

Advanced PFAS precursor digestion methods for biosolids
Samuel Hutchinson A B , Tarsha Rieck A and XiangLan Wu A
A B , Tarsha Rieck A and XiangLan Wu A
A Urban Utilities, SAS Laboratory, 180 Ashridge Road, Darra, Qld 4076, Australia.
B Corresponding author. Email: Samuel.Hutchinson@urbanutilities.com.au
Environmental Chemistry 17(8) 558-567 https://doi.org/10.1071/EN20008
Submitted: 16 January 2020 Accepted: 25 August 2020 Published: 11 September 2020
Journal Compilation © CSIRO 2020 Open Access CC BY-NC-ND
Environmental context. The majority of biosolids produced in Australia from wastewater treatment processes are applied to agricultural land for beneficial use. We have demonstrated, through improvements to the analytical method, that levels of PFAS in biosolids are significantly higher than historically understood. The land application of biosolids could result in sensitive environments being exposed to PFAS at levels higher than previously anticipated.
Abstract. The current industry standard for per- and polyfluoroalkyl substances (PFAS) analysis is for the measurement of only 28 PFAS, even though there are greater than 4700 PFAS known to be in existence. The total oxidisable precursor (TOP) assay, originally published by Houtz and Sedlak, is widely used as an estimate of the total perfluoro alkyl acids (PFAA) content of a sample, particularly in wastewater and biosolid matrices. The total PFAA content is an important measure of potential environmental contamination, which assists in the inference of potential harm that may occur from both well characterised PFAS, such as perfluorooctanesulfonic acid (PFOS) and perfluorooctanoic acid (PFOA), as well as lesser known precursor compounds and their breakdown products. With the majority of Australian biosolids beneficially applied to land, it is important to understand the future PFAS capacity before they are land applied to maintain the preservation of our agricultural and environmental assets. Our investigation of the TOP method and its application to biosolids involves a comparison of the Houtz and Sedlak method with a modified version coupled with a hydrogen peroxide pretreatment. The underperformance of the previously published method is demonstrated by its inability to sufficiently digest PFAS within biosolids. Therefore, the Houtz and Sedlak method significantly underestimated the levels of PFAS compared with the modified method, which showed a 10-fold increase in the measured PFAS after digestion. Further improvement of this modified method may provide a greater degree of accuracy for the TOP assay. The significant underestimation of the total PFAS load and, therefore, potential environmental harm has significant implications for public and agricultural health and compliance with regulatory limits.
Introduction
Per- and polyfluorinated alkyl substances (PFAS) are a group of manufactured chemicals used in many industrial and consumer applications as a result of their properties that make products non-stick, water repellent, and fire, weather and stain resistant (HEPA 2018). PFAS also exhibit properties that allow them to persist in the environment and bio-accumulate; although to date, only perfluorooctanesulfonic acid (PFOS) and perfluorooctanoic acid (PFOA) have been added to the Stockholm Convention on Persistent Organic Pollutants (POP) (UNEP 2009). Perfluorohexanesulfonic acid (PFHxS), its salts and related chemicals were nominated for addition to this treaty in 2017 (HEPA 2018). While conclusive evidence of their toxicity to humans is contentious, research has linked PFAS fetal and post-natal exposure to some developmental defects (Liew et al. 2018; Sunderland et al. 2019) and has demonstrated a detrimental impact on multiple aquatic species (Giesy et al. 2010; Wei et al. 2009). PFAS contain durable carbon-fluorine bonds, exhibit both hydrophilic and lipophilic behaviours, and span a broad range of molecular weights (Schultz et al. 2006). These compounds have been dispersed globally owing to these properties and have the potential to present in even the most pristine environments (Hamid and Li 2016).
The number of compounds characterised as PFAS continues to grow, with the current count being over 4700 molecules (OECD 2018). However, only a select few of these compounds are able to be quantified against analytical standards. This is, in part, a result of the proprietary nature of many compounds and mixtures, which has prevented the development of analytical grade standards for them. Analysis for only 28 of these compounds has become the standard in Australia; however, this still leaves the vast majority of compounds unmeasured.
Precursors are often larger PFAS molecules that can be degraded chemically or biologically into smaller perfluoroalkyl structures (Liu and Avendano 2013). In an effort to better understand the total detectable precursor content, the total oxidisable precursor (TOP) assay method was developed and shown to digest precursors from urban runoff samples into select measurable PFAS (Houtz and Sedlak 2012). The TOP assay allows for the analysis of perfluoro alkyl acids (PFAA) and PFAA precursors by measuring the levels of product compounds before and after digestion. The TOP assay achieves this by using a persulfate digestion that is strong enough to convert a wide array of PFAA precursor compounds to primarily per-fluorinated carboxylic acids (PFCA); other fluorinated acids can also be generated (Houtz and Sedlak 2012). These acids are selectively analysed by liquid chromatography with tandem mass spectrometry (LC-MS/MS). The TOP assay method is controlled to limit the breakdown of all PFAS within the sample, as increasingly aggressive conditions have been shown to completely digest PFAS through mineralisation (Dombrowski et al. 2018). These results may not indicate environmental breakdown patterns; however, the technique demonstrates the PFAA content of a sample and can be useful when acting to prevent unnecessary exposure to sensitive environments or to manage levels of PFAS entering a sewage treatment plant.
The recent introduction of regulatory limits applied to PFAS has given a clear motive to fully understand the analytical techniques used to measure them. The Queensland Government, through the Department of Environment and Science, has prescribed limits on biosolids based on contaminant concentrations of total organofluorine (TOF), with the TOP assay acting as an acceptable substitute (DES 2019). The TOP assay is also included in the PFAS National Environment Management Plan (NEMP) as an advanced analytical method for establishing the presence of precursor compounds in a sample and informing on appropriate risk management (HEPA 2018). It is therefore imperative to have a standardised method that is universally shown to analyse and digest each matrix sufficiently and be reproducible through a TOP assay.
In a wastewater treatment plant (WWTP), solids are separated from the effluent stream through many successive treatment processes before being digested (if applicable) and dewatered into biosolids. Many WWTPs include sources that historically have shown high levels of PFAS contamination, which include landfill leachates, domestic waste and industrial waste (Alder and van der Voet 2015). The collection and concentration of these sources via the sewerage system and WWTP processes pose an important question as to the level of PFAS measurable in WWTP outputs. WWTP outputs include treated effluent released to receiving environments or supplied to third parties as recycled water, and biosolids which are predominantly applied to land for beneficial use. Previous estimates of PFAS in WWTPs have shown higher levels of measured PFAS exiting the treatment process than entering (Gallen et al. 2018). This evidence suggests that precursors are digested during treatment of biosolids and sludges, similar to digestion demonstrated within a TOP assay.
Biosolids are high in organic matter, a characteristic which highly impacts oxidisation reactions. This, along with the demonstrated presence of large precursor molecules such as fluorotelomer phosphate diesters (DiPAP), causes problems when oxidising the matrix (Kim Lazcano et al. 2019). It is not unreasonable to assume that biosolids will bind other precursor compounds of similar sizes. This occurrence is also indiscriminate to the type of wastewater treatment process, where biosolids collected from three different treatment types were shown to all contain precursor compounds (Kim Lazcano et al. 2019). The oxidation and removal of the high amount of organic content within biosolid samples is theorised here to allow for greater precursor oxidation. It is therefore crucial that care is taken to properly understand the performance of the oxidisation and assess the completion of precursor conversion.
In Australia, 83 % of biosolids produced are diverted to beneficial land applications, improve soil and agricultural use (Department of the Environment and Energy 2018). To understand the total measurable PFAS content in biosolids, it is important to not only look at the easily measured compounds but also attempt to measure the potential content represented by precursor compounds. Conventional analysis of nine PFCA and perfluorosulfonic acids (PFSA) in Australian biosolids gives measured PFAS sums of ~100 µg kg−1; however, with little to no accessible data on the TOP assay results for these biosolids, further investigation is needed to understand the true PFAS content in the biosolids applied to land (Gallen et al. 2018).
In this paper, a method comparison was undertaken to better understand the level of PFAS in biosolids and to adapt the TOP assay process to the biosolids matrix. Four methods of digestion were trialled, which included that reported by Houtz et al. (2013) where aquifer soils were tested. Along with Houtz’s method, a purpose-built biosolids digestion method was used to better suit the matrix in question. Concentrations of the three variables in the digestion (persulfate, sodium hydroxide and the final volume of water) were determined by the addition of a uniform volume of extract from an aqueous film forming foam (AFFF) impacted site. The combination of variables that gave the highest final PFCA sum was chosen as our purpose built biosolids digestion method. A hydrogen peroxide pretreatment was used in an attempt to remove the non-PFAS organic matter seen in biosolids. The thorough digestion and analysis of biosolids will aid in the protection of environmental assets by providing a greater understanding on the potential level of PFAS contamination in biosolids beneficially applied to land in South-East Queensland.
Experimental
Materials
Mass-labelled standards (Appendix 1) sourced from Wellington Laboratories were used for internal and surrogate standardisation. All solvents and reagents used were sourced from Merck at pesticide-grade or higher where possible. Ammonium hydroxide, used for sample extraction, was sourced as 28–30 % (NH3 basis) ammonium hydroxide and diluted as 0.1 % of the 30 % solution.
Sample collection and storage
The biosolid sample used in this study was collected as a grab sample from a WWTP in South-East Queensland according to the sampling procedures outlined in EPA 537.1 (Determination of selected per- and polyfluorinated alkyl substances into drinking water by solid phase extraction and liquid chromatography/tandem mass spectrometry (LC/MS/MS)). After collection, the sample was stored at ≤4 °C.
Sample preparation
The collected biosolid sample was subsampled and freeze-dried for 24 h. The dried sample was homogenised by shaking into a fine powder, and 15 portions of 0.1 g were weighed out into polypropylene (PP) centrifuge tubes and separated into five groups. Sample Group A (non-digested) contained three samples spiked with internal standard and surrogate standards before extraction (Fig. 1). Three Group B (Houtz) samples were treated with a TOP assay digestion following the method outlined by Houtz et al. (2013) (Fig. 1). Three samples were separated into Group C (working method) and treated with the adapted TOP assay method detailed below (Fig. 1). Six samples were pre-treated with 5 mL of hydrogen peroxide and heated at 85 °C for 24 h (Fig. 1). Three of these samples were subjected to the Houtz et al. (2013) method and three were digested using the adapted TOP assay method, being of Groups D (Houtz H2O2) and E (working H2O2) respectively (Fig. 1).

|
All biosolids were extracted according to Houtz et al. (2013), using three successive extractions of 2.5 mL of 0.1 % ammonium hydroxide in methanol that were collected after centrifuging at 3007 g for 5 min. The extracts were combined and evaporated to dryness under a nitrogen stream at 45 °C. Samples were resuspended in 1.5 mL of 0.1 % acetic acid in methanol and transferred to a centrifuge tube containing 25 µg of ENVI-CARB. Extracts were centrifuged at 3007 g for 30 min and supernatants were removed and evaporated to dryness under nitrogen. Samples initially spiked with internal and surrogate standards in Group A were resuspended in a 60 : 40 mixture of 5 mM ammonium acetate in water and 5 mM ammonium acetate in methanol, transferred to a vial containing a PP liner and PP-lined cap, and analysed by LC-MS/MS direct injection to give a baseline of the PFAS concentration.
TOP assay method
After extraction and ENVI-CARB clean-up, Group B and Group D samples were resuspended in 6 mL of 60 mM potassium persulfate and 75 µL of 10 M sodium hydroxide was added to reach a final concentration of 0.125 M (Fig. 1). These samples were sonicated for 15 min to resuspend all extracted material and held until heating was required. Group C and Group E samples were resuspended in 10 mL of MilliQ water and sonicated for 15 min. Six portions of 4.365 g of potassium persulfate were weighed out into 150-mL high-density polyethylene bottles and the sonicated sample extracts of Groups C and E were then added (Fig. 1). The centrifuge tubes of the Groups C and E samples were rinsed a further nine times with 10 mL of MilliQ water to make a total sample volume of 100 mL in each container (Fig. 1). 5 mL of 10 M sodium hydroxide was added to each sample, and the sample bottles were briefly shaken to ensure dissolution of the potassium persulfate. All Group B, C, D and E samples were placed into an 85 °C water bath for six hours (Fig. 1). After cooling, digested samples were neutralised with concentrated hydrochloric acid and spiked with internal and surrogate standards matching those added to Group A.
Sample clean-up
Kenisis WAX SPE Cartridges 100 mg/3 mL were conditioned in an extraction manifold using 2 mL of 3 % ammonium hydroxide in methanol, followed by 2 mL each of methanol and water. Samples were introduced to the cartridges with no vacuum applied and the columns were dried with a vacuum for one hour after sample addition was complete. Samples were eluted into PP centrifuge tubes from the cartridges using 2 mL of 3 % ammonium hydroxide in methanol and evaporated to dryness under a gentle flow of nitrogen at 45 °C. Each sample was reconstituted in a 60 : 40 mixture of 5 mM ammonium acetate in water and 5 mM ammonium acetate in methanol, vortexed and sonicated for 15 min. Samples were transferred to vials containing PP liners and PP-lined caps.
Sample analysis
Analysis was conducted on a Sciex LC-MS/MS 5500 system using a Gemini C18 analytical column and an Eclipse Plus C18 solvent delay column. For complete instrument parameters, see Table A1, Appendix 1.
Results
Analysis of digested samples
All samples involved in this method comparison were analysed for PFCA, PFSA, and PFAS precursor compounds such as fluorotelomersulfonates (FTS) and perfluorooctanesulfonamides (FOSA). Averages and standard deviations for each replicate analysis are shown in Table 1.

|
FOSA precursor compounds were detected in all Houtz-based methods with the sums for the Houtz and Houtz H2O2 methods being 4.07 ± 0.2 µg kg−1 and 2.21 ± 0.1 µg kg−1 respectively (Table 1). Non-digested samples also showed positive FOSA numbers with a result of 3.85 ± 0.2 µg kg−1 (Table 1). Working method samples did not contain any analysed FOSA compounds. Many FOSA compounds were not detected in any samples, including perfluorooctanesulfonamide (PFOSA), N-methyl perfluorooctane sulfonamide (MeFOSA), N-ethyl perfluorooctane sulfonamide (EtFOSA), N-methylperfluorooctane sulfonamido ethanol (MeFOSE), and N-ethylperfluorotane sulfonamido ethanol (EtFOSE).
FTS precursor compounds were highest in the Houtz H2O2 method sample with a sum of 14.54 ± 1.4 µg kg−1 after sample analysis (Table 1). All other method treatments showed similar levels of FTS compounds with the working, working H2O2, Houtz and no digest method samples resulting in levels of 1.30 ± 0.2 µg kg−1, 1.72 ± 0.0 µg kg−1, 1.48 ± 0.1 µg kg−1 and 1.29 ± 0.1 µg kg−1 (Table 1) respectively. 4 : 2 FTS was not detected in any of the method treatments.
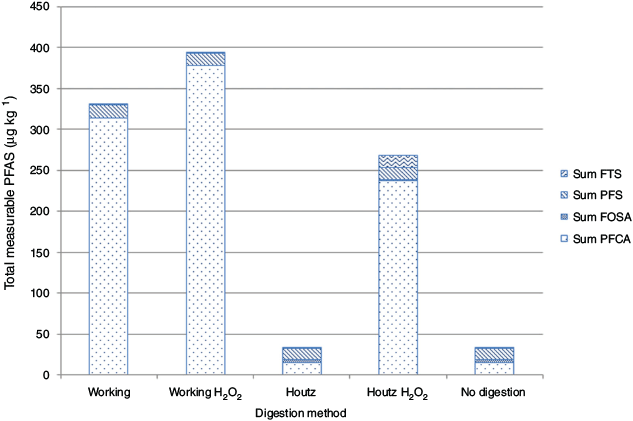
The lowest level of PFCA was seen in the undigested samples at an average of 14.83 ± 0.3 µg kg−1 and the Houtz method samples showed a very similar level of 15.07 ± 1.1 µg kg−1 (Table 1). The highest concentration of PFCA was seen in the working H2O2 method with an average result of 378.99 ± 8.0 µg kg−1 for the replicate samples (Table 1). Perfluorohexanoic acid (PFHxA) had the largest concentration of all PFCA in this method treatment with an average of 79.39 ± 2.8 µg kg−1 (Table 1). The Houtz H2O2 method resulted in a PFCA level of 237.13 ± 17.5 µg kg−1, also showing the highest concentration of PFHxA at 58.48 ± 3.0 µg kg−1 (Table 1). The sum of PFCA concentrations analysed in the working method were averaged to 313.96 ± 15.1 µg kg−1 with perfluoropentanoic acid (PFPeA) having the highest concentration of 69.58 ± 4.4 µg kg−1 (Table 1).
Analysed PFSA compounds were fairly consistent over all methods with the working method, working H2O2 method, Houtz method, Houtz H2O2 method and no digest method samples containing 15.72 ± 0.8 µg kg−1, 14.03 ± 0.5 µg kg−1, 13.77 ± 0.5 µg kg−1, 14.73 ± 0.5 µg kg−1 and 13.84 ± 0.3 µg kg−1 respectively (Table 1). PFOS had the highest concentration of all PFSA compounds for all treatment methods, making up 80–98 % of the PFSA sum (Table 1). Perfluoropentanesulfonic acid (PFPeS) and perfluorodecane sulfonate (PFDS) were not detected in any of the digestion methods analysed.
During sample extraction, significant changes in sample colour were observed between the different treatment methods. Samples treated with hydrogen peroxide showed a significant elimination of colour in the methanol extraction process (Fig. 2). Colour difference persisted through all steps of extraction, with ENVI-CARB clean-up showing no change in sample colour (Fig. 2). Changes in the sample colour were reflected after SPE clean-up (Fig. 3).

|

|
Discussion
Analysis and quality data
Sample concentration factors were corrected for the amount of sample collected at each extraction step. Samples were quantified based on the peak area against a calibration of known concentration. Results were adjusted for internal standard recoveries. Internal quality control procedures were followed, including digest blanks, instrument blanks and spiked samples digested using the working method to demonstrate PFCA recoveries. Where the uncertainty was greater for the results reporting less than the limit of reporting (LOR = 5 µg kg−1), positive detection was verified by peak shape and second ion confirmation; however, results for these compounds should be considered as approximations at best. The limit of reporting was calculated based on recoveries of biosolids spiked with 2 µg kg−1of each compound measured.
All quality control data is available in the supplementary material.
Method comparison
Variable changes between each method include the dilution of the sample, differences in persulfate and sodium hydroxide concentration, and the addition of a hydrogen peroxide pre-digest. Method controls and deviations occurred at critical points with extraction and digestion parameters being controlled while reagent concentration and dilution were varied (Fig. 1). All samples were extracted according to the method of Houtz et al. (2013) in triplicate to demonstrate the effect of variables on digestion and the difference in results not attributed to inconsistencies in extraction procedures. The conditions in the working method combined with the hydrogen peroxide pre-digest achieved the highest rate of precursor digestion (Table 1).
In the method outlined by Houtz et al. (2013), no drying step was used on the solids; however, the working method outlined here included freeze drying. To properly homogenise and increase the active surface area of a biosolid sample, a method of drying is often used (Huset and Barry 2018; Myers et al. 2012). All samples used in this comparison were initially freeze-dried before being weighed for treatment and extraction.
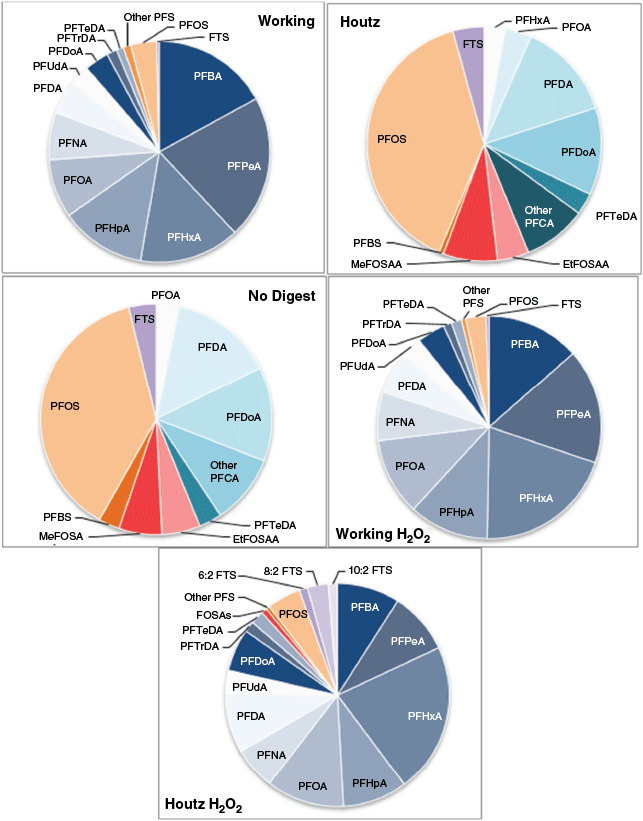
The TOP assay is built on the foundation of digesting all PFAA precursor compounds into analysable PFCA; therefore, the samples with the greatest increase in PFCA compounds have digested the most precursor compounds. The working method samples with hydrogen peroxide pre-digestion showed the highest level of PFCA out of all treatment methods; therefore, digestion was most complete with this treatment method (Fig. 4). Proportions of PFCA were larger in all samples treated with hydrogen peroxide pre-digestion and the working method, which demonstrated a further precursor conversion when using these techniques (Fig. 5). Levels of PFAS in the undigested biosolids place this sample at the low end of the Australian average with the levels of PFAS in land applied biosolids possibly significantly higher than anticipated (Gallen et al. 2016).
|
|
As headspace was not controlled during the experiment, it is possible that the formation of volatile PFAS compounds may have been favoured in one method over another and contribute to the differences in results.
Hydrogen peroxide digest
As a methanol extraction was used in these methods to capture PFAS into an actionable matrix, all methanol soluble compounds will also be contained within the extract. Owing to the high organic content of biosolids compared with soil (Wijesekara et al. 2016; Zhao 2019), the concentration of these co-extractable compounds will be much higher. The significant contribution of organic material in the extract was demonstrated by the colour changes (Fig. 3). The subsequent digestion of these sample extracts will have a limited capacity to react with PFAS precursors as a portion of produced radicals will be consumed by oxidising the non-PFAS extracted organics. This may possibly be an issue for all other matrixes with high levels of co-extractable compounds.
·OH radicals produced by hydrogen peroxide decay are capable of breaking down organic matter; however, current research does not support the breakdown of PFAS precursors with this oxidisation technique (Yang et al. 2014). Even if hydrogen peroxide is directly contributing to the breakdown of precursors, the difference between the working H2O2and the Houtz H2O2 methods showed that the predigest alone was not sufficient to digest all organic matter and precursors (Fig. 4). Additionally, the similarity between the working and the working H2O2 methods demonstrated that the working method was able to breakdown both the organic matter and the PFAS precursors more than the other methods.
The change of colour in the hydrogen peroxide digested samples was indicative of the breakdown of organic compounds by the ·OH radical (Fig. 3). With these compounds broken down in advance, the persulfate digest is free to attack PFAA precursors. This colour change reflected the greater final PFCA concentration, with the working method being able to digest the colour compounds just as readily as the hydrogen peroxide predigest and gave a comparable final concentration (Fig. 4). In comparison, the Houtz methods colouration was very similar to the undigested sample as was its total measured PFCA concentration (Fig. 4).
Precursor presence
AFFFs produced using fluorotelemerisation contain FTS precursor compounds, and oxidation or hydrolysis of the precursor compounds within these AFFFs has been demonstrated as being able to produce FTS compounds (Fang et al. 2015; Houtz et al. 2016). The Houtz H2O2 method has the highest levels of FTS compounds, which demonstrates that the pre-oxidation of organic matter with hydrogen peroxide has allowed the Houtz digest method to begin the breakdown of some PFAA precursors, but it is still not sufficient to digest all the precursor compounds within the sample (Fig. 4). The working and working H2O2 methods had the lowest levels of FTS compounds and it is likely that the digest was more complete and that the initial source of FTS precursors in the samples had been depleted. The limited capacity of the reagents present in the Houtz methods did not allow for complete digestion, and altering the method variables enabled the conversion of more precursor compounds in the TOP assay. However, as all methods contained detectable precursor compounds, none of them completely digested the samples. This indicates that further research should be undertaken to properly optimise the TOP assay for biosolids by focussing on the removal of the non-PFAS organic compounds that are supressing the oxidisation through the use of pre-oxidation clean-up treatments that have limited impact on PFAS such as H2O2.
FOSA compounds characteristically break down into PFOA during persulfate digestion; however, the concentration of PFOA was increased by a low 0.17 µg kg−1 between the undigested sample and the Houtz method, which demonstrated a limited FOSA conversion (Houtz et al. 2013) (Table 1). The presence of these compounds in digested samples indicates that minimum digestion had occurred (Table 1). However, select precursor compounds have been demonstrated to be biodegraded into these measured FOSA; therefore, an increase in these compounds could demonstrate partial digestion (Benskin et al. 2013).
NEMP guidelines for a TOP assay dictate the absence of all precursor compounds and that no decrease in PFCA and PFSA shows a successful digestion (HEPA 2018). The measurable precursors were very low in concentration in the non-digested sample and were all below LOR, meaning that the reported values had a significantly higher uncertainty (Table 1). Proportionally, measured precursor compounds made up only 16 % of the total PFAS content detected in the Houtz digest samples (Fig. 5). If these compounds were not present at or below the detection limit, results from the Houtz methodology would meet the requirements set out by the Queensland Government and the NEMP despite having not digested the unmeasurable precursor compounds. What is significant in the context of biosolids is the absence of measured precursors in undigested samples; therefore, minimal to no digestion will comply with the NEMP guidelines for successful digestion. Further development of the working method has demonstrated that the significant lack of precursor digestion can be attributed to methods not being tailored to specific matrices. The likelihood, rather than the absence, of precursors is better at describing the success or failure of digestion.
Land application and environmental context
In accordance with the End of Waste Code for Biosolids issued by the Queensland Government, biosolids must contain less than 0.39 mg kg−1 of TOF to be land applied for agricultural use and contain less than 0.005 mg kg−1 of TOF once incorporated into the soil. When analysing biosolids using the best performing method described here, 0.395 mg kg−1 of PFAS is measured, which, when adjusted for fluorine (F), equates to 0.259 mg kg−1 of TOF equivalent, putting the PFAS content at 66 % of the proposed limit compared with the 7 % of the limit that the Houtz method detects. Monitoring of PFAS contents in biosolids is important to minimise unforeseen exposure and ensure the protection of environmental assets. When using methods similar to that of Houtz et al. (2013), PFAS contents are significantly underestimated and this increases the risk of biosolids being applied to land above the regulatory limits.
The uptake of precursor compounds, such as DiPAPs, into agricultural crops and vegetation has been demonstrated, posing a significant environmental consequence for irresponsible monitoring of PFAS (Bizkarguenaga et al. 2016). The demonstrated presence of DiPAP compounds in biosolids, along with the subsequent uptake, conversion to PFCA and bioaccumulation, represents significant risks to land application of contaminated biosolids (Lee et al. 2014). Further, the mobility of these breakdown products leads to slow leaching of PFAS into the surrounding environment. It has been demonstrated that the mobility of PFAS within soils is inversely related to the chain length; therefore, the breakdown of larger PFAS precursors into smaller compounds will increase the amount of PFAS leached from the soil (Bizkarguenaga et al. 2016). This may result in a prolonged release of PFAS from fields amended with biosolids as the precursor compounds breakdown over time.
The potential for PFAS precursors in biosolids to be degraded into more mobile smaller PFAS paired with the failure of the Houtz method to properly measure the levels of PFAA precursors in biosolids gives motivation to properly understand PFAS contents. To protect sensitive environments from potential PFAS exposure through the direct application of biosolids to land or the subsequent breakdown and leaching of precursors, we must truly understand the full PFAS content of these biosolids through correctly applied methods.
Conclusion
The results reported here demonstrate that the total PFAA and PFAA precursor content detected in WWTP biosolids is significantly underestimated using the currently accepted methodology. The difference in results between the working method and working H2O2 method indicates that the working method is failing to adequately digest all of the PFAA precursors present in the sample. The working H2O2 method results in a sum of measured PFAS that is almost 400 % greater than the average of the conventional analysis for Australian biosolids. The undigested results for this WWTP are significantly lower than the national average and so it is possible that the precursor content is also significantly lower than the precursor content seen in other WWTPs. The working H2O2 method would also put this WWTP very near the proposed Queensland limit for PFAS and severely limit the ability of the WWTP to beneficially apply the biosolids to land. The land application of these analysed biosolids may have serious implications for future PFAS uptake, leaching and bioaccumulation from agricultural use. Future reviews of farmland where biosolids are applied is necessary to understand the true extent of any PFAS contamination.
A standardised methodology is needed to protect environmental assets from PFAS contamination from land applied biosolids. Currently, no standard TOP assay methodology has been adopted for biosolids and commercial laboratories are free to offer testing according to individually developed procedures. The inconsistencies between methods offered by different laboratories is a significant risk, as demonstrated here where a published method used on solids is adapted to biosolids but does not effectively analyse the matrix. Future investigation is needed into the validity of method variables, as larger PFAA contents may be seen using more extensive and tailored extraction and digestion.
Supplementary material
Quality control information, including recoveries of fortified samples, and additional digestion data for biosolids from different treatment plants are available on the Journal’s website.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
We would like to acknowledge Townsville laboratory service for their collaboration and the initial duplication of our working methods results. We would also like to acknowledge Tony Bradshaw and Timothy Dubber for proof reading and providing feedback. This paper was funded wholly by Queensland Urban Utilities.
References
Alder AC, van der Voet J (2015). Occurrence and point source characterisation of perfluoroalkyl acids in sewage sludge. Chemosphere 129, 62–73.| Occurrence and point source characterisation of perfluoroalkyl acids in sewage sludgeCrossref | GoogleScholarGoogle Scholar | 25176581PubMed |
Benskin JP, Ikonomou MG, Gobas FAPC, Begley TH, Woudneh MB, Cosgrove JR (2013). Biodegradation of N-ethyl perfluorooctane sulfonamido ethanol (EtFOSE) and EtFOSE-based phosphate diester (SAmPAP diester) in marine sediments. Environmental Science & Technology 47, 1381–1389.
| Biodegradation of N-ethyl perfluorooctane sulfonamido ethanol (EtFOSE) and EtFOSE-based phosphate diester (SAmPAP diester) in marine sedimentsCrossref | GoogleScholarGoogle Scholar |
Bizkarguenaga E, Zabaleta I, Prieto A, Fernandez LA, Zuloaga O (2016). Uptake of 8:2 perfluoroalkyl phosphate diester and its degradation products by carrot and lettuce from compost-amended soil. Chemosphere 152, 309–317.
| Uptake of 8:2 perfluoroalkyl phosphate diester and its degradation products by carrot and lettuce from compost-amended soilCrossref | GoogleScholarGoogle Scholar | 26991379PubMed |
Department of Environment and Science (DES) (2019). End of waste code Biosolids (ENEW07359617) (Queensland Government: Brisbane). Available at https://environment.des.qld.gov.au/assets/documents/regulation/wr-eowc-approved-biosolids.pdf [verified 7 November 2019]
Department of the Environment and Energy (2018). National waste report 2018 (Australian Government: Canberra). Available at https://www.environment.gov.au/system/files/resources/7381c1de-31d0-429b-912c-91a6dbc83af7/files/national-waste-report-2018.pdf [verified 7 November 2019]
Dombrowski PM, Kakarla P, Caldicott W, Chin Y, Sadeghi V, Bogdan D, Barajas-Rodriguez F, Chiang S (2018). Technology review and evaluation of different chemical oxidation conditions on treatability of PFAS. Remediation 28, 135–150.
| Technology review and evaluation of different chemical oxidation conditions on treatability of PFASCrossref | GoogleScholarGoogle Scholar |
Fang C, Megharaj M, Naidu R (2015). Chemical oxidisation of some AFFFs leads to the formation of 6:2FTS and 8:2FTS. Environmental Toxicology and Chemistry 34, 2625–2628.
| Chemical oxidisation of some AFFFs leads to the formation of 6:2FTS and 8:2FTSCrossref | GoogleScholarGoogle Scholar | 26076996PubMed |
Gallen C, Drage D, Kaserzon S, Baduel C, Gallen M, Banks A, Broomhall S, Mueller JF (2016). Occurrence and distribution of brominated flame retardants and perfluoroalkyl substances in Australian landfill leachate and biosolids. Journal of Hazardous Materials 312, 55–64.
| Occurrence and distribution of brominated flame retardants and perfluoroalkyl substances in Australian landfill leachate and biosolidsCrossref | GoogleScholarGoogle Scholar | 27016666PubMed |
Gallen C, Eaglesham G, Drage D, Hue Nguyen T, Mueller JF (2018). A mass estimate of perfluoroalkyl substance (PFAS) release from Australian wastewater treatment plants. Chemosphere 208, 975–983.
| A mass estimate of perfluoroalkyl substance (PFAS) release from Australian wastewater treatment plantsCrossref | GoogleScholarGoogle Scholar | 30068041PubMed |
Giesy JP, Naile JE, Khim JS, Jones PD, Newsted JL (2010). Aquatic Toxicology of Perfluorinated compounds. Reviews of Environmental Contamination and Toxicology 202, 1–52.
| Aquatic Toxicology of Perfluorinated compoundsCrossref | GoogleScholarGoogle Scholar | 19898760PubMed |
Hamid H, Li LY (2016). Role of wastewater treatment plant in environmental cycling of poly- and perfluoroalkyl substances. Ecocycles 2, 43–53.
| Role of wastewater treatment plant in environmental cycling of poly- and perfluoroalkyl substancesCrossref | GoogleScholarGoogle Scholar |
Heads of EPAs Australia and New Zealand (HEPA) (2018). PFAS national environmental management plan. Available at https://www.epa.vic.gov.au/for-community/environmental-information/pfas/pfas-national-environmental-management-plan [verified 31 August 2020]
Houtz EF, Sedlak DL (2012). Oxidative conversion as a means of detecting precursors to perfluoroalkyl acids in urban runoff. Environmental Science & Technology 46, 9342–9349.
| Oxidative conversion as a means of detecting precursors to perfluoroalkyl acids in urban runoffCrossref | GoogleScholarGoogle Scholar |
Houtz EF, Higgins CP, Field JA, Sedlak DL (2013). Persistence of perfluoroalkyl acid precursors in AFFF-impacted groundwater and soil. Environmental Science and Technology 47, 8187–8195.
| Persistence of perfluoroalkyl acid precursors in AFFF-impacted groundwater and soilCrossref | GoogleScholarGoogle Scholar | 23886337PubMed |
Houtz EF, Sutton R, Park JS, Sedlak M (2016). Poly- and perfluoroalkyl substances in wastewater: Significance of unknown precursors, manufacturing shifts, and likely AFFF impacts. Water Research 95, 142–149.
| Poly- and perfluoroalkyl substances in wastewater: Significance of unknown precursors, manufacturing shifts, and likely AFFF impactsCrossref | GoogleScholarGoogle Scholar | 26990839PubMed |
Huset CA, Barry KM (2018). Quantitative determination of perfluoroalkyl substances (PFAS) in soil, water, and home garden produce. MethodsX 5, 697–704.
| Quantitative determination of perfluoroalkyl substances (PFAS) in soil, water, and home garden produceCrossref | GoogleScholarGoogle Scholar | 29998069PubMed |
Kim Lazcano R, de Perre C, Mashtare ML, Lee LS (2019). Per‐ and polyfluoroalkyl substances in commercially available biosolid‐based products: The effect of treatment processes. Water Environment Research
| Per‐ and polyfluoroalkyl substances in commercially available biosolid‐based products: The effect of treatment processesCrossref | GoogleScholarGoogle Scholar | 31260167PubMed |
Lee H, Tevlin AG, Mabury SA, Mabury SA (2014). Fate of polyfluoroalkyl phosphate diesters and their metabolites in biosolids-applied soil: Biodegradation and plant uptake in greenhouse and field experiments. Environmental Science & Technology 48, 340–349.
| Fate of polyfluoroalkyl phosphate diesters and their metabolites in biosolids-applied soil: Biodegradation and plant uptake in greenhouse and field experimentsCrossref | GoogleScholarGoogle Scholar |
Liew Z, Goudarzi H, Oulhote Y (2018). Developmental exposures to perfluoroalkyl substances (PFASs): An update of associated health outcomes. Current Environmental Health Reports 5, 1–19.
| Developmental exposures to perfluoroalkyl substances (PFASs): An update of associated health outcomesCrossref | GoogleScholarGoogle Scholar | 29556975PubMed |
Liu J, Avendano SM (2013). Microbial degradation of polyfluoroalkyl chemical in the environment: A review. Environment International 61, 98–114.
| Microbial degradation of polyfluoroalkyl chemical in the environment: A reviewCrossref | GoogleScholarGoogle Scholar | 24126208PubMed |
Myers AL, Crozier PW, Helm PA, Brimacombe C, Furdui VI, Reiner EJ, Burniston D, Marvin CH (2012). Fate, distribution, and contrasting temporal trends of perfluoroalkyl substances (PFASs) in Lake Ontario, Canada. Environment International 44, 92–99.
| Fate, distribution, and contrasting temporal trends of perfluoroalkyl substances (PFASs) in Lake Ontario, CanadaCrossref | GoogleScholarGoogle Scholar | 22406021PubMed |
Organisation for Economic Co-operation and Development (OECD) (2018). Toward a new comprehensive global database of per- and polufluoroalkyl substances (PFASs): Summary report on updating the OECD 2007 list of per- and polyfluoroalkyl substance (PFASs). Available at http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=ENV-JM-MONO(2018)7&doclanguage=en [verified 7 November 2019]
Schultz MM, Higgins CP, Huset CA, Luthy RG, Barofsky DF, Field JA (2006). Fluorochemical mass flows in a municipal wastewater treatment facility. Environmental Science & Technology 40, 7350–7357.
| Fluorochemical mass flows in a municipal wastewater treatment facilityCrossref | GoogleScholarGoogle Scholar |
Sunderland EM, Hu XC, Dassuncao C, Tokranov AK, Wagner CC, Allen JG (2019). A review of the pathways of human exposure to poly- and perfluoroalkyl substances (PFASs) and present understanding of health effects. Journal of Exposure Science & Environmental Epidemiology 29, 131–147.
| A review of the pathways of human exposure to poly- and perfluoroalkyl substances (PFASs) and present understanding of health effectsCrossref | GoogleScholarGoogle Scholar |
United Nations Environment Programme (UNEP) (2009). Listing of perfluorooctane sulfonic acid, its salts and perfluorooctane sulfonyl fluoride (Secretariat of the Stockholm Convention: Geneva). Available at: http://chm.pops.int/Implementation/IndustrialPOPs/PFOS/Overview/tabid/5221/Default.aspx [verified 7 November 2019]
Wei Y, Shi X, Zhang H, Wang J, Shou B, Dai J (2009). Combined effects of polyfluorinated and perfluorinated compounds on primary cultured hepatocytes from rare minnow (Gobiocypris rarus) using toxicogenomic analysis. Aquatic Toxicology 95, 27–36.
| Combined effects of polyfluorinated and perfluorinated compounds on primary cultured hepatocytes from rare minnow (Gobiocypris rarus) using toxicogenomic analysisCrossref | GoogleScholarGoogle Scholar | 19712982PubMed |
Wijesekara H, Bolan NS, Kumarathilaka P, Geekiyanage N, Kunhikrishnan A, Seshadri B, Saint C, Surapaneni A, Vithanage M (2016). Biosolids enhance mine site rehabilitation and revegetation. In ‘Environmental materials and waste resource recovery and pollution prevention’. (Eds MNV Prasad, K Shih) pp. 45–71. (Academic Press: Cambridge)
Yang X, Huang J, Zang K, Yu G, Deng S, Wang B (2014). Stability of 6:2 fluorotelomer sulfonate in advanced oxidation processes: degradation kinetics and pathway. Environmental Science and Pollution Research International 21, 4634–4642.
| Stability of 6:2 fluorotelomer sulfonate in advanced oxidation processes: degradation kinetics and pathwayCrossref | GoogleScholarGoogle Scholar | 24352540PubMed |
Zhao Y (2019). ‘Pollution control technology for leachate from municipal solid waste.’ (Butterworth-Heinemann: Oxford)
Appendix 1
Mass-labelled standards:
Mass-labelled perfluoroalkylcarboxylic acids
-
Perfluoro-n-13C4 butanoic acid (cat: MPFBA)
-
Perfluoro-n-13C5 pentanoic acid (cat: M5PFPeA)
-
Perfluoro-n-13C5 hexanoic acid (cat: M5PFHxA)
-
Perfluoro-n-13C4 heptanoic acid (cat: M4PFHpA)
-
Perfluoro-n-13C8-octanoic acid (cat: M8PFOA)
-
Perfluoro-n-13C9 nonanoic acid (cat: M9PFNa)
-
Perfluoro-n-13C6 decanoic acid (cat: M6PFDA)
-
Perfluoro-n-13C7 undecanoic acid (cat: M7PFUdA)
-
Perfluoro-n-n-13C2 dodecanoic acid (cat: MPFDoA)
-
Perfluoro-n-13C2 tetradecanoic acid (cat: M2PFTeDA)
Mass-labelled perfluorooctanesulfonamides
-
M8FOSA-1(Perfluoro-1–13C8-octanesulfonamide)
-
D-N-MeFOSA-M (N-methyl-d3-perfluoro-1-octanesulfonamide)
-
d-N-EtFOSA-M (N-ethyl-d5-perfluoro-1-octanesulfonamide)
Mass-labelled perfluorooctanesulfonamidoethanols
-
D7-N-MeFOSE-M (2-(N-methyl-d3-perfluro-1-octane-sulfonamido) ethan-D4-ol)
-
D9-N-EtFOSE-M (2-(N-ethyl-d5-perfluro-1-octane-sulfonamido) ethan-D4-ol)
Mass-labelled perfluorooctanesulfonamidoacetic acids
-
D3-N-MeFOSAA (N-methyl-d3-perfluoro-1-octane-sulfonamidoacetic acid)
-
D5-N-EtFOSAA (N-ethyl-d5-perfluoro-1-octane-sulfonamidoacetic acid)
Mass-labelled perfluoroalkylsulfonates
-
M3PFBS (Sodium perfluoro-1–13C3-butanesulfonate)
-
M3PFHxS (perfluoro-1–13C3-hexanesulfonate)
-
M8PFOS (Sodium perfluoro-1–13C8-octanesulfonate)
Mass-labelled telomere sulfonates
-
M2–4 : 2FTS (Sodium 1H,1H,2H,2H-perfluoro-1–13C2-hexanesulfonate (4 : 2))
-
M2–6 : 2FTS (Sodium 1H,1H,2H,2H-perfluoro-1–13C2-octanesulfonate (6 : 2))
-
M2–8 : 2FTS (Sodium 1H,1H,2H,2H-perfluoro-1–13C2-decanesulfonate (8 : 2))

|