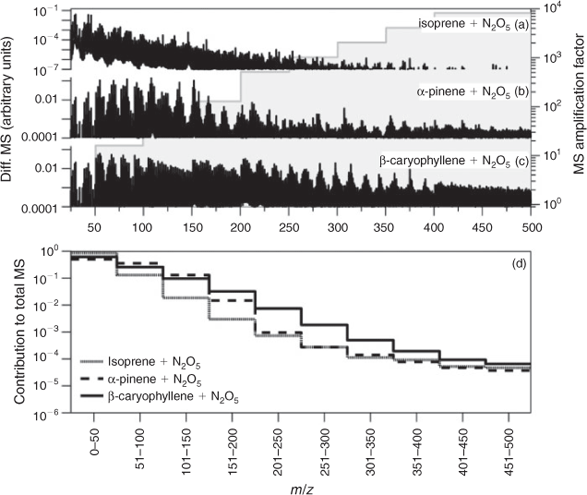
|
The GC-MS analysis of β-caryophyllene SOA generated from either photooxidation or ozonolysis shows the presence of several significant peaks and a large number of smaller peaks. Most of these peaks were identified from the ozonolysis of β-caryophyllene.[23,30,38] Fig. 4 shows four CI mass spectra associated with peaks from PFBHA, BSTFA, PFBHA–BF3 and PFBHA–BSTFA derivatisations of aerosol extracts from the photooxidation of β-caryophyllene. The BSTFA CI mass spectrum of this compound shows characteristic fragment ions at m/z 73, 255 ([M – 15]+), 181 ([M – 89]+) and 153 ([M – 117]+), and adducts at [M + 1]+, [M + 29]+ and [M + 41]+. These fragments and adducts are consistent with the presence of one OH group and a MW of the derivatised compound of 270 Daltons (all masses hereafter given as Daltons) and 198 for the underivatised compound. The presence of a peak at m/z 153 ([M – 117]+) is consistent with a compound bearing an organic acid group. The PFBHA CI spectrum of this compound (Fig. 4a) shows strong fragments at m/z 181 and 394 ([M + 1]+), consistent with the presence of one CO group and giving a MW of 393 for the derivatised compound and 198 for the underivatised compound. The reaction of BF3–methanol with carboxylic acids forms methyl esters with characteristic ions at 59, [M – 59]+, [M – 31]+ and [M + 1]+, and weak adducts at [M + 15]+, [M + 29]+ and [M + 41]+. Double derivatisations result in adducts and fragments that include characteristic ions from each derivatisation. The mass spectrum associated with PFBHA and BSTFA (Fig. 4b) shows fragment ions at m/z 73, 181, 376 ([M – 89]+), 450 ([M – 15]+) and adducts at [M + 1]+, [M + 29]+ and [M + 41]+. The mass spectrum for PFBHA–BF3 (Fig. 4c) shows fragment ions at m/z 300 ([M – 107]+), 376 ([M – 31]+) and 408 ([M – 1]+), and adducts at [M + 29]+ and [M + 41]+. These mass spectra are consistent with the presence of one OH and one CO group.

|
The compound associated with these peaks was tentatively identified as 3,3-dimethyl-2-(3-oxobutyl)cyclobutanecarboxylic acid (C11H18O3). Other β-caryophyllene photooxidation compounds tentatively identified in the particle phase at very low levels include 3-(4-acetyl-7–7-dimethyl-3-oxabicyclo[4,2,0]oct-4-en-2yl)propanoic acid (C14H20O4) and 2,3-dihydroxy-4-[2,(4-hydroxy-3-oxobutyl]-4-oxobutanoic acid (C14H22O7). These compounds were reported first by Li et al.[30] from the ozonolysis of β-caryophyllene. In this study, they were detected in SOA formed from photooxidation of β-caryophyllene, as well as from ozonolysis, but not from the NO3 reaction. As mentioned earlier, particles were observed early in the reaction due mainly to ozone reaction. These compounds may be formed following similar mechanistic pathways reported by Li et al. involving reaction of ozone with first generation products.[30]
Fig. 5 shows CI mass spectra associated with peaks for BSTFA derivatisation of aerosol extracts from the photooxidation of α-humulene and α-cedrene. Literature information about compound identification exists for α-humulene photooxidation[39] and α-cedrene ozonolysis[40] only. This study focuses also on the largely unreported α-humulene ozonolysis and α-cedrene photooxidation reactions in order to provide a more thorough dataset. The mass spectra shown are those for α-humuladionic acid (MW 254), a diacid similar to pinic acid bearing two carboxylic groups; α-nohumulonic acid (MW 254), a compound bearing one carboxylic and two ketone groups; α-cedrenic acid (MW 254), a diacid similar to pinic acid and finally α-cedronic acid (MW 252), a compound similar to pinonic acid. α-Nohumulonic acid is detected for the first time in this study in both ozonolysis and photooxidation experiments but not from NO3 reaction.

|
Fig. 6a and 6b respectively show total ion chromatograms (13–26 min) from BSTFA derivatisation of SOA extracts from α-farnesene–NOx and α-farnesen–NO3 systems. A variety of OH-bearing compounds were observed in both systems reflected by the presence of the trimethylsilyl group (m/z 73) in the BSTFA-derivatised compounds. Similar to α-humulene systems, a detailed analysis of spectra associated with each peak reveals that the majority of peaks eluted (using BSTFA derivatisation) in both chromatograms were not the same, suggesting that the chemistry involved in both systems is different. The number of peaks observed in NO3–α-farnesene oxidation is higher than in the α-humulene–NO3 system possibly due to the physical characteristics of α-farnesene compared with α-humulene (linear with four double bonds for farnesene compared with cyclic and three double bonds for humulene: Table 1). Spectra associated with NO3–α-farnesene reactions are consistent with compounds bearing OH groups, however, they are difficult to interpret following BSTFA derivatisation rules, mainly to get structural information about the derivative MW of the compound. Most spectra are dominated by odd fragment ions making it more difficult to know if the compound associated with a peak contains nitrate moieties. Therefore, all spectra originated in this study involving NO3 reactions are not presented. Clearly, additional research with a focus on understanding the BSTFA derivatisation of compounds bearing nitrates groups needs to be conducted.

|
However, analysis of spectra originated from SOA extracts from α-farnesene–NOx is consistent with BSTFA derivatisation rules and their interpretation leads in general to tentative structural identification when authentic standards are missing. As an example Fig. 6c–d show two mass spectra associated with two compounds tentatively identified for the first time as 3,7,11-trimethyldodeca-1,3,5,10-tetraen-7-ol (or isomers) (Fig. 6c) still preserving the α-farnesene carbon backbone and all four double bonds and methylene succinic acid (itaconic acid: Fig. 6d), a diacid bearing one ethylene group. The M+, [M – 15]+ and [M – 89]+ fragment ions of the silylated 3,7,11-trimethyldodeca-1,3,5,10-tetraen-7-ol occurred at m/z 293, 277 and 203. Other key ions included those at m/z 187 ([M – 105]+), 321 ([M + 29]+), 333 ([M + 41]+), 365 ([M + 73]+) and 73 (Fig. 6c). For methylene succinic acid, a similar fragmentation pattern was observed. The CI BSTFA mass spectrum (Fig. 6d) shows the presence of strong fragmentation ions at m/z 259 ([M – 15]+), 185 ([M – 89]+) and 169 ([M – 105]+), and the [M + 1]+, [M + 29]+ and [M + 41]+ adducts, all of which are consistent with the presence of a BSTFA derivative of MW 274 formed from a compound of MW 130 containing two OH groups.
Summary
Several recent studies have suggested that secondary PM can be formed under a wide range of conditions through reactions of organic compounds with O3, OH and NO3 radicals. This study investigates the formation of SOA through reaction of a series of SQTs, including α-cedrene, β-caryophyllene, α-humulene and α-farnesene, with NOx, O3 and NO3 radicals under a wide range of conditions. These hydrocarbons, emitted by terrestrial vegetation, are oxidised in the ambient atmosphere and lead to the formation of SOA, ground-level ozone and other pollutants. Experiments were conducted with each SQT individually in a 9-m3 or a 14.5-m3 smog chamber operated in either dynamic mode, to ensure adequate collection of the aerosol at reasonably low reactant concentrations, or static mode, in which temporal evolution of the reactants and products could be obtained.
The results of this study suggest that the nature of the oxidant (O3 v. NO3 v. OH radicals) is an important factor influencing the chemical characteristics of the particle phase. Although similar reaction products were observed during SQT photooxidation in the presence of NOx and ozonolysis, the SOA formed from NO3 oxidation shows different chemical characteristics. The SOA chemical characteristics tended to depend primarily on the daytime v. night-time chemistry (NO3). The chemical characteristics of the SOA from SQTs were found to evolve over the course of the static reactions in the chamber as reflected by the time profile of the integrated SMPS volume and OC. This may be due to the oxidation of less volatile first generation products leading to polar second generation products that reside mainly in the particle phase.[32] This study also suggests that SOA from ozonolysis experiments may adequately represent ambient aerosol in areas with SQT emissions.
Several aerosol parameters were determined and compared with literature data when available. The OM/OC ratio, for example, is an important parameter for converting SOA into SOC in air quality models and was found in this study to give an average value of 1.8 for ozonolysis and photoxidarion reactions and 1.6 for the dark reaction involving NO3 radicals. The effective enthalpy of vaporisation for the aerosol generated in this study was measured for β-caryophyllene–NOx, β-caryophyllene–O3, β-caryophyllene–NO3, α-humulene–NOx and α-farnesene–NOx systems and found to be 43.9, 41.1, 44.9, 48.2 and 27.7 kJ mol–1. The average SOA yields were found to be 0.53, 0.55 and 1.19 in this work for ozonolysis, photooxidation and NO3 reaction, in good agreement with literature data.
Reactions of β-caryophyllene, α-humulene and α-farnesene with NO3 have been shown to form considerable amounts of SOA. MS analyses of filter-based and AMS data show that the condensable oxidised products have far fewer nitrogen moieties than SOA from other terpenes (e.g. isoprene–NO3 system). Although SQTs are thought to be emitted mainly during daylight hours, given their high emission rate even with their short atmospheric lifetime mainly with ozone, substantial concentrations are likely to be present in the early evening when the NO2 + O3 reaction producing NO3 is likely to be most pronounced.
GC-MS data analysis was employed in this study to determine the degree to which unique organic tracers indicative of these systems might be formed during reactions and are represented in the SOA. One of the most prevalent SQT tracers observed in ambient PM2.5, β-caryophyllinic acid, was readily produced in this study during the photooxidation in the presence of NOx and ozonolysis of β-caryophyllene. It was detected at very low levels, however, during the night-time oxidation involving NO3 radicals. Although a series of polar compounds are observed in the SOA of SQTs, a preliminary survey suggests that only β-caryophyllinic acid can be found in ambient aerosol.
MS analyses of filter-based and AMS data from systems involving reaction with NO3 show that the condensable oxidised products have far fewer nitrogen moieties than SOA from other terpenes (e.g. isoprene–NO3 system). The analysis also shows that condensable products observed in the photooxidation and ozonolysis experiments are similar reflecting similar chemistry involved in their oxidation in both systems. O3 seems to play an important role early in photooxidation reactions contrary to those observed for other terpenes. This is consistent with ambient data showing that SQTs act as effective ozone sinks within the plant canopy.
Acknowledgements
The US Environmental Protection Agency through its Office of Research and Development funded and collaborated in the research described here under Contract EP-D-10-070 to Alion Science and Technology. The manuscript has been subjected to external peer review and has been cleared for publication. Mention of trade names or commercial products does not constitute endorsement or recommendation for use.
References
[1] A. Guenther, C. N. Hewitt, D. Erickson, R. Fall, C. Geron, T. Graedel, P. Harley, L. Klinger, M. Lerdau, W. A. McKay, T. Pierce, B. Scholes, R. Steinbrecher, R. Tallamraju, J. Taylor, P. Zimmerman, A global model of natural volatile organic compound emissions. J. Geophys. Res. 1995, 100, 8873.| A global model of natural volatile organic compound emissions.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXmvFKrsb0%3D&md5=f8ec60b8d0c502e237993887de9c1a69CAS |
[2] T. R. Duhl, D. Helmig, A. Guenther, Sesquiterpene emissions from vegetation: a review. Biogeosciences 2008, 5, 761.
| Sesquiterpene emissions from vegetation: a review.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtVCqsLrK&md5=27487476e2ca021c6fe360d1c76055dfCAS |
[3] F. W. Went, Blue hazes in the atmosphere. Nature 1960, 187, 641.
| Blue hazes in the atmosphere.Crossref | GoogleScholarGoogle Scholar |
[4] M. Kanakidou, J. H. Seinfeld, S. N. Pandis, I. Barnes, F. J. Dentener, M. C. Facchini, R. Van Dingenen, B. Ervens, A. Nenes, C. J. Nielsen, E. Swietlicki, J. P. Putaud, Y. Balkanski, S. Fuzzi, J. Horth, G. K. Moortgat, R. Winterhalter, C. E. L. Myhre, K. Tsigaridis, E. Vignati, E. G. Stephanou, J. Wilson, Organic aerosol and global climate modeling: a review. Atmos. Chem. Phys. 2005, 5, 1053.
| Organic aerosol and global climate modeling: a review.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXktlyrtbw%3D&md5=9658c4771c5ada0b0161d82d71086b1fCAS |
[5] M. Hallquist, J. C. Wenger, U. Baltensperger, Y. Rudich, D. Simpson, M. Claeys, J. Dommen, N. M. Donahue, C. George, A. H. Goldstein, J. F. Hamilton, H. Herrmann, T. Hoffmann, Y. Iinuma, M. Jang, M. E. Jenkin, J. L. Jimenez, A. Kiendler-Scharr, W. Maenhaut, G. McFiggans, Th. F. Mentel, A. Monod, A. S. H. Prevot, J. H. Seinfeld, J. D. Surratt, R. Szmigielski, J. Wildt, The formation, properties and impact of secondary organic aerosol: current and emerging issues. Atmos. Chem. Phys. 2009, 9, 5155.
| The formation, properties and impact of secondary organic aerosol: current and emerging issues.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsFGhs77M&md5=92e7020caa93e2e140c369b93a625364CAS |
[6] J. F. Sisler, W. C. Malm, The relative importance of soluble aerosols to spatial and seasonal trends of impaired visibility in the United States. Atmos. Environ. 1994, 28, 851.
| The relative importance of soluble aerosols to spatial and seasonal trends of impaired visibility in the United States.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2cXjtVKhsL8%3D&md5=f2f1f98d9f9d1213c6975e21c4665b5eCAS |
[7] R. J. Charlson, S. E. Chwartz, J. M. Hales, R. D. Cess, J. A. Coakley, J. E. Hansen, D. J. Hoffman, Climate forcing by anthropogenic aerosols. Science 1992, 255, 423.
| Climate forcing by anthropogenic aerosols.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK38XhtFSntbk%3D&md5=8c11d15ca6762712cc223408fd8741aeCAS | 17842894PubMed |
[8] C. A. Pope, M. Ezzati, D. W. Dockery, Fine particulate air pollution and life expectancy in the United States. New Engl. J. Med. 2009, 360, 376.
| Fine particulate air pollution and life expectancy in the United States.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtVSltr8%3D&md5=8b7bb10b8ce70ab3950b8eda8a7ef5bdCAS | 19164188PubMed |
[9] I. Faloona, D. Tan, W. Brune, J. Hurst, D. Barket, T. L. Couch, P. Shepson, E. Apel, D. Riemer, T. Thornberry, M. A. Carroll, S. Sillman, G. J. Keeler, J. Sagady, D. Hooper, K. Paterson, Nighttime observations of anomalously high levels of hydroxyl radicals above a deciduous forest canopy. J. Geophys. Res. 2001, 106, 24315.
| Nighttime observations of anomalously high levels of hydroxyl radicals above a deciduous forest canopy.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXoslWhtLo%3D&md5=0e78df41ee8763ec4a917c39f2e41249CAS |
[10] R. A. Holzinger, K. T. Lee, U. Paw, A. H. Goldstein, Observations of oxidation products above a forest imply biogenic emissions of very reactive compounds. Atmos. Chem. Phys. 2005, 5, 67.
| Observations of oxidation products above a forest imply biogenic emissions of very reactive compounds.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXktlyqsbw%3D&md5=60696723abd34581464222423a7a2002CAS |
[11] A. H. Goldstein, I. E. Galbally, Known and unexplored organic constituents in the Earth’s atmosphere. Environ. Sci. Technol. 2007, 41, 1514.
| Known and unexplored organic constituents in the Earth’s atmosphere.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXit1Cnuro%3D&md5=bb559320fbd5146133774d732702004cCAS | 17396635PubMed |
[12] P. Di Carlo, W. H. Brune, M. Martinez, H. Harder, R. Lesher, X. Ren, T. Thornberry, M. A. Carroll, V. Young, P. B. Shepson, D. Riemer, E. Apel, C. Campbell, Missing OH reactivity in a forest: evidence for unknown reactive biogenic VOCs. Science 2004, 304, 722.
| Missing OH reactivity in a forest: evidence for unknown reactive biogenic VOCs.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXjsV2gt7c%3D&md5=f2365626c80deda0d0930b47dccd11bbCAS | 15118159PubMed |
[13] A. H. Goldstein, M. McKay, M. R. Kurpius, G. W. Schade, A. Lee, R. Holzinger, R. A. Rasmussen, Forest thinning experiment confirms ozone deposition to forest canopy is dominated by reaction with biogenic VOCs. Geophys. Res. Lett. 2004, 31, L22106.
| Forest thinning experiment confirms ozone deposition to forest canopy is dominated by reaction with biogenic VOCs.Crossref | GoogleScholarGoogle Scholar |
[14] D. K. Farmer, R. C. Cohen, Observations of HNO3, ΣAN, ΣPN and NO2 fluxes: evidence for rapid HOx chemistry within a pine forest canopy. Atmos. Chem. Phys. 2008, 8, 3899.
| Observations of HNO3, ΣAN, ΣPN and NO2 fluxes: evidence for rapid HOx chemistry within a pine forest canopy.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtFeiu7fO&md5=8775939cfa4ca4a9570028d738ecabe6CAS |
[15] K. Jardine, A. Yañez Serrano, A. Arneth, L. Abrell, A. Jardine, J. van Haren, P. Artaxo, L. V. Rizzo, F. Y. Ishida, T. Karl, J. Kesselmeier, S. Saleska, T. Huxman, Within-canopy sesquiterpene ozonolysis in Amazonia. J. Geophys. Res. 2011, 116, D19301.
| Within-canopy sesquiterpene ozonolysis in Amazonia.Crossref | GoogleScholarGoogle Scholar |
[16] P. Ciccioli, E. Brancaleoni, M. Frattoni, V. Di Palo, R. Valentini, G. Tirone, G. Seufert, N. Bertin, U. Hansen, O. Csiky, R. Lenz, M. Sharma, Emission of reactive terpene compounds from orange orchards and their removal by within-canopy processes. J. Geophys. Res. 1999, 104, 8077.
| Emission of reactive terpene compounds from orange orchards and their removal by within-canopy processes.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXjtFyltLw%3D&md5=1f6ed0a493cd0e8605cec5cfde60c4e7CAS |
[17] C. E. Vickers, J. Gershenzon, M. T. Lerdau, F. Loreto, A unified mechanism of action for volatile isoprenoids in plant abiotic stress. Nat. Chem. Biol. 2009, 5, 283.
| A unified mechanism of action for volatile isoprenoids in plant abiotic stress.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXks12ksbg%3D&md5=6f5ed8a9361ac5513ed4ba104342ca5aCAS | 19377454PubMed |
[18] T. Sakulyanontvittaya, T. Duhl, C. Wiedinmyer, D. Helmig, S. Matsunaga, M. Potosnak, J. Milford, A. Guenther, Monoterpene and sesquiterpene emission estimates for the United States. Environ. Sci. Technol. 2008, 42, 1623.
| Monoterpene and sesquiterpene emission estimates for the United States.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXoslyitg%3D%3D&md5=e8dd68288bf6c57def8b3a4b9508bb69CAS | 18441812PubMed |
[19] R. T. Griffin, D. R. Cocker, R. C. Flagan, J. H. Seinfeld, Organic aerosol formation from the oxidation of biogenic hydrocarbons. J. Geophys. Res. 1999, 104, 3555.
| Organic aerosol formation from the oxidation of biogenic hydrocarbons.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXitFWjs7Y%3D&md5=fd5c8df4f3d60bc615965ab6e935be6aCAS |
[20] Y. Shu, R. Atkinson, Rate constants for the gas-phase reactions of O3 with a series of terpenes and OH radical formation for the O3 reactions with sesquiterpenes at 296 K. Int. J. Chem. Kinet. 1994, 26, 1193.
| Rate constants for the gas-phase reactions of O3 with a series of terpenes and OH radical formation for the O3 reactions with sesquiterpenes at 296 K.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXitF2ksb8%3D&md5=f249a1c859d3a57e8ca2a7954b8a14b6CAS |
[21] Y. Shu, R. Atkinson, Atmospheric lifetimes and fates of a series of sesquiterpenes. J. Geophys. Res. 1995, 100, 7275.
| Atmospheric lifetimes and fates of a series of sesquiterpenes.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXmt1Chsbc%3D&md5=82602c194881102fb1e2cb5a75916dc7CAS |
[22] R. Atkinson, J. Arey, Atmospheric degradation of volatile organic compounds. Chem. Rev. 2003, 103, 4605.
| Atmospheric degradation of volatile organic compounds.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXosVChtrs%3D&md5=fef3fb034520ef1e5691a65da2ee5a53CAS | 14664626PubMed |
[23] M. Jaoui, S. Leungsakul, R. M. Kamens, Gas and particle products distribution from the reaction of β-caryophyllene with ozone. J. Atmos. Chem. 2003, 45, 261.
| Gas and particle products distribution from the reaction of β-caryophyllene with ozone.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXks1Cnt7w%3D&md5=46e37340a873c17fc0a6f6328a8697a4CAS |
[24] A. Lee, A. H. Goldstein, M. D. Keywood, S. Gao, V. Varutbangkul, R. Bahreini, N. L. Ng, R. C. Flagan, J. H. Seinfeld, Gas-phase products and secondary aerosol yields from the ozonolysis of ten different terpenes. J. Geophys. Res. 2006, 111, D07302.
| Gas-phase products and secondary aerosol yields from the ozonolysis of ten different terpenes.Crossref | GoogleScholarGoogle Scholar |
[25] A. Lee, A. H. Goldstein, J. H. Kroll, N. L. Ng, V. Varutbangkul, R. C. Flagan, J. H. Seinfeld, Gas-phase products and secondary aerosol yields from the photooxidation of 16 different terpenes. J. Geophys. Res. 2006, 111, D17305.
| Gas-phase products and secondary aerosol yields from the photooxidation of 16 different terpenes.Crossref | GoogleScholarGoogle Scholar |
[26] N. L. Ng, P. S. Chhabra, A. W. H. Chan, J. D. Surratt, J. H. Kroll, A. J. Kwan, D. C. McCabe, P. O. Wennberg, A. Sorooshian, S. M. Murphy, N. F. Dalleska, R. C. Flagan, J. H. Seinfeld, Effect of NOx level on secondary organic aerosol (SOA) formation from the photooxidation of terpenes. Atmos. Chem. Phys. 2007, 7, 5159.
| Effect of NOx level on secondary organic aerosol (SOA) formation from the photooxidation of terpenes.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhtlOnsrvL&md5=517b2a279527b665dd8010097481df58CAS |
[27] T. Hoffmann, J. R. Odum, F. Bowman, D. Collins, D. Klockow, R. C. Flagan, J. H. Seinfeld, Formation of organic aerosols from the oxidation of biogenic hydrocarbons. J. Atmos. Chem. 1997, 26, 189.
| Formation of organic aerosols from the oxidation of biogenic hydrocarbons.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXjvVKmsLg%3D&md5=2b59106c295ae7dfd6ad53fd29e85a85CAS |
[28] B. Kanawati, F. Herrmann, S. Joniec, R. Winterhalter, G. K. Moortgat, Mass spectrometric characterization of β-caryophyllene ozonolysis products in the aerosol studied using an electrospray triple quadrupole and time-of-flight analyzer hybrid system and density functional theory. Rapid Commun. Mass Spectrom. 2008, 22, 165.
| Mass spectrometric characterization of β-caryophyllene ozonolysis products in the aerosol studied using an electrospray triple quadrupole and time-of-flight analyzer hybrid system and density functional theory.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXitVSlsrk%3D&md5=3b397f514807a7fcf8b7b891a9da7b12CAS | 18069749PubMed |
[29] R. Winterhalter, F. Herrmann, B. Kanawati, T. L. Nguyen, J. Peeters, L. Vereecken, G. K. Moortgat, The gas-phase ozonolysis of β-caryophyllene (C15H24). Part I: an experimental study. Phys. Chem. Chem. Phys. 2009, 11, 4152.
| The gas-phase ozonolysis of β-caryophyllene (C15H24). Part I: an experimental study.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXmtFCitrY%3D&md5=29fda3a5a8ab1b04c209f3ff4ae79e44CAS | 19458818PubMed |
[30] Y. J. Li, Q. Chen, M. I. Guzman, C. K. Chan, S. M. Martin, Second-generation products contribute substantially to the particle-phase organic material produced by β-caryophyllene ozonolysis. Atmos. Chem. Phys. 2011, 11, 121.
| Second-generation products contribute substantially to the particle-phase organic material produced by β-caryophyllene ozonolysis.Crossref | GoogleScholarGoogle Scholar |
[31] T. L. Nguyen, R. Winterhalter, G. K. Moortgat, B. Kanawati, J. Peeters, L. Vereecken, The gas-phase ozonolysis of b-caryophyllene (C15H24). Part II: a theoretical study. Phys. Chem. Chem. Phys. 2009, 11, 4173.
| The gas-phase ozonolysis of b-caryophyllene (C15H24). Part II: a theoretical study.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXmtFCit74%3D&md5=2a41ebf9e9358f63b31b4d492e48da13CAS | 19458819PubMed |
[32] M. N. Chan, J. D. Surratt, A. W. H. Chan, K. Schilling, J. H. Offenberg, M. Lewandowski, E. O. Edney, T. E. Kleindienst, M. Jaoui, E. S. Edgerton, R. L. Tanner, S. L. Shaw, M. Zheng, E. M. Knipping, J. H. Seinfeld, Influence of aerosol acidity on the chemical composition of secondary organic aerosol from β-caryophyllene. Atmos. Chem. Phys. 2011, 11, 1735.
| Influence of aerosol acidity on the chemical composition of secondary organic aerosol from β-caryophyllene.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXlvFyltbw%3D&md5=86c66711febc6e5e61c7eabe23c18dd6CAS |
[33] M. Jaoui, M. Lewandowski, T. E. Kleindienst, J. H. Offenberg, E. O. Edney, β-Caryophyllinic acid: an atmospheric tracer for β-caryophyllene secondary organic aerosol. Geophys. Res. Lett. 2007, 34, L05816.
| β-Caryophyllinic acid: an atmospheric tracer for β-caryophyllene secondary organic aerosol.Crossref | GoogleScholarGoogle Scholar |
[34] M. R. Alfarra, J. F. Hamilton, K. P. Wyche, N. Good, M. W. Ward, T. Carr, M. H. Barley, P. S. Monks, M. E. Jenkin, G. B. McFiggans, The effect of photochemical ageing and initial precursor concentration on the composition and hygroscopic properties of β-caryophyllene secondary organic aerosol. Atmos. Chem. Phys. 2012, 12, 6417.
| The effect of photochemical ageing and initial precursor concentration on the composition and hygroscopic properties of β-caryophyllene secondary organic aerosol.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhslSnu7vE&md5=a01968422654289f49db878261a3de04CAS |
[35] Q. Chen, Y. J. Li, A. McKinney, M. Kuwata, S. T. Martin, Particle mass yield from β-caryophyllene ozonolysis. Atmos. Chem. Phys. 2012, 12, 3165.
| Particle mass yield from β-caryophyllene ozonolysis.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XptFelsrY%3D&md5=772217567da5b69ecb74245f9fabdcc5CAS |
[36] T. E. Kleindienst, M. Jaoui, M. Lewandowski, J. H. Offenberg, C. W. Lewis, P. V. Bhave, E. O. Edney, Estimates of the contributions of biogenic and anthropogenic hydrocarbons to secondary organic aerosol at a southeastern US location. Atmos. Environ. 2007, 41, 8288.
| Estimates of the contributions of biogenic and anthropogenic hydrocarbons to secondary organic aerosol at a southeastern US location.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhtlWntrjJ&md5=0dcfb3871c51e5d4a661c177c693e912CAS |
[37] J. Parshintsev, J. Nurmi, I. Kilpelainen, K. Hartonen, M. Kulmala, M. L. Riekkola, Preparation of β-caryophyllene oxidation products and their determination in ambient aerosol samples. Anal. Bioanal. Chem. 2008, 390, 913.
| Preparation of β-caryophyllene oxidation products and their determination in ambient aerosol samples.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXmsVyjuw%3D%3D&md5=b572baf857cd9f02766958bfc4ab56d0CAS | 18080117PubMed |
[38] M. E. Jenkin, K. P. Wyche, C. J. Evans, T. Carr, P. S. Monks, M. R. Alfarra, M. H. Barley, G. B. McFiggans, J. C. Young, A. R. Rickard, Development and chamber evaluation of the MCM v3.2 degradation scheme for β-caryophyllene. Atmos. Chem. Phys. 2012, 12, 5275.
| Development and chamber evaluation of the MCM v3.2 degradation scheme for β-caryophyllene.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xhtlaltb3P&md5=14a7e01bdb19eccee4e25a0cc9a10e60CAS |
[39] M. Jaoui, R. M. Kamens, Gas and particulate products distribution from the photooxidation of α-humulene in the presence of NOx, natural atmospheric air and sunlight. J. Atmos. Chem. 2003, 46, 29.
| Gas and particulate products distribution from the photooxidation of α-humulene in the presence of NOx, natural atmospheric air and sunlight.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXls1Smu7o%3D&md5=c646ec5c4d04ab88dfc0100c5db8ba66CAS |
[40] M. Jaoui, K. G. Sexton, R. M. Kamens, Reaction of α-cedrene with ozone: mechanism, gas and particulate products distribution. Atmos. Environ. 2004, 38, 2709.
| Reaction of α-cedrene with ozone: mechanism, gas and particulate products distribution.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXjtFCksro%3D&md5=0433ee930a1582288b92a8efb8d4e16dCAS |
[41] I. Kourtchev, I. Bejan, J. R. Sodeau, J. C. Wenger, Gas-phase reaction of (E)-β-farnesene with ozone: rate coefficient and carbonyl products. Atmos. Environ. 2009, 43, 3182.
| Gas-phase reaction of (E)-β-farnesene with ozone: rate coefficient and carbonyl products.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXlvFGjurs%3D&md5=0242013b649127470a79ce51416c8773CAS |
[42] I. Kourtchev, I. Bejan, J. R. Sodeau, J. C. Wenger, Gas phase reaction of OH radicals with (E)-β-farnesene at 296 ± 2 K: rate coefficient and carbonyl products. Atmos. Environ. 2012, 46, 338.
| Gas phase reaction of OH radicals with (E)-β-farnesene at 296 ± 2 K: rate coefficient and carbonyl products.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsFGks7vK&md5=36c2aed13d385016e0645eef4621e4e1CAS |
[43] A. G. Carlton, B. J. Turpin, K. E. Altieri, S. Seitzinger, A. Reff, H. J. Lim, B. Ervens, Atmospheric oxalic acid and SOA production from glyoxal: results of aqueous photooxidation experiments. Atmos. Environ. 2007, 41, 7588.
| Atmospheric oxalic acid and SOA production from glyoxal: results of aqueous photooxidation experiments.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXht1yis7vJ&md5=3f93353ef136482c3558eac9b47e7e72CAS |
[44] J. Liggio, S. M. Li, R. McLaren, Reactive uptake of glyoxal by particulate matter. J. Geophys. Res. 2005, 110, D10304.
| Reactive uptake of glyoxal by particulate matter.Crossref | GoogleScholarGoogle Scholar |
[45] R. Volkamer, F. San Martini, L. T. Molina, D. Salcedo, J. L. Jimenez, M. Molina, A missing sink for gas-phase glyoxal in Mexico City: formation of secondary organic aerosol. Geophys. Res. Lett. 2007, 34, L19807.
| A missing sink for gas-phase glyoxal in Mexico City: formation of secondary organic aerosol.Crossref | GoogleScholarGoogle Scholar |
[46] J. H. Kroll, N. L. Ng, S. M. Murphy, V. Varutbangkul, R. C. Flagan, J. H. Seinfeld, Chamber studies of secondary organic aerosol growth by reactive uptake of simple carbonyl compounds. J. Geophys. Res. 2005, 110, D23207.
| Chamber studies of secondary organic aerosol growth by reactive uptake of simple carbonyl compounds.Crossref | GoogleScholarGoogle Scholar |
[47] W. E. Wilson, E. L. Merryman, A. Levy, H. R. Taliaferro, Aerosol formation in photochemical smog. I. Effect of stirring. J. Air Pollut. Control Assoc. 1971, 21, 128.
| Aerosol formation in photochemical smog. I. Effect of stirring.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE3MXhtlSjtb8%3D&md5=5c2932948df39b2b9ead77abefe04b02CAS |
[48] T. E. Kleindienst, E. O. Edney, M. Lewandowski, J. H. Offenberg, M. Jaoui, Secondary organic carbon and aerosol yields from the irradiations of isoprene and α-pinene in the presence of NOx and SO2. Environ. Sci. Technol. 2006, 40, 3807.
| Secondary organic carbon and aerosol yields from the irradiations of isoprene and α-pinene in the presence of NOx and SO2.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XksValt78%3D&md5=89cae7b34a25aad1077e0599f33f7cf0CAS | 16830546PubMed |
[49] D. W. Fahey, C. S. Eubank, G. Hubler, F. C. Fehsenfeld, A calibrated source of N2O5. Atmos. Environ. 1985, 19, 1883.
| A calibrated source of N2O5.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL28XjvV2isw%3D%3D&md5=fde763192715e45b51344027cb1fd210CAS |
[50] A. Fried, B. E. Henry, J. G. Calvert, M. Mozurkewich, The reaction probability of N2O5 with sulfuric-acid aerosols at stratospheric temperatures and compositions. J. Geophys. Res. 1994, 99, 3517.
| The reaction probability of N2O5 with sulfuric-acid aerosols at stratospheric temperatures and compositions.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2cXkvFSgtr4%3D&md5=53941118e9b9ea3ac92600d4867f49e1CAS |
[51] M. Hallquist, D. J. Stewart, S. K. Stephenson, R. A. Cox, Hydrolysis of N2O5 on sub-micron sulfate aerosols. Phys. Chem. Chem. Phys. 2003, 5, 3453.
| Hydrolysis of N2O5 on sub-micron sulfate aerosols.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXlvFakt7Y%3D&md5=094f5d3f555546ec8ce38e8c8cc84f8aCAS |
[52] C. L. Badger, P. T. Griffiths, I. George, J. P. D. Abbatt, R. A. Cox, Reactive uptake of N2O5 by aerosol particles containing mixtures of humic acid and ammonium sulfate. J. Phys. Chem. A 2006, 110, 6986.
| Reactive uptake of N2O5 by aerosol particles containing mixtures of humic acid and ammonium sulfate.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XhtFegsbY%3D&md5=f82dec10a12710dda679b0c571faeca6CAS | 16722713PubMed |
[53] J. Blunden, V. P. Aneja, W. A. Lonneman, Characterization of non-methane volatile organic compounds at swine facilities in eastern North Carolina. Atmos. Environ. 2005, 39, 6707.
| Characterization of non-methane volatile organic compounds at swine facilities in eastern North Carolina.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXhtFarsLnL&md5=a7372335b0597f747359b2ab64d815d5CAS |
[54] J. Offenberg, M. Lewandowski, E. O. Edney, T. E. Kleindienst, M. Jaoui, Investigation of a systematic offset in the measurement of organic carbon with a Semicontinuous analyzer. J. Air Waste Manage. Assoc. 2007, 57, 596.
| Investigation of a systematic offset in the measurement of organic carbon with a Semicontinuous analyzer.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXmvVWhu7k%3D&md5=7d3cebbaab16105b1586b9df3ed254beCAS |
[55] J. H. Offenberg, T. E. Kleindienst, M. Jaoui, M. Lewandowski, E. O. Edney, Thermal properties of secondary organic aerosol. Geophys. Res. Lett. 2006, 33, L03816.
| Thermal properties of secondary organic aerosol.Crossref | GoogleScholarGoogle Scholar |
[56] K. S. Docherty, M. Jaoui, E. Corse, J. L. Jimenez, J. H. Offenberg, M. Lewandowski, T. E. Kleindienst, Collection efficiency of the aerosol mass spectrometer for chamber-generated secondary organic aerosols. Aerosp. Sci. Technol. 2013, 47, 294.
| Collection efficiency of the aerosol mass spectrometer for chamber-generated secondary organic aerosols.Crossref | GoogleScholarGoogle Scholar |
[57] P. F. DeCarlo, J. R. Kimmel, A. Trimborn, M. J. Northway, J. T. Jayne, A. C. Aiken, Foeld-deployable, high-resolution, time-of-flight aerosol mass spectrometer. Anal. Chem. 2006, 78, 8281.
| Foeld-deployable, high-resolution, time-of-flight aerosol mass spectrometer.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XhtFynurzI&md5=6fb0ab8e0c2142eb156596bc104fb972CAS | 17165817PubMed |
[58] M. Jaoui, T. E. Kleindienst, M. Lewandowski, E. O. Edney, Identification and quantification of aerosol polar oxygenated compounds bearing carboxylic and/or hydroxyl groups. 1. Method development. Anal. Chem. 2004, 76, 4765.
| Identification and quantification of aerosol polar oxygenated compounds bearing carboxylic and/or hydroxyl groups. 1. Method development.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXmsFSgs70%3D&md5=5c7b8debb635466f5507fe08d96f1442CAS | 15307788PubMed |
[59] M. Jaoui, R. M. Kamens, Mass balance of gaseous and particulate products analysis from α-pinene/NOx/air in the presence of NOx. J. Geophys. Res. 2001, 106, 12 541.
| Mass balance of gaseous and particulate products analysis from α-pinene/NOx/air in the presence of NOx.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXltlegtbY%3D&md5=613a9305c186802c2e46ac289dfa4b9dCAS |
[60] M. Jaoui, T. E. Kleindienst, J. H. Offenberg, M. Lewandowski, W. A. Lonneman, SOA formation from the atmospheric oxidation of 2-methyl-3-buten-2-ol and its implications for PM2.5. Atmos. Chem. Phys. 2012, 12, 2173.
| SOA formation from the atmospheric oxidation of 2-methyl-3-buten-2-ol and its implications for PM2.5.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xms1eisL4%3D&md5=3c38de1f3e163fb8d6d455184750628eCAS |
[61] E. O. Edney, T. E. Kleindienst, T. S. Conver, C. D. McIver, E. W. Corse, W. S. Weathers, Polar organic oxygenates in PM2.5 at a southeastern site in the United States. Atmos. Environ. 2003, 37, 3947.
| Polar organic oxygenates in PM2.5 at a southeastern site in the United States.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXlvFejsbY%3D&md5=6fa88f8d7a200b592881a27d9e698df6CAS |
[62] J. H. Offenberg, M. Lewandowski, E. O. Edney, T. E. Kleindienst, M. Jaoui, Influence of aerosol acidity on the formation of secondary organic aerosol from biogenic precursor hydrocarbons. Environ. Sci. Technol. 2009, 43, 7742.
| Influence of aerosol acidity on the formation of secondary organic aerosol from biogenic precursor hydrocarbons.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtFGiu7rI&md5=3ee15418e8d662ad33c434b7f742e506CAS | 19921888PubMed |
[63] A. Calogirou, D. Kotzias, A. Kettrup, Products analysis of the gas phase reaction of β-caryophyllene with ozone. Atmos. Environ. 1997, 31, 283.
| Products analysis of the gas phase reaction of β-caryophyllene with ozone.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK28XntVykt7g%3D&md5=37c6de16744011dcbc7fc6a329395e9eCAS |