

Halogenated hydrocarbon formation in a moderately acidic salt lake in Western Australia – role of abiotic and biotic processes
A. Ruecker A , P. Weigold A , S. Behrens A , M. Jochmann B , X. L. Osorio Barajas B and A. Kappler A CA Geomicrobiology, Centre for Applied Geosciences, University of Tuebingen, Sigwartstraße 10, D-72076 Tuebingen, Germany.
B Instrumental Analytical Chemistry, Faculty of Chemistry, University of Duisburg-Essen, Universitätsstraße 5, D-45141 Essen, Germany.
C Corresponding author. Email: andreas.kappler@uni-tuebingen.de
Environmental Chemistry 12(4) 406-414 https://doi.org/10.1071/EN14202
Submitted: 27 September 2014 Accepted: 23 December 2014 Published: 2 June 2015
Environmental context. Volatile halogenated organic compounds (VOX) contribute to ozone depletion and global warming. Here we demonstrate that acidic salt lake sediments in Western Australia contribute to the global natural emission of these compounds and that the emissions are primarily of biotic origin. Elucidating major sources and sinks of VOX is a key task in environmental chemistry because their formation and degradation have major effects on atmospheric chemistry and thus earth climate.
Abstract. Volatile organohalogen compounds (VOX) are known environmental pollutants and contribute to stratospheric ozone depletion. Natural formation of VOX has been shown for many environments from the deep sea to forest soils and Antarctica. Recently, we showed that VOX are emitted from pH-neutral salt lakes in Western Australia and that they are mainly of biotic origin. To which extent this biotic organohalogen formation in salt lakes is pH-dependent and whether VOX are also formed under acidic conditions are unknown. Therefore, we quantified VOX emissions from an acidic salt lake in Western Australia (Lake Orr) in biotic and abiotic (γ ray-irradiated) microcosm experiments under controlled laboratory conditions. The experiments revealed that biotic halogenation processes also occurred under acidic conditions (pH range 3.8–4.8), though the emissions were approximately one order of magnitude lower (nanogram per kilogram dry sediment range) than from pH-neutral lake sediments. Among the detected substances were brominated, e.g. tribromomethane, as well as chlorinated compounds (e.g. trichloromethane). The addition of lactate and acetate, and ferrihydrite showed no stimulation of VOX formation in our microcosms. Hence, the stimulation of Fe-metabolising microorganisms and their potential effect on the formation of reactive Fe species did not promote VOX emissions, suggesting a direct enzymatic formation of the emitted compounds.
Introduction
Until the 1970s, the formation and emissions of volatile halogenated organic compounds (VOX) were mainly quantified with emphasis on anthropogenic sources.[1] Thereafter, first studies indicated that there is also natural production of VOX compounds.[2] Today, there is evidence of more than 5000 naturally produced VOX, with geogenic sources ranging from biomass burning and volcanic activities, soils and sediments to biotic sources such as marine algae, fungi, microorganisms and insects.[3]
For the natural formation of VOX compounds in soils and sediments, both biotic and abiotic processes have been suggested to contribute to total emissions. Although some studies provide experimental evidence for an abiotic formation pathway by a ‘Fenton-like’ reaction,[4] others suggest direct biotic formation by halogenating enzymes.[5] Different types of halogenating enzymes have been identified and the mechanisms by which they form volatile halogenated hydrocarbons have been intensively studied in the last years.[6–8] The abiotic ‘Fenton-like’ reaction suggested that FeIII is reduced to FeII during the oxidative degradation of organic matter in soil in the presence of a halide ion (e.g. Cl–). In a second step, the oxidised organic compound is halogenated. Laboratory experiments using catechol as a redox-sensitive aromatic model compound for phenolic groups in humic substances, FeIII and a halide ion confirmed this reaction pathway.[9] Moreover, it was shown that the addition of hydrogen peroxide (H2O2) as a strong oxidising agent increased the release of methyl halides from the set-ups by more than 100 %, supporting the suggested abiotic formation pathway.[9] However, hydrogen peroxide is also relevant in the enzymatic formation of VOX compounds by peroxidases because these enzymes use, for example, chloride and H2O2 as substrates to form halogenated organic compounds.[10] For forest soils, it has been shown that both, abiotic as well as biotic processes, are involved in the formation of volatile halogenated hydrocarbons.[11–15] Although the net emissions and the formation mechanisms of halogenated hydrocarbons from forest soils have been extensively studied, emissions of VOX compounds from saline environments have mainly been quantified for the open ocean,[16–18] and to a lesser extent for coastal areas or inland saline environments.[19,20] Considering that high salt concentrations up to the solubility limit of, for example, NaCl found in hypersaline lakes might promote high rates of organic-matter halogenation, this is surprising.[21,22]
One of the first pieces of evidence for terrestrial chloromethane (CH3Cl) and bromomethane (CH3Br) emissions in the presence of high salt concentrations was presented for coastal salt marshes.[19] The authors found seasonal patterns, with higher emissions during March to September and concluded that plants and plant-associated microorganisms are the main sources of VOX compounds in salt marsh environments. Emissions from marshes reached up to 42 µmol m–2 day–1 (166 µg m–2 h–1) for CH3Br and up to 570 µmol m–2 day–1 (1199 µg m–2 h–1) for CH3Cl.[19] The direct enzymatic formation of chloromethane and bromomethane might have been caused by salt-marsh plants that express methyltransferases.[23] Although salt marshes cover only 0.1 % of the Earth’s surface, they may produce up to 10 % of the global annual fluxes of CH3Cl and CH3Br.[19] Hence, salt marshes may be, apart from tropical rain forests, the largest natural terrestrial source of chloromethane and bromomethane.[24] Very recently, significant emissions of chloromethane and bromomethane have also been reported for salt marshes in southern Texas, supporting that subtropical salt marsh environments are important sources of halogenated hydrocarbons.[25] However, it has to be considered that there could be strong regional variations in the emissions of VOX compounds from salt marshes and that the conclusions drawn from the study by Rhew et al.[19] are only rough estimates. For comparison, emissions from salt marshes in Scotland were lower, with a mean CH3Br emission of 250 ng m–2 h–1 whereas the CH3Br fluxes reported for the marshes in California correspond to an average emission flux of 4200 ng m–2 h–1.[26] The authors also quantified chloromethane fluxes and the mean CH3Cl emissions reached 662 ± 266 ng m–2 h–1 whereas the emissions from Californian salt marsh ecosystems were up to two orders of magnitude higher, reaching 51 100 ng m–2 h–1.[26] Based on these data, the authors estimated that Scottish salt marches contribute up to 3.2 % to the annual global fluxes of CH3Br and up to 0.33 % to the CH3Cl emissions.[26] Because removal of the plants from the soil plots stopped the emissions, these studies confirm the importance of the biotic emissions of methyl halides from salt marshes.[25,26] The idea that vegetation plays a major role in the VOX emissions from salt marshes was confirmed by the significantly higher emissions from salt marshes in the tropics compared with the lower emissions from salt marshes in Scotland, where the vegetative period is much shorter and thus the potential for VOX formation was lower.[26,27]
Apart from salt marshes, emissions of VOX have also been described for pH-neutral hypersaline salt lake sediments. It was shown that salt lake sediments from a field site in southern Russia contribute to the global annual fluxes of C1 and C2 hydrocarbons.[20] Emissions were in the nanogram per gram fresh-weight sediment range and highest emissions were found for tetrachloroethene (C2Cl4) and trichloromethane (CHCl3). The highest emissions from those sediments coincided with a high activity of halophilic microorganisms and with high concentrations of dissolved iron. In particular, the latter observation led to the conclusion that microbial Fe-redox transformations might play a role in the formation of chlorinated hydrocarbons in the investigated sediments. Because salt lakes are globally distributed under semiarid and arid conditions, this study added an important natural source to the known global natural organohalogen sources. A few years later, the organohalogen emissions from salt lakes and salt-affected soils were investigated on a broader scale by combining microcosm experiments and remote sensing.[28] For an area of 15 000 km2 in the southern Aral Sea basin, fluxes of 16 873 g day–1 in the cover area for C2H2Cl2 (dichloroethene) were estimated. Recently, it was shown that pH-neutral to alkaline hypersaline lake sediments from Western Australia also contribute to the global emissions of halogenated hydrocarbons and that biotic processes dominate the emissions.[29] For forest soils, it is known that acidic conditions stimulate the VOX emissions by a ‘Fenton-like’ reaction.[4,12,30] How far these abiotic reactions may also occur in salt lakes and to what extent they contribute to the overall emissions from acidic salt lake sediments is currently unknown. This is of particular interest for Western Australian salt lakes because many of them are acidic, reaching pH values as low as 3.[21,31] Therefore, the goals of the present study were (i) to quantify and identify organohalogen emissions from acidic salt lake sediments in Western Australia; (ii) to quantify the biotic and abiotic contribution to the overall emissions; and (iii) to determine to what extent the VOX formation is dependent on organic carbon availability, the presence of Fe and the formation of hydroxyl radicals.
Materials and methods
Field site and sampling procedure
Sediment samples were taken from Lake Orr (33°8′1.51″S 119°9′47.14″E), a hypersaline, moderately acidic and carbon-poor lake in Western Australia in April 2012. The sampling site is located within the Western Australian wheat belt, with an average rainfall of 363 mm year–1,[32] and is characterised by a high number of salt lakes with varying geochemical conditions (Fig. 1). Three different sediment horizons could be distinguished visually. A whitish salt crust (0–2 cm), a dark, blackish layer (2–8 cm) and a brighter greyish layer (>8 cm). For geochemical analyses, six sediment cores (30 cm length) ranging from the top layer to the deepest layer were taken. Before pulling the cores from the sediment, the top was closed with a butyl rubber stopper. After extraction from the sediment, the bottom of the tube was closed with a stopper as well. For the microcosm experiments, we additionally took bulk material from all three depth zones. Samples were immediately cooled, transported at 8 °C to the laboratory and stored for ~10 days at 8 °C until further analysis and the start of the incubation experiments.

|
Set-up of sediment microcosm experiments
Emissions of volatile organohalogens from the sediments were quantified in microcosm experiments using sediments from the three distinguishable depth zones. Microcosms were set up in triplicates in 20-mL gas chromatography (GC) vials, each filled with 3.5 g of field-fresh sediment and 8.5 mL of incubation solution and closed with PTFE (polytetrafluorethylene) silicone septa. The incubation solution was an NaCl solution of varying concentrations, corresponding to the different salt concentrations measured in water extracts. These pore water extracts were obtained by suspending 1 g of dried, powdered sediment in 20 mL of deionised H2O in a 50-mL centrifuge tube, followed by shaking for 24 h at 150 rpm on a horizontal shaker. Subsequently, samples were centrifuged for 5 min at 3750g at room temperature. The supernatant was diluted 1 : 20 in deionised water and filtered through a cellulose ester filter with a pore size of 0.45 µm. Major ions were quantified using ion chromatography.[29] Based on the analyses of these extracts, concentrations of Cl– ranged from 127 g L–1 in the incubations with sediment from the top 2 cm to 58 g L–1 in set-ups with sediments from >8 cm. Although chloride was the predominant halide ion quantified in pore waters, bromide was identified as well. Sediment microcosms were prepared and incubated under oxic conditions on a horizontal shaker at 25 rpm for 60 min, 24 h or 72 h at 30 °C in the dark. Temperature measurements at the field site in southern Western Australia demonstrated that 30 °C is representative for the sediment temperatures within the salt lakes.[32] Several additives were used to study their effects on VOX emissions from the microcosms: (i) 10 mM H2O2; (ii) 2.5 mM catechol; (iii) 10 mM H2O2 + 2.5 mM catechol; (iv) 425 µL of a 500 mM ferrihydrite suspension; (v) 100 µL of a stock solution containing 500 mM each of sodium lactate and sodium acetate; (vi) both ferrihydrite + lactate and acetate; and (vii) sterilised set-ups. Sterilisation was achieved by gamma-irradiation at 35 kGy (Beta-Gamma-Service (BGS), Wiehl, Germany). Volatile halogenated organic compounds were quantified in the headspace using a Trace GC Ultra coupled to a DSQII single-quadrupole mass spectrometer (Thermo Fisher Scientific, Waltham, MA) at the University of Duisburg-Essen.
GC-MS instrumental parameters and ITEX method
VOX were quantified in the headspace using a Trace GC Ultra coupled to a DSQ II single-quadrupole mass spectrometer (Thermo Fisher Scientific). Details of the GC-MS used and the In-Tube Extraction (ITEX) were described in 2010.[33] For our analyses, the injection temperature was set to 200 °C and the cryotrap was cooled to –165 °C with a hold–transfer time of 2 min and a constant column flow of 1.5 mL min–1 He (Air Liquide, Oberhausen, Germany). Compound separation was performed on a Rtx-VMS column, 60 m, 0.32-mm inner diameter and 1.8-µm film thickness (Restek GmbH, Bad Homburg, Germany). The GC oven start temperature was set to 40 °C for 2 min and then heated up to 200 °C at 10 °C min–1 and with a hold time of 5 min. The ion source temperature was 200 °C and the mass spectrometer (MS) was operated in full scan mode with 5.7 scans s–1 and a mass range m/z from 45 to 200. The Combi-PAL autosampler (CTC Analytics, Zwingen, Switzerland) containing the samples was kept at 6 °C. Before analysis, each sample was kept for 15 min at 60 °C. Fifty extraction strokes with an aspirating and dispensing volume of 1 mL were performed with a flow rate of 100 µL s–1. The ITEX trap temperature was 30 °C and the syringe was heated to 60 °C to avoid condensation of water. Desorption temperature was 300 °C with a desorption flow of 50 µL s–1 and a total injection volume of 200 µL.
Geochemical sediment analysis, standard preparation and data analysis
For pH measurements, 10 g of air-dried sediment were suspended in 25 mL of a 0.01 M CaCl2 solution and pH was measured after 2 h. Total organic carbon was determined from milled sediment samples that were dried at 60 °C until constant mass was reached using an Elementar Vario EL element analyser. Soluble organic carbon was analysed in artificial pore water as described earlier.[22,29] Concentrations of the identified halogenated hydrocarbons were calculated from a standard calibration curve using the EPA 624 standard calibration mix (100 µg mL–1 of each component in methanol) as well as trichloromethane and chloromethane, which were purchased from Sigma–Aldrich. Standards were prepared in 20-mL GC headspace vials in 10 mL of incubation solution. Final concentrations ranged from 1 to 1000 ng L–1.
All other used substances and solutions were set up in reagent water from an analytical water purification system (Merck Milllipore, Darmstadt, Germany). The ferrihydrite mineral suspension was prepared as described earlier.[34,35] Student’s t-test was applied to determine if differences between experimental set-ups were significant.
Results
Geochemical sediment characterisation
The three visually distinguishable sediment zones varied considerably regarding their geochemistry. Table 1 gives an overview of the geochemical properties of Lake Orr sediments. Concentrations of soluble organic carbon increased with increasing sediment depth and ranged from 1.7 mg L–1 in the top layer (salt crust) to 9.6 mg L–1 in the layer >8 cm. Total organic carbon (TOC), similarly to soluble organic carbon, increased with increasing sediment depths. However, because the highest TOC concentrations were only 0.8 %, the sediment investigated can be considered as carbon-poor. The pH of the sediments was acidic to slightly acidic and was lowest in the top 2 cm of the sediment profile (3.8). The dark, blackish zone from 2 to 8 cm had a pH of 4.5. The pH was 4.8 in sediments >8 cm. Sediment water content showed the same trend and increased from 11.4 % in the salt crust to 46.4 % in the layer >8 cm. The relative amount of Fe (%) was quantified by X-ray fluorescence (XRF) and increased from the top layer (0.2 wt-%) to the deepest sediment layer (2.4 wt-%). Concentrations of chlorine in percent decreased with increasing sediment depth.

|
Organohalogen formation in Lake Orr sediments
Our experiments demonstrated that acidic Lake Orr sediments are a natural source of volatile halogenated hydrocarbons. Among the detected substances were chlorinated, e.g. trichloromethane, as well as brominated compounds, such as bromomethane and tribromomethane (Fig. 2). Generally, the emissions were higher in the deeper sediment zones that were rich in carbon in comparison with the carbon-poor, halide-rich top layer (salt crust) from 0 to 2 cm (Fig. 2a, c, d, f). The only exception is trichloromethane, for which the highest emissions were found in the salt crust after 60 min of incubation (Fig. 2e). However, with increasing incubation time, the importance of the deeper, carbon-richer sediments also became evident for trichloromethane (Fig. 2c).

|
Dependence of organohalogen formation in Lake Orr sediments on catechol and H2O2
The addition of catechol to sediments from all three layers led to a slight increase in trichloromethane emissions after 72 h of incubation (Fig. 2a). The effect was strongest in the sediment depth zone >8 cm where the emissions increased from 67.8 ± 15.9 to 93.8 ± 10.1 ng kg–1 dry sediment. Although a stimulation of VOX emission by catechol addition was found for all sediment zones, statistical analysis revealed that the differences between the set-ups with and without catechol were not significant.
In addition to determining the effect of catechol, we quantified trichloromethane formation after H2O2 addition and after addition of a combination of catechol and H2O2. Although the addition of H2O2 as a strong oxidising agent had only little effect on VOX emission from the deeper, halide-poorer sediment zones after 72 h of incubation in the dark, H2O2 addition showed a very strong promoting effect on CHCl3 emission from the zone from 0 to 2 cm (salt crust), which was found to be statistically significant (P < 0.001) (Fig. 2b). In this layer, the CHCl3 emission increased from 8.7 ± 2.3 ng kg–1 dry sediment in the set-ups without H2O2 addition to 923.0 ± 123.4 ng kg–1 dry sediment in the presence of H2O2. Although VOX emissions from the salt crust were very low without any amendments, this layer seems to have a high halogenation potential. The addition of both H2O2 and catechol had only a weak promoting effect on the trichloromethane emissions from the sediment zones from 2 to 8 cm and >8 cm (Fig. 2c). However, the addition of H2O2 and catechol led to a statistically significant increase in the trichloromethane emissions from the salt crust (P < 0.01). Emissions increased from 8.7 ± 2.3 ng kg–1 dry sediment in the set-up without any amendments to 22.9 ± 3.5 ng kg–1 dry sediment in the set-ups with H2O2 and catechol (Fig. 2c).
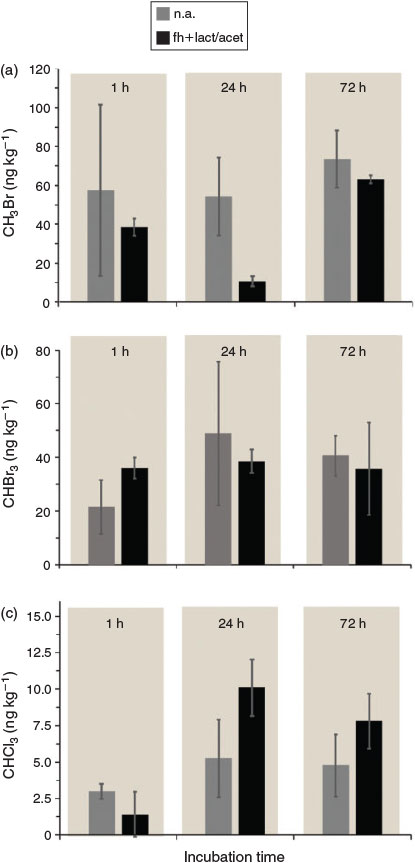
Dependence of organohalogen formation in Lake Orr sediments on ferrihydrite and lactate and acetate
Set-ups amended with lactate and acetate in combination with ferrihydrite were prepared to evaluate the potential stimulation of microbial activity in general (by addition of easily available carbon) and particularly by stimulation of Fe-metabolising microorganisms. However, in all set-ups tested, we did not see any stimulation or inhibition of the emissions of organohalogen compounds. Regardless of the depth zone, no clear effect could be observed for bromomethane emissions from Lake Orr sediments after the addition of ferrihydrite in combination with lactate and acetate (Fig. 3a). Similar results were observed for tribromomethane (Fig. 3b). The results for trichloromethane looked slightly different and after 24 and 72 h of incubation, an increase in the emissions was observed (Fig. 3c) After 24 h of incubation, emissions for set-ups without any amendments were 5.3 ± 1.9 ng kg–1 dry sediment, whereas emissions for the set-up with both ferrihydrite and lactate–acetate reached 10.1 ± 2.7 ng kg–1 dry sediment and thus were almost twice as high (Fig. 3c).
|
Biotic v. abiotic organohalogen formation in Lake Orr sediments
To distinguish the contribution of biotic v. abiotic VOX formation, sterilisation using gamma-irradiation was performed for the sediment zones from 2 to 8 cm and >8 cm because these zones showed generally higher emissions than the top sediment zone from 0 to 2 cm (salt crust). A clear effect of sterilisation on VOX emissions was observed for several halogenated compounds. Although the microbial active sediments from 2 to 8 cm and >8 cm depth emitted 139.3 ± 37.5 and 93.9 ± 6.0 ng kg–1 dry sediment tribromomethane (CHBr3) respectively, the concentrations of released CHBr3 were below the detection limit for the sterilised set-ups (Fig. 2d), indicating a strong biotic contribution to CHBr3 formation in the sediments. The results for trichloromethane and bromomethane showed the same trend, with a clear decrease in the emissions in the sterilised set-ups in comparison with the biotically active ones (Fig. 2e, f). Trichloromethane emissions from the sediment depth zone from 2 to 8 cm were significantly reduced from 24.0 ± 13.5 ng kg–1 in the biotic set-ups to 1.5 ± 2.6 ng kg–1 dry sediment in the gamma-irradiated set-ups (P < 0.05) (Fig. 2e). Emissions of trichloromethane from the depth zone >8 cm decreased from 10.1 ± 8.6 to 0.8 ± 1.4 ng kg–1, though the differences were, owing to the large standard deviations, statistically not significant (Fig. 2e). Bromomethane emissions for the zone from 2 to 8 cm were reduced from 57.4 ± 44.0 to 9.1 ± 1.3 ng kg–1 dry sediment. For the depth zone >8 cm, emissions of bromomethane decreased from 89.6 ± 8.1 to 25.1 ± 11.5 ng kg–1 dry sediment (Fig. 2f).
Discussion
Role of biotic processes in VOX emissions from salt lake sediments
Our study suggests that Western Australian acidic salt lake sediments are a potential source of chlorinated as well as brominated hydrocarbons found in the atmosphere. However, it has to be considered that conclusions regarding global emissions cannot simply be drawn from our laboratory incubation experiments. This would require additional on-site flux measurements. Highest emissions from our laboratory experiments were found from the biotic and thus microbiologically active set-ups, thus confirming the importance of biotic processes in the formation of VOX compounds in salt lake sediments. Although γ-irradiation stopped the emission of CHBr3 (tribromomethane) in the depth zone from 2 to 8 as well as >8 cm completely (Fig. 2d), CHCl3 (trichloromethane) and CH3Br (bromomethane) could still be detected in the sterilised set-ups (Fig. 2e, f) although their overall extent was strongly reduced: γ-irradiation reduced the chloroform emissions by 93.7 % in the zone from 2 to 8 cm and by 91.8 % in the depth zone >8 cm after 60 min of incubation (Fig. 2e). Effects on bromomethane formation were slightly weaker and reduced the emissions by 84.4 % in the zone from 2 to 8 cm and 36.6 % in the sediment >8 cm (Fig. 2f). Although γ-irradiation is a widely used sterilisation technique in environmental microbiology, it alters the physicochemical properties of the sediment, which may affect VOX emissions.[36] Owing to the destruction and subsequent new formation of aggregates by γ-radiation,[37] new sorption sites may form. Hence, VOX previously stabilised within the sediments might be released into the atmosphere during the radiation process, thus contributing to a minor extent to the observed later decrease in the emissions in the sterilised experimental set-ups compared with the biotic set-ups. Nevertheless, compared with other sterilisation techniques such as autoclaving, the alteration of the physicochemical properties of the sediments is probably significantly lower or even negligible.[37] Overall, our data confirmed the suitability of γ-irradiation as a sterilisation technique in sediment microcosm experiments and suggests that γ-irradiation can be used to distinguish between abiotic and biotic processes in the sediment.
Our findings are comparable with the emissions of chlorinated hydrocarbons from a pH-neutral salt lake in Western Australia, which were also primarily of biotic origin.[29] Generally, the emissions from acidic Lake Orr were one order of magnitude lower than the emissions from pH-neutral Lake Strawbridge and also lower than emissions from pH-neutral to alkaline salt lakes in southern Russia.[20] Because biotic processes are obviously the main source of VOX at the two pH-neutral and acidic Australian field sites, the lower emissions at Lake Orr may among other factors be explained by a lower microbial diversity under acidic conditions compared with pH-neutral environments.[38] Although the net emissions from acidic Lake Orr sediments are one order of magnitude lower than from Lake Strawbridge, concentrations found in our microcosm experiments are above the atmospheric background concentrations for all detected halogenated hydrocarbons.[39–43] This emphasises the importance of salt lakes and their sediments in the global budgets of halogenated hydrocarbons. Particularly as atmospheric concentrations of many halogenated hydrocarbons (e.g. bromomethane) are decreasing as a result of the 1987 Montreal Protocol, the identification of new natural sources and the quantification of their individual contribution to the global budgets are of importance.
Effects of H2O2, catechol and ferrihydrite plus lactate and acetate on VOX emissions
In 2000, the importance of Fe in the formation of methyl halides from a temperate forest soil was shown for the first time.[12] The authors suggested that organic material is oxidatively degraded with FeIII as electron acceptor in the presence of halide ions. In a second step, the oxidised organic material is halogenated. Whether and to what extent Fe-metabolising microorganisms may stimulate this reaction by the formation of reactive iron species or by microorganisms that use natural organic matter (NOM) as electron acceptor and form NOM radicals is currently unknown. In order to quantify the effects of Fe on VOX emissions at our field site, we set up microcosm experiments and amended them with 425 µL of a 500 mM ferrihydrite suspension and 100 µL of a 500 mM stock solution containing lactate and acetate as an easily accessible carbon source. Depending on the TOC present in the respective sediment layer, the amount of added lactate and acetate added corresponded to 10–46 % carbon of the TOC already present in the sediments. The added amount of ferrihydrite corresponded to 32–62 % of the total Fe initially quantified in the different sediment zones. With the concentrations of amended ferrihydrite and organics used in our experiments, we did not observe a stimulating effect of the ferrihydrite and lactate and acetate additions on the emissions of bromomethane or bromoform (Fig. 3a, b). These results suggest that a ‘Fenton-like’ reaction or the activity of Fe- and humic substance-metabolising microorganisms may not be major contributors to the emissions of CH3Br and CHBr3 at the field site investigated here. Thus, an enzymatic formation by methyltransferases (methylbromide) and bromoperoxidases (bromoform) seems to be conceivable. This is in line with results from a pH-neutral hypersaline lake from the same area in Western Australia.[29] The results for chloroform were slightly different and a weak stimulation of the CHCl3 emissions was observed with prolonged incubation time (after 24 and 72 h), suggesting that Fe plays a minor role in the chloroform emissions from the field site (Fig. 3c). However, it remains to be elucidated whether this promoting effect is due to a ‘Fenton-like’ reaction or whether Fe stimulates the formation of halogenated hydrocarbons by other, yet unknown, mechanisms as was suggested for Russian salt lake sediments.[20]
The addition of ferrihydrite as potential electron acceptor in combination with lactate and acetate as additional carbon source was intended to stimulate microbial activity in our microcosms[44] and thus potentially the biotic formation of halogenated hydrocarbons in the microcosms. However, the addition of catechol and H2O2 may promote both, the abiotic as well as the enzymatic formation of VOX compounds.[9,17] Catechol has previously been used as a model compound for phenolic groups in humic substances promoting the formation of vinyl chloride.[9] Owing to its redox activity, it may leverage the formation of OH radicals during its oxidation and simultaneous reduction of FeIII to FeII, which may again stimulate the formation of volatile organohalogen compounds and their precursors.[9] Our experiments showed a stimulating effect of catechol on the chloroform emissions from all sediment depth zones after 3 days of incubation (Fig. 2a). This is probably due to the chemical reactivity of catechol as mentioned before, and confirms the promoting effect of catechol on VOX formation.
The effects of H2O2 on the emissions of VOX compounds have been reported repeatedly for temperate forest soils and confirm the importance of a ‘Fenton-like’ formation of halogenated hydrocarbons in forest soil samples.[4,9] Although the formation of H2O2 has mainly been shown to happen photochemically with subsequent transportation to soils by precipitation,[45] recently, a mechanism that shows the formation of H2O2 in the dark by the oxidation of reduced humics by oxygen has been postulated.[46] This suggests that H2O2 may also be formed in Lake Orr subsurface sediments. The strongest effect of the H2O2 addition was found in the salt crust, with an increase from 8.7 ± 2.3 ng kg–1 dry sediment without H2O2 to 923.0 ± 123.4 ng kg–1 dry sediment in the set-up amended with H2O2 after 72 h of incubation. Although the effects were not statistically significant for the other sediment depth zones, slightly elevated VOX emission after H2O2 addition were also observed for these sediments. Despite the fact that the overall emissions from the salt crust in the set-ups without any amendments were the lowest, these results suggest that the halogenation potential of the salt crust is very high. This is highly probable as the highest chlorine concentrations were also found in this layer. However, it cannot be ruled out that the newly formed VOX compounds from the depth zones >2 cm are degraded faster than those in the salt crust,[47–49] explaining to some extent the differences between the salt crust and the other sediment depth zones.
The simultaneous addition of catechol and H2O2 stimulated chloroform emissions from the salt crust and the layer >8 cm to a lesser extent when compared with the set-ups that were amended with only H2O2 or catechol. The VOX emissions from the zone between 2 and 8 cm were not affected by the simultaneous addition of H2O2 and catechol when compared with the set-ups without any amendments. This might be caused by the oxidation of catechol to o-quinone by H2O2 rather than by the weaker oxidant FeIII. Thus the next step, namely the formation of OH radicals by a ‘Fenton-like’ reaction between FeII and H2O2 as described, for example, by Keppler et al. cannot take place,[9] because the H2O2 has already reacted with catechol and thus no FeII is formed, which is one of the prerequisites for the abiotic ‘Fenton-like’ VOX formation.
Environmental implications and conclusions
Our study showed for the first time that acidic salt lakes and their sediments potentially contribute to the global emissions of important halogenated hydrocarbons such as bromomethane (CH3Br), tribromomethane (CHBr3) and trichloromethane (CHCl3) to the atmosphere, and that the compounds formed are mainly of biotic origin. Compared with pH-neutral salt lakes from the same area in the Western Australian wheat belt, the emissions were one order of magnitude lower and were in the nanogram per kilogram dry sediment range, suggesting that pH-neutral hypersaline lakes are more important in the global VOX budgets than acidic hypersaline lakes. As reported before, ~37 % of the studied salt lakes in the area of Esperance (Western Australia) are acidic to strongly acidic and therefore most of the salt lakes (63 %) in this area are pH-neutral to alkaline,[21] and may act as a potential important source of VOX compounds to the atmosphere. The predominance of pH-neutral and alkaline lakes in our study area in Western Australia has recently been empirically verified, although the geochemistry of most analysed salt lakes was very diverse and pH values were as low as 4.2.[31] Although our data suggest that acidic salt lake sediments have a lower effect on the global VOX emissions in comparison with pH-neutral or alkaline salt lake sediments, increasing salinisation under arid conditions, the acidification of existing salt lakes and the progressive transition of freshwater to saline lakes may further affect the global budgets of VOX compounds.[21,50–52] This is of particular importance because freshwater lakes seem to act as a sink for halogenated hydrocarbons whereas salt lakes and their sediments are a net source of VOX compounds.[53] In Western Australia, for example, 80 % of all lakes are salt lakes,[21,54] potentially emitting halogenated hydrocarbons. Thus, primary natural salinisation and secondary or anthropogenic salinisation are of major concern in Western Australia, particularly in the Western Australian wheat belt.[52,55] In the future, areas undergoing salinisation will increase;[56] thus, potentially more VOX will be emitted to the atmosphere. For the Western Australian wheat belt, detailed models on the future progression of salinisation have been put forward. These models assume that 33 % of the land in this area will suffer from salinisation by 2050.[57] The proportion of affected wetlands and thus the transition from freshwater to salt lakes will be much higher, because water bodies contribute only to a minor part to the land-cover in Western Australia.[55] Therefore, a thorough quantification of VOX emissions from salt lakes is of importance, because these environments will continuously expand in the next decades and thus their contribution to the global budgets of halogenated hydrocarbons will increase.
Hence, more studies are needed that quantify the contribution of salt lakes to the global budgets of relevant halogenated hydrocarbons such as chloroform, bromoform and bromomethane. Particularly interesting would be to not only study water bodies and sediments, but also the emissions and fluxes from halophytic vegetation surrounding the salt lakes in flux-chamber experiments. It was shown, for example for salt-marsh vegetation, that the emissions from halophytic plants were up to 10 times higher than the emissions from the surrounding sediments. This suggests that not only saline and hypersaline sediments but also halophytic plants emit high concentrations of methyl halides such as chloromethane and bromomethane.[26,58] Very recently, seagrass meadows have been shown to emit significant amounts of bromomethane (CH3Br), chloromethane (CH3Cl), iodomethane (CH3I) and bromoform (CHBr3), supporting the fact that not only oceans,[59] salt lakes sediments[29] and salt marshes,[19] but also seagrass meadows contribute to the global emissions of halogenated hydrocarbons from saline environments.[60,61]
Acknowledgements
The authors thank H. Taubald for XRF analyses, E. Struve for total inorganic carbon, total organic carbon, ion chromatography and leachable organic carbon measurements, and J. Laaks for assistance during the GC measurements. This study was supported by the research unit 763 ‘Natural Halogenation Processes in the Environment – Atmosphere and Soil’ funded by the German Research Foundation (DFG). We also thank H. F. Schöler, C. Zetzsch, J. Ofner, T. Krause and K. Kamilli for support during sampling in Australia.
References
[1] R. J. Cicerone, R. S. Stolarski, S. Walters, Stratospheric ozone destruction by man-made chlorofluoromethanes. Science 1974, 185, 1165.| Stratospheric ozone destruction by man-made chlorofluoromethanes.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE2cXlslGntrs%3D&md5=e644397d76220439cbb4182ad954c8b7CAS | 17835469PubMed |
[2] J. E. Lovelock, Natural halocarbons in the air and in the sea. Nature 1975, 256, 193.
| Natural halocarbons in the air and in the sea.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE28Xnslahsg%3D%3D&md5=bda8c3c8bb9c9946bef93cca7cc9e230CAS | 1152986PubMed |
[3] G. W. Gribble, Naturally Occurring Organohalogen Compounds – A Comprehensive Update (Eds A. D. Kinghorn, H. Falk, J. Koboyashi) 2010 (Springer Vienna: Vienna).
[4] S. G. Huber, K. Kotte, H. F. Schöler, J. Williams, Natural abiotic formation of trihalomethanes in soil: results from laboratory studies and field samples. Environ. Sci. Technol. 2009, 43, 4934.
| Natural abiotic formation of trihalomethanes in soil: results from laboratory studies and field samples.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXmtVGnsbY%3D&md5=878e371b694f0cd74e15b81156df73f5CAS | 19673288PubMed |
[5] C. Aeppli, D. Bastviken, P. Andersson, Ö. Gustafsoon, Chlorine isotope effects and composition of naturally produced organochlorines from chloroperoxidases, flavin-dependent halogenases, and in forest soil. Environ. Sci. Technol. 2013, 47, 6864.
| 1:CAS:528:DC%2BC3sXns1egsQ%3D%3D&md5=e23c5b8f31ca5b7044527e72a61d1d0aCAS | 23320408PubMed |
[6] C. S. Neumann, D. G. Fujimori, C. T. Walsh, Halogenation strategies in natural product biosynthesis. Chem. Biol. 2008, 15, 99.
| Halogenation strategies in natural product biosynthesis.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXitlKhsrg%3D&md5=07c0f3522eece895813260763b8ca6ffCAS | 18291314PubMed |
[7] C. Wagner, M. El Omari, G. M. Ko, Biohalogenation: Nature’s way to synthesize halogenated metabolites. J. Nat. Prod. 2009, 72, 540.
| Biohalogenation: Nature’s way to synthesize halogenated metabolites.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXisVKkurg%3D&md5=dca249ca5f56cb3c1bed5925aad494b6CAS | 19245259PubMed |
[8] F. Breider, D. Hunkeler, Investigating chloroperoxidase-catalyzed formation of chloroform from humic substances using stable chlorine isotope analysis. Environ. Sci. Technol. 2014, 48, 1592.
| Investigating chloroperoxidase-catalyzed formation of chloroform from humic substances using stable chlorine isotope analysis.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXitVWmtL3M&md5=24f20d44b72b06197ace9a13cf6a65b1CAS | 24377317PubMed |
[9] F. Keppler, R. Borchers, J. Pracht, S. Rheinberger, H. F. Schöler, Natural formation of vinyl chloride in the terrestrial environment. Environ. Sci. Technol. 2002, 36, 2479.
| Natural formation of vinyl chloride in the terrestrial environment.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XivVGgtLo%3D&md5=810e268850daecade50562ef616034a3CAS | 12075808PubMed |
[10] R. Wever, M. A. van der Horst, The role of vanadium haloperoxidases in the formation of volatile brominated compounds and their impact on the environment. Dalton Trans. 2013, 42, 11 778.
| The role of vanadium haloperoxidases in the formation of volatile brominated compounds and their impact on the environment.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXhtF2isLbI&md5=18f4e6b4812f7c334909bb89e13730faCAS |
[11] E. J. Hoekstra, E. W. B. de Leer, U. A. T. Brinkman, Natural formation of chloroform and brominated trihalomethanes in soil. Environ. Sci. Technol. 1998, 32, 3724.
| Natural formation of chloroform and brominated trihalomethanes in soil.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXmsFWksL8%3D&md5=bc59f0cb00d48c535e2d9bcb6b9e28f2CAS |
[12] F. Keppler, R. Eiden, V. Niedan, J. Pracht, H. F. Schöler, Halocarbons produced by natural oxidation processes during degradation of organic matter. Nature 2000, 403, 298.
| Halocarbons produced by natural oxidation processes during degradation of organic matter.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXns1ChsA%3D%3D&md5=c99967dc51e17d29c8d517dfdab9200dCAS | 10659846PubMed |
[13] N. Clarke, K. Fuksová, M. Gryndler, Z. Lachmanová, H.-H. Liste, J. Rohlenová, S. Schroll, P. Schroeder, M. Matucha, The formation and fate of chlorinated organic substances in temperate and boreal forest soils. Environ. Sci. Pollut. Res. Int. 2009, 16, 127.
| The formation and fate of chlorinated organic substances in temperate and boreal forest soils.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXitlGmtrY%3D&md5=6dcbede8d376072935b8fe9987fd7120CAS | 19104865PubMed |
[14] D. Bastviken, T. Svensson, S. Karlsson, P. Sandén, G. Oberg, Temperature sensitivity indicates that chlorination of organic matter in forest soil is primarily biotic. Environ. Sci. Technol. 2009, 43, 3569.
| Temperature sensitivity indicates that chlorination of organic matter in forest soil is primarily biotic.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXjvFKksb0%3D&md5=4957016aff66af1fb629c95e715d380cCAS | 19544856PubMed |
[15] C. N. Albers, O. S. Jacobsen, É. M. M. Flores, J. S. F. Pereira, T. Laier, Spatial variation in natural formation of chloroform in the soils of four coniferous forests. Biogeochemistry 2011, 103, 317.
| Spatial variation in natural formation of chloroform in the soils of four coniferous forests.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXisVKiurc%3D&md5=60346cbbfc889b0fdcf363e677388960CAS |
[16] P. M. Gschwend, J. K. MacFarlane, K. A. Newman, Volatile halogenated organic compounds released to seawater from temperate marine macroalgae. Science 1985, 227, 1033.
| Volatile halogenated organic compounds released to seawater from temperate marine macroalgae.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2MXhsFCnsrc%3D&md5=18174824e00a0911a3e756c90e0b947eCAS | 17794227PubMed |
[17] K. Ballschmiter, Pattern and sources of naturally produced organohalogens in the marine environment: biogenic formation of organohalogens. Chemosphere 2003, 52, 313.
| Pattern and sources of naturally produced organohalogens in the marine environment: biogenic formation of organohalogens.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXjsVGnsLg%3D&md5=61e3cf9aaf5aa9aa8ec0b25ff4eefa02CAS | 12738255PubMed |
[18] C. Paul, G. Pohnert, Production and role of volatile halogenated compounds from marine algae. Nat. Prod. Rep. 2011, 28, 186.
| Production and role of volatile halogenated compounds from marine algae.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtVWisrg%3D&md5=aeb551123c725a3be66345aea7eb1be0CAS | 21125112PubMed |
[19] R. C. Rhew, B. R. Miller, R. F. Weiss, Natural methyl bromide and methyl chloride emissions from coastal salt marshes. Nature 2000, 403, 292.
| Natural methyl bromide and methyl chloride emissions from coastal salt marshes.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXns1Chsg%3D%3D&md5=71a507101c661b0ddf5b379fb1d47bf0CAS | 10659844PubMed |
[20] L. Weissflog, C. A. Lange, A. Pfennigsdorff, K. Kotte, N. Elansky, L. Lisitzyna, E. Putz, G. Krueger, Sediments of salt lakes as a new source of volatile highly chlorinated C1/C2 hydrocarbons. Geophys. Res. Lett. 2005, 32, L01401.
| Sediments of salt lakes as a new source of volatile highly chlorinated C1/C2 hydrocarbons.Crossref | GoogleScholarGoogle Scholar |
[21] B. V. Timms, Study of the saline lakes of the Esperance hinterland, Western Australia, with special reference to the roles of acidity and episodicity. Nat. Resour. Env. Iss. 2009, 15, 1.
[22] M. Emmerich, A. Bhansali, T. Lösekann-Behrens, C. Schröder, A. Kappler, S. Behrens, Abundance, distribution, and activity of Fe(II)-oxidizing and Fe(III)-reducing microorganisms in hypersaline sediments of Lake Kasin, southern Russia. Appl. Environ. Microbiol. 2012, 78, 4386.
| Abundance, distribution, and activity of Fe(II)-oxidizing and Fe(III)-reducing microorganisms in hypersaline sediments of Lake Kasin, southern Russia.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XpvFKgt78%3D&md5=062428e74387b1e18baf098fe8dc8537CAS | 22504804PubMed |
[23] A. M. Wuosmaa, L. P. Hager, Methyl chloride transferase: a carbocation route for biosynthesis of halometabolites. Science 1990, 249, 160.
| Methyl chloride transferase: a carbocation route for biosynthesis of halometabolites.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3cXls1Oktbc%3D&md5=96e30f3bd640a13fca0e651ae62bd880CAS | 2371563PubMed |
[24] R. Rhew, O. Mazéas, Gross production exceeds gross consumption of methyl halides in northern California salt marshes. Geophys. Res. Lett. 2010, 37, L18813.
| Gross production exceeds gross consumption of methyl halides in northern California salt marshes.Crossref | GoogleScholarGoogle Scholar |
[25] R. C. Rhew, M. E. Whelan, D. H. Min, Large methyl halide emissions from south Texas salt marshes. Biogeosciences Discuss. 2014, 11, 9451.
| Large methyl halide emissions from south Texas salt marshes.Crossref | GoogleScholarGoogle Scholar |
[26] E. Blei, M. R. Heal, K. V. Heal, Long-term CH3Br and CH3Cl flux measurements in temperate salt marshes. Biogeosciences 2010, 7, 3657.
| Long-term CH3Br and CH3Cl flux measurements in temperate salt marshes.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXntlKkt7w%3D&md5=a67ce21335ecee343367b354fc7c22b0CAS |
[27] J. Drewer, M. R. Heal, K. V. Heal, K. A. Smith, Temporal and spatial variation in methyl bromide flux from a salt marsh. Geophys. Res. Lett. 2006, 33, L16808.
| Temporal and spatial variation in methyl bromide flux from a salt marsh.Crossref | GoogleScholarGoogle Scholar |
[28] K. Kotte, F. Löw, S. G. Huber, T. Krause, I. Mulder, H. F. Schöler, Organohalogen emissions from saline environments – spatial extrapolation using remote sensing as most promising tool. Biogeosciences 2012, 9, 1225.
| Organohalogen emissions from saline environments – spatial extrapolation using remote sensing as most promising tool.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xhs1Gks7rO&md5=c6e4c3c07f3491cdc925585d2719331fCAS |
[29] A. Ruecker, P. Weigold, S. Behrens, M. Jochmann, J. Laaks, A. Kappler, Predominance of biotic over abiotic formation of halogenated hydrocarbons in hypersaline sediments in Western Australia. Environ. Sci. Technol. 2014, 48, 9170.
| Predominance of biotic over abiotic formation of halogenated hydrocarbons in hypersaline sediments in Western Australia.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXhtFyns7jN&md5=acef6f91cff86cf6dd527ba4a3a988a0CAS | 25073729PubMed |
[30] I. J. Fahimi, F. Keppler, H. F. Schöler, Formation of chloroacetic acids from soil, humic acid and phenolic moieties. Chemosphere 2003, 52, 513.
| Formation of chloroacetic acids from soil, humic acid and phenolic moieties.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXjsVGntrk%3D&md5=ca850a8b72d3eb0a3fc45390cf1e8d8dCAS | 12738276PubMed |
[31] T. Krause, C. Tubbesing, K. Benzing, H. F. Schöler, Model reactions and natural occurrence of furans from hypersaline environments. Biogeosciences Discuss. 2013, 10, 17 439.
| Model reactions and natural occurrence of furans from hypersaline environments.Crossref | GoogleScholarGoogle Scholar |
[32] T. Krause, Natural Occurrence of Volatile Mono-/Polyhalogenated and Aromatic/Heteroaromatic Hydrocarbons from Hypersaline Environments 2014, Ph.D. thesis, University of Heidelberg, Germany.
[33] J. Laaks, M. A. Jochmann, B. Schilling, T. C. Schmidt, In-tube extraction of volatile organic compounds from aqueous samples: an economical alternative to purge-and-trap enrichment. Anal. Chem. 2010, 82, 7641.
| In-tube extraction of volatile organic compounds from aqueous samples: an economical alternative to purge-and-trap enrichment.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtVCksL7O&md5=32a2e82ca608f6e215ff5b4cb0b192f1CAS | 20722393PubMed |
[34] U. Schwertmann, R. M. Cornell, Ferrihydrite, in Iron Oxides in the Laboratory 2007, pp. 103–112 (Wiley-VCH Verlag GmbH: Weinheim, Germany).
[35] K. Amstaetter, T. Borch, A. Kappler, Influence of humic substance-imposed changes of ferrihydrite aggregation on microbial Fe(III) reduction. Geochim. Cosmochim. Acta 2012, 85, 326.
| Influence of humic substance-imposed changes of ferrihydrite aggregation on microbial Fe(III) reduction.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XlvVGgs78%3D&md5=c9a6207b582a107c3810127eeb9958c7CAS |
[36] J. T. Trevors, Sterilization and inhibition of microbial activity in soil. J. Microbiol. Methods 1996, 26, 53.
| Sterilization and inhibition of microbial activity in soil.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK28XltFOnt7Y%3D&md5=a6d6cbd7437fc5b6f179dabec137cf4cCAS |
[37] A. E. Berns, H. Philipp, H.-D. Narres, P. Burauel, H. Vereecken, W. Tappe, Effect of gamma-sterilization and autoclaving on soil organic matter structure as studied by solid-state NMR, UV and fluorescence spectroscopy. Eur. J. Soil Sci. 2008, 59, 540.
| Effect of gamma-sterilization and autoclaving on soil organic matter structure as studied by solid-state NMR, UV and fluorescence spectroscopy.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXnvVOisrs%3D&md5=d64c989c910ad7a5006bb3ca302e6617CAS |
[38] N. Fierer, R. B. Jackson, The diversity and biogeography of soil bacterial communities. Proc. Natl. Acad. Sci. USA 2006, 103, 626.
| The diversity and biogeography of soil bacterial communities.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XhtVOiurY%3D&md5=20692fe32ac6827e9a21e45a1b5d0686CAS | 16407148PubMed |
[39] R. J. Cicerone, L. E. Heidt, W. H. Pollock, Measurements of atmospheric methyl bromide and bromoform. J. Geophys. Res. Atmos. 1988, 93, 3745.
| Measurements of atmospheric methyl bromide and bromoform.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL1cXkt1Krurg%3D&md5=16643e810f495e7f39d2d000b0c7163fCAS |
[40] K. D. Goodwin, R. K. Varner, P. M. Crill, R. S. Oremland, Consumption of tropospheric levels of methyl bromide by C1 compound-utilizing bacteria and comparison to saturation kinetics. Appl. Environ. Microbiol. 2001, 67, 5437.
| Consumption of tropospheric levels of methyl bromide by C1 compound-utilizing bacteria and comparison to saturation kinetics.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXovFehsLg%3D&md5=a87a2be26677ef17bca86efef45a0ca0CAS | 11722890PubMed |
[41] R. C. Rhew, B. R. Miller, R. F. Weiss, Chloroform, carbon tetrachloride and methyl chloroform fluxes in southern California ecosystems. Atmos. Environ. 2008, 42, 7135.
| Chloroform, carbon tetrachloride and methyl chloroform fluxes in southern California ecosystems.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtFSqsr7F&md5=2d516153b7c151df49dcb037a2f4ebacCAS |
[42] N. Yassaa, A. Wishkerman, F. Keppler, J. Williams, Fast determination of methyl chloride and methyl bromide emissions from dried plant matter and soil samples using HS-SPME and GC-MS: method and first results. Environ. Chem. 2009, 6, 311.
| Fast determination of methyl chloride and methyl bromide emissions from dried plant matter and soil samples using HS-SPME and GC-MS: method and first results.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVSlu77N&md5=0d8a6fe2197e80f29b84df382f119413CAS |
[43] B. Kuyper, C. Labuschagne, R. Philibert, N. Moyo, H. Waldron, C. Reason, C. Palmer, Development of a simplified, cost effective GC-ECD methodology for the sensitive detection of bromoform in the troposphere. Sensors 2012, 12, 13583.
| Development of a simplified, cost effective GC-ECD methodology for the sensitive detection of bromoform in the troposphere.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xhs1OisLjP&md5=422427e102c07259b7f8955acc21142fCAS | 23202011PubMed |
[44] E. D. Melton, E. D. Swanner, S. Behrens, C. Schmidt, A. Kappler, The interplay of microbially mediated and abiotic reactions in the biogeochemical Fe cycle. Nat. Rev. Microbiol. 2014, 12, 797.
| The interplay of microbially mediated and abiotic reactions in the biogeochemical Fe cycle.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXhslOktL%2FF&md5=33faef1de8cde934836b470a0b15c6a4CAS | 25329406PubMed |
[45] R. G. Zepp, B. C. Faust, J. Hoigne, Hydroxyl radical formation in aqueous reactions (pH 3–8) of iron(II) with hydrogen peroxide: the photo-Fenton reaction. Environ. Sci. Technol. 1992, 26, 313.
| Hydroxyl radical formation in aqueous reactions (pH 3–8) of iron(II) with hydrogen peroxide: the photo-Fenton reaction.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK38XksFyjsw%3D%3D&md5=a5a61719bafa3c18bed93ddc86a1f20cCAS |
[46] S. E. Page, M. Sander, W. A. Arnold, K. McNeill, Hydroxyl radical formation upon oxidation of reduced humic acids by oxygen in the dark. Environ. Sci. Technol. 2012, 46, 1590.
| Hydroxyl radical formation upon oxidation of reduced humic acids by oxygen in the dark.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhs1OjtLjL&md5=4534c335e90767bb6c3cad370cbb8e98CAS | 22201224PubMed |
[47] K. Pecher, S. B. Haderlein, R. P. Schwarzenbach, Reduction of polyhalogenated methanes by surface-bound Fe(II) in aqueous suspensions of iron oxides. Environ. Sci. Technol. 2002, 36, 1734.
| Reduction of polyhalogenated methanes by surface-bound Fe(II) in aqueous suspensions of iron oxides.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XhslOgtL4%3D&md5=b4a4eb8eeff40364a23f0e8e8347fd07CAS | 11993871PubMed |
[48] A. Kappler, S. B. Haderlein, Natural organic matter as reductant for chlorinated aliphatic pollutants. Environ. Sci. Technol. 2003, 37, 2714.
| Natural organic matter as reductant for chlorinated aliphatic pollutants.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXjs1Gltrc%3D&md5=2de347b8595093bbc16058908c9979c8CAS | 12854710PubMed |
[49] B. Kjellerup, C. Naff, S. J. Edwards, U. Ghosh, J. E. Baker, K. R. Sowers, Effects of activated carbon on reductive dechlorination of PCBs by organohalide respiring bacteria indigenous to sediments. Water Res. 2014, 52, 1.
| Effects of activated carbon on reductive dechlorination of PCBs by organohalide respiring bacteria indigenous to sediments.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXjtVGgt7c%3D&md5=857f5cc657673a77ef2368eac1f3426dCAS | 24440760PubMed |
[50] W. D. Williams, Anthropogenic salinisation of inland waters. Hydrobiologia 2001, 466, 329.
| Anthropogenic salinisation of inland waters.Crossref | GoogleScholarGoogle Scholar |
[51] C. J. Clarke, R. J. George, R. W. Bell, T. J. Hatton, Dryland salinity in south-western Australia: its origins, remedies, and future research directions. Soil Res. 2002, 40, 93.
| Dryland salinity in south-western Australia: its origins, remedies, and future research directions.Crossref | GoogleScholarGoogle Scholar |
[52] S. A. Halse, J. K. Ruprecht, A. M. Pinder, Salinisation and prospects for biodiversity in rivers and wetlands of south-west Western Australia. Aust. J. Bot. 2003, 51, 673.
| Salinisation and prospects for biodiversity in rivers and wetlands of south-west Western Australia.Crossref | GoogleScholarGoogle Scholar |
[53] W. Huang, X. Bu, L. Nguyen, R. H. Gammon, Production and consumption of methyl halides in a freshwater lake. Limnol. Oceanogr. 2000, 45, 1537.
| Production and consumption of methyl halides in a freshwater lake.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXosFymt70%3D&md5=635de5b5b9b55a5b26ba063fadd66b4dCAS |
[54] B. V. Timms, Salt lakes in Australia: present problems and prognosis for the future. Hydrobiologia 2005, 552, 1.
| Salt lakes in Australia: present problems and prognosis for the future.Crossref | GoogleScholarGoogle Scholar |
[55] S. A. Halse, M. N. Lyons, A. M. Pinder, R. J. Shiel, Biodiversity patterns and their conservation in wetlands of the Western Australian wheat-belt. Rec. West. Aust. Museum 2004, 67, 337.
[56] R. V. Schofield, M. J. Kirkby, Application of salinization indicators and initial development of potential global soil salinization scenario under climatic change. Global Biogeochem. Cycles 2003, 17, 1078.
| Application of salinization indicators and initial development of potential global soil salinization scenario under climatic change.Crossref | GoogleScholarGoogle Scholar |
[57] R. J. Short, C. McConnell, Extent and impacts of dryland salinity. Resource Management Technical Report 202 2001 (Department of Agriculture and Food: Perth, WA).
[58] S. L. Manley, N.-Y. Wang, M. L. Walser, R. J. Cicerone, Coastal salt marshes as global methyl halide sources from determinations of intrinsic production by marsh plants. Global Biogeochem. Cycles 2006, 20, GB3015.
| Coastal salt marshes as global methyl halide sources from determinations of intrinsic production by marsh plants.Crossref | GoogleScholarGoogle Scholar |
[59] H. Hepach, B. Quack, F. Ziska, S. Fuhlbruegge, E. L. Atlas, K. Krüger, I. Peeken, D. W. R. Wallace, Drivers of diel and regional variations of halocarbon emissions from the tropical north-east Atlantic. Atmos. Chem. Phys. 2014, 14, 1255.
| Drivers of diel and regional variations of halocarbon emissions from the tropical north-east Atlantic.Crossref | GoogleScholarGoogle Scholar |
[60] I. Weinberg, E. Bahlmann, W. Michaelis, R. Seifert, Determination of fluxes and isotopic composition of halocarbons from seagrass meadows using a dynamic flux chamber. Atmos. Environ. 2013, 73, 34.
| Determination of fluxes and isotopic composition of halocarbons from seagrass meadows using a dynamic flux chamber.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXnsVyktr4%3D&md5=c72566fc4073bec8916d38258f26ff00CAS |
[61] I. Weinberg, E. Bahlmann, T. Eckhardt, W. Michaelis, R. Seifert, A halocarbon survey from a seagrass-dominated subtropical lagoon, Ria Formosa (Portugal): flux pattern and isotopic composition. Biogeosciences Discuss. 2014, 11, 10 605.
| A halocarbon survey from a seagrass-dominated subtropical lagoon, Ria Formosa (Portugal): flux pattern and isotopic composition.Crossref | GoogleScholarGoogle Scholar |